

Abstract
Protein cages are hollow spherical proteins assembled from a defined number of subunits. Because they are extremely homogeneous in size and structure, their interior cavities can serve as ideal templates to encapsulate and synthesize well-defined nanoparticles. Here, we describe the exemplary synthesis of a hard and a soft material in two representative protein cages, i.e., magnetite nanoparticles in ferritin and a poly(2-aminoethyl)methacrylate inside a viral capsid derived from the bacteriophage P22.
Keywords: Protein cages, Ferritin, Viral capsid, Biomineralization, Atom transfer radical polymerization (ATRP)
1. Introduction
Biology has provided inspiration for material scientists for years. One of these examples is the synthesis of nano-sized particles [1,2], nucleated and constrained within a protein cage-like architecture. Nanoparticles have made a significant impact in material science for the past few decades, because they exhibit unique properties (e.g., magnetic, photonic, and catalytic) different from bulk materials. One of the critical challenges in the preparation of nanoparticles is the control of their size and morphology and, because the emergent properties of nanoparticles are intimately related to their dimension, the homogeneity of the size distribution is critical.
Through millions of years of evolution, nature has developed nano-size cage-like protein assemblies that are designed to encapsulate guest molecules inside and protect them from the external environment. Ferritins that sequester iron as iron oxide particles [3] and viral capsids that encapsulate their nucleic acid genome [4] are examples of protein cages in this category. Material scientists have been inspired from the natural function of protein cages and exploited them as platforms for the development of a wide range of nanomaterials [4–7]. Cage-like protein structures are ideal templates to synthesize nanoparticles because they have homogeneous size and their interior cavity can provide a confined environment for the selectively directed synthesis or for entrapment of guest molecules. In this chapter, we describe two examples of nanoparticle synthesis in protein cages, the synthesis of magnetite (or maghemite) nanoparticles (hard inorganic materials) in ferritin (Fig. 1a) and the synthesis of a 2-aminoethyl methacrylate (AEMA) polymer, via atom transfer radical polymerization (ATRP), in a viral capsid derived from the bacteriophage P22 (Fig. 2).
Fig. 1.
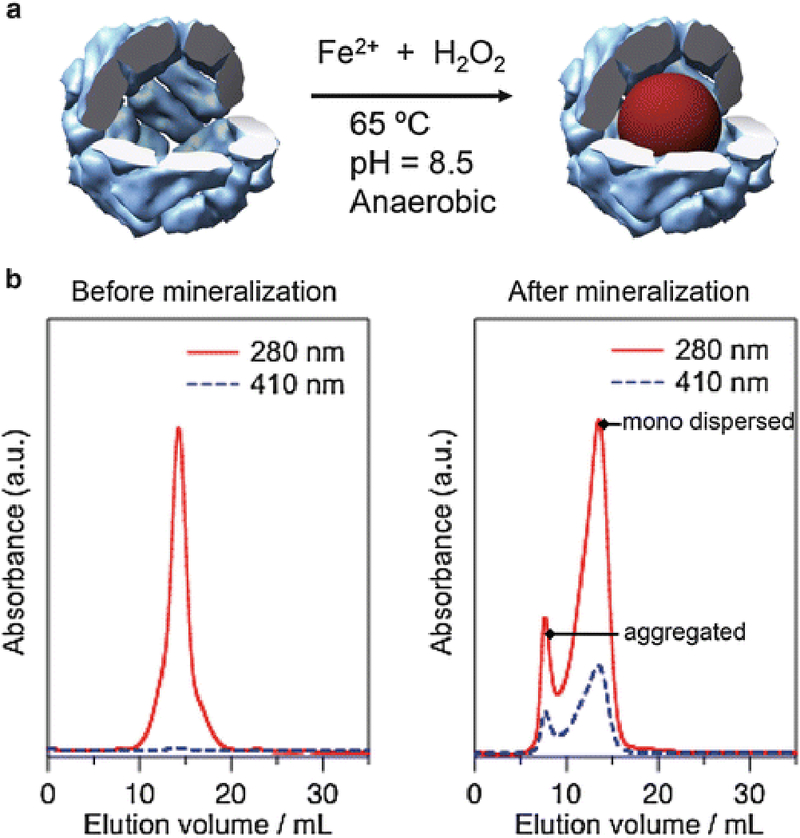
(a) Schematic illustration of magnetite (or maghemite) nanoparticle synthesis in HFn. (b) SEC profiles of HFn before and after mineralization reaction with 3,000Fe/cage. Co-elution profile of HFn (280 nm) and iron oxide (410 nm) after the mineralization reaction indicates the composite nature of the mineralized HFn
Fig. 2.
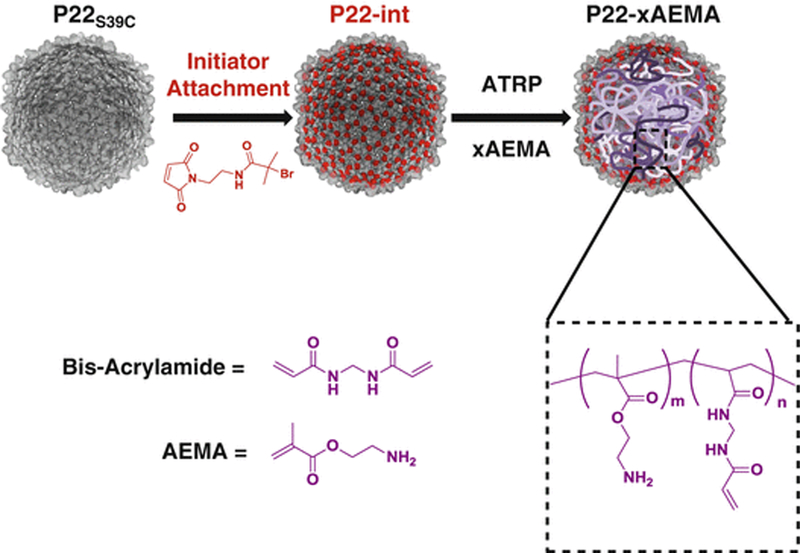
Reaction scheme for the synthesis of P22 protein-polymer hybrid. P22 is first modified with a cysteine reactive ATRP initiator which is used as a macroinitiator to start the growth of an ATRP polymer on the inside of the virus-like particle (VLP)
2. Materials
All solutions should be prepared using ultrapure water (resistivity of 18.2 MΩ cm at 25 °C) unless otherwise noted.
2.1. Synthesis of Magnetite Nanoparticle in Human Ferritin
The apo form of human heavy-chain ferritin (HFn) is heterologously expressed in, and purified from, E. coli (BL21 (DE3)). The cloning, expression, and purification procedure has been described in previous papers [8, 9]. The purified HFn can be stored at ‒80 °C in a standard buffer such as 50 mM Tris, 100 mM NaCl, pH 7.5, until further use.
10× reaction solution: 1 M NaCl. Weigh 58.44 g NaCl and add to 900 mL water. Dissolve the salt and add water up to a final volume of 1.0 L. Filter through 0.22 μm membrane filter. Store at room temperature.
Degassed water and 100 mM NaCl: Degas about 30 mL of ultrapure water and 10 mL of 100 mM NaCl (for use in Subheading 3.1, step 3) by bubbling nitrogen (or argon) gas for at least 30 min. Degas the solutions immediately before magnetite synthesis and keep them under a positive pressure of the inert gas.
Ammonium iron (II) sulfate hexahydrate (12.5 mM) solution in degassed water: Weigh 49.0 mg ammonium iron (II) sulfate hexahydrate (NH4)2Fe(SO4)2 6H2O and prepare a 10 mL solution using degassed water. Prepare the solution just prior to magnetite synthesis and maintain under a positive pressure ofN2.
Hydrogen peroxide (4.17 mM) solution in degassed water: Dilute 4.7 μL of 30 % hydrogen peroxide solution (8.82 M) to 10 mL using degassed water. Prepare the solution just prior to magnetite synthesis and maintain under a positive pressure ofN2.
NaOH (100 mM) solution. Weigh 4.00 g NaOH and add to about 900 mL water. Once the NaOH is well dissolved, add 20 Masaki Uchida et al. water to a final volume of 1.0 L. Filter through 0.22 μm membrane filter. Store at room temperature. Degas the solution by gently bubbling nitrogen gas through the solution just before magnetite synthesis and maintain under a positive pressure of N2.
Sodium citrate (300 mM, pH 7.0) solution: Weigh 8.82 g of sodium citrate dihydrate and add to about 90 mL of water. Adjust the pH with 2 M HCl and make up to 100 mL with water. Filter through 0.22 μm membrane filter. Store at room temperature.
Dulbecco’s phosphate buffered saline (DPBS): Weigh 19.2 g of premixed chemical reagents of DPBS purchased from Sigma-Aldrich and dissolve into 1,900 mL of water. Adjust the pH to 7.4 and make up to a final volume of 2.0 L with water. Filter through 0.22 μm membrane filter. Store at room temperature.
2.2. Synthesis of Cross-Linked AEMA (x-AEMA) in P22 Viral Capsid
Phosphate buffered saline (PBS, 100 mM sodium phosphate, 50 mM sodium chloride): Three different PBS buffers, each having a different pH (i.e., 7.0, 7.6, and 8.0), are used during the synthesis. Weigh 31.2 g sodium phosphate monobasic dihydrate and 5.84 g sodium chloride and add to 1,900 mL water. Dissolve well and adjust to the target pH with 2 M NaOH. Make up to 2.0 L with water. Filter through 0.22 μm membrane filter. Store at room temperature.
0.5 M guanidine-HCl in PBS, pH 7.0: Weigh 47.8 g of guanidine-HCl and add to about 900 mL of PBS, pH 7.0. Dissolve well and make up to 1.0 L with water.
ATRP polymer is initiated from a genetically introduced cysteine residue mutated from serine at position 39, which is on the interior of the capsid of wild-type P22 coat protein as described [10]. According to the available P22 structural models, this site is expected to be exposed to the interior cavity [11] and is therefore suitable for attachment of the ATRP initiator (see Note 1) [10, 12]. The procapsid form (PC) of P22S39C is heterologously expressed in and purified from E. coli (BL21 (DE3)). The cloning, expression, and purification procedure has been previously described [10, 13]. The PC of P22S39C is transformed to the expanded form (EX) as previously described [10] (see Note 2).
2-Bromoisobutyryl aminoethyl maleimide: The ATRP initiator, 2-bromoisobutyryl aminoethyl maleimide, is synthesized by a modification of the procedures reported previously [14, 15]. Mix N-2-aminoethylmaleimide (250 mg, 0.98 mmol) with dry triethylamine (300 μl, 2.2 mmol) in 5 ml dry dichloromethane on ice. Add 2-bromo-2-methylpropionylbromide (200 μl, 1.6 mmol) to the reaction dropwise. Allow the reaction to warm to room temperature and then extract three times with dichloromethane and water, followed by drying over anhydrous sodium sulfate. Purify the crude product using column chromatography (silica gel, 10 % ethyl acetate in dichloromethane).
Metal catalyst solution for ATRP: Degas ultrapure water in advance by gently bubbling argon gas through the solution for 30 min. Weigh 19.2 mg CuBr (0.134 mmol), 29.9 mg CuBr2 (0.134 mmol), and 83.5 mg 2,2’-bipyridine (0.535 mmol) into a 10 mL crimp-top vial, seal the vial, and degas with argon gas flow. After 30 min, add 10 mL of degassed water to the vial and sonicate for 5 min. Repeat as necessary until solution turns dark brown to black in color (see Note 3). The solution is prepared just prior to the polymerization experiment with P22.
High-salt buffer solution (250 mM sodium sulfate, 100 mM sodium phosphate, pH 8.0): Weigh 35.5 g sodium sulfate and 15.6 g sodium phosphate monobasic dihydrate and add to about 900 mL water. Dissolve well and adjust to the target pH with 2 M NaOH. Adjust the volume to 1.0 L with water. Filter through a 0.22 μm membrane filter. Store at room temperature.
3. Methods
3.1. Synthesis of Magnetite Nanoparticle in Human Ferritin
Make 2 × 1.0 L solutions of 0.1 M NaCl solution. Dialyze HFn into 1.0 L of 100 mM NaCl for buffer exchange. After 6 h, replace the dialysis solution with fresh 0.1 M NaCl solution and dialyze for an additional 12 h. Filter the HFn through a 0. 1 μm syringe filter to remove dust and adjust the concentration to 2 mg/mL with 100 mM NaCl.
Insert a pH electrode connected to a pH auto titrator (Brinkmann 718 STAT Titrino) into the mouth of a jacketed (and temperature-controlled) reaction vessel pre-equilibrated at 65 °C. Use a PTFE/silicone septum to seal the mouth of the vessel.
Add 7.5 mL of degassed 100 mM NaCl and 2.5 mL of the HFn solution prepared above to the vessel under an Ar atmosphere (i.e., final HFn concentration is 0.5 mg/mL) (see Notes 4 and 5). Stir the solution gently with a magnetic stirrer.
Load two disposable syringes with 12.5 mM ammonium iron (II) sulfate hexahydrate solution and 4.17 mM hydrogen peroxide solution, respectively. The volumes of the solutions needed depend on the size of the magnetite particle (i.e., iron-loading factor per HFn cage) (see Note 6). Insert the syringes, with injection tubes, into a syringe pump and insert the tubes into the reaction vessel.
Connect the inlet and outlet hoses of the jacketed reaction vessel to a water bath set at 65 °C so that temperature of the vessel is brought up to and maintained at 65 °C during the mineralization reaction by circulating water through the jacketed flask.
Raise the pH of the solution slowly to 8.5 with 100 mM NaOH using the auto titrator. When the pH reaches 8.5, start the simultaneous injection of ammonium iron (II) sulfate hexahydrate and hydrogen peroxide solutions at a constant rate of 100 Fe/(cage · min) (i.e., 79 μL/min) using the syringe pump. H+ generated during the reaction is titrated using 100 mM NaOH to maintain the pH at 8.5. The reaction is considered to be completed 5 min after the addition of ammonium iron (II) sulfate hexahydrate and hydrogen peroxide solutions necessary for the targeted iron-loading factor (see Note 7).
Add 200 μL of 300 mM sodium citrate solution to the completed reaction in order to chelate any free iron in the solution. Dialyze the mineralized HFn solution against 1.0 L of DPBS.
Purify the mineralized HFn away from any aggregated products by fast protein liquid chromatography (FPLC) equipped with a Superose 6 size-exclusion column (SEC) while detecting absorbance at 280 and 410 nm to monitor elution of protein and mineral, respectively. Typical SEC profiles of HFn before and after mineralization reaction are shown in Fig. 1b.
3.2. Synthesis of x-AEMA in P22 Viral Capsid
Dissolve 2-bromoisobutyryl aminoethyl maleimide (4.63 mg, 016 mmol) in DMSO (400 μL) to a concentration of 40 mM.
Add 322 μL of the initiator solution (prepared above) to 24 mL ofP22S39C EX (5 mg/mL) in PBS, pH 7.6 (this corresponds to a fivefold excess per P22 subunit). Gently shake the mixture to react for three hours at room temperature. After 3 h, quench the reaction with DTT (322 μL, 40 mM in water). Pellet the P22S39C by ultracentrifugation at approximately 209,000 x g (45,000 rpm) (F50L-8×39 rotor, Piramoon Technologies) for 50 min, to remove any excess of the soluble initiator and DTT, and then resuspend the pellet in 1.0 mL PBS, pH 7.6.
Dissolve 160 mg of 2-aminoethyl methacrylate (AEMA) and 40 mg of bis-acrylamide in 4 mL of PBS pH 8.0. Adjust back to pH 8.0 with 2 M NaOH and adjust the volume to 5.0 mL. Spin down the mixture for 1 min at 4,000 × g to pellet and remove any aggregate.
Transfer 4 mL of the AEMA/Bis-acrylamide solution to a 10 mL crimp-top vial and add 2.2 mL of PBS, pH 8.0. Add mL of 8.0 mg/mL P22S39c with the initiator molecule prepared in step 2 to this mixture. Seal the crimp-top vial, followed by a cycle of pumping with a vacuum and back filling with argon gas four times to deaerate the mixture (see Note 8).
Add the metal catalyst solution (0.6 mL) to the monomer-P22S39C mixture anaerobically (under N2 or Ar) and cover the entire vial in foil, to avoid any photochemical reactions, and maintain the temperature at 23 °C for the duration of the polymerization reaction (180 min).
Quench the reaction by exposure to air and spin for 5 min at 17,000 × g to remove any aggregates formed during polymerization. Purify the P22S39C-polymer composite away from any unreacted monomer and the metal catalyst by pelleting the protein as described previously by ultracentrifugation and resuspend in 1 mL of PBS, pH 8.0. Add 2 mL of the high salt buffer (250 mM sodium sulfate, 100 mM sodium phosphate, pH 8.0) and agitate gently for 3 h at 4 °C to remove any AEMA and bis-acrylamide monomer which might be non-covalently associated with the P22. Pellet the protein again by ultracentrifugation and resuspend in 1.0 mL PBS, pH 8.0. Analyze the obtained material as previously described [10, 12].
4. Notes
The location of the initiator on a protein cage is critical to achieve confined polymerization inside of the cage cavity.
The scaffolding protein of P22S39C is removed using 0.5 M guanidine-HCl. Ultracentrifuge the P22S39C at approximately 209,000 × g for 50 min to pellet the capsid. Resuspend the pellet in 0.5 M guanidine-HCl by rocking gently for 1–2 h at 4 °C. Ultracentrifuge the protein again at approximately 209,000 × g for 50 min [16]. Repeat the cycle of pelleting and resuspension four times. Dialyze the empty-shell (ES) form of P22 against PBS, pH 7.0, overnight. Heat-treat the ES form of P22 for 20 min at 65 °C to transform the protein cage into expanded shell (EX) form. Purify the heat treated sample by ultracentrifugation pelleting as above, followed by resuspension into PBS, pH 7.6. Analyze the transformation of the P22 from PC form to EX form as previously described [10, 13, 17].
The catalyst solution should be dark brown to black in color. Slight blue color suggests insufficient deaeration and could cause poor polymerization.
The typical HFn concentration we have described is 0.5 mg/mL, but protein concentrations in the range of 0.1–1 mg/mL work equally well for the synthesis.
A critical key to the successful synthesis of magnetite (or maghemite) is keeping the reaction vessel atmosphere anaerobic. Continuous gentle flow of Ar (or N2) to the reaction vessel through narrow-bore tubing (exterior diameter 1/16 in., interior diameter 0.02 in.) helps to maintain anaerobic conditions.
The iron loading in HFn can be successfully achieved up to 5,000 Fe per HFn cage. When 5 mg of HFn is used for the synthesis of 5,000 Fe per HFn cage, the volume of ammonium iron (II) sulfate hexahydrate solution (12.5 mM) is 3,951 μL and the volume of hydrogen peroxide solution (4.17 mM) is 3,951 μL as well. For lower loadings, the required volumes are scaled accordingly.
The color of the mineralized HFn solution should be dark brown if magnetite (or maghemite) has been successfully synthesized in HFn. A bright orange/brown color suggests contamination of the product with ferrihydrite (or other iron oxides) formation. Definitive analysis can be achieved with electron diffraction equipped with transmission electron microscope or X-ray diffraction.
Tap the vial during the vacuum pumping cycle to remove air bubbles.
Acknowledgment
This work was supported with grants from the US Department of Energy, Office of Basic Energy Sciences, Division of Materials Science and Engineering DE-FG02–07ER46477 (for the inorganic nanoparticle work) and the National Institutes of Health NIAID R01AI104905 (for the polymer work).
References
- 1.Douglas T, Young M (1998) Host-guest encapsulation of materials by assembled virus protein cages. Nature 393:152–155 [Google Scholar]
- 2.Meldrum FC, Wade VJ, Nimmo DL, Heywood BR, Mann S (1991) Synthesis of inorganic nanophase materials in supramolecular protein cages. Nature 349:684–687 [Google Scholar]
- 3.Harrison PM, Arosio P (1996) Ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta 1275:161–203 [DOI] [PubMed] [Google Scholar]
- 4.Douglas T, Young M (2006) Viruses: making friends with old foes. Science 312:873–875 [DOI] [PubMed] [Google Scholar]
- 5.Manchester M, Steinmetz NF (2009) Viruses and nanotechnology, vol 327, Current topics in microbiology and immunology. Springer, Berlin: [PubMed] [Google Scholar]
- 6.Uchida M, Klem MT, Allen M, Flenniken ML,Gillitzer E, Varpness Z, Suci P, Young MJ, Douglas T (2007) Protein cage architecture: containers as templates for materials synthesis. Adv Mater 19:1025–1042 [Google Scholar]
- 7.Witus LS, Francis MB (2011) Using synthetically modified proteins to make new materials. Acc Chem Res 44:774–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uchida M, Flenniken ML, Allen M, Willits DA,Crowley BE,Brumfield S, Willis AF,Jackiw L, Jutila M, Young MJ, Douglas T (2006) Targeting of cancer cells with ferrimagnetic ferritin cage nanoparticles. J Am Chem Soc 128:16626–16633 [DOI] [PubMed] [Google Scholar]
- 9.Uchida M, Terashima M, Cunningham CH, Suzuki Y, Willits DA, Willis AF, Yang PC, Tsao PS,McConnell MV, Young MJ, Douglas T (2008) A human ferritin iron oxide nanocomposite magnetic resonance contrast agent. Magn Reson Med 60:1073–1081 [DOI] [PubMed] [Google Scholar]
- 10.Lucon J, Qazi S, Uchida M, Bedwell GJ, LaFrance B, Prevelige PE Jr, Douglas T (2012) Use of the interior cavity of the P22 capsid for site-specific initiation of atomtransfer radical polymerization with high- density cargo loading. Nat Chem 4:781–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen D-H, Baker ML, Hryc CF, DiMaio F, Jakana J, Wu W, Dougherty M, Haase-Pettingell C, Schmid MF, Jiang W, Baker D, King JA, Chiu W (2011) Structural basis for scaffolding-mediated assembly and maturation of a dsDNA virus. Proc Natl Acad Sci U S A 108:1355–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lucon J, Edwards E, Qazi S, Uchida M, Douglas T (2013) Atom transfer radical polymerization on the interior ofthe P22 capsid and incorporation of photocatalytic monomer crosslinks. Eur Polym J 49:2976–2985 [Google Scholar]
- 13.Kang S, Uchida M, O’Neil A, Li R,Prevelige PE, Douglas T (2010) Implementation of P22 viral capsids as nanoplatforms. Biomacromolecules 11:2804–2809 [DOI] [PubMed] [Google Scholar]
- 14.Mantovani G, Lecolley F, Tao L, Haddleton DM, Clerx J, Cornelissen JJ, Velonia K (2005) Design and synthesis of N-maleimido-functionalized hydrophilic polymers via copper-mediated living radical polymerization: a suitable alternative to PEGylation chemistry. J Am Chem Soc 127:2966–2973 [DOI] [PubMed] [Google Scholar]
- 15.Heredia KL, Bontempo D, Ly T, Byers JT, Halstenberg S, Maynard HD (2005) In situ preparation of protein-“smart” polymer conjugates with retention of bioactivity. J Am Chem Soc 127:16955–16960 [DOI] [PubMed] [Google Scholar]
- 16.Prevelige PE, Thomas D, King J (1988) Scaffolding protein regulates the polymerization of P22 coat subunits into icosahedral shells in vitro. J Mol Biol 202:743–757 [DOI] [PubMed] [Google Scholar]
- 17.Teschke CM, McGough A, Thuman-Commike PA (2003) Penton release from P22 heat-expanded capsids suggests importance of stabilizing penton-hexon interactions during capsid maturation. Biophys J 84:2585–2592 [DOI] [PMC free article] [PubMed] [Google Scholar]