

Abstract
ABCB5 is an ABC transporter that was shown to confer low-level multidrug resistance in cancer. In this study, we show that ABCB5 was mutated in 13.75% of the 640 melanoma samples analyzed. Besides nonsense mutations, two mutation hotspots were found in the ABCB5 protein, in the drug-binding pocket and the nucleotide-binding domains. Four mutations, which are representative of the mutation pattern, were selected. ATPase assays showed that these mutations resulted in a decrease in basal ATP hydrolysis by ABCB5. To select informative melanoma cell lines, mutational profiles of the clinical samples were further analyzed. This study showed mutations in the tumor suppressor CDKN2A gene and the NRAS oncogene in 62.5% and 75%, respectively of the samples that had mutations in the ABCB5 gene. No mutation was found in the tumor suppressor PTEN gene, whereas the activating V600E mutation in the BRAF oncogene was found in 25% of the samples with a mutated ABCB5 gene. Studies in four melanoma cell lines with various genetic backgrounds showed an increase in the proliferation and migration capacity of mutant ABCB5-expressing cells, suggesting that ABCB5 plays a role in the development of melanoma as a tumor suppressor gene.
INTRODUCTION
ABCB5, an ABC transporter closely related to the multidrug transporter ABCB1 (P-glycoprotein), is predominantly expressed in pigment-producing cells. Previous studies have suggested that it is a marker of melanoma-initiating cells (Schatton et al., 2008) and is linked to the development of multidrug resistance in melanoma (Chartrain et al., 2012; Frank et al., 2005). However, those studies were based on the expression of a transcript variant of the ABCB5 gene, namely ABCB5β, which encodes a truncated transporter (Chen et al., 2005; Frank et al., 2003). The functionality of this transporter is controversial and has yet to be directly demonstrated (Kawanobe et al., 2012; Keniya et al., 2014). More recently, the Sugimoto group reported the cloning of a transcript that encodes a typical full ABC transporter, which is composed of two transmembrane and two nucleotide-binding domains (Kawanobe et al., 2012). They showed the involvement of this more typical transporter in resistance to doxorubicin, paclitaxel, and docetaxel (Kawanobe et al., 2012).
ABC transporters have been extensively studied for their role in the mechanisms of multidrug resistance in cancer. Nonetheless, transcriptomic studies carried out to characterize these mechanisms in clinical samples indicated that these transporters may also have a major role in tumor biology (Gillet et al., 2011). Indeed, there is a growing body of evidence that their loss or inhibition have an impact on the malignant potential of cancer cells both in vitro and in vivo (Fletcher et al., 2016). ABCB5 mutations appeared to be common in melanoma (Krauthammer et al., 2015), and we hypothesized that this transporter contributes to the development of melanoma in addition to its role as a drug-efflux transporter.
Here, we report the results of the systematic analysis of the nature and function of mutations in the ABCB5 gene. Exome sequencing of the ABCB5 gene was performed in 153 human melanoma samples including matched normal DNA. These data were combined with mutational data from published studies, resulting in a data set generated from 640 human melanoma samples. Mutations were mapped in a three-dimensional predicted model of the protein, and two mutation hotspots in the ABCB5 protein were found. The impact of four mutations, representative of the mutation pattern, was further studied in four melanoma cell lines.
RESULTS AND DISCUSSION
Exome sequencing
To identify somatic mutations in ABCB5, we analyzed the coding regions of ABCB5 in 29 melanoma samples and corresponding normal DNA from a previously published study by Gartner et al. (2013). Our initial analysis of these samples showed six mutations, including three nonsynonymous mutations, two nonsense, and one synonymous mutation in five of our 29 samples (17%). The 5:1 nonsynonymous to synonymous ratio (20%) is considerably higher than the ratio predicted for nonselected passenger mutations of 2.5:1 (Sjoblom et al., 2006). Further sequencing of ABCB5 in an additional panel of 25 untreated samples showed four additional mutations in three different samples, including a third nonsense mutation. Taken together, these two analyses showed that ABCB5 contained 10 mutations (eight nonsynonymous) in eight of 54 samples (14.5%; P < 0.001) (Table 1). A validation was performed on six of these 10 mutations with digital PCR, which confirmed their presence at the RNA level. Thus, melanoma cells with mutant ABCB5 also contain wild-type mRNAs, resulting in coexpression of mutant and wild-type ABCB5 (data not shown).
Table 1.
ABCB5 mutations in untreated samples (results of Whole Exome, Whole Genome, and Sanger sequencing in a total of 54 untreated melanoma samples shown)
| Mutations | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | Mutation | Mutation Type | Het/Homo | Type of Screen | SIFT Score | Nonsynonymous | Synonymous | Total | CDKN2A Status | PTEN Status | NRAS Status | BRAF Status |
| 17T | Q187* | Nonsynonymous | Het | Genome | N/A | 438 | 246 | 684 | WT | WT | Q61K | WT |
| 55T | E520D | Nonsynonymous | Het | Exome | 0.21 | 1,555 | 835 | 2,390 | WT | WT | WT | WT |
| 44T | R587* | Nonsynonymous | Het | Prevalence | N/A | N/A | N/A | N/A | 2:64L>L/P, 83H>H/Y | 9:102350G>A (−2 intronic) | Q61K | WT |
| 83T | V827I | Nonsynonymous | Het | Prevalence | 1 | N/A | N/A | N/A | Deleted | WT | S39F | V600E |
| 12T | I828I | Synonymous | Het | Genome | 1 | 1,061 | 533 | 1,594 | Deleted | WT | Q61K | WT |
| 83T | S830F | Nonsynonymous | Het | Prevalence | 0 | N/A | N/A | N/A | Deleted | WT | S39F | V600E |
| 105T | L840L | Synonymous | Het | Prevalence | 0.7 | N/A | N/A | N/A | Deleted | WT | WT | V600E |
| 32T | S1184P | Nonsynonymous | Het | Genome | 0 | 1091 | 556 | 1,647 | WT | WT | WT | L597Q |
| 24T | S1091F | Nonsynonymous | Het | Exome | 0.01 | 415 | 213 | 628 | Deleted | WT | Q61R | WT |
| 55T | Q1098* | Nonsynonymous | Het | Exome | N/A | 1,555 | 835 | 2,390 | WT | WT | WT | WT |
Abbreviations: Het, heterogenous; Homo, homogenous; N/A, not applicable; WT, wild type.
We extended the study to an additional cohort containing 99 melanoma samples, which showed 14 additional mutations (nine nonsynonymous and five synonymous). Of these 24 total mutations observed, 17 were nonsynonymous, and four were nonsense mutations affecting 9.8% of our samples (15 of 153).
We also reviewed the mutational data from published studies (Berger et al., 2012; Hodis et al., 2012; Nikolaev et al., 2011; Stark et al., 2011; Wei et al., 2011), which were combined with exome data from The Cancer Genome Atlas, resulting in 487 published melanoma samples. Analysis of ABCB5 in these 487 published melanoma samples showed 85 coding mutations in 73 of 487 samples, or 14.9%. The mutations include 60 nonsynonymous mutations, one splice site mutation, six nonsense mutations, and 18 silent mutations (see Supplementary Table S1 online). In summary, we show that the gene ABCB5 was mutated in 13.75% of the 640 melanoma samples analyzed.
Effect of the mutations on the transporter activity
Mutations were localized on a two- and three-dimensional predicted models constructed based on the sequence alignment of full-length ABCB5 to mouse ABCB1, for which experimental structures are known (Figure 1a and b) (Esser et al., 2017; Li et al., 2014). Two mutation hotspots were found in the predicted drug binding pocket and the nucleotide binding domains of the ABCB5 protein. The impact of four mutations was further investigated based on their localization in the mutation hotspots and the likely deleterious effect on the transporter activity (Table 1). Besides a nonsense mutation (Q187*), a nonsynonymous mutation was located in the predicted drug binding pocket (S830F), and two were in the second nucleotide binding domain (S1091F, S1184P) (Figure 1c and d).
Figure 1. Molecular model of ABCB5.

An atomic model of ABCB5 was constructed based on the sequence alignment of full-length ABCB5 to mouse ABCB1 or P-glycoprotein, for which experimental structures are known. (a) Schematic of the ABCB5 protein topology with the conserved domains indicated as blocks, including transmembrane domains (TMD) and nucleotide binding domains (NBD). Somatic alterations are represented by arrowheads. A total of 71 different alterations were identified, and for clarity, only some amino acid changes were indicated. Underlined alterations were functionally assessed. Red triangles represent deleterious alterations, blue triangles represent missense mutations, the orange triangle represents splice site, and purple triangles represent silent mutations. (b) Ribbon diagram of the full-length ABCB5 model, with the N-terminal transmembrane domain, or TMD1 in red and the C-terminal TMD2 in cyan. The N-terminal NBD1 and the C-terminal NBD2 are shown in yellow and green, respectively. Residues where mutations were found are displayed as ball models. (c) Residues S830 in the TM8 is shown in magenta, and residues S1091 and S1184 in NBD2 are colored in orange. (d) Magnified view of NBD2 showing detailed structural environment in the vicinity of the two mutation sites in ball models in orange. The Walker A and B motifs are colored light orange and pink, respectively. The signature motif is in purple. The two intracellular helical motifs are shown in cyan and red, respectively.
The effect of the mutations on the transporter activity was assessed by an ATPase assay. All mutants were expressed in High Five insect cells at the same level as wild-type WT protein (Figure 2a). The ATPase assays confirmed that these mutations resulted in a decrease in basal ATP hydrolysis by ABCB5 (Figure 2b). Based on these data, we hypothesized that ABCB5 may play a role in the tumor biology, perhaps as a tumor suppressor.
Figure 2. ATPase activity of ABCB5 WT and mutants.
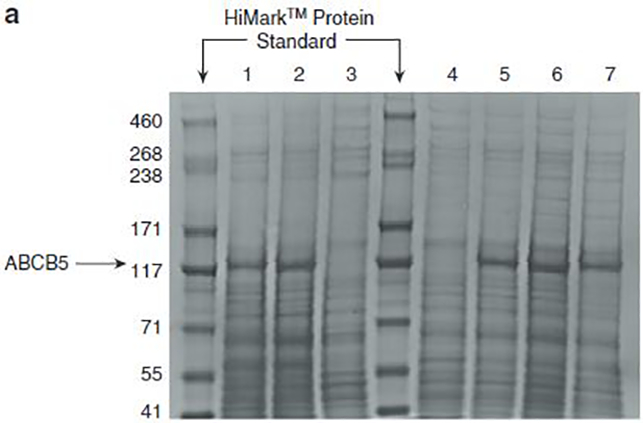
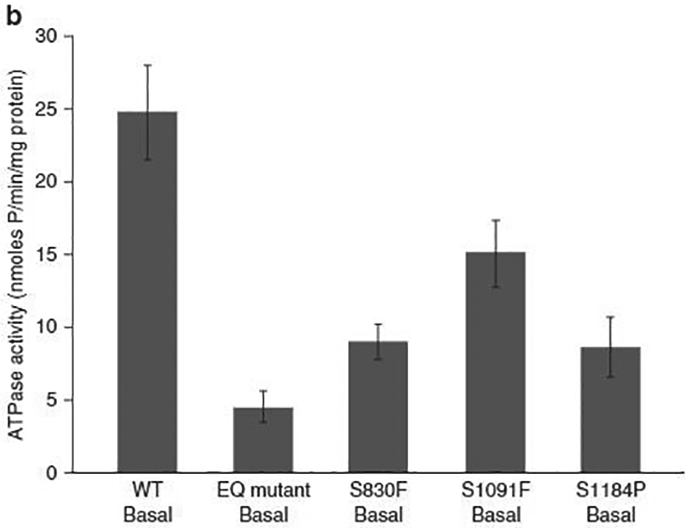
(a) ABCB5 WT and mutants were expressed in High Five insect cells, and 30 mg isolated crude membrane proteins were run in a Nupage Tris-acetate gel, along with a HiMark protein standard. All mutants were expressed in High Five insect cells to the same level as WT protein. (1) ABCB5 WT. (2) ABCB5 E1181Q mutant (nonfunctional transporter, negative control). (3) Crude membranes (negative control). (4) ABCB5-FLAG Q187*. (5) ABCB5-FLAG S830F. (6) ABCB5-FLAG S1091F. (7) ABCB5-FLAG S1184P. (b) ATPase activity of ABCB5 WT and ABCB5 mutants (E1181Q, S830F, S1091F, and S1184P) in High Five cell crude membranes was measured by endpoint Pi assay. WT and mutant ABCB5-specific ATPase activities were recorded as beryllium fluoride-sensitive ATPase activity. (Error bars denote standard deviation; n= 3). For the same expression level, we observed a decrease in the ATPase activity for the cells overexpressing ABCB5 mutants compared with the cells overexpressing ABCB5 WT. EQ, E1181Q; WT, wild type.
Impact of the mutations on the proliferation ability of human melanoma cell lines
To explore the deleterious effect of the mutations in vitro, we wanted to determine the genetic background of the ABCB5 mutated melanoma. Further studies on the first set of 54 human melanoma samples analyzed showed mutations in the tumor suppressor CDKN2A gene and the NRAS oncogene in 62.5% and 75% of the samples, respectively, that had mutations in the ABCB5 gene. No mutation was found in the tumor suppressor PTEN gene, whereas the activating V600E mutation in the BRAF oncogene was found in 25% of the samples with a mutated ABCB5 gene.
We have chosen the 17T and 63T human melanoma cell lines, both of which harbor the activated mutant NRAS Q61K. The 17T cell line also harbors the heterozygous nonsense mutant ABCB5 Q187*, and WT BRAF, PTEN, and CDKN2A. The 63T cell line carries WT ABCB5, WT BRAF, nonsense mutant PTEN R130*, and knockout mutant CDKN2A del. Ex 1, 2, 3. SK-Mel-28 and A375 human melanoma cell lines were also chosen to test the impact of the ABCB5 mutants in a mutated BRAF genetic background. Both cell lines carry WT ABCB5, WT NRAS, and mutated BRAF. Furthermore, SK-Mel-28 cells carry WT CDKN2A, while A375 cells carry a partial deletion of CDKN2A (Poliseno et al., 2011).
Clones of human melanoma cell lines 17T and 63T were produced that stably overexpressed either WT ABCB5 or mutants of ABCB5 (Q187*, S830F, S1091F, S1184P). To investigate the possible effects of ABCB5 on melanoma cell growth, in vitro proliferation on plastic was first examined. All 17T clones overexpressing ABCB5 mutants have a statistically highly significant increased proliferation rate compared with the WT ABCB5 clone (Figure 3a). The impact of the ABCB5 mutations is lower in the 63T cells except for S1184P, for which the increase of the proliferation rate is highly statistically significant compared with the parental WT 63T cells (Figure 3b).
Figure 3. Effects of ABCB5 mutations on proliferation of melanoma cells.

(a) Proliferation rates of 17T clones were assayed over 3 days. All 17T clones overexpressing ABCB5 mutants had a significant increase of their proliferation rate compared with the ABCB5 WT-overexpressing clone for each time point. (b) Proliferation rates of 63T ABCB5 clones were measured 3 and 4 days after seeding. The proliferation rate of the S1184P mutant was found to be very highly statistically significant only when compared with the parental WT 63T cells. *P < 0.05, **P < 0.01, ***P <0.001,****P < 0.0001. n = 4. Values are the mean ± standard error of the mean. WT, wild type.
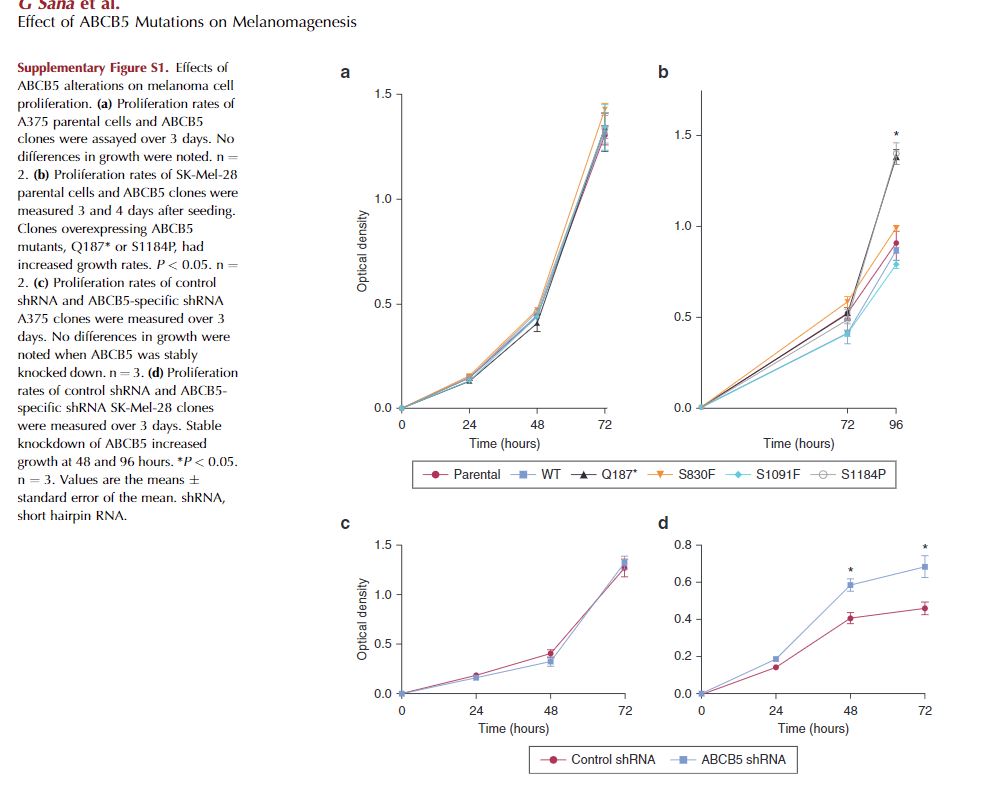
The proliferation rates of A375 WT ABCB5 and mutant clones were identical to parental cells (see Supplementary Figure S1a online). SK-Mel-28 clones overexpressing either the Q187* or S1184P mutations had significantly increased proliferation rates compared with parental cells and the other clones (see Supplementary Figure S1b). Additionally, the Q187* and S1184P clones displayed a reduced adhesion capacity and did not fully attach to plastic until 48 hours after seeding. Stable knockdown of ABCB5 in A375 cells had no effect on cell proliferation (see Supplementary Figure S1c). In contrast, stable knockdown of ABCB5 in SK-Mel-28 cells resulted in a significantly increased proliferation rate (see Supplementary Figure S1d).
A proliferation assay indicates a difference in terms of the proliferation capacity of the cells, but it has the disadvantage in that the proliferation of the cells is influenced by their ability to adhere to plastic. Anchorage-independent growth, which is considered a hallmark of carcinogenesis, was next assayed with a standard soft agar assay (Figure 4). For both cell lines, colony number was significantly higher for cells expressing ABCB5 mutants compared with the WT ABCB5-expressing cells. Again for this assay, we observed a greater effect for the 17T cell line (P < 0.0001 for each mutation except for the mutation S1091F, for which P < 0.001) (Figure 4a) than for the 63T cell line (P < 0.0001 for S830F and S1091F, P < 0.05 for the mutations Q187* and S1184P) (Figure 4b).
Figure 4. Effects of ABCB5 mutations on anchorage-independent growth of melanoma cells.
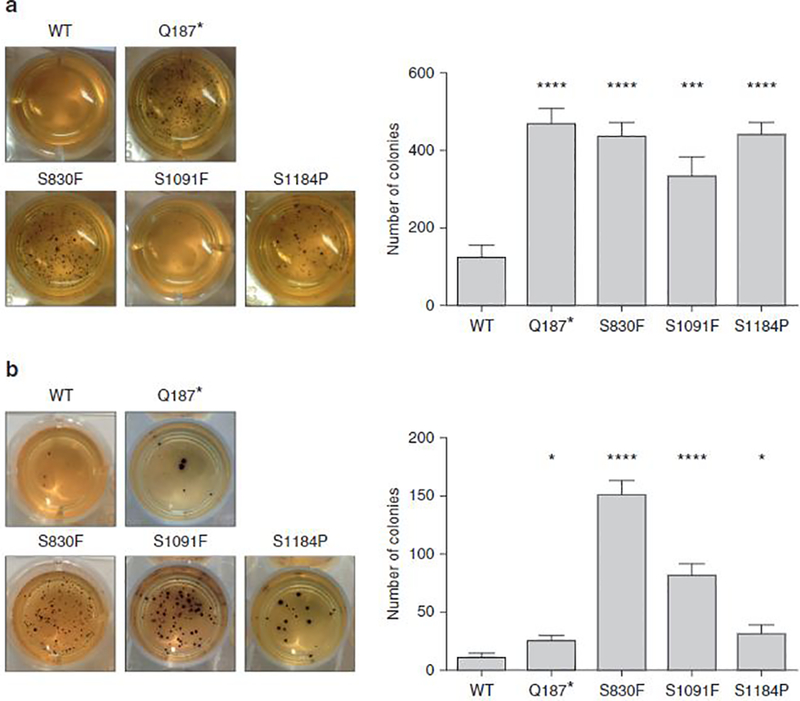
(a) All 17T ABCB5 mutant clones had a highly significant increase in number of colonies compared with the 17T ABCB5 WT clone. (b) The 63T mutant clones showed a highly significant increase in number of colonies only for the mutations S830F and S1091F, whereas the effect was lower for the Q187* and S1184P mutations. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. n = 3. Values are the mean ± standard error of the mean. WT, wild type.
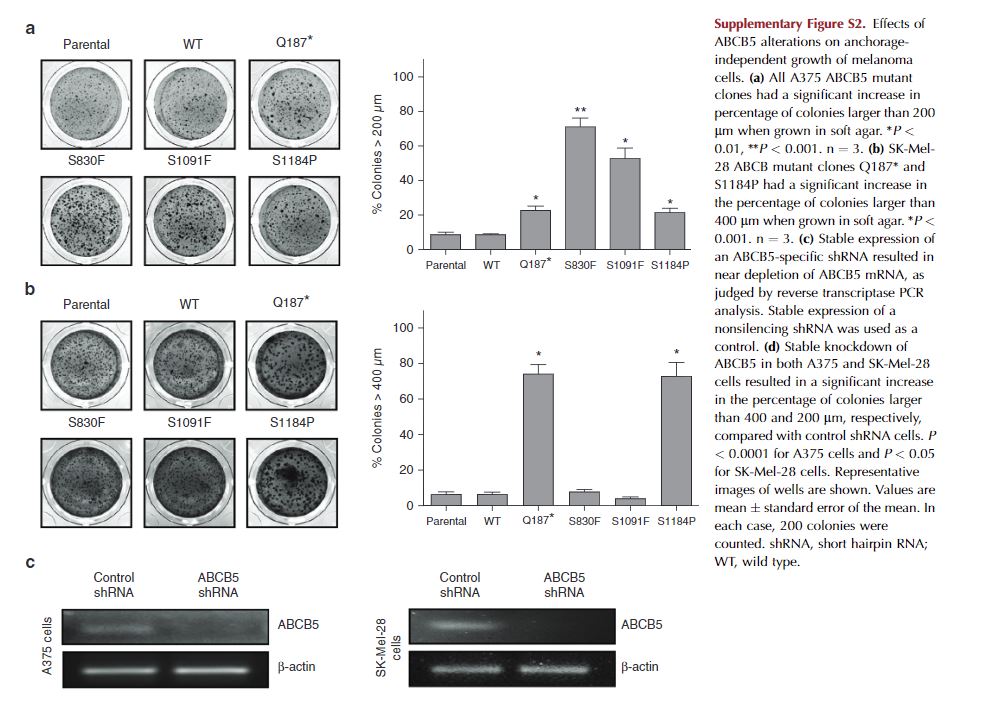
In both the SK-Mel-28 and A375 cell lines examined, colony numbers were not different, but colony sizes varied. In the A375 cell line, all four ABCB5 mutant clones formed significantly larger colonies compared with parental cells (see Supplementary Figure S2a online). When we examined SK-Mel-28 cells, only the Q187* and S1184P mutant clones had significantly larger colonies compared with parental cells (see Supplementary Figure S2b). In addition to overexpression of various ABCB5 clones, A375 and SK-Mel-28 clonal cell lines were produced that stably expressed either a nontargeting short hairpin RNA (shRNA) or an ABCB5-specific shRNA (see Supplementary Figure S2c). In both cell lines, stable knockdown of ABCB5 resulted in significantly larger colonies in soft agar, corroborating the overexpression data (see Supplementary Figure S2d).
Overall, we observed that the impact of the ABCB5 mutant expression on the proliferation capacities of melanoma cells is greater in cell lines that carry the activated mutant NRAS Q61K compared with those carrying the activated mutant BRAF. Furthermore, the impact of ABCB5 mutant expression was greater in 17T cells compared with 63T. This can be explained by the expression of heterozygous nonsense mutant ABCB5 Q187* in 17 cells, whereas 63T cells carry a homozygous WT ABCB5.
Impact of the mutations on development of metastases
A possible role of ABCB5 mutants in melanoma cell migration and invasion was then examined. For this, cells were seeded in a Boyden chamber in serum-free medium and were allowed to migrate through the pores to the other side of the semipermeable membrane in a serum-enriched medium compartment. In the invasion assay, cells must be able to digest the three-dimensional Matrigel (Corning Inc, Corning, NY) layer located at the bottom of the Boyden chamber. The cells were stained and counted after a 24-hour period. The 17T ABCB5 clones showed a higher migration ability for the mutations Q187* and S1091F, whereas the cells over-expressing ABCB5 S830F were not able to migrate through the pores of the Boyden chamber membrane (Figure 5a). The migration ability of 63T mutant ABCB5 clones was also affected and was significant for the mutations S830F and S1091F (Figure 5b). Both 17T (Figure 5c) and 63T cells (Figure 5d) were not able to invade through the Matrigel layer, except for the 63T S830F mutant.
Figure 5. Effects of ABCB5 mutations on migratory and invasive abilities of melanoma cells.
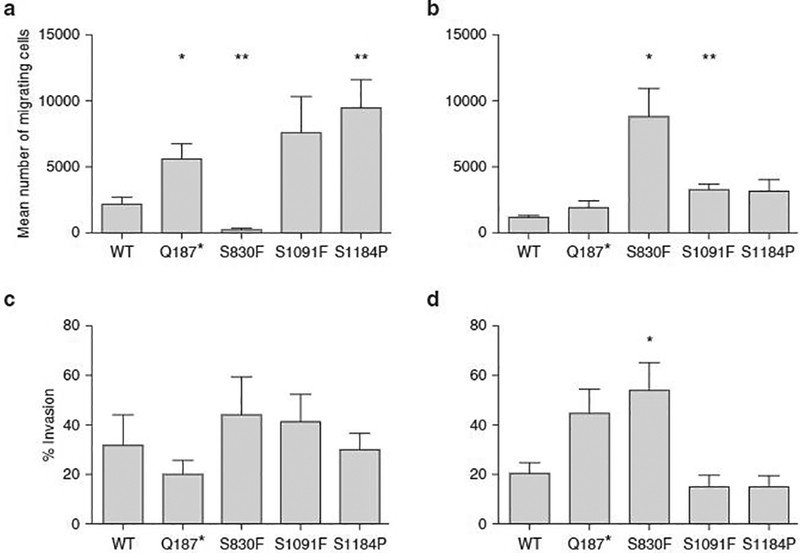
(a) 17T ABCB5 Q187*, S1091F, and S1184P mutant clones showed a higher migration ability, whereas the ABCB5 S830F mutant clone was not able to migrate from the upper side of the Boyden chamber to the lower one. (b) The migration ability of 63T ABCB5 mutant clones was also affected and was significant for the mutations S830F and S1091F. (c) The invasive ability of 17T ABCB5 WT and mutant clones was assessed, but no significant increase in invasion was observed. The P values (Student t test) are 0.4187 for Q187*, 0.5458 for S830F, 0.5856 for S1091F, and 0.8927 for the mutation S1184P. (d) The invasive ability of 63T ABCB5 WT and mutant clones was assessed, but no significant increase in invasion was observed, except for the S830F mutation. The P values (Student t test) are 0.0885 for Q187*, 0.0458 for S830F, 0.4291 for S1091F, and 0.4215 for S1184P. *P <0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. n = 3. Values are the mean ± standard error of the mean.
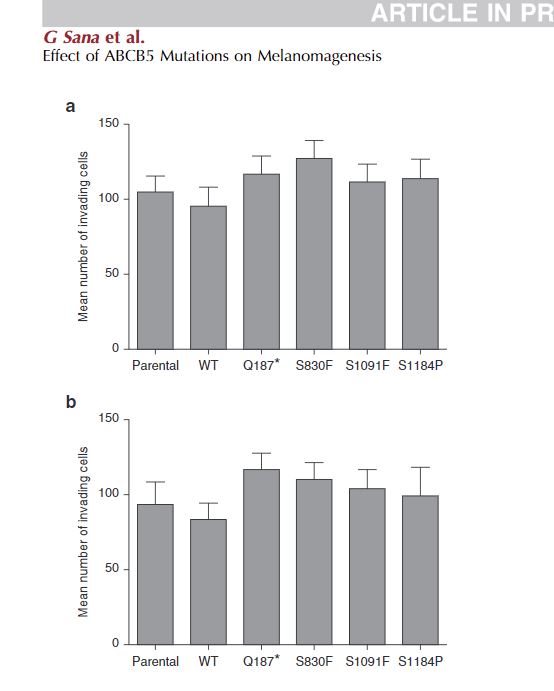
In both A375 (see Supplementary Figure S3a online) and SK-Mel-28 (see Supplementary Figure S3b) cell lines, there was a slight trend toward increased invasion of the ABCB5 mutant clones, along with a trend toward reduced invasion in WT ABCB5 clones. For both the A375 and SK-Mel-28 cell lines, stable knockdown of ABCB5 resulted in a significant increase in invasive capacity (see Supplementary Figure S3c).
In summary, the combination of genetic, biochemical, and cellular data presented here suggests that loss of ABCB5 gene function accelerates the development of melanoma. Indeed, in experiments performed in four separate melanoma cell lines, either mutation of ABCB5 or loss of ABCB5 expression resulted in increased proliferative capacities, which were higher in cells with a mutated NRAS genetic background compared with a mutated BRAF background. However, the invasive capacities of ABCB5 mutant clones was observed only in mutated BRAF cell lines. In an NRAS background, ABCB5 mutant clones were able to migrate only through the membrane pores. The ABCB5 mutations are presumed to reduce ABCB5 transporter activity in the melanoma lines, with the possibility that some mutations may have dominant negative effects. Overall, these data indicate that ABCB5 is a potential tumor suppressor in melanoma. The physiological role of ABCB5 remains to be unraveled. However, we can speculate that once this transporter is no longer functional, there are physiological changes that affect cell metabolism and/or mutagenesis related to toxic reactive oxygen species (Kondo et al., 2015). BRAF inhibitors alone or in combination with MEK inhibitors exert immunomodulatory effects on the tumor and its microenvironment (Deken et al., 2016; Hu-Lieskovan et al., 2015). It would be interesting to assess combined BRAF-MEK inhibition in vitro, and also with PD-1 blockade in vivo, in mice recapitulating these genetic alterations.
MATERIALS AND METHODS
Tumor tissues
All human samples were obtained after written informed patient consent was given. Tissue and melanoma cell lines used for the Discovery and Prevalence Screen in this study were described previously (Palavalli et al., 2009). Tissues used for validation set 1 were fresh frozen melanoma tumors obtained from the National Cancer Institute Surgery Branch (see Supplementary Table S2 online). Tissue was collected at the NCI Medical Center, under institutional review board protocols. DNA was isolated from enriched macrodissected tumor isolates as previously described (see http://www.riedlab.nci.nih.gov). Tissue processing and storage were previously described by Morente et al. (2006). Tissues used for validation set 2 of melanomas were obtained from optimum cutting temperature-embedded frozen clinical specimens from the Melanoma Informatics, Tissue Resource, and Pathology Core (MelCore) at The University of Texas MD Anderson Cancer Center under institutional review board-approved protocols. DNA isolation from the tumor-enriched isolates has been described previously (Davies et al., 2009). Tissue was further collected, and cell lines were established at Queensland Institute of Medical Research (41 stage III and 46 stage IV (American Joint Committee on Cancer) early-passage metastatic melanoma cell lines). All cell lines were established as described previously (Castellano et al., 1997; Dutton-Regester et al., 2012; Pavey et al., 2004) with written informed patient consent under a protocol approved by the Queensland Institute of Medical Research Human Research Ethics Committee.
DNA extraction
DNA was extracted with a DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany), following the manufacturer’s instructions. DNA was eluted in 35 μl of elution buffer. DNA measurements were made with a ND-1000 UV-Vis spectrophotometer from NanoDrop Technologies (Wilmington, DE).
Whole-exome and genome sequencing
Whole-exome and genome sequencing was described previously (Gartner et al., 2013), and results have been deposited in the dbSNP, ClinVar database with the batch identification 1057273.
Sanger sequencing
PCR and sequencing primers were designed with Primer 3 (http://www-genome.wi.mit.edu/cgibin/primer/primer3_www.cgi) and synthesized by Integrated DNA Technologies (Coralville, IA) (see Supplementary Table S3 online). PCR amplification was performed as previously described (Samuels et al., 2004), and PCR products were purified with exonuclease (Epicentre Biotechnologies, Madison, WI) and shrimp alkaline phosphatase (USB Corporation, Cleveland, OH). Products were purified with rehydrated Sephadex G-50 powder (GE Healthcare, Piscataway, NJ), and cycle sequencing was carried out with a BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems, Foster City, CA). Sequence data were collected on an ABI3730xl (Applied Biosystems). Sequence traces of the secondary screen were analyzed with the Mutation Surveyor software package (SoftGenetics, State College, PA).
ATPase assay
ATPase activity of ABCB5 WT and mutated ABCB5 (E1181Q, S830F, S1091F, and S1184P) in High Five cell crude membranes was measured by an endpoint inorganic phosphate assay as described by Ambudkar (1998). The E1181Q mutation is found in the second nucleotide binding domain and renders the ABCB5 transporter nonfunctional. This mutant was produced as a negative control. A FLAG-tag was inserted in the first extracellular loop. ABCB5- and mutated ABCB5-specific ATPase activities were recorded as beryllium fluoride-sensitive ATPase activity as described by Ambudkar (1998).
Cell culture
The human melanoma cell lines 17T and 63T were cultured in RPMI media supplemented with 10% fetal clone serum, 1% penicillin/streptomycin, 1% L-Glut and HEPES (Life Technologies, Carlsbad, CA). The human melanoma cell lines A375 and SK-Mel-28 were cultured in RPMI media supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Cells were maintained at 37 °C in a humidified atmosphere of 5% CO2.
Lentiviral ABCB5 WT and mutated ABCB5 production
Lentiviral ABCB5-FLAG tag plasmid DNAs (ABCB5-FLAG tag WT - M01, ABCB5-FLAG Q187* - M02, ABCB5-FLAG S830F - M03, ABCB5-FLAG S1091F - M04, and ABCB5-FLAG S1184P - M05) were supplied by the Protein Expression Laboratory Cloning and Optimization Group (Frederick National Laboratory for Cancer Research, Frederick, MD). The HEK293T cells were cotransfected with the lentiviral envelope plasmid (pMD2.G, Addgene number 12259), the lentiviral packaging plasmid (psPAX2, Addgene number 12260), and one of the five lentiviral ABCB5-FLAG tag plasmid to generate lentivirus particles. The melanoma cell lines were infected with these lentivirus particles to overexpress either WT ABCB5 or one of the four ABCB5 mutats. Cells were selected by using 5 μg/ml puromycin. Sequencing was performed to assess the presence of WT or mutated ABCB5.
Proliferation assay
A total of 3,000 cells per well of A375 cells, 4,000 cells per well of SK-Mel-28 cells, or 4,500 cells per well of 17T and 63T cells were seeded into 96-well plates in complete RPMI media. At 24-hour time points (0, 24, 48, and 72 hours), growth media was removed and replaced with complete RPMI media solution containing a working concentration of 0.5 mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and incubated for 3 hours. Media with MTT was removed, and cells were solubilized in DMSO. Absorbance was measured at 570 nm on a SpectraMax i3 plate reader (Molecular Devices, San Jose, CA).
Soft agar colony formation assay
In each well of a 24-well plate, 4,500 cells (A375, SK-Mel-28) and 4,000 cells (17T, 63T) for all cell line clones were suspended in0.33% Bacto-Agar (Sigma-Aldrich, St. Louis, MO), diluted in complete RPMI media. This layer was plated on top of a layer of 0.5% Bacto-Agar, diluted in complete RPMI media. Plates were maintained at 37 °C in a humidified atmosphere of 5% CO2 for 3 weeks. Colonies were stained with 2 mg/ml MTT for 3 hours and then counted.
Transwell migration and Matrigel invasion assay
A total of 5 × 104 cells for A375 and SK-Mel-28 cell lines and clones and 12.5 × 103 cells for 17T and 63T cell lines were suspended in serum-free RPMI and pipetted into a Transwell insert (BD Biosciences, San Jose, CA) to assess their migration ability. The insert was placed into a well of a 24-well plate containing complete RPMI media and incubated for 24 hours at 37 °C in a humidified atmosphere of 5% CO2. The same manipulation was performed for the invasion assay with a BioCat Matrigel Invasion chamber (Corning Inc). The inserts were washed with phosphate-buffered saline (to remove the nonmigrating and noninvading cells from the interior of the inserts) and stained with a Hema3 staining kit (Thermo Fisher Scientific, Waltham, MA). The migrating and invading cells were counted under a light microscope, and the percent invasion was calculated by the ratio between the mean number of invading cells and the mean number of migrating cells.
Statistical analysis
Data analysis was done by an unpaired Student t test or Welch t test when samples had unequal variances. Values are the means ± standard error of the mean. P values less than 0.05 were considered statistically significant. Statistics and graphing were done with Prism software (GraphPad, La Jolla, CA).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKOWLEDGMENTS
This work was supported by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, National Cancer Institute. YS is supported by the Israel Science Foundation (grant no. 696/17), the European Research Council under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 770854), and the Minerva Foundation Grant. The authors acknowledge receiving the melanoma cells from the Biospecimen Core of the Yale SPORE in Skin Cancer, funded by the National Cancer Institute, US National Institutes of Health, under award number 1 P50 CA121974. The authors also thank George Leiman for editorial assistance, Robert Rutledge (LCB, National Cancer Institute, National Institutes of Health, Bethesda, MD) and Michel Jadot (Molecular Physiology Research Unit, NAmur Research Institute for LIfe Sciences, University of Namur, Belgium) for critical comments and discussion. Finally, the authors are grateful to Laurent Duvivier and Florence Gaudray for their technical support on the migration/invasion assay.
Abbreviation:
- shRNA
short hairpin RNA
Footnotes
CONFLICT OF INTEREST
C-HL is an employee and shareholder of Natera. The other authors state no conflict of interest.
SUPPLEMENTARY MATERIALS
Supplementary material is linked to the online version of the paper at www.jidonline.org, and at https://doi.org/10.1016/j.jid.2019.01.036.
REFERENCES
- Ambudkar SV. Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzymol 1998;292: 504–14. [DOI] [PubMed] [Google Scholar]
- Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 2012;485(7399):502–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano M, Pollock PM, Walters MK, Sparrow LE, Down LM, Gabrielli BG, et al. CDKN2A/p16 is inactivated in most melanoma cell lines. Cancer Res 1997;57:4868–75. [PubMed] [Google Scholar]
- Chartrain M, Riond J, Stennevin A, Vandenberghe I, Gomes B, Lamant L, et al. Melanoma chemotherapy leads to the selection of ABCB5-expressing cells. PLoS One 2012;7(5):e36762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KG, Szakács G, Annereau J-P, Rouzaud F, Liang X-J, Valencia JC, et al. Principal expression of two mRNA isoforms (ABCB 5α and ABCB 5β) of the ATP-binding cassette transporter gene ABCB 5 in melanoma cells and melanocytes. Pigment Cell Res 2005;18:102–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies MA, Stemke-Hale K, Lin E, Tellez C, Deng W, Gopal YN, et al. Integrated molecular and clinical analysis of AKT activation in metastatic melanoma. Clin Cancer Res 2009;15:7538–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deken MA, Gadiot J, Jordanova ES, Lacroix R, van Gool M, Kroon P, et al. Targeting the MAPK and PI3K pathways in combination with PD1 blockade in melanoma. Oncoimmunology 2016;5(12):e1238557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutton-Regester K, Aoude LG, Nancarrow DJ, Stark MS, O’Connor L, Lanagan C, et al. Identification of TFG (TRK-fused gene) as a putative metastatic melanoma tumor suppressor gene. Genes Chromosomes Cancer 2012;51:452–61. [DOI] [PubMed] [Google Scholar]
- Esser L, Zhou F, Pluchino KM, Shiloach J, Ma J, Tang WK, et al. Structures of the multidrug transporter P-glycoprotein reveal asymmetric ATP binding and the mechanism of polyspecificity. J Biol Chem 2017;292:446–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher JI, Williams RT, Henderson MJ, Norris MD, Haber M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist Updat 2016;26:1–9. [DOI] [PubMed] [Google Scholar]
- Frank NY, Margaryan A, Huang Y, Schatton T, Waaga-Gasser AM, Gasser M, et al. ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res 2005;65:4320–33. [DOI] [PubMed] [Google Scholar]
- Frank NY, Pendse SS, Lapchak PH, Margaryan A, Shlain D, Doeing C, et al. Regulation of progenitor cell fusion by ABCB5 P-glycoprotein, a novel human ATP-binding cassette transporter. J Biol Chem 2003;278:47156–65. [DOI] [PubMed] [Google Scholar]
- Gartner JJ, Parker SC, Prickett TD, Dutton-Regester K, Stitzel ML, Lin JC, et al. Whole-genome sequencing identifies a recurrent functional synonymous mutation in melanoma. Proc Natl Acad Sci USA 2013;110:13481–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet JP, Wang J, Calcagno AM, Green LJ, Varma S, Bunkholt Elstrand M, et al. Clinical relevance of multidrug resistance gene expression in ovarian serous carcinoma effusions. Mol Pharm 2011;8:2080–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A landscape of driver mutations in melanoma. Cell 2012;150:251–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu-Lieskovan S, Mok S, Homet Moreno B, Tsoi J, Robert L, Goedert L, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAFV600E melanoma. Sci Transl Med 2015;7(279):279ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawanobe T, Kogure S, Nakamura S, Sato M, Katayama K, Mitsuhashi J, et al. Expression of human ABCB5 confers resistance to taxanes and anthracyclines. Biochem Biophys Res Commun 2012;418:736–41. [DOI] [PubMed] [Google Scholar]
- Keniya MV, Holmes AR, Niimi M, Lamping E, Gillet JP, Gottesman MM, et al. Drug resistance is conferred on the model yeast Saccharomyces cerevisiae by expression of full-length melanoma-associated human ATP-binding cassette transporter ABCB5. Mol Pharm 2014;11:3452–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S, Hongama K, Hanaya K, Yoshida R, Kawanobe T, Katayama K, et al. Upregulation of cellular glutathione levels in human ABCB5- and murine Abcb5-transfected cells. BMC Pharmacol Toxicol 2015;16:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauthammer M, Kong Y, Bacchiocchi A, Evans P, Pornputtapong N, Wu C, et al. Exome sequencing identifies recurrent mutations in NF1 and RAS-opathy genes in sun-exposed melanomas. Nat Genet 2015;47:996–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Jaimes KF, Aller SG. Refined structures of mouse P-glycoprotein. ProteinSci 2014;23:34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morente MM, Mager R, Alonso S, Pezzella F, Spatz A, Knox K, et al. TuBaFrost 2: standardising tissue collection and quality control procedures for a European virtual frozen tissue bank network. Eur J Cancer 2006;42: 2684–91. [DOI] [PubMed] [Google Scholar]
- Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet 2011;44:133e9. [DOI] [PubMed] [Google Scholar]
- Palavalli LH, Prickett TD, Wunderlich JR, Wei X, Burrell AS, Porter-Gill P, et al. Analysis of the matrix metalloproteinase family reveals that MMP8 is often mutated in melanoma. Nat Genet 2009;41:518–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavey S, Johansson P, Packer L, Taylor J, Stark M, Pollock PM, et al. Microarray expression profiling in melanoma reveals a BRAF mutation signature. Oncogene 2004;23:4060–7. [DOI] [PubMed] [Google Scholar]
- Poliseno L, Haimovic A, Christos PJ, Vega y Saenz de Miera EC, Shapiro R, Pavlick A, et al. Deletion of PTENP1 pseudogene in human melanoma. J Invest Dermatol 2011;131:2497–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004;304(5670):554. [DOI] [PubMed] [Google Scholar]
- Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M, et al. Identification of cells initiating human melanomas. Nature 2008;451(7176):345–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006;314(5797):268–74. [DOI] [PubMed] [Google Scholar]
- Stark MS, Woods SL, Gartside MG, Bonazzi VF, Dutton-Regester K, Aoude LG, et al. Frequent somatic mutations in MAP3K5 and MAP3K9 in metastatic melanoma identified by exome sequencing. Nat Genet 2011;44: 165–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Walia V, Lin JC, Teer JK, Prickett TD, Gartner J, et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat Genet 2011;43:442–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.