

Abstract
Sestrin 3 (Sesn3) belongs to a small protein family that has been implicated in multiple biological processes including anti-oxidative stress, anti-aging, cell signaling, and metabolic homeostasis. However, the role of Sesn3 in hepatocellular carcinoma (HCC) remains unclear. Here we generated a Sesn3 knockout mouse model and induced HCC development by a combination of a single dose of diethylnitrosamine and chronic feeding of a choline deficient-high fat diet. After 6 months of the dietary treatment, Sesn3 knockout mice developed more severe HCC with higher levels of alpha-fetoprotein, arginase 1, and cytokeratin 19, but also higher metastatic rates than wild-type mice. Histological analysis revealed elevated extracellular matrix and cancer stem cell markers including Acta2, Cd44, and Cd133. Signaling analysis showed activated IL6-Stat3 and Akt pathways. Biochemical and microscopic analyses uncovered a novel inhibitory regulation of Gli2, a downstream transcription factor of the hedgehog signaling, by Sesn3. Two of the Gli2-regulated genes – Pdgfrb and Cd44 were upregulated in the Sesn3-deficient liver tissue. In conclusion, our data suggest that Sesn3 plays a critical tumor suppressor role in the liver partly through the inhibition of the hedgehog signaling.
Keywords: Sestrin, liver cancer, hepatocellular carcinoma, hedgehog signaling, cancer stem cell
1. Introduction
Sestrins (Sesns) belong to a small family of evolutionally conserved proteins that do not share domain structures with any other eukaryotic proteins [1]. Sestrins have been implicated in multiple biological functions including protection against oxidative and genotoxic stresses, anti-aging, and metabolic homeostasis [1–3]. Mammals have three sestrin genes - Sesn1/2/3 - which are regulated differently [3]. Sesn1 and Sesn2 are activated by p53 transcription factor in response to genotoxic stress [4, 5]. Sesn2 can also be induced by oxidative stress, endoplasmic reticulum stress, starvation, and high-fat diets [4–14]. Interestingly under certain pathophysiological conditions, SESN3 gene expression is upregulated in leg muscle biopsies from patients with type 2 diabetes whereas it downregulated in ethanol-treated mouse livers and nonalcoholic steatohepatitis human livers [15–17]. Regarding the role of Sesn3 in the liver, only alcoholic and nonalcoholic fatty liver disease and hepatic metabolic regulation have been reported [14, 16–18]. However, it remains unknown what role of Sesn3 plays in the development of hepatocellular carcinoma (HCC).
Liver cancer is one of the most common cancers in the world. In 2016, there were 1.0 million new cases of liver cancer and 829,000 deaths globally [19]. HCC is the most frequent primary liver cancer. In 2012, HCC was ranked as the 6th most common neoplasm and the third leading cause of cancer death [20]. Multiple signaling pathways including IL6-STAT3 and hedgehog signaling have been implicated in the development of HCC [21]. A recent report suggests that Stat3 but not Stat1 is responsible for the obesity-triggered HCC pathogenesis [22]. In mammals, the core components of the hedgehog signaling pathway include: three ligands (sonic hedgehog, desert hedgehog, and Indian hedgehog), Patched1 receptor, Smoothened (a G-protein-coupled receptor), and three transcription factors GLI1, GLI2, and GLI3 [23]. The oncogenic role of GLI2 in HCC has been shown in vivo as well [24]. Cancer stem cells (CSCs) have been identified as an early lesion of numerous cancer types including HCC [25]. A number of cell markers including CD44, CD133, EpCAM, and CK-19 have been used to identify CSCs [26]. In general, those CSCs may represent early transdifferentiated cells within the initial tumor lesion [25]. In this study, we investigated the role of Sesn3 in HCC development in a preclinical mouse model.
2. Materials and Methods
2.1. HCC mouse model
Sesn3 knockout (KO) mice were generated by crossing a floxed strain as reported previously [18] and a CMV-Cre line [27] from the Jackson Laboratory. Mice were housed in the animal facility with controlled temperature (22 ± 2°C), humidity (60 ± 5%), and regular 12:12 light/dark cycle. To induce HCC, we used a protocol of combination of a single dose of diethylnitrosamine (DEN) and chronic feeding a choline-deficient high-fat diet (CD-HFD). Wild type (WT) and Sesn3 KO mice were intraperitoneally injected with DEN at a dose of 25 mg/kg at 2 weeks of age. At 4 weeks of age, the mice were fed with a CD-HFD (Research Diets #D05010402, containing 20 kcal% protein, 35.1 kcal% carbohydrate, and 44.9 kcal% fat) for 24 weeks. Body weights were measured twice weekly. On the final day of experiment, mice were sacrificed under an anesthesia condition. Blood samples were collected and major organs including liver, lungs, and spleen were dissected and weighed. Tissue samples were fixed in 10% neutral formalin or immediately frozen in liquid nitrogen for further analysis. All animal care and experimental procedures performed in this work were approved by the Institutional Animal Care and Use Committee of Indiana University School of Medicine in accordance with National Institutes of Health guidelines for the care and use of laboratory animals.
2.2. Liver tumor measurements
Hepatic tumor nodules were counted after the animals were euthanized on the final day of experiment. The tumor nodule size cutoff was 2 mm in diameter. Tumor volumes were measured using a Vernier caliper and calculated according to the following formula: (A×B2)/2, where A is the larger and B is the smaller of the two-dimensional measurements [28].
2.3. Serum biochemistry and hepatic lipid analysis
Serum alanine aminotransferase (ALT) levels were serially measured at time points of 4, 8, 12, and 24 weeks of CD-HFD treatment, respectively. Hepatic total cholesterol (TC) and triglycerides (TG) were extracted as previously described [29], and analyzed using commercial assay kits (Thermo Fisher and Wako USA) following the manufacture’s manuals.
2.4. Histological analysis
Liver tissue samples were fixed in 10% formalin then processed for embedding and sectioning at the Histology Core of Indiana University School of Medicine. Liver sections (5 μm thickness) were stained using hematoxylin and eosin (H&E) following the standard protocol. Immunofluorescence (IF) analysis was performed for detecting general HCC markers including Arginase-1 (Arg1, rabbit mAb, Cat. No. 93668S, Cell Signaling Technology), alpha-fetoprotein (AFP, mouse mAb, sc-8399, Santa Cruz Biotechnology), and cytokeratin 19 (CK-19, rat mAb, TROMA-III, DSHB), ECM molecules such as Collagen 1 and 3 (Col1, rabbit anti-Collagen I antibody, #ab21286; Col3, rabbit anti-Collagen III antibody, #ab7778, Abcam), and alpha-smooth muscle actin (α-SMA, rabbit anti-αSMA antibody, #ab5694, Abcam), and cancer stem cell markers like CD44 (mouse anti-CD44 antibody, #sc-7297, Santa Cruz Biotechnology) and CD 133 (rabbit anti-CD133 antibody, #NBP244250, Novus Biologicals). Fluorescence-conjugated secondary antibodies (Goat anti-Rabbit IgG (H+L) secondary antibody Alexa Fluor 488, #A11034, and goat anti-mouse IgG (H&L) secondary antibody Alexa 594, #A11032, Invitrogen) were incubated. After washing, tissue sections were mounted with DAPI for counterstaining.
Images for H&E were captured using a Leica DM750 microscope equipped with an EC3 digital camera and LAS EZ software (x40 to x100 magnification). IF images were obtained by a ZEISS fluorescence microscope using an AxioVison Rel 4.8 software (x200 to x640 magnification). Positive IF signals were quantified from the randomly selected sections at least five fields per sample using Image J 1.52 software (NIH, Bethesda, MD).
2.5. Real-time PCR, RNA-seq, and bioinformatic data analysis
Total RNAs from tissues and cells were isolated using TRI reagent (Sigma) as previously described [30]. cDNA was synthesized using a cDNA synthesis kit (Invitrogen). Real-time PCR analysis was performed using a SYBR Green qPCR kit (Applied Biosystems). Primer sequences are described in the Supplementary Table S1. RNA samples were also subjected to RNA-seq at the Center for Medical Genomics of Indiana University School of Medicine. The sequencing data were deposited to the Gene Expression Omnibus (GEO) database of the National Center for Biotechnology Information. The accession number is GSE133441. RNA-seq data analysis was performed as we previously described [17]. We also analyzed survival probability of liver cancer patients in correlation with SESN3 gene expression using the Cancer Genome Atlas (TCGA) dataset as previously described [31].
2.6. Protein analysis
Western blot, ELISA, and co-immunoprecipitation (Co-IP) were performed as previously described [18]. For Western blot analysis, protein samples were resolved on an SDS-PAGE gel and transferred to nitrocellulose membrane, followed by incubation with a primary antibody and an HRP-conjugated secondary antibody. Antibodies used in this work were the followings: Sesn3 (Rabbit pAb, PIPA522220, Fisher Scientific), Actinin (Mouse mAb, sc-17829, Santa Cruz Biotechnology), Pdgfrb (Rabbit mAb, #3169S, Cell Signaling Technology), phospho-Stat3 (Tyr705) (Rabbit mAb, #9145S, Cell Signaling Technology), Stat3 (Mouse mAb, #9139S, Cell Signaling Technology), phospho-Akt (Thr308) (Rabbit mAb, #4056S, Cell Signaling Technology), phospho-Akt (Ser473) (Rabbit mAb, #4058S, Cell Signaling Technology), and Akt (Rabbit mAb, #4685S, Cell Signaling Technology), To measure hepatic levels of IL-6, we used a commercial ELISA kit for mouse IL-6 (BD Biosciences) following the manufacturer’s instruction. Co-IP was performed in HEK 293 cells after co-transfection of indicated DNA constructs. IP and immunoblots were performed using tag antibodies: monoclonal anti-FLAG M2 affinity gel (Sigma) and HA tag antibody (Rabbit mAb, #3724S, Cell Signaling Technology).
2.7. Immunocytochemistry analysis
Human hepatoma cell line (Huh7) was cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin (Thermo Fisher). To monitor Gli2 localization, cells were seeded in a glass-bottom 35-mm dish at a density of 1 χ 105 cells/dish. After an overnight incubation, cells were transfected with different combinations of plasmids including vector, mouse Flag-Sesn3, and mouse HA-Gli2 (Addgene #37671, a gift from Dr. Philip Beachy [32]), in the presence or absence of a smoothened agonist (SAG, 100 nM, Sigma). In addition, CD44 was also analyzed using the similar imaging approach. After 24 hrs of incubation, cells were washed twice with PBS, and fixed with 4% of paraformaldehyde for 1 hr at RT, then washed three times with PBS. Blocking buffer (0.25% normal goat serum) was added to the dish. Primary antibodies (rabbit-HA tag antibody from Cell Signaling Technology, mouse-DYKDDDDK tag antibody (FG4R) from Invitrogen, or mouse-CD44 antibody, sc-7297, Santa Cruz Biotechnology) were incubated with the cells at 4 °C for overnight. Next day, primary antibodies were washed out with 0.05% PBST twice and PBS once, then secondary antibodies (Goat anti-Rabbit IgG (H+L) secondary antibody Alexa Fluor 488, #A11034, and goat anti-mouse IgG (H&L) goat anti-mouse IgG (H&L) secondary antibody Alexa 594, #A11032, Invitrogen) were incubated for 1 hr. Finally, cells were washed with PBST and PBS. Nuclei were stained with DAPI in the mounting medium. Fluorescence images were captured using a ZEISS fluorescence microscope using an AxioVison Rel 4.8 software (x640 magnification).
2.8. Statistical analysis
Data were presented as mean ± standard error (SEM). Comparisons between groups were performed using two-tailed unpaired t-test (GraphPad, La Jolla, CA). Differences with a p value < 0.05 were considered statistically significant.
3. Results
3.1. Sesn3 deficiency promotes carcinogen- and high-fat diet-induced HCC development
As little is known about the role of SESN3 in liver cancer, we analyzed correlation of hepatic SESN3 gene expression with survival probability in human liver cancer patients using the TCGA dataset. Our analysis revealed that patients with higher hepatic SESN3 mRNA levels had a tendency of better survival than those with lower SESN3 levels although the p value did not reach a statistical significance (Fig. 1A). To further investigate the pathophysiological function of Sesn3, we generated Sesn3 global knockout mice using a floxed Sesn3 strain and a CMV-Cre line. The gene knockout efficiency was analyzed by both Western blot and qPCR analysis (Fig. 1B–D). Hepatic Sesn3 protein and mRNA levels were decreased to approximately 21% and 7% of the WT liver levels, respectively, but the gene was not completely ablated due to an incomplete deletion by the CMV-Cre (Fig. 1B–D). To assess the role of Sesn3 in HCC development, we adopted a protocol of combinatory treatments with a single dose of DEN (25 mg/kg) at 2 weeks of age and chronic feeding of CD-HFD at 4 weeks of age (Fig. 1E). During the dietary treatment, Sesn3 KO mice were consistently heavier than control WT mice (Fig. 1F). At the end of the experiment, even though liver weights were not different, liver to body weight ratios were significantly lower in the Sesn3 KO group owing to heavier body weights (Suppl. Fig. 1A, B). We also examined other tissues including spleen, lung, and white adipose tissue, and only spleen was significantly increased in the Sesn3 KO mice (Suppl. Fig. 1C–H). Biochemical analysis of hepatic triglycerides and total cholesterol did not reveal any significant difference between WT and Sesn3 KO mice (Suppl. Fig. 1I, J).
Fig 1. Sesn3 knockout mice are more susceptible to carcinogen-induced hepatocellular carcinoma.
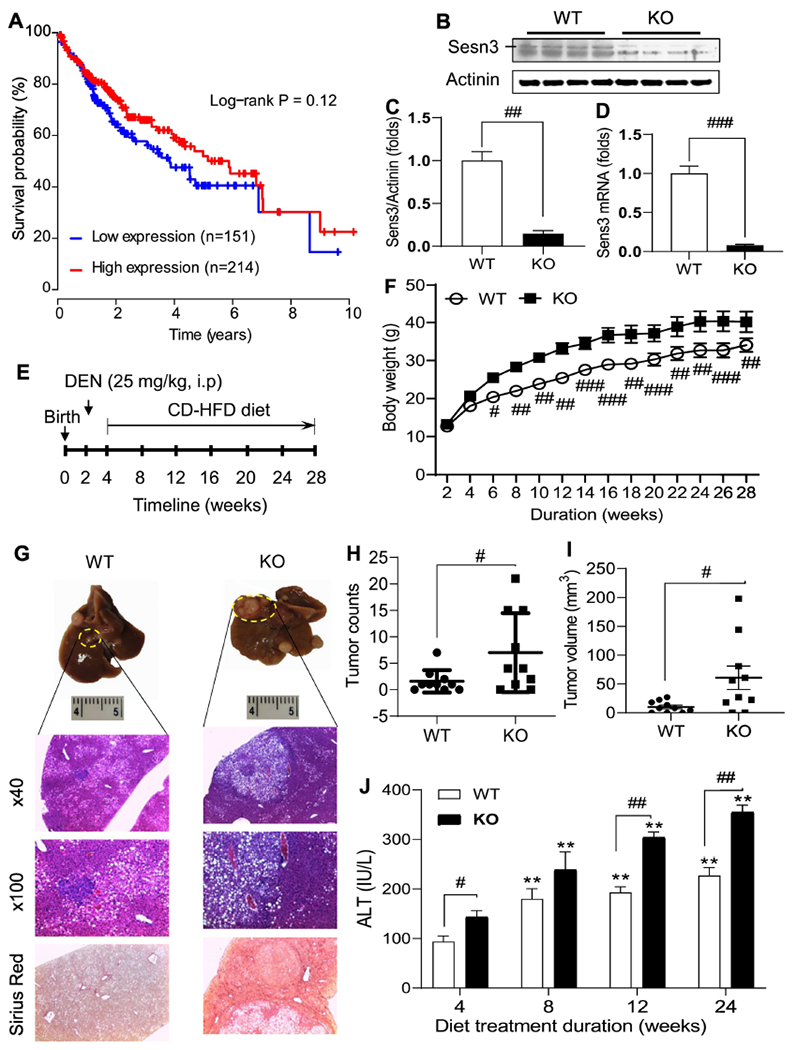
Kaplan-Meier survival curve was generated using the TCGA liver cancer dataset (A). Sesn3 KO in liver tissues was confirmed by immunoblot analysis (B), protein quantification (C, n=4), and real-time PCR mRNA analysis (D, n=4). Experimental scheme for the pre-clinical HCC model in Sesn3 KO mice (E). Mouse body weights during the experimental period (F, n=10). Macroscopic and microscopic analysis of tumor-bearing liver tissue (G). Tumor nodule counts (H, n=10) and tumor volume (I, n=10) in the mouse hepatic tissue. Serum ALT measurements at multiple time points during the HCC development (J, n=10). Data are presented as mean ± SEM. #p < 0.05, ##p < 0.01, and ###p < 0.001 for KO vs. WT. **p < 0.05 for comparisons vs. 4 weeks in panel J.
At the end of CD-HFD treatment, we observed a much more severe tumor phenotype in the Sesn3 KO mice than that in the WT mice according to both macroscopic and microscopic analyses (Fig. 1G). Total tumor counts and tumor volumes were both significantly increased in the Sesn3 KO mice compared to the WT group (Fig. 1H, I). Serum ALT levels were consistently higher in the Sesn3 KO mice throughout the CD-HFD treatment (Fig. 1J). Ascites were observed in both WT and Sesn3 KO groups. Whereas 10% WT mice had ascites, 40% Sesn3 KO mice had ascites (Suppl. Table S2). Additionally, metastasis from liver to other organs including lung, stomach, and intestine also happened in 30% Sesn3 KO mice, but no cancer metastasis was observed in the WT group (Suppl. Table S2).
3.2. Sesn3 is required to maintain normal hepatocellular status
To investigate what led to the tumorigenesis in the Sesn3 KO liver, we first performed immunofluorescence analysis of liver sections. Our data showed that Sesn3 KO caused a dramatic increase in ECM molecules including Acta2, Col1, and Col3 (Fig. 2A, B). The upregulated ECM genes like Acta2, Col1a1, and Pdgfrb in the KO livers were also confirmed by either real-time PCR or immunoblot analysis (Fig. 2C, D). We also characterized the HCC tumors by staining for a few common HCC and CSC markers. AFP, CK-19, and Arg1 were significantly elevated in the lesions of the Sesn3 KO liver. Interestingly, two CSC markers – Cd133 and Cd44 were also significantly increased in the Sesn3 KO liver sections (Fig. 2E, F).
Fig 2. Sesn3 deficiency promotes HCC by enhancing the production of ECM and induction of CSC markers.
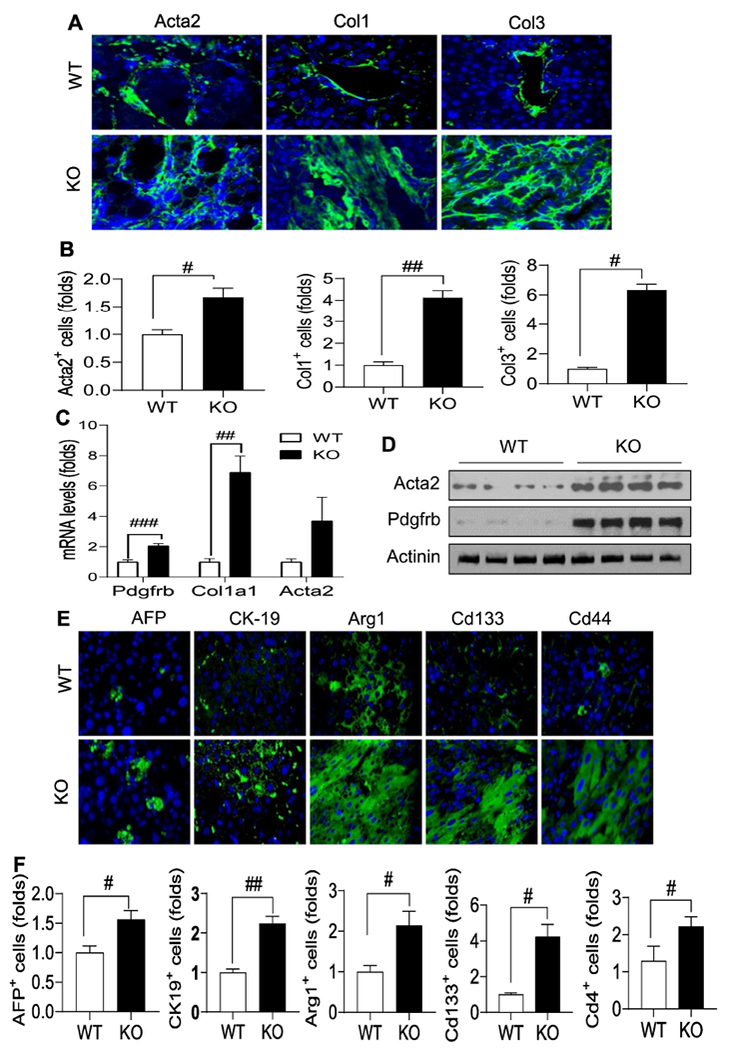
Representative immunofluorescence analysis of Acta2 (α-SMA), Col1, and Col3 (A). Analysis of fluorescence intensity for Acta2, Col1, and Col3 (B). Hepatic mRNA levels of Pdgfrb, Col1a1, and Acta2 (C). Immunoblot analysis of hepatic Acta2 and Pdgfrb (D). Representative immunofluorescence analysis of hepatic AFP, CK-19, Arg-1, Cd133, and Cd44 (E). Analysis of fluorescence intensity of hepatic AFP, CK-19, Argl, Cd133, and Cd44 (F). Data are expressed as mean ± SEM (n = 4 to 6 each group). #p < 0.05 and ##p < 0.01 for KO vs. WT. Immunofluorescence image analyses were performed using a fluorescence microscope (ZEISS, x200 magnification).
3.3. Sesn3 regulates cancer stemness partly through the control of Hedgehog signaling
To profile gene expression changes in the Sesn3 KO liver during the HCC development, we performed RNA-seq analysis. As compared to the WT livers, KO livers had 155 genes upregulated and 114 genes downregulated shown in a volcano plot (Fig. 3A and Suppl. Tables S3, S4). Among those upregulated genes were Col1a1, Col3a1, Cd44, Cd63, Sgk1, and Syk whereas fatty acid and cholesterol biosynthetic genes were downregulated (Fig. 3B and Suppl. Tables S3, S4). In the upregulated genes, acute-phase response, immune pathways, and integrin-mediated signaling pathway were among the top 10 biological processes (Fig. 3C). In the downregulated genes, lipid metabolic processes including cholesterol and fatty acid biosynthesis were among the top 10 biological processes (Fig. 3D).
Fig 3. Sesn3 deficiency induced multiple cancer signaling pathways including the hedgehog pathway.
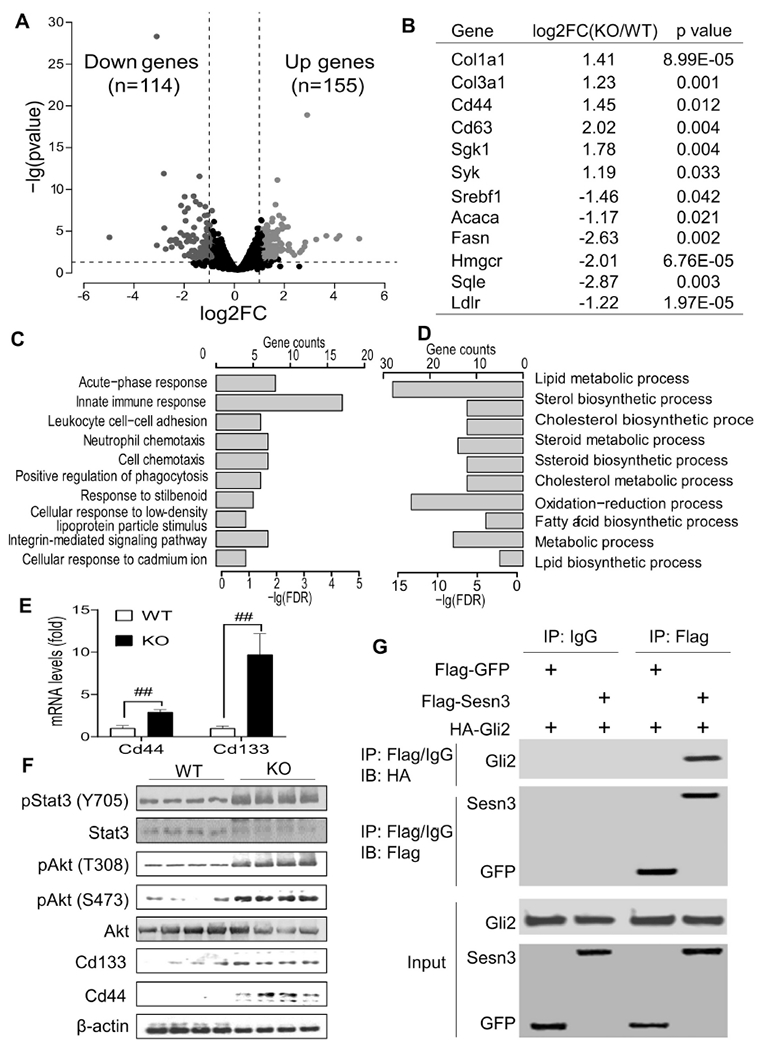
Differentially expressed genes (DEGs) from the RNA-seq analysis of WT and Sens3-KO livers were presented by the volcano plot (A). A selected list of DEGs were presented in the table (B). Top 10 biological processes were over-represented in the significantly upregulated (C) and downregulated (D) genes. Hepatic Cd44 and Cd133 mRNA analysis by real-time qPCR (E). Immunoblot analysis of cancer signaling-related proteins in the liver of WT and Sesn3 KO mice (F). Co-IP analysis of Sesn3 and Gli2 interaction in HEK 293T cells (G). Data are expressed as mean ± SEM (n=4 for the immunoblot analysis, n=10 for ELISA, n=6 for qPCR analysis, respectively). #p < 0.05 and ##p < 0.01 for KO vs. WT.
We were particularly interested in those CSC marker genes as they could play a significant role in the early stage of HCC development. Thus, we performed real-time PCR and immunoblot to confirm the upregulation of Cd133 and Cd44 genes in the liver tissue of Sesn3 KO mice. Indeed, our data showed that both Cd44 and Cd133 genes were highly induced in the Sesn3 KO livers (Fig. 3E, F). In addition, our protein analysis also revealed elevated levels of Stat3 and Akt phosphorylation (Fig. 3F), commonly activated in cancers by cytokines including IL-6 and growth factors, respectively. Serum IL-6 and hepatic IL-6 gene expression were both increased in the Sesn3 KO mice compared to WT counterparts (Suppl. Fig. 2A, B).
As the hedgehog signaling has been implicated in the regulation of CSCs [26], we tested whether Sesn3 interacts with Gli2. Our Co-IP analysis revealed that Sesn3 indeed interacted with Gli2 in HEK 293T cells (Fig. 3G). To further examine the functional implication of the Sesn3-Gli2 interaction, we performed immunofluorescence analysis in an HCC cell line Huh7 after transfection with Sesn3 and Gli2 plasmid DNA (Suppl. Fig. 3A, B). In the absence of a Smoothened agonist SAG, Gli2 had very little nuclear localization (Fig. 4A); however, upon stimulation with SAG, Gli2 was mostly localized in the nucleus of the empty vector-transfected cells whereas only a small fraction of Gli2 was in the nucleus of the Sesn3-transfected cells (Fig. 4B and Suppl. Fig. 3C), suggesting an inhibitory function of Sesn3 against Gli2 nuclear translocation. As the CD44 gene is known to be regulated by Gli2 [33, 34], we also observed that CD44 expression was significantly suppressed in the Sesn3-transfected cells compared to the empty vector-transfected cells (Fig. 4C). Real-time PCR analysis also showed that both GLI2 and CD44 mRNA levels were remarkably increased by the SAG treatment, but the SAG inductive effect was strongly suppressed by the Sesn3 overexpression (Fig. 4D, E), suggesting an inhibitory effect on the hedgehog signaling by Sesn3.
Fig 4. Sesn3 suppressed the Gli2 nuclear localization and CD44 gene expression.
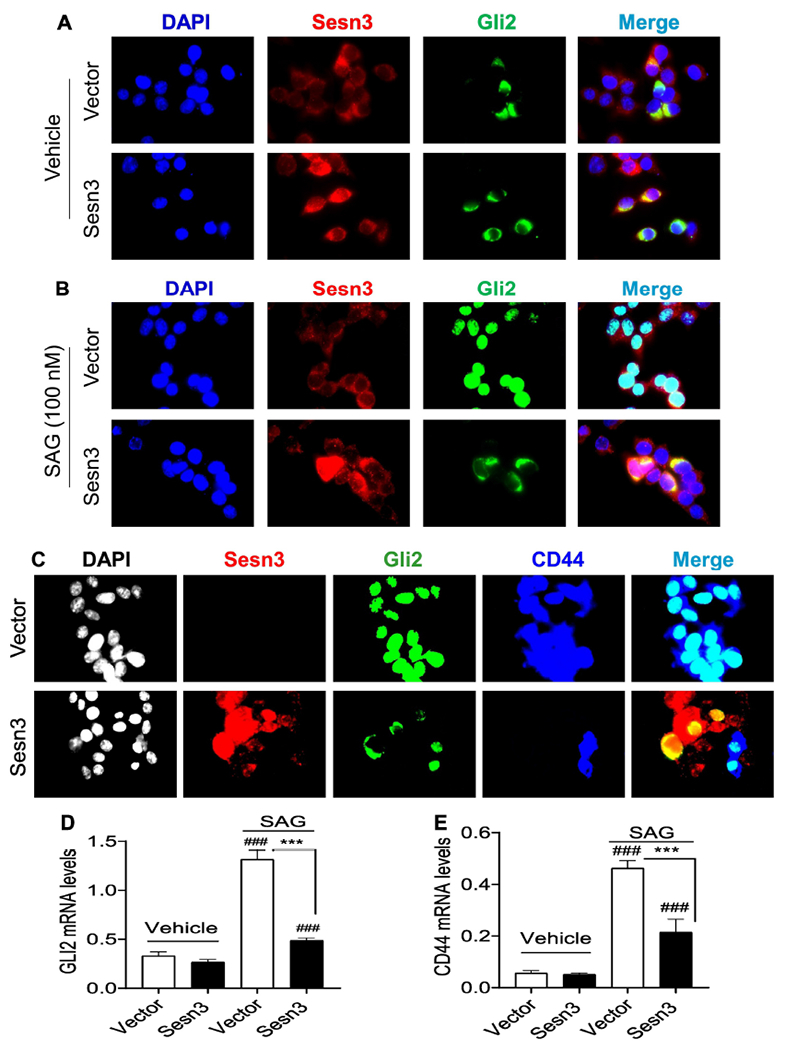
Representative immunofluorescent images of Sesn3 and Gli2 after vector, Flag-Sesn3, and HA-Gli2 plasmid DNAs were transfected to Huh7 cells in the absence or presence of 100 nM SAG. Sesn3 and Gli2 were detected using anti-Sesn3 or anti-HA antibody followed by Alexa 594 and Alexa 488 secondary antibody, respectively (A, B). Vector or Flag-Sesn3 plus HA-Gli2 were transfected to Huh7 cells in the presence of 100 nM SAG. Sesn3, Gli2, and CD44 were detected using anti-Flag, anti-HA, and anti-CD44 antibody followed by Alexa 594, Alex 488, and Cy5 secondary antibody, respectively (C). Fluorescence images were captured using a Zeiss fluorescence microscope at x640 magnification. Real-time PCR analysis of endogenous GLI2 and CD44 mRNAs in vector or Sesn3 transfected Huh7 cells treated with vehicle or SAG (100 nM) for 24 hours (D, E). Data are expressed as mean ± SEM (n = 3). ###p < 0.001 for vehicle vs. SAG for vector or Sesn3 transfection, and ***p < 0.001 for vector vs. Sesn3 transfection after the SAG treatment.
4. Discussion
In this work, we have reported that Sesn3 may serve as a tumor suppressor for the liver cancer development, especially HCC. To generate a useful preclinical HCC mouse model, we have taken advantage of the combinatory treatment with a single dose of DEN and chronic feeding with CD-HFD. This model has allowed us to investigate the molecular pathogenesis of HCC and the role of Sesn3 in the liver cancer development. From this study, we have revealed several Sesn3 functions in the primary HCC development. First, Sesn3 plays a significant role in ECM remodeling. Deficiency of Sesn3 leads to the accumulation of ECM and the impairment of tissue repair in the liver. Second, Sesn3 is required to restrict CSCs. When Sesn3 becomes deficient, the number of CSCs in the liver tumors has expanded significantly. Third, Sesn3 inhibits the IL6-Stat3 signaling. Fourth, Sesn3 inhibits Gli2 activity by blocking its nuclear translocation.
Although Sesn3 has been shown to regulate hepatic glucose and lipid metabolism via activation of AMPK and mTORC2, inhibition of mTORC1, and stimulation of autophagy [2, 14–16, 18, 35–38], the tumor suppressor role of Sesn3 in HCC has not been previously reported. The tumor phenotype in the Sesn3 KO mice represents the overall consequence of whole-body Sesn3 deficiency. Future study on cell-type-specific Sesn3 function will be needed to dissect the contribution of cell types involved in the HCC development, including hepatocytes, hepatic stellate cells, and Kupffer cells and other immune cells circulated to the liver. Previously, we have reported that Sesn3 activates Akt via mTORC2 in the mouse liver under the physiological conditions [18]. In this work, we have shown that Akt phosphorylation is increased the Sesn3 KO livers during the HCC development. This suggests that Sesn3 might play a different role in the regulation of Akt under tumorigenic conditions or different microenvironments. In addition, it is likely that the Sesn3 function in anti-oxidative stress and mTORC1 inhibition also plays a role in the tumor suppressor activity.
In general, hepatocytes have been thought to be the main source of early HCC lesions as they are often insulted by both endogenous and exogenous harmful molecules including carcinogens [39]. Tumor microenvironment also makes a significant contribution to the HCC development [39]. In our data, elevated ECM markers support the ECM effect on the tumor development. Upregulated Pdgfrb gene expression may play a significant role in the ECM production. With regard to cancer cell signaling, we have identified IL6-Stat3 and hedgehog pathways as potential contributors to the HCC tumorigenesis as they both have been implicated in the regulation of CSCs [21, 26, 40]. As Sesn3 is primarily a cytosolic protein, physical interaction between Sesn3 and Gli2 may hinder the Gli2 nuclear translocation. However, we do not know the exact fate of the Sesn3-Gli2 complex. They could rapidly degrade as we have observed low levels of Gli2 cytosolic staining. Those are intriguing questions to be answered in future studies.
In summary, we have demonstrated a tumor suppressor role of Sesn3 in a preclinical HCC mouse model. Our data suggest that Sesn3 deficiency makes the liver more susceptible to carcinogen and diet-induced HCC. Mechanistically, regulation of ECM and the IL6-Stat3 and hedgehog signaling underlies some of the Sesn3 tumor suppressor function.
Supplementary Material
Highlights.
Sesn3 protects liver from carcinogen-induced hepatocellular carcinoma.
Sesn3 deficiency leads to an increase in hepatic extracellular matrix.
Sesn3 inhibits the hedgehog signaling by blocking Gli2 nuclear translocation.
Sesn3 suppresses expression of the Cd44 gene, a cancer stem cell marker.
Acknowledgements
We thank Dr. Yang Zhang, Dr. Xiaolin Zhong, and Zhigang Fang for the technical and experimental assistance. We thank Dr. Guoli Dai for a kind gift of Huh7 cells.
Funding: This work was supported in part by the US National Institutes of Health (National Institute on Alcohol Abuse and Alcoholism R21AA024550 to Xiaocheng Charlie Dong; National Institute of Diabetes and Digestive and Kidney Diseases R01DK107682 to Suthat Liangpunsakul and Xiaocheng Charlie Dong and R01DK091592 to Xiaocheng Charlie Dong), Showalter Scholar Award (Xiaocheng Charlie Dong), Indiana Diabetes Research Center grant NIH P30DK097512, and Indiana Clinical and Translational Sciences Institute funded from the NIH NCATS CTSA UL1TR002529. Yunjian Liu was also supported by a visiting research scholarship from Jiangxi Province Department of Education.
Abbreviations:
- Acta2
smooth muscle actin alpha 2
- AFP
alpha fetoprotein
- Arg1
arginase 1
- CD44
CD44 antigen
- CD133
CD133 antigen
- CD-HFD
choline-deficient high-fat diet
- CK-19
cytokeratin 19
- Col1
collagen 1
- Col3
collagen 3
- CSC
cancer stem cell
- ECM
extracellular matrix
- Gli2
GLI family member zinc finger 2
- HCC
hepatocellular carcinoma
- IL-6
interleukin 6
- PDGFRB
platelet derived growth factor receptor beta
- SAG
smoothened agonist
- Sesn3
Sestrin 3
- Stat3
signal transducer and activator of transcription 3
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: None.
References
- [1].Ho A, Cho CS, Namkoong S, Cho US, Lee JH, Biochemical Basis of Sestrin Physiological Activities, Trends Biochem Sci, 41 (2016) 621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Dong XC, The potential of sestrins as therapeutic targets for diabetes, Expert Opin Ther Targets, 19 (2015) 1011–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Budanov AV, Lee JH, Karin M, Stressin’ Sestrins take an aging fight, EMBO molecular medicine, 2 (2010) 388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Budanov AV, Karin M, p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling, Cell, 134 (2008) 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM, Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD, Science, 304 (2004) 596–600. [DOI] [PubMed] [Google Scholar]
- [6].Jegal KH, Park SM, Cho SS, Byun SH, Ku SK, Kim SC, Ki SH, Cho IJ, Activating transcription factor 6-dependent sestrin 2 induction ameliorates ER stress-mediated liver injury, Biochim Biophys Acta Mol Cell Res, 1864 (2017) 1295–1307. [DOI] [PubMed] [Google Scholar]
- [7].Shi X, Doycheva DM, Xu L, Tang J, Yan M, Zhang JH, Sestrin2 induced by hypoxia inducible factor1 alpha protects the blood-brain barrier via inhibiting VEGF after severe hypoxic-ischemic injury in neonatal rats, Neurobiol Dis, 95 (2016) 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jegal KH, Ko HL, Park SM, Byun SH, Kang KW, Cho IJ, Kim SC, Eupatilin induces Sestrin2-dependent autophagy to prevent oxidative stress, Apoptosis, 21 (2016) 642–656. [DOI] [PubMed] [Google Scholar]
- [9].Garaeva AA, Kovaleva IE, Chumakov PM, Evstafieva AG, Mitochondrial dysfunction induces SESN2 gene expression through Activating Transcription Factor 4, Cell Cycle, 15 (2016) 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ding B, Parmigiani A, Divakaruni AS, Archer K, Murphy AN, Budanov AV, Sestrin2 is induced by glucose starvation via the unfolded protein response and protects cells from non-canonical necroptotic cell death, Scientific reports, 6 (2016) 22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Park HW, Park H, Ro SH, Jang I, Semple IA, Kim DN, Kim M, Nam M, Zhang D, Yin L, Lee JH, Hepatoprotective role of Sestrin2 against chronic ER stress, Nat Commun, 5 (2014) 4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bae SH, Sung SH, Oh SY, Lim JM, Lee SK, Park YN, Lee HE, Kang D, Rhee SG, Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage, Cell Metab, 17 (2013) 73–84. [DOI] [PubMed] [Google Scholar]
- [13].Shin BY, Jin SH, Cho IJ, Ki SH, Nrf2-ARE pathway regulates induction of Sestrin-2 expression, Free Radic Biol Med, 53 (2012) 834–841. [DOI] [PubMed] [Google Scholar]
- [14].Lee JH, Budanov AV, Talukdar S, Park EJ, Park HL, Park HW, Bandyopadhyay G, Li N, Aghajan M, Jang I, Wolfe AM, Perkins GA, Ellisman MH, Bier E, Scadeng M, Foretz M, Viollet B, Olefsky J, Karin M, Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3, Cell Metab, 16 (2012) 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nascimento EB, Osler ME, Zierath JR, Sestrin 3 regulation in type 2 diabetic patients and its influence on metabolism and differentiation in skeletal muscle, Am J Physiol Endocrinol Metab, 305 (2013) E1408–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kang X, Petyaykina K, Tao R, Xiong X, Dong XC, Liangpunsakul S, The inhibitory effect of ethanol on Sestrin3 in the pathogenesis of ethanol-induced liver injury, Am J Physiol Gastrointest Liver Physiol, 307 (2014) G58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Huang M, Kim HG, Zhong X, Dong C, Zhang B, Fang Z, Zhang Y, Lu X, Saxena R, Liu Y, Zhang C, Liangpunsakul S, Dong XC, Sesn3 protects against diet-induced nonalcoholic steatohepatitis in mice via suppression of the TGFbeta signal transduction, Hepatology, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tao R, Xiong X, Liangpunsakul S, Dong XC, Sestrin 3 Protein Enhances Hepatic Insulin Sensitivity by Direct Activation of the mTORC2-Akt Signaling, Diabetes, 64 (2015) 1211–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Global Burden of Disease Cancer C, Fitzmaurice C, Akinyemiju TF, Al Lami FH, Alam T, Alizadeh-Navaei R, Allen C, Alsharif U, Alvis-Guzman N, Amini E, Anderson BO, Aremu O, Artaman A, Asgedom SW, Assadi R, Atey TM, Avila-Burgos L, Awasthi A, Ba Saleem HO, Barac A, Bennett JR, Bensenor IM, Bhakta N, Brenner H, Cahuana-Hurtado L, Castaneda-Orjuela CA, Catala-Lopez F, Choi JJ, Christopher DJ, Chung SC, Curado MP, Dandona L, Dandona R, das Neves J, Dey S, Dharmaratne SD, Doku DT, Driscoll TR, Dubey M, Ebrahimi H, Edessa D, El-Khatib Z, Endries AY, Fischer F, Force LM, Foreman KJ, Gebrehiwot SW, Gopalani SV, Grosso G, Gupta R, Gyawali B, Hamadeh RR, Hamidi S, Harvey J, Hassen HY, Hay RJ, Hay SI, Heibati B, Hiluf MK, Horita N, Hosgood HD, Ilesanmi OS, Innos K, Islami F, Jakovljevic MB, Johnson SC, Jonas JB, Kasaeian A, Kassa TD, Khader YS, Khan EA, Khan G, Khang YH, Khosravi MH, Khubchandani J, Kopec JA, Kumar GA, Kutz M, Lad DP, Lafranconi A, Lan Q, Legesse Y, Leigh J, Linn S, Lunevicius R, Majeed A, Malekzadeh R, Malta DC, Mantovani LG, McMahon BJ, Meier T, Melaku YA, Melku M, Memiah P, Mendoza W, Meretoja TJ, Mezgebe HB, Miller TR, Mohammed S, Mokdad AH, Moosazadeh M, Moraga P, Mousavi SM, Nangia V, Nguyen CT, Nong VM, Ogbo FA, Olagunju AT, Pa M, Park EK, Patel T, Pereira DM, Pishgar F, Postma MJ, Pourmalek F, Qorbani M, Rafay A, Rawaf S, Rawaf DL, Roshandel G, Safiri S, Salimzadeh H, Sanabria JR, Santric Milicevic MM, Sartorius B, Satpathy M, Sepanlou SG, Shackelford KA, Shaikh MA, Sharif-Alhoseini M, She J, Shin MJ, Shiue I, Shrime MG, Sinke AH, Sisay M, Sligar A, Sufiyan MB, Sykes BL, Tabares-Seisdedos R, Tessema GA, Topor-Madry R, Tran TT, Tran BX, Ukwaja KN, Vlassov VV, Vollset SE, Weiderpass E, Williams HC, Yimer NB, Yonemoto N, Younis MZ, Murray CJL, Naghavi M, Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2016: A Systematic Analysis for the Global Burden of Disease Study, JAMA Oncol, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Forner A, Reig M, Bruix J, Hepatocellular carcinoma, Lancet, 391 (2018) 1301–1314. [DOI] [PubMed] [Google Scholar]
- [21].Blagotinsek K, Rozman D, Targeting Signalling Pathways in Hepatocellular Carcinoma, Curr Pharm Des, 23 (2017) 170–175. [DOI] [PubMed] [Google Scholar]
- [22].Grohmann M, Wiede F, Dodd GT, Gurzov EN, Ooi GJ, Butt T, Rasmiena AA, Kaur S, Gulati T, Goh PK, Treloar AE, Archer S, Brown WA, Muller M, Watt MJ, Ohara O, McLean CA, Tiganis T, Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC, Cell, 175 (2018) 1289–1306 e1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Pak E, Segal RA, Hedgehog Signal Transduction: Key Players, Oncogenic Drivers, and Cancer Therapy, Dev Cell, 38 (2016) 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Riordan JD, Feddersen CR, Tschida BR, Beckmann PJ, Keng VW, Linden MA, Amin K, Stipp CS, Largaespada DA, Dupuy AJ, Chronic liver injury alters driver mutation profiles in hepatocellular carcinoma in mice, Hepatology, 67 (2018) 924–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Prager BC, Xie Q, Bao S, Rich JN, Cancer Stem Cells: The Architects of the Tumor Ecosystem, Cell Stem Cell, 24 (2019) 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Saygin C, Matei D, Majeti R, Reizes O, Lathia JD, Targeting Cancer Stemness in the Clinic: From Hype to Hope, Cell Stem Cell, 24 (2019) 25–40. [DOI] [PubMed] [Google Scholar]
- [27].Schwenk F, Baron U, Rajewsky K, A cre-transgenic mouse strain for the ubiquitous deletion of loxPflanked gene segments including deletion in germ cells, Nucleic Acids Res, 23 (1995) 5080–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Faustino-Rocha A, Oliveira PA, Pinho-Oliveira J, Teixeira-Guedes C, Soares-Maia R, da Costa RG, Colaco B, Pires MJ, Colaco J, Ferreira R, Ginja M, Estimation of rat mammary tumor volume using caliper and ultrasonography measurements, Lab Anim (NY), 42 (2013) 217–224. [DOI] [PubMed] [Google Scholar]
- [29].Tao R, Xiong X, DePinho RA, Deng CX, Dong XC, Hepatic SREBP-2 and cholesterol biosynthesis are regulated by FoxO3 and Sirt6, J Lipid Res, 54 (2013) 2745–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tao R, Xiong X, DePinho RA, Deng CX, Dong XC, FoxO3 transcription factor and Sirt6 deacetylase regulate low density lipoprotein (LDL)-cholesterol homeostasis via control of the proprotein convertase subtilisin/kexin type 9 (Pcsk9) gene expression, J Biol Chem, 288 (2013) 29252–29259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zheng S, Luo X, Dong C, Zheng D, Xie J, Zhuge L, Sun Y, Chen H, A B7-CD28 family based signature demonstrates significantly different prognoses and tumor immune landscapes in lung adenocarcinoma, Int J Cancer, 143 (2018) 2592–2601. [DOI] [PubMed] [Google Scholar]
- [32].Kim J, Kato M, Beachy PA, Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus, Proc Natl Acad Sci U S A, 106 (2009) 21666–21671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yang N, Zhou TC, Lei XX, Wang C, Yan M, Wang ZF, Liu W, Wang J, Ming KH, Wang BC, Xu BL, Liu Q, Inhibition of Sonic Hedgehog Signaling Pathway by Thiazole Antibiotic Thiostrepton Attenuates the CD44+/CD24-Stem-Like Population and Sphere-Forming Capacity in Triple-Negative Breast Cancer, Cell Physiol Biochem, 38 (2016) 1157–1170. [DOI] [PubMed] [Google Scholar]
- [34].Wang JX, Zhou JF, Huang FK, Zhang L, He QL, Qian HY, Lai HL, GLI2 induces PDGFRB expression and modulates cancer stem cell properties of gastric cancer, European review for medical and pharmacological sciences, 21 (2017) 3857–3865. [PubMed] [Google Scholar]
- [35].Chantranupong L, Wolfson RL, Orozco JM, Saxton RA, Scaria SM, Bar-Peled L, Spooner E, Isasa M, Gygi SP, Sabatini DM, The Sestrins interact with GATOR2 to negatively regulate the amino-acidsensing pathway upstream of mTORC1, Cell reports, 9 (2014) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Parmigiani A, Nourbakhsh A, Ding B, Wang W, Kim YC, Akopiants K, Guan KL, Karin M, Budanov AV, Sestrins inhibit mTORC1 kinase activation through the GATOR complex, Cell reports, 9 (2014) 1281–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Peng M, Yin N, Li MO, Sestrins Function as Guanine Nucleotide Dissociation Inhibitors for Rag GTPases to Control mTORC1 Signaling, Cell, 159 (2014) 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Peng M, Yin N, Li MO, SZT2 dictates GATOR control of mTORC1 signalling, Nature, 543 (2017) 433–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M, From NASH to HCC: current concepts and future challenges, Nat Rev Gastroenterol Hepatol, 16 (2019) 411–428. [DOI] [PubMed] [Google Scholar]
- [40].Sonbol MB, Ahn DH, Bekaii-Saab T, Therapeutic Targeting Strategies of Cancer Stem Cells in Gastrointestinal Malignancies, Biomedicines, 7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.