

To the Editor: Oculocutaneous albinism (OCA) is an autosomal recessive disorder caused by a group of mutations related to the regulation of melanin synthesis and melanosome biogenesis. The prevalence of OCA worldwide is approximately 1 in 17,000.[1] Other than symptomatic treatment, there is no effective treatment for albinism. Due to the variability in clinical phenotypes, it is difficult to classify sub-types simply by clinical features; therefore, molecular and genetic analyses are the most reliable methods for confirming diagnosis, carrier screening, and pre-natal diagnosis.
With the development of gene detection technology, most albinism patients can be diagnosed and classified by Sanger sequencing or high-throughput sequencing (“next-generation” sequencing technology [NGS]).[2,3] However, these traditional detection methods cannot detect the second mutation in some patients, which are called uncharacterized mutations. One source of these uncharacterized mutations is the gene copy number variations (CNVs) caused by a large deletion or duplication of genomic segments. Multiplex ligation-dependent probe amplification (MLPA) was first reported in 2002 to detect CNVs, including small intra-genic rearrangements.[4]
A total of 12 unrelated patients with OCA were selected for this study who had only one allelic point mutation detectable by Sanger sequencing or NGS in either the tyrosinase (TYR) or oculocutaneous albinism II (OCA2) gene. In some cases, the NGS data hinted at large heterozygous deletions. All patients in this study exhibited different degrees of hypopigmentation in the skin, hair, and iris, accompanied by nystagmus, photophobia, and visual impairment. The unaffected family members presented with normal pigmentation.
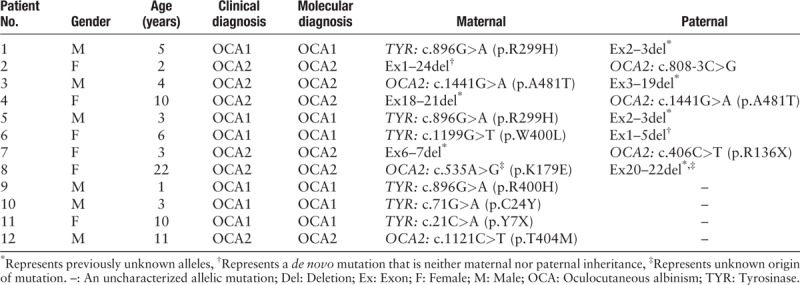
MLPA was utilized to detect potential large heterozygous deletions in these patients. The MLPA results revealed that eight cases contained large heterozygous deletions of one or more exons (Ex), and the detection rate of mutation was 66.7%. Seven kinds of large heterozygous deletions were found among these patients: two were found in the TYR gene and five were found in the OCA2 gene. Except for the Ex1–24 deletion in OCA2 found in Patient 2 and Ex1–5 deletion in TYR found in Patient 6, and other mutations were not previously reported in the Human Gene Mutation Database or 1000 Genomes Database. All mutations were graded as pathogenic variations according to the standards and guidelines for the interpretation of sequence variants published by the American College of Medical Genetics and Genomics,[5] and two of these were de novo mutations [Table 1].
Table 1.
Genotypes of the 12 patients with oculocutaneous albinism.
OCA1 (MIM 203100) is caused by mutations in the TYR gene that lead to the complete or partial loss of tyrosinase activity. The most severe type of OCA, OCA1A, is characterized by white skin and hair from birth and a complete lack of melanin throughout the patient's life.[1] Due to residual tyrosinase enzyme activity, patients with OCA1B have little or no pigmentation at birth but pigments can accumulate with age. OCA2 (MIM 203200) is caused by mutations of the OCA2 gene (previously known as the P gene). Research suggests that the OCA2 protein may regulate the pH of melanosomes and maintain tyrosinase activity.[6] Typically, patients with OCA2 have white skin, yellow hair, and blue/light or brown/light or brown irises that are accompanied by nystagmus, photophobia, and impaired visual acuity. The vision of patients with OCA2 can improve with age.[7]
Due to the variation in OCA phenotypes, it is difficult to classify OCA into sub-types based on clinical features alone. Molecular analyses can provide confirmative diagnoses and guide genetic counseling and pre-natal screenings for families at risk. However, second mutations may not be identified by Sanger sequencing or NGS in some patients with OCA. CNVs include entire or partial gene deletions or duplications, which account for 5.5% of all previously reported disease-causing mutations.[8] CNVs can be predicted by Sanger sequencing or NGS data and confirmed by MLPA or real-time quantitative polymerase chain reaction. MLPA can only detect CNVs at specific loci (target region), that is, the region or gene where a CNV is likely to occur. The resolution is determined by the probe settings. For NGS, CVNs results are generally obtained by analyzing differences in sequencing depth on the basis of sequencing data. In general, 100 kilobytes of CNVs are not always detectable. Using NGS to detect CNVs, we do not need to select candidate genes in advance, which is suitable for primary screening. MLPA has the advantages of high sensitivity, high specificity, and ease of use, while the limitation is that the kits need to be specially designed to detect candidate genes. In summary, MLPA is a complementary tool for the genetic diagnosis of OCA.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patients have given their consent for their images and other clinical information to be reported in the journal. The patients understand that the names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Funding
This work was partially supported by grants from the National Natural Science Foundation of China (No. 81472871) and High-level Professional Talents of Beijing Healthcare System (No. 2015-3-011).
Conflicts of interest
None.
Footnotes
How to cite this article: Zhang YZ, Bai DY, Qi Z, Zhao SZ, Yang XM, Li W, Wei AH. Application of multiplex ligation-dependent probe amplification in the genetic testing of oculocutaneous albinism. Chin Med J 2019;00:00–00. doi: 10.1097/CM9.0000000000000356
References
- 1.Wei AH, Yang XM, Lian S, Li W. Genetic analyses of Chinese patients with digenic oculocutaneous albinism. Chin Med J 2013; 126:226–230. doi: 10.3760/cma.j.issn.0366-6999.20121104. [PubMed] [Google Scholar]
- 2.Wei A, Yang X, Lian S, Li W. Implementation of an optimized strategy for genetic testing of the Chinese patients with oculocutaneous albinism. J Dermatol Sci 2011; 62:124–127. doi: 10.1016/j.jdermsci.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Wei A, Yuan Y, Bai D, Ma J, Hao Z, Zhang Y, et al. NGS-based 100-gene panel of hypopigmentation identifies mutations in Chinese Hermansky-Pudlak syndrome patients. Pigment Cell Melanoma Res 2016; 29:702–706. doi: 10.1111/pcmr.12534. [DOI] [PubMed] [Google Scholar]
- 4.Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 2002; 30:e57.doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brilliant MH. The mouse p (pink-eyed dilution) and human P genes, oculocutaneous albinism type 2 (OCA2), and melanosomal pH. Pigment Cell Res 2001; 14:86–93. doi: 10.1034/j.1600-0749.2001.140203.x. [DOI] [PubMed] [Google Scholar]
- 7.Carden SM, Boissy RE, Schoettker PJ, Good WV. Albinism: modern molecular diagnosis. Br J Ophthalmol 1998; 82:189–195. doi: 10.1136/bjo.82.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee C, Iafrate AJ, Brothman AR. Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat Genet 2007; 39:S48–S54. doi: 10.1038/ng2092. [DOI] [PubMed] [Google Scholar]