

Abstract
Virus-like particles are unique platforms well suited for the construction of nanomaterials with broad-range applications. The research presented here describes the development of a modular approach for the covalent attachment of protein domains to the exterior of the versatile bacteriophage P22 virus-like particle (VLP) via a sortase-mediated ligation strategy. The bacteriophage P22 coat protein was genetically engineered to incorporate an LPETG amino acid sequence on the C-terminus, providing the peptide recognition sequence utilized by the sortase enzyme to catalyze peptide bond formation between the LPETG-tagged protein and a protein containing a polyglycine sequence on the N-terminus. Here we evaluate attachment of green fluorescent protein (GFP) and the head domain of the influenza hemagglutinin (HA) protein by genetically producing polyglycine tagged proteins. Attachment of both proteins to the exterior of the P22 VLP was found to be highly efficient as judged by SDS-PAGE densitometry. These results enlarge the tool kit for modifying the P22 VLP system and provide new insights for other VLPs that have an externally displayed C-terminus that can use the described strategy for the modular modification of their external surface for various applications.
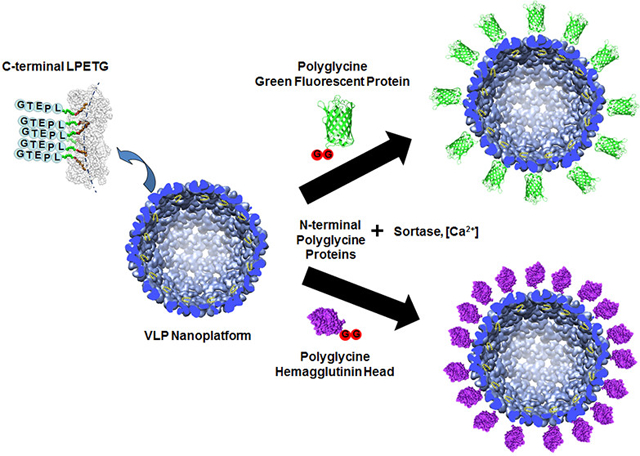
Graphical Abstract
Virus-like particles (VLPs) have been the focus of intense interest in the past decade for use in constructing advanced nanomaterials for a range of applications.1–9 Interest stems from their unique size, typically in the 10–100 nm range, and often homogeneous structures formed via biologically encoded assembly of proteins into regular, well-defined structures. The size of VLPs makes them particularly attractive for biomedical applications, where they can easily travel through circulatory systems and interact with cells. In addition, VLPs are produced without pathogenic components found in their infectious viral analogs, eliminating the chance of infection in biomedical applications. Their regular structures are often well characterized down to the molecular level, allowing VLPs to be used as scaffolds that can be engineered, through rational molecular design, to construct nanomaterials containing properties not inherent in the VLP structure alone. Through rational design approaches, researchers have utilized VLPs as size constrained reaction chambers for synthesizing inorganic nanoparticles,10 as carriers of drug molecules for drug delivery,8 to encapsulate enzymes to produce catalytic nanoreactors,10 as scaffolds for constructing nanoelectronics,11 and as platforms for the generation of immunotherapeutic nanomaterials.12–14Furthermore, VLPs are produced through recombinant heterologous protein expression, exploiting the power of biological systems to genetically produce exact copies of homogeneous nanoparticles. Continued research in developing VLPs as scaffolds that can be modified through modular strategies to produce nanomaterials with desired properties is particularly intriguing and of significant interest.
The P22 VLP derived from the Salmonella typhimurium bacteriophage P22 has been shown to be a particularly robust and versatile VLP platform due to a combination of easy heterologous expression, tolerance to modification, relatively large size, and stability. The P22 VLP assembles from 420 copies of a 47 kDa coat protein (CP) to form a T = 7, 58 nm nanoparticle referred to as the procapsid (PC).15 Assembly of the P22 VLP requires a scaffolding protein (SP), which templates assembly of the CP into the PC P22 VLP, with the SP trafficked to the interior of the P22 VLP. For assembly, it has been found that the N-terminus of the SP can be extensively modified and severely truncated, requiring only the last 18 amino acids on the C-terminus, with no deleterious effects on assembly.16,17 This feature of the P22 system has allowed the use of truncated SP, which has N-terminal amino acids removed yielding empty space in the interior of the P22 VLP, as a vehicle for directing the encapsulation of a wide array of protein cargoes on the interior of the assembled P22 VLP.18–27 For encapsulation, genes encoding desired cargo proteins are fused to the gene encoding the truncated SP, which upon expression leads to the production of a cargo protein-SP fusion protein. Coproduction of the cargo protein-SP fusion with CP in the cell leads to in vivo encapsulation of the cargo protein-SP on the interior of the P22 VLP. This method has proven to be very robust with the ability to encapsulate fusion proteins up to 180 kDa in size reported thus far, while maintaining the P22 VLP structure.19 In addition to the unique ability to genetically encapsulate protein cargoes, the P22 VLP is able to undergo a series of morphological change-s that alter the overall size and porosity of the VLP structure.28–31 Heating of the PC form at 60 °C for 15 min causes a rearrangement in the CP monomers to form a more angular structure termed the expanded shell form (EX), which has an increased diameter of 60 nm and an internal volume almost double that of the PC form.32 Additionally, heating of the PC or EX forms at 70 °C for 20 min leads to the transformation of the EX form to the wiffleball form (WB), which results from the dissociation of CP pentons at each of the fivefold vertices in the EX form, leading to the formation of 10 nm pores in the VLP structure. Access to these morphologies allow the end user to manipulate internal volume and capsid porosity and solute access to the VLP interior.
To expand the utility of the P22 VLP as a platform for constructing nanomaterials, developing modular methodology for the modification of the exterior of the VLP is desirable. Decoration of the exterior could be used to attach protein domains which can direct the assembly of higher order VLP materials in a specified manner, incorporate new catalytic or functional activities, and integrate immune stimulating antigens for constructing immunotherapeutic materials. Modification of the exterior can be performed either via noncovalent association of proteins to the surface or via direct covalent modification and attachment. Genetic approaches are particularly desirable, which require relatively little processing and yield superior homogeneity. For P22, it has been shown that the exterior can be readily modified noncovalently by the decorator protein (Dec) from phage L.33,34 Dec is a trimeric protein that selectively binds to the EX P22 VLP at quasi-threefold sites of symmetry with high affinity (Kd = 10–50 nM).35 The N-terminal regions of the subunits comprising Dec bind to the surface P22, with the C-termini projecting outward from the P22 surface. Attachment of proteins to the C-termini of Dec proteins, by construction of gene fusion and expression of Dec fusion proteins, allows proteins to be attached to the surface of EX P22 VLP through Dec mediated noncovalent association. Large protein domains (17 kDa) have been shown to be attached using this method,32 which provides a unique modular approach for attachment of proteins of significant size. However, using the trimeric Dec protein favors attachment of proteins that form trimeric or noninteracting monomeric structures, which can only bind to the EX (or WB) form of P22 and the noncovalent nature does not ensure permanent connection of a protein of interest to the surface. Covalent attachment to the surface of P22 VLPs has also been shown for short peptide sequences that can be genetically added to the C-terminus of P22, which has been shown to project to the exterior of the VLP structure.36 While attachment of short peptides to the exterior is useful for some applications, attachment of larger proteins and protein domains is desired, particularly for applications in immunotherapeutics where the stimulation of antibodies must be to a specific epitope of an antigen whose conformation is dependent on the larger protein framework that cannot be minimized down to a short peptide fragment. A modular approach that enables covalent attachment of larger proteins and protein fragments to the exterior surface of the P22 VLP is thus desired.
The research presented here examines a modular approach for the covalent attachment of large proteins/protein domains to the exterior of the P22 VLP. A number of synthetic chemical approaches have been described for the external modification of protein cages, such as incorporation of cysteine residues on the exterior which can be reacted with maleimide functional groups, incorporation of azide/alkyne functional groups for click chemistry, EDC/NHS cross-linking reactions with lysine residues, and other amino acid side change reactions.1,4,8,37,38 Our aim was to develop a totally genetic approach that would minimize overall processing and also enable the use of synthetic strategies, as noted, which could be used for attachment of nonprotein molecules. The strategy developed is to exploit the C-terminus of P22 CP as an attachment site for large proteins by utilizing sortase-mediated ligation to form a covalent peptide bond between the exposed P22 CP and a protein of interest (Figure 1). Sortase is an enzyme that catalyzes peptide bond formation between a protein containing a C-terminal LPETG amino acid sequence and a protein containing an N-terminal polyglycine sequence (two or more glycines on the N-terminus).39–41 Peptide bond formation is between the threonine (T) of the LPETG containing protein and the N-terminal glycine of the polyglycine containing protein. Sortase mediated ligation has been used to effectively cross-link a number of proteins, including a recent example exploiting the process for encapsulation of proteins on a VLP interior.42 By genetically modifying the P22 CP to contain a C-terminal LPETG sequence, proteins containing N-terminal polyglycines can be covalently added to exterior of the P22 VLP via sortase through the exposed LPETG peptide on the P22 VLP surface. The use of the C-terminus of the VLP for display of the LPETG enables polyglycine tags, which are less intrusive than the LPETG sequence, to be appended to the accessory proteins that one wishes to attach to the surface. Because glycines are also part of some common protease cleavage sites (i.e., TEV protease), proteins for attachment to the VLP surface can be produced as recombinant fusions with proteins that aid their production (i.e., improve expression levels, solubility, folding, etc.), which can be removed with proteases to generate N-terminal glycines. This is a unique feature of the research presented here in comparison with past examples of protein cage or VLPs, which focus on placing the polyglycine tags on the VLP,42,43 although other VLP or protein cage systems exist with externally displayed C-termini that can be modified in a similar manner as described here. This provides a completely genetic approach; all components are biologically produced and require no synthetic modification, for the covalent attachment of proteins to P22. In addition, the work presented here expands the versatility of the P22 system, whereby sortase-mediated ligation can be used in combination with the noncovalent Dec approach developed, as well as the encapsulation of proteins to produce a multivalent platform for the display of multiple proteins outside or inside the P22 VLP with molecular control over placement, which has not been described for other systems. For the studies presented here, the attachment of two different proteins to the P22 VLP was examined, green fluorescent protein (GFP) and the head domain from the influenza hemagglutinin protein (HAhead). GFP provides a useful protein for visualizing attachment to the P22 VLP surface for verifying proof of concept and attachment of HAhead provides supporting proof to the modularity of the approach by incorporating an important antigenic protein domain.
Figure 1.

Attachment of proteins to the P22 VLP via sortase. Construction of the P22 VLP containing a C-terminal LPETG amino acid sequence is predicted to allow modular attachment of N-terminal polyglycine (polyG; red circles) containing proteins (purple) via sortase catalyzed peptide bond formation between the N-terminal glycine of the polyG-protein and C-terminal threonine of the P22 coat proteins.
RESULTS AND DISCUSSION
Previous studies have shown that the C-terminus of P22 CP is exposed on the exterior of the P22 VLP and additional amino acid sequences were incorporated into the P22 CP gene to provide a C-terminal LPETG sortase recognition sequence.36 Coexpression of the P22 CP-LPETG and SP resulted in P22-LPETG VLPs with yields and behavior similar to that of the wild type P22 VLP. SDS-PAGE showed a band of slightly higher molecular weight than wild type P22 CP, at the expected molecular weight of CP-LPETG (48.3 kDa) and TEM images showed particles of ~58 nm as expected for the PC P22 (Supporting Information Figure S1). Dynamic light scattering showed an increased diameter for P22-LPETG of 64.24 ± 0.41 nm in comparison to unmodified P22 VLP (59.79 ± 1.06 nm), consistent with appendage of the peptide on the exterior of the P22 VLP, and polydispersity indexes (PDIs) ranging from 0.006 to 0.042 for all samples, indicating that both P22-LPETG and P22 VLPs are monodisperse (values <0.05 are considered monodisperse). Particles were further evaluated by size exclusion chromatography (SEC) coupled to inline multiangle light scattering (MALS) and refractive index detectors which showed hydrodynamic radii of 26.02 ± 0.208 nm and molar mass values of 26.73 ± 0.03 MDa for the intact PC P22-LPETG VLPs, consistent with unmodified PC P22 VLPs examined previously. PC P22-LPETG was transformed by heating to obtain EX P22-LPETG and the resulting VLPs were analyzed by TEM and SEC-MALS, which showed particles having a slight increase in size, as evidenced by a shift in the elution time and increased hydrodynamic radius to 26.89 ± 0.089 nm. In addition, the molecular weight determined by SEC-MALS showed a slight loss in molecular weight, 25.69 ± 0.221 MDa, consistent with transformation to EX enabling SP detachment and release from the P22 VLP interior. For initial studies, GFP containing N-terminal polyglycine (polyG-GFP) was also produced to probe attachment of proteins to the exterior of P22-LPETG via sortase-mediated ligation. Both polyG-GFP and sortase were produced via heterologous expression in E. coli and purified by nickel affinity chromatography in good yield to produce highly pure protein for ligation reactions as evidenced by SDS-PAGE (Supporting Information Figure S1).
Initially, small scale sortase mediated reactions were evaluated to determine optimal incubation time, ratios of polyG-GFP to CP-LPETG, and ratio of sortase to protein substrates. Screens were run on both the PC and EX forms of P22 and allowed to incubate from 1 h to overnight, evaluating various time increments between by SDS-PAGE analysis (Figure 2 and Supporting Information Figure S2). Cross-linking of polyG-GFP to CP-LPTG was observed for all reaction times, with the best cross-linking found after 4–6 h, which did not appear to improve with longer incubation periods. The best cross-linking in general was found for the higher ratios of 0.5–2 sortase to P22-LPETG/polyG-GFP in all cases. Comparison between 10:1 and 1:1 molar ratios of GFP to P22-LPETG showed relatively minor differences in ability to cross-link, with densitometry indicating best cross-linking ratios of 2.39 and 2.31 CP-LPETG to CP-GFP fusion product, respectively, for the 6 h incubation time as shown in Figure 2. Such differences may be negligible in cases where the protein being attached is precious and lower amounts can be used with good result. The densitometry indicates that the GFP is attached at GFP: P22-LPETG CP ratios lower than 1:1, with overall reaction efficiency of 15% for the best 6 h incubation sample (1:1 molar ratios of reactants), with lower efficiencies for all other samples. This is not surprising since the C-termini of the CP are arranged in close proximity at the fivefold and sixfold centers of symmetry, the VLP is composed of pentamer and hexamer assemblies of the CP, and attachment of GFP would induce steric hindrance of additional GFP proteins. Estimates from densitometry suggest attachment of roughly 140 copies (ratio of CP-LPETG:CP-GFP of 2.0) of GFP per capsid, equivalent to 2 GFP proteins attached at each fivefold (12 pentamers in total) and sixfold (60 hexamers in total) symmetry centers on the T= 7 P22 VLP. This number compares well with the number of proteins that can be attached noncovalently by the Dec protein (240 monomers),32 but provides a robust covalent bond that precludes dissociation of the attached protein, which might be beneficial and essential for some applications.
Figure 2.

Screening conditions for the sortase mediated ligation of polyG-GFP to P22 CP-LPETG. SDS-PAGE gels of samples incubated at 42 °C for times of 1, 2, 4, and 6 h, and overnight (22 h). Reactions that evaluated ratios of sortase ranging from 0.02 to 2 of sortase to reactants (polyGGFP and P22 CP-LPETG) for ligation and 10:1 and 1:1 ratios of reactants were evaluated.
Results from the initial small scale screen reactions were used as a guide in performing scaled up reactions for producing larger amounts of cross-linked material. Scaled up reactions were prepared at the 10:1 ratio of polyG-GFP to P22-LPETG CP and were incubated for 5 h. During the course of the reaction an increase in turbidity was observed, consistent with growth in nanoparticle size leading to increased light scattering observable by eye (Supporting Information Figure S3). SDS-PAGE confirmed covalent attachment of polyG-GFP to P22-LPETG, with higher reaction efficiency than observed for the smaller scale reactions (Figure 3A). For initial scale up reactions, P22-GFP VLPs were purified by ultracentrifugation, which for P22-LPETG results in an opaque pellet that can be resuspended in buffer with gentle rocking. Ultracentrifugation of the scaled up reaction mixture resulted in a green supernatant and a green pellet (Figure 3B), suggesting that GFP was attached to and pelleted with the dense P22 VLPs. SDS-PAGE showed that the pellet contained bands corresponding to cross-linked P22 CP-GFP and P22 CP-LPETG, in addition to some free sortase and GFP while the supernatant contained polyG-GFP and sortase only (Figure 3A). Interestingly, additional bands were also observed at molecular weights higher than expected for CP-GFP alone (75 kDa). Since no bands of higher molecular weight were seen in the starting materials, these likely are the result of additional cross-linking catalyzed by sortase. It has been previously observed that sortase shows some pliability in amino acids in the LPETG sequence, particularly at high substrate concentrations and extended incubation periods as described here,44 and performs a noncanonical isopeptide bond cross-linking reaction at the ε-amino group of lysine residues in specific sequences.45 Although no additional consensus sequences is apparent in the CP-LPETG or polyG-GFP, several glycine residues are found at the C-termini of both proteins, which are essential amino acids for both sortase mediated reactions and may contribute to the additional reactivity observed. Upon addition of buffer to the pellet and overnight resuspension by gentle rocking, only a small amount of the total pellet resuspended, unlike unmodified P22/P22-LPETG which almost completely resuspends. SDS-PAGE was used to evaluate the VLP resuspension and the aggregate that did not resuspend, with results showing that both the resuspension and aggregate contained cross-linked P22 CP-GFP. Ultracentrifugation of the resuspended P22 VLP through a cesium chloride gradient produced a green colored band where unmodified P22 VLP is normally observed (Figure 3C). SDS-PAGE confirmed polyGGFP cross-linking to the P22 CP-LPETG in the cesium chloride band corresponding to P22 VLP (Figure 3D) with densitometry showing a ratio of 1.39 CP to CP-GFP, corresponding to 176 GFPs attached per capsid (more than two at each five- and sixfold center of symmetry). Material at the top of the cesium chloride gradient corresponded to free polyG-GFP (Figure 3C,D), in agreement with its lower expected density than the 20 MDa P22 VLPs, and indicating that free polyG-GFP also pelleted with the P22 during the initial ultracentrifugation purification step. Attempts were made to bypass the initial ultracentrifugation pelleting by performing ultracentrifugation of ligation reactions through cesium chloride. However, upon cesium chloride ultracentrifugation a band containing a solid disk structure was observed where the P22-GFP VLP band had been observed previously (Figure 4A). SDS-PAGE analysis showed that the disk was composed of cross-linked CP-GFP and TEM images of the disk verified that it was comprised of tightly packed (but not ordered) P22 VLPs (Figure 4B,C and Supporting Information Figure S3). The disk was not observed to resuspend when transferred to buffers, even after several months. Together the results for ultra-centrifugation indicate that attachment of GFP to CP leads to strong interparticle interactions. In addition, free polyG-GFP was observed to copurify with P22-GFP VLPs, suggesting that GFP has a strong interaction with the P22-GFP VLPs. GFP has been reported to form dimers, with values for the dissociation constant (KD) of the GFP dimer of 100 μM.46 The results suggests that the observed interparticle interaction is therefore the result of the concentrating action of ultracentrifugation, which results in noncovalent cross-linking between the GFP monomers on the VLP surface. Interestingly, the results show that a majority of the P22-LPETG was conjugated to polyGGFP, as indicated by the absence of significant white banding for the soluble P22-LPETG in the region of the gradient where the P22-GFP disk was found.
Figure 3.

Analysis of scaled up cross-linking reaction between polyG-GFP and P22-LPETG. (A) SDS-PAGE gel evaluating the cross-linking in the polyG-GFP/P22-LPETG ligation reaction mixture after 4 h and pellet and supernatant fractions resulting from ultracentrifugation of the reaction mixture. (B) Image showing the P22 VLP pellet from ultracentrifugation after sortase mediated ligation with polyG-GFP. (C) Comparison of CsCl gradient results for P22 without GFP and resuspended P22-GFP from ultracentrifugation pellet. (D) SDS-PAGE results from the CsCl purification of the P22-GFP bands from the cesium chloride gradient of the ligation reaction in panel C.
Figure 4.

Purification and characterization of P22-GFP Sortase fusion product by cesium chloride and dialysis approaches. (A) Side by side comparison of results from cesium chloride purification of P22-LPETG VLPs without sortase added (left) and with sortase added (right) to the ligation reaction mixture. For the reaction containing sortase, a noticeable solid “disk” was observed (arrow pointing to zoomed in view) with a greenish hue. (B) SDS-PAGE was run on a sample of the disk and the gel shows that the disk is composed of cross-linked P22-GFP. (C) TEM images taken of the disk indicate that it is composed of P22 VLPs. (D) Solid “disks” were collected and evaluated by SAXS, but showed no long-range organization beyond the unaltered P22-LPETG. (E) Dialysis results for free polyG-GFP (left) and sortase ligation reaction mixtures (right) with 50 kDa MWCO and 100 kDa MWCO dialysis tubing, respectively. (F) SDS-PAGE gel evaluation of dialysis samples with lanes: (1) ladder, (2) GFP 50 MWCO dialysis, (3) GFP 100 kDa MWCO, (4) P22/GFP ligation reaction 50 kDa MWCO, and (5) P22/GFP ligation reaction 100 kDa MWCO. Larger MWCO allowed removal of free GFP.
To obtain soluble P22-GFP free from aggregation, removal of excess polyG-GFP from P22-GFP from quenched ligation reactions was evaluated using dialysis with tubing of 50 kDa and 100 kDa molecular weight cutoffs (MWCOs). Controls indicated that free polyG-GFP diffused from the 100 kDa MWCO dialysis tubing, but that polyG-GFP largely stayed inside the 50 kDa MWCO dialysis tubing at the high concentrations of GFP used in the experiment (Supporting Information Figure S5). These results are consistent with the fact that the dimer of GFP has an expected molecular weight of 56 kDa, large enough to be retained by the 50 kDa MWCO dialysis tubing, but easily diffusible through the 100 kDa MWCO dialysis tubing. P22-GFP samples dialyzed against the 50 kDa MWCO showed almost full retention of GFP in the dialysis tube with no detectable GFP in the dialysate, in agreement with the free polyG-GFP findings. However, P22-GFP samples dialyzed using the 100 kDa MWCO tubing sample retained green color (Figure 4E) but also showed free GFP in the dialysate, supporting conjugation of polyG-GFP to the P22-LPETG. UV–vis spectra of the samples after dialysis confirmed the differences in UV–vis absorption, which were initially identical before dialysis. Samples were then evaluated by SDS-PAGE to determine the amount of free GFP retained in the P22-GFP sample after dialysis (Figure 4F), which were consistent with previously observed conjugation results. Evaluation of the dialyzed P22-GFP sample by DLS showed a diameter of 96.43 ± 0.15 nm, an increase of 30 nm from the unmodified P22-LPETG VLP confirming attachment to the exterior. Based on crystal structure measurements, GFP is approximately 5 nm long and therefore addition of 10 nm to the P22 is predicted; however, some amount of P22-GFP dimers are likely present in solution that contribute to the scattering measurement to give the slightly higher value observed. PDI values of P22-GFP ranged from 0.178 to 0.199 indicating a modest amount of heterogeneity in the samples, as expected for the cross-linking method. Thus, the results indicate that dialysis can be used to remove nonconjugated proteins from the P22 VLPs, which may be a necessary step for proteins such as GFP that show interparticle oligomerization at the concentrations used in the ligation reactions that provide effective cross-linking or during purification by banding during ultracentrifugation.
The results showing the noncovalent association of the covalently conjugated P22-GFP to form disks suggested the potential for utilizing this approach for the formation of highly organized soft materials. To further evaluate this finding, disks were prepared by ultracentrifugation through a cesium chloride gradient. P22-GFP disks were gently separated from cesium chloride by removing the solution from the centrifuge tube with a Pasteur pipet and rinsing the pellet gently with deionized water. Disks were found to fracture easily, but they were not found to show any appreciable dissociation. To determine if the disks were the result of long-range, ordered organization of P22-GFP particles, the disks were analyzed by small-angle X-ray scattering (SAXS). Results of the P22-GFP disk showed no difference in SAXS profile patterns in comparison to either P22-LPETG or mixtures of P22-GFP and GFP (Figure 4D and Supporting Information Figure S4). These results indicate that the P22-GFP disk assembly does not have long-range organization, but is amorphous in its structural arrangement. This is not surprising since cross-linking by sortase does not guarantee regular placement of GFP on the P22 VLP platform and additionally some covalently cross-linked GFP subunits can likely form dimers with free GFP, as observed by bands in SDS-PAGE results corresponding to free GFP from the P22-GFP disks. However, the results suggest that this approach has the potential for developing organized materials with further design and development of an assembly strategy to provide additional control.
A major motivation for developing an approach for covalent attachment of proteins to the exterior of the P22 VLP is for the construction of immunotherapeutic nanomaterials. P22 has been shown to stimulate protective immune responses to human pathogens, and covalent attachment of antigens to the exterior is expected to provide stable nanoparticles that can provide multivalent display of antigens for directed immune responses.21,32,47 To further explore the efficacy of the approach toward incorporating antigen to the P22 surface, attachment of the head portion of the hemagglutinin protein (HAhead) from influenza was evaluated. HAhead exists as a monomer in solution and does not form higher order assemblies as observed for GFP. Initially, a plasmid containing the gene encoding HAhead containing an N-terminal poly glycine was constructed; however, this construct was not found to express measurable amounts of protein. To circumvent this problem the polyG-HAhead gene was inserted into a plasmid containing the gene encoding maltose binding protein (MBP) to produce a MBP-polyG-HAhead fusion protein upon expression. MBP is known to aid in improving expression levels and solubility of proteins in E. coli when produced as a fusion protein with proteins that are otherwise not easily produced. Expression of the MBP-HAhead in BL21(DE3) Clear Coli, a genetically modified strain of E. coli that does not produce LPS, yielded soluble protein that was purified in good quantity via nickel affinity chromatography. For removal of MBP a TEV protease sequence (ENLYFQG) was included between MBP and HAhead. Treatment of MBP-polyG-HAhead with TEV protease yielded predominantly free polyG-HAhead (Supporting Information Figure S6). Some precipitation was observed after TEV treatment; however, protein precipitate was refolded on a nickel affininty chromatography column and combined with nonprecipitated polyG-HAhead.
After producing polyG-HAhead, ligation reactions were set up based on the results found for GFP sortase ligation. GFP ligation reactions showed little difference between 10:1 and 1:1 ratios, and since protein antigens are typically produced in lower quantities, it was decided to evaluate a 1:1 ratio of polyGHAhead to P22-LPETG CP for the sortase mediated ligation. Indeed, 1:1 ratios of polyG-HAhead to P22-LPETG CP showed cross-linking efficiency consistent with those found for GFP when examined by SDS-PAGE (Figure 5). Densitometry of the bands in the SDS-PAGE indicated a CPLPETG to CP-HAhead of 1.3, indicating an attachment of ~183 HAhead proteins per capsid, consistent with more than two protein subunits at every sixfold and fivefold center of symmetry. Bands of slightly higher molecular weight than CPHAhead were also observed by SDS-PAGE, as observed for the polyG-GFP reactions, indicating that a similar cross-reaction is also likely at play. For purification, EDTA quenched reactions were run through a cesium chloride gradient by ultra-centrifugation to purify cross-linked P22-HAhead constructs from sortase and free polyG-HAhead. Unlike GFP experiments, the high concentrations of P22-HAhead did not result in the formation of solid protein disks, but rather white bands of soluble protein corresponding to P22 VLPs (Figure 5). SDS-PAGE of the bands from cesium chloride purification showed that purified VLPs contained covalently attached CP-HAhead with conjugation levels similar to those observed in the unpurified reaction mixture. These results provide support that the inability to resuspend a majority of the P22-GFP pellet and the formation of disks by P22-GFP resulted from interparticle GFP dimerization, which does not occur with the HAhead construct. Evaluation of the purified P22-HAhead sample by DLS showed a diameter of 124.9 ± 0.59 nm, an increase of 60 nm from the unmodified P22-LPETG VLP confirming attachment to the exterior. Based on crystal structure measurements, the dimensions of HAhead are slightly larger than GFP at approximately 7 nm at its longest axis, and therefore addition of 14 nm to the P22 is predicted. The larger value from expected suggests that some larger assemblies are likely present in solution that contribute to the scattering measurement to give the slightly higher value observed. PDI values of P22-GFP ranged from 0.157 to 0.172 showing some heterogeneity in the samples, consistent with values observed for GFP cross-linking. Analysis by SEC-MALS was also carried out; however, elution of the protein from the SEC column was not observed, suggesting the surface modification to P22 caused a change in the stability of the P22-HAhead relative to P22-LPETG under the SEC conditions examined leading to retention of the protein on the SEC resin. Overall, the results support the use of the sortase mediated ligation of protein antigens to P22-LPETG VLP scaffold, especially monomeric protein subunits or peptide/protein fragments, allowing for multivalent display which has been shown to improve the avidity of immune responses generated.
Figure 5.

Evaluation of sortase mediated cross-linking of the polyG-HAhead to P22-LPETG VLP. (A) SDS-PAGE showing the characterization of individual components used in the ligation experiments. (B) Purification of P22-HAhead fusion VLPs by ultracentrifugation through a cesium chloride gradient. (C) SDS-PAGE analysis of the band corresponding to the P22 VLP from cesium chloride purification; in A and C, SP identifies the scaffolding protein of the P22 VLP.
CONCLUSIONS
The results show that the P22 VLP is readily amendable to external bioconjugation by covalent attachment of proteins to the exposed C-terminus of the P22 CP. Modification of the C-terminus to install a LPETG sequence facilitated the attachment of N-terminal polyglycine containing proteins via sortase mediated ligation, with overall efficient conjugation. The results indicate that special considerations may be required for the purification of P22 VLPs decorated with proteins, particularly when the decorating protein can form a concentration dependent oligomeric structure. However, sortase mediated ligation of proteins to the surface of P22 was found to be general, with attachment of two different proteins producing similar cross-linking results, suggesting that this can be used as a modular approach for externally modifying P22. The P22 VLP has previously been shown to be a robust scaffold for constructing diverse catalytic and therapeutic nanomaterials. Results outlined here provide additional versatility for engineering the P22 VLP in further developing enhanced nanomaterials.
MATERIALS AND METHODS
Materials.
DNA modifying enzymes were purchased from New England Biolabs (Ipswich, MA). DNA primers were purchased from Eurofins MWG Operon (Huntsville, AL). E. coli BL21(λDE3) and 10G E. cloni electrocompotent cells were purchased from Lucigen (Madison, WI). QIAquick gel extraction kit, MinElute Enzyme Reaction Cleanup kit, and QIAprep Spin Miniprep kit were purchased from Qiagen (Valencia, CA). InstantBlue protein stain was purchased from Expedeon (San Deigo, CA). All chemical reagents were purchased from Fisher Scientific (Hampton, NH).
Molecular Biology.
The gene encoding GFP was amplified from a plasmid containing GFP using the primers 5’-AAAAGCCCATGggcggaggaggcAAGGGGGTGAAGGAAG-3’and 5’-AAAAGCGCGGATCCCTTGTTCTTCTCAGGCAG-CAG-3’ incorporating a polyglycine encoding sequence (lower case) and the restriction enzyme cut sites for NcoI and BamHI (underlined). The polyG-GFP gene and a pRSFDuet plasmid was then digested with NcoI/BamHI and purified using a Qiagen MinElute Enzyme Reaction Cleanup Kit to remove restriction enzymes and DNA fragments. The cut polyG-GFP and pRSFDuet plasmid were then mixed and ligated using NEB T4 DNA ligase according to the manufacturer’s directions and subsequently transformed into electrocomponent 10G E. cloni cells (Lucigen) and plated for selection on a Agar plate containing Kanamycin (50 μg/mL). Colonies were selected from plates, grown up in 5 mL overnights, DNA extracted from cells the following day via QIAprep Spin Miniprep kit, and the extracted DNA sent out for sequence verification (Eurofins Genomics). A 6xHistidine purification tag was added to the GFP containing plasmid by codigestion with BamHI/SacI restriction sites, purification using the ERC MinElute kit, and ligation with annealed gene fragments purchased from Eurorfins containing the correct sticky ends (encoding 5’-GATCGGCGGACTGGTGCCGCGCGGCAGCGGACAT-CACCATCATCACCACTAAGAGCT-3’ and complementary 5’-CTTAGTGGTGATGATGGTGATGTCCGCTGCCGCGCGGCACCAGTCCGCCG-3’). For constructing the coat protein containing the C-terminal LPETG sortase recognition sequence (CP-LPETG) the primer 5’-TTTTGCCGCTCGAG-TTAGCTACCGCCGGTTTCCGGCAG-GGTACCTCCT-CCGGCACCTCCGCCGCCTGCAGATCTCGCAGTCT-GACCAGGCAGGCCAACACC-3’ was constructed allowing amplification of the CP containing a C-terminal polyglycine spacer flanked by the LPETG and insertion into the pRSFDuet vector using the NdeI/XhoI restriction enzymes sites according to the same procedure noted above. The primers also encoded restriction enzyme sites for BglII and KpnI (underlined) for removal and modification of the spacer in the future if necessary. For obtaining the MBP-polyG-HAhead construct the primers 5′-AAAGGTACCGAGCTCGGAAAATCTGTA-CTTTCAAGGTGGCGGCGGTGCAAGCAAGGGC-3′ and 5′-AAAGCGGATCCTTAATGATGATGATGATGATGCT-CGAGACTGGTGATAATGCCCGAACC-3′ were used to amplify the HAhead gene by PCR and digested with SacI and BamHI for insertion into the pRK793 plasmid containing MBP. The pRK793 plasmid was digested with SacI and BamHI and gel purified to remove the gene insert removed by digestion. After purification of pRK793 vector and polyGHAhead gene, the two constructs were mixed and ligated with T4 DNA ligase, subsequently transformed, and sequences of DNA resulting from cell growth screened as described previously. All constructs were transformed into BL21(DE3) or Clear Coli upon confirmation of sequence for subsequent protein expression.
Protein Expression and Purification.
E. coli strains harboring expression constructs were grown in LB medium at 37 °C in the presence of ampicillin (0.1 μg/mL) or kanamycin (0.5 μg/m L) depending on plasmid to maintain selection for the plasmid. In addition, to the P22-LPETG, polyG-GFP, and MBP-polyG-HAhead constructs, sortase and TEV protease were also produced from plasmids previously obtained. The sortase gene was the generous gift of Dr. Robert T. Clubb (UCLA). Expression of the genes were induced by addition of isopropyl β-D-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM once the cells reached mid log phase (OD600 = 0.8). For P22-LPETG, polyG-GFP, and Sortase, cultures were grown for 4 h after addition of IPTG, and then the cells were harvested by centrifugation and cell pellets were stored at −20 °C. For TEV and MBP-HAhead the temperature was lowered at induction to 30 °C and protein expression allowed overnight, with subsequent harvesting and storage as noted for the other constructs.
Cell pellets were resuspended in Lysis buffer (50 mM Tris, 150 mM sodium chloride, pH 7.4) and the cell suspension was lysed by sonication. Cell debris was removed by centrifugation at 12 000g for 45 min at 4 °C. The supernatant was decanted and spun an additional 45 min at 12 000g to remove any excess cellular debris. P22-LPETG was purified from the supernatant by ultracentrifugation over a 35% (w/v) sucrose cushion on a Sorvall wX+ Ultra Series centrifuge (Thermoscientific) at 38 000 rpm using a Fiberlite F50L-8 × 39 rotor. The resulting viral pellet was resuspended in PBS (50 mM sodium phosphate, 25 mM sodium chloride, pH 7.0) and then purified over an S-500 Sephadex size exclusion column using an Biorad NGC FLPC. Flow rate for SEC purification was 1 mL/min of PBS. Fractions taken from SEC containing P22-LPETG were concentrated by ultracentrifugation and the resulting viral pellet was resuspended in an adequate volume of Tris Buffer (50 mM Tris, 150 mM sodium chloride, pH 8.0). The polyGGFP, MBP-polyG-HAhead, Sortase, and TEV protease constructs were purified by nickel affinity chromatography using PerfectPro Ni-NTA resin (5 Prime) according to standard procedures using Wash Buffer (50 mM Tris, 300 mM sodium chloride, 10% glycerol, pH 7.4) and Elution Buffer (50 mM Tris, 300 mM sodium chloride, 250 mM Imidazole, 10% glycerol, pH 7.4) to perform washes and elution using a step gradient. After Ni-affinity purification, polyG-GFP and sortase were dialyzed against Tris Buffer. To remove MBP from polyG-HAhead, TEV digests were performed by mixing of MBP-HAhed and TEV in a 10:1 ratio with addition of DTT (1 mM) and EDTA (0.5 mM) and overnight incubation at 4 °C. After incubation and dialysis against Wash Buffer, polyGHAhead was separated from cleaved MBP via Ni-affinity chromatography. Concentrations for the protein constructs were determined by UV absorption measured at 280 nm assuming molar extinction coefficients of CP-LPETG ε280 = 44 380 M−1 cm−1, SP ε280 = 6400 M−1 cm–1, polyG-GFP ε280 = 26 860 M−1 cm−1, polyG-HAhead ε280 = 49 500−1 cm−1, and Sortase ε280 = 13 370 M−1 cm−1 (calculated using Protein Calculator v 3.3, Chris Putnam, Scripps).
Screening Conditions for Sortase Mediated Ligation.
Ligation reactions were set up based on previously reported reaction conditions for protein–protein fusion.48 P22-LPETG and polyG-GFP were mixed at either 1:1 or 1:10 ratios of the CP-LPETG to polyG-GFP protein subunit concentration to a final concentration of 60 μM protein in Tris Buffer supplement with calcium chloride (6 mM). Sortase was added to mixtures to give Sortase:polyG-GFP/CP-LPETG ratios ranging from 0.02 to 2. Reactions were incubated at 42 °C and samples removed from reaction mixtures at 1, 2, 4, and 6 h and overnight (16–22 h), quenched with EDTA, and kept at 4 °C for SDS-PAGE analysis described below.
Scaled Up Cross-Linking Reactions.
For scaled up reactions, polyG-GFP (120 μM) was mixed with P22 CPLPETG (12 μM) at a 10:1 ratio and incubated at 42 °C for 5 h. Reactions were quenched and placed at 4 °C to await purification. Initial runs of polyG-GFP ligation to P22-LPETG evaluated purification by ultracentrifugation over a 35% (w/v) sucrose cushion as described above. Later, purification was evaluated by ultracentrifugation through a cesium chloride gradient (X–Y) at 38 000 rpm on a Sorvall wX+ Ultra Series centrifuge (Thermoscientific) using a TH-641 rotor, which resulted in protein disks. Subsequently it was determined that P22-GFP fusion ligation reactions were best purified from nonligated proteins (polyG-GFP and sortase) by dialysis. Dialysis was performed with either 50 000 or 100 000 Da MWCO Spectra/Por dialysis membrane dialysis tubing (Spectrum Laboratories) against Tris Buffer (two exchanges in 0.5–1 L each). Reactions for attachment of polyG-HAhead utilized a polyG-HAhead (20 μM) was mixed with P22 CPLPETG (20 μM) at a 1:1 ratio incubated at 42 °C for 5 h. Purification of the P22-HAhead fusion was performed by ultracentrifugation of ligation reaction mixtures through a cesium chloride gradient (0.26–0.54 g CsCl/m Lin Tris buffer) at 38 000 rpm on a Sorvall wX+ Ultra Series centrifuge (Thermoscientific) using a TH-641 rotor. Bands corresponding to P22-HAhed were removed from the centrifuge tube by pipetting and combined fractions containing P22-HAhead were run through a Zeba Spin Desalting Column, 40 000 Da MWCO, equilibrated with Tris Buffer according to the manufacturer’s directions to remove cesium chloride in exchange for the desired buffer. All constructs were evaluated by SDS-PAGE to determine the extent of Sortase mediated cross-linking.
SDS-PAGE.
Protein samples were mixed with 4× loading buffer containing DTT and heated in a boiling water bath for 10 min, and subsequently spun down on a benchtop centrifuge. Samples were separated on a gel containing a 4% polyacrylamide stacking gel and a 16% polyacrylamide running/separating gel using a constant current of 38 mA for approximately 1 h. Gels were stained with Coomassie Brillant Blue stain for 20–30 min and subsequently destained overnight or stained with InstantBlue Stain (Expedeon) for 1 h. Images were taken on an AlphaImager Mini (Protein Simple) and analyzed using the AlphaView SA software.
Transmission Electron Microscopy.
Samples (10 μL, 0.1 mg/mL protein) were applied to Formvar coated grids and incubated for 30 s and excess liquid was removed with filter paper. Grids were then washed with 10 μL of distilled water, liquid was removed with filter paper shortly after addition, and then stained with 5 μL 1% uranyl acetate after which excess stain was removed with filter paper. Images were taken on a JEOL 1010 transmission electron microscope at accelerating voltage of 80 keV.
Dynamic Light Scattering.
Samples were prepared as described for the scaled up reactions. After reactions, samples were centrifuged at 10 000g on a benchtop microcentrifuge and subsequently sterile filtered through a 0.22 μm syringe filter. Samples were analyzed on a Zetasizer Nano ZS (Malvern Instruments) for their z-average diameters and polydispersity index.
Multiangle Light Scattering.
Samples were separated over a WTC-0200S (Wyatt Technologies) size exclusion column utilizing an Agilent 1200 HPLC to apply and maintain a 0.7 mL/min flowrate of 50 mM phosphate, pH 7.2 buffer containing 100 mM sodium chloride and 200 ppm sodium azide. Samples of 25 μL were injected onto the column and total run time was 30 min. Samples were detected using a UV – vis detector (Agilent), a Wyatt HELEOS Multi Angle Laser Light Scattering (MALS) detector, a quasi-elastic light scattering detector (QELS), and an Optilab rEX differential refractometer (Wyatt Technology Corporation). The number-average molecular weight, Mn, was calculated with Astra 5.3.14 software (Wyatt Technology Corporation) based on the molecular weight distribution.
Small Angle X-ray Scattering.
SAXS measurement was done at the 12ID-B beamline at the Advanced Photon Source (APS). The measurements were carried out at 14 keV and the scattering data were collected with a Pilatus 2 M detector. Silver behenate was used as a standard to calibrate the system. One-dimensional scattering data were obtained by averaging two-dimensional scattering patterns.
Supplementary Material
ACKNOWLEDGMENTS
B.W., M.H., and M.T. were supported by the Welch Foundation Grant BP-0037. B.S. was supported by the Department of Defense (DoD) Air Force Office of Sponsored Research through the National Defense Science & Engineering Graduate Fellowship (NDSEG) Program. This research was supported by a grant (to T.D.) from the National Institutes of Health (R01 AI104905 547) and the National Science Foundation (NSF-BMAT DMR-1507282).
ABBREVIATIONS
- VLPs
Virus like particles
- GFP
Green fluorescent protein
- HA
Hemagglutinin
- CP
Coat protein
- PC
Procapsid
- EX
Expanded shell
- WB
Wiffleball
- SDS-PAGE
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- Dec
Decorator protein
- PDI
polydispersity index
- SAXS
Small angle X-ray scattering
- polyG
poly glycine
- MWCO
Molecular weight cutoff
- SEC
Size exclusion chromatography
- MALS
multiangle light scattering
- DLS
dynamic light scattering
- QELS
Quasi-elastic light scattering
- MBP
Maltose binding protein
- TEM
transmission electron microscopy
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconjchem.7b00296.
Nucleotide, protein sequences, and supplementary figures (PDF)
REFERENCES
- (1).Smith MT, Hawes AK, and Bundy BC (2013) Reengineering viruses and virus-like particles through chemical functionalization strategies. Curr. Opin. Biotechnol 24, 620–626. [DOI] [PubMed] [Google Scholar]
- (2).Rodríguez-Limas WA, Sekar K, and Tyo KE (2013) Virus-like particles: the future of microbial factories and cell-free systems as platforms for vaccine development. Curr. Opin. Biotechnol 24, 1089–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Frietze KM, Peabody DS, and Chackerian B (2016) Engineering virus-like particles as vaccine platforms. Curr. Opin. Virol 18, 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Rother M, Nussbaumer MG, Renggli K, and Bruns N (2016) Protein cages and synthetic polymers: a fruitful symbiosis for drug delivery applications, bionanotechnology and materials science. Chem. Soc. Rev 45, 6213–6249. [DOI] [PubMed] [Google Scholar]
- (5).Maassen SJ, van der Ham AM, and Cornelissen JJ (2016) ACS Macro Lett. 5, 987–994. [DOI] [PubMed] [Google Scholar]
- (6).Uchida M, Klem MT, Allen M, Suci P, Flenniken M, Gillitzer E, Varpness Z, Liepold LO, Young M, and Douglas T (2007) Biological containers: protein cages as multifunctional nanoplatforms. Adv. Mater 19, 1025–1042. [Google Scholar]
- (7).Pushko P, Pumpens P, and Grens E (2013) Development of virus-like particle technology from small highly symmetric to large complex virus-like particle structures. Intervirology 56, 141–165. [DOI] [PubMed] [Google Scholar]
- (8).Ma Y, Nolte RJ, and Cornelissen JJ (2012) Virus-based nanocarriers for drug delivery. Adv. Drug Delivery Rev. 64, 811–825. [DOI] [PubMed] [Google Scholar]
- (9).Cormode DP, Jarzyna PA, Mulder WJ, and Fayad ZA (2010) Modified natural nanoparticles as contrast agents for medical imaging. Adv. Drug Delivery Rev 62, 329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Maity B, Fujita K, and Ueno T (2015) Use of the confined spaces of apo-ferritin and virus capsids as nanoreactors for catalytic reactions. Curr. Opin. Chem. Biol 25, 88–97. [DOI] [PubMed] [Google Scholar]
- (11).Soto CM, and Ratna BR (2010) Virus hybrids as nanomaterials for biotechnology. Curr. Opin. Biotechnol 21, 426–438. [DOI] [PubMed] [Google Scholar]
- (12).Rynda-Apple A, Patterson DP, and Douglas T (2014) Virus-like particles as antigenic nanomaterials for inducing protective immune responses in the lung. Nanomedicine 9, 1857–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Jennings GT, and Bachmann MF (2008) The coming of age of virus-like particle vaccines. Biol. Chem 389, 521–536. [DOI] [PubMed] [Google Scholar]
- (14).Al-Barwani F, Donaldson B, Pelham SJ, Young SL, and Ward VK (2014) Antigen delivery by virus-like particles for immunotherapeutic vaccination. Ther. Delivery 5, 1223–1240. [DOI] [PubMed] [Google Scholar]
- (15).Botstein D, Waddell CH, and King J (1973) Mechanism of head assembly and DNA encapsulation in Salmonella phage P22: I. Genes, proteins, structures and DNA maturation. J. Mol. Biol 80, 669 IN19-IN21, 705–731. [DOI] [PubMed] [Google Scholar]
- (16).Parker MH, Casjens S, and Prevelige PE (1998) Functional domains of bacteriophage P22 scaffolding protein. J. Mol. Biol 281, 69–79. [DOI] [PubMed] [Google Scholar]
- (17).Weigele PR, Sampson L, Winn-Stapley D, and Casjens SR (2005) Molecular genetics of bacteriophage P22 scaffolding protein’s functional domains. J. Mol. Biol 348, 831–844. [DOI] [PubMed] [Google Scholar]
- (18).Patterson DP, Prevelige PE, and Douglas T (2012) Nanoreactors by programmed enzyme encapsulation inside the capsid of the bacteriophage P22. ACS Nano 6, 5000–5009. [DOI] [PubMed] [Google Scholar]
- (19).Patterson DP, Schwarz B, Waters RS, Gedeon T, and Douglas T (2014) Encapsulation of an enzyme cascade within the bacteriophage P22 virus-like particle. ACS Chem. Biol 9, 359–365. [DOI] [PubMed] [Google Scholar]
- (20).Patterson DP, Schwarz B, El-Boubbou K, van der Oost J, Prevelige PE, and Douglas T (2012) Virus-like particle nano-reactors: programmed encapsulation of the thermostable CelB glycosidase inside the P22 capsid. Soft Matter 8, 10158–10166. [Google Scholar]
- (21).Patterson DP, Rynda-Apple A, Harmsen AL, Harmsen AG, and Douglas T (2013) Biomimetic antigenic nanoparticles elicit controlled protective immune response to influenza. ACS Nano 7, 3036–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Jordan PC, Patterson DP, Saboda KN, Edwards EJ, Miettinen HM, Basu G, Thielges MC, and Douglas T (2015) Self-assembling biomolecular catalysts for hydrogen production. Nat. Chem 8, 179–185. [DOI] [PubMed] [Google Scholar]
- (23).Patterson DP, LaFrance B, and Douglas T (2013) Rescuing recombinant proteins by sequestration into the P22 VLP. Chem. Commun 49, 10412–10414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Patterson DP, McCoy K, Fijen C, and Douglas T (2014) Constructing catalytic antimicrobial nanoparticles by encapsulation of hydrogen peroxide producing enzyme inside the P22 VLP. J. Mater. Chem. B 2, 5948–5951. [DOI] [PubMed] [Google Scholar]
- (25).O’Neil A, Prevelige PE, Basu G, and Douglas T (2012) Coconfinement of fluorescent proteins: spatially enforced communication of GFP and mCherry encapsulated within the P22 capsid. Biomacromolecules 13, 3902–3907. [DOI] [PubMed] [Google Scholar]
- (26).O’Neil A, Prevelige PE, and Douglas T (2013) Stabilizing viral nano-reactors for nerve-agent degradation. Biomater. Sci 1, 881–886. [DOI] [PubMed] [Google Scholar]
- (27).Qazi S, Miettinen HM, Wilkinson RA, McCoy K, Douglas T, and Wiedenheft B (2016) Programmed Self-Assembly of an Active P22-Cas9 Nanocarrier System. Mol. Pharmaceutics 13, 1191–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Teschke CM, McGough A, and Thuman-Commike PA (2003) Penton release from P22 heat-expanded capsids suggests importance of stabilizing penton-hexon interactions during capsid maturation. Biophys. J 84, 2585–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kang S, Uchida M, O’Neil A, Li R, Prevelige PE, and Douglas T (2010) Implementation of p22 viral capsids as nanoplatforms. Biomacromolecules 11, 2804–2809. [DOI] [PubMed] [Google Scholar]
- (30).Parent KN, Khayat R, Tu LH, Suhanovsky MM, Cortines JR, Teschke CM, Johnson JE, and Baker TS (2010) P22 coat protein structures reveal a novel mechanism for capsid maturation: stability without auxiliary proteins or chemical crosslinks. Structure 18, 390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Casjens S, Adams M, Hall C, and King J (1985) Assembly-controlled autogenous modulation of bacteriophage P22 scaffolding protein gene expression. Journal of Virology 53, 174–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Schwarz B, Madden P, Avera J, Gordon B, Larson K, Miettinen HM, Uchida M, LaFrance B, Basu G, Rynda-Apple A, et al. (2015) Symmetry Controlled, Genetic Presentation of Bioactive Proteins on the P22 Virus-like Particle Using an External Decoration Protein. ACS Nano 9, 9134–9147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gilcrease EB, Winn-Stapley DA, Hewitt FC, Joss L, and Casjens SR (2005) Nucleotide sequence of the head assembly gene cluster of bacteriophage L and decoration protein characterization. J. Bacteriol 187, 2050–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Tang L, Gilcrease EB, Casjens SR, and Johnson JE (2006) Highly discriminatory binding of capsid-cementing proteins in bacteriophage L. Structure 14, 837–845. [DOI] [PubMed] [Google Scholar]
- (35).Parent KN, Deedas CT, Egelman EH, Casjens SR, Baker TS, and Teschke CM (2012) Stepwise molecular display utilizing icosahedral and helical complexes of phage coat and decoration proteins in the development of robust nanoscale display vehicles. Biomaterials 33, 5628–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Servid A, Jordan P, O’Neil A, Prevelige P, and Douglas T (2013) Location of the bacteriophage P22 coat protein C-terminus provides opportunities for the design of capsid-based materials. Biomacromolecules 14, 2989–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Wen AM, and Steinmetz NF (2016) Design of virus-based nanomaterials for medicine, biotechnology, and energy. Chem. Soc. Rev 45, 4074–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Koudelka KJ, Pitek AS, Manchester M, and Steinmetz NF (2015) Virus-based nanoparticles as versatile nanomachines. Annu. Rev. Virol 2, 379–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Tsukiji S, and Nagamune T (2009) Sortase-Mediated Ligation: A Gift from Gram-Positive Bacteria to Protein Engineering. ChemBioChem 10, 787–798. [DOI] [PubMed] [Google Scholar]
- (40).Proft T (2010) Sortase-mediated protein ligation: an emerging biotechnology tool for protein modification and immobilisation. Biotechnol. Lett 32, 1. [DOI] [PubMed] [Google Scholar]
- (41).Mao H, Hart SA, Schink A, and Pollok BA (2004) Sortase-mediated protein ligation: a new method for protein engineering. J. Am. Chem. Soc 126, 2670–2671. [DOI] [PubMed] [Google Scholar]
- (42).Schoonen L, Pille J, Borrmann A, Nolte RJ, and van Hest JC (2015) Sortase A-mediated N-terminal modification of cowpea chlorotic mottle virus for highly efficient cargo loading. Bioconjugate Chem. 26, 2429–2434. [DOI] [PubMed] [Google Scholar]
- (43).Chen Q, Sun Q, Molino NM, Wang S-W, Boder ET, and Chen W (2015) Sortase A-mediated multi-functionalization of protein nanoparticles. Chem. Commun 51, 12107–12110. [DOI] [PubMed] [Google Scholar]
- (44).Kruger RG, Otvos B, Frankel BA, Bentley M, Dostal P, and McCafferty DG (2004) Analysis of the substrate specificity of the Staphylococcus aureus sortase transpeptidase SrtA. Biochemistry 43, 1541–1551. [DOI] [PubMed] [Google Scholar]
- (45).Bellucci JJ, Bhattacharyya J, and Chilkoti A (2015) A Noncanonical Function of Sortase Enables Site-Specific Conjugation of Small Molecules to Lysine Residues in Proteins. Angew. Chem 127, 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Chalfie M, and Kain SR (2005) Green fluorescent protein: properties, applications and protocols, Vol. 47, John Wiley & Sons. [Google Scholar]
- (47).Schwarz B, Morabito KM, Ruckwardt TJ, Patterson DP, Avera J, Miettinen HM, Graham BS, and Douglas T (2016) Viruslike Particles Encapsidating Respiratory Syncytial Virus M and M2 Proteins Induce Robust T Cell Responses. ACS Biomater. Sci. Eng 2, 2324–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Levary DA, Parthasarathy R, Boder ET, and Ackerman ME (2011) Protein-protein fusion catalyzed by sortase A. PLoS One 6, e18342. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.