

Abstract
Podocytes have limited ability to recover from injury. Here, we demonstrate that increased mitochondrial biogenesis, to meet the metabolic and energy demand of a cell, accelerates podocyte recovery from injury. Analysis of events induced during podocyte injury and recovery showed marked upregulation of PGC-1α, a transcriptional co-activator of mitochondrial biogenesis, and key components of the mitochondrial electron transport chain. To evaluate our hypothesis that increasing mitochondrial biogenesis enhanced podocyte recovery from injury, we treated injured podocytes with formoterol, a potent, specific, and long-acting β2-adrenergic receptor agonist that induces mitochondrial biogenesis in-vitro and in-vivo. Formoterol increased mitochondrial biogenesis, restored mitochondrial morphology and the injury-induced changes to the organization of the actin cytoskeleton in podocytes. Importantly, β2-adrenergic receptors were found to be present on podocyte membranes. Their knockdown attenuated formoterol-induced mitochondrial biogenesis. To determine the potential clinical relevance of these findings mouse models of acute nephrotoxic serum nephritis and chronic (adriamycin) glomerulopathy were used. Mice were treated with formoterol post-injury when glomerular dysfunction was established. Strikingly, formoterol accelerated the recovery of glomerular function by reducing proteinuria and ameliorating kidney pathology. Furthermore, formoterol treatment reduced cellular apoptosis and increased the expression of the mitochondrial biogenesis marker PGC-1α and multiple electron transport chain proteins. Thus, our results support β2-adrenergic receptors as novel therapeutic targets and formoterol as a therapeutic compound for treating podocytopathies.
Keywords: albuminuria, focal segmental glomerulosclerosis, glomerulus, glomerulonephritis, podocyte
INTRODUCTION
Glomerular function is highly dependent on specialized cells known as podocytes. Podocytes are critical components of the glomerular filtration system and their loss results in progressive renal failure1,2. Podocytes are terminally differentiated cells and consist of a unique architecture, consisting of primary, secondary and tertiary interdigitating foot processes that surround glomerular capillaries2. While podocyte injury is a common denominator in many glomerular diseases, including focal and segmental glomerulosclerosis (FSGS)2,3, specific drugs that can restore injury-induced loss of podocyte structure and function remain unknown. Although much of the research in the field of podocyte biology has focused on preventing injury to podocytes, this does not represent the majority of clinical situations in which podocyte injury is identified after the injury has occurred.
Many glomerular diseases including FSGS affect podocyte structure and function, which is characterized by scarring in scattered regions of glomeruli4,5 and is frequently associated with nephrotic syndrome (NS) in adults and children5,6. Although multiple etiologies drive FSGS and include primary/idiopathic or secondary causes, the dysfunction of podocytes is the central feature in all cases7,8. Immunosuppressive therapy is the most common treatment option for FSGS patients, though not all patients respond to such therapy9–11. Moreover, growing concerns indicate that immunosuppressive agents need further scrutiny due to their side effects and the inability to target immunological mechanisms that may affect podocyte health9,11,12. With increased understanding of FSGS pathogenesis and other glomerular diseases, it is likely that a defined target-based treatment focused on an underlying mechanism will result into novel therapies for treating FSGS patients.
Mitochondrial dysfunction is one of the major mechanisms involved in podocyte injury and death13–15, where energy depletion may lead to irreversible cellular injury16,17. Many patients with mitochondrial mutations including MTTL118, COQ219, COQ620 and PDSS221 present with FSGS symptoms. Mice lacking genes such as mTORC122, ROCK123 and Arg524, which indirectly affect mitochondrial function, also show FSGS-like symptoms. Interestingly, podocyte specific knockout of Pdss2 in mice results in mitochondrial Co-Q deficiency and exhibits proteinuria and podocyte foot process effacement25. Deletions in mitochondrial DNA have been noted previously in ~60% of primary FSGS patients26. Collectively, these findings suggest that mitochondrial dysfunction participates in the pathogenesis of podocyte injury and regulating podocyte energy metabolism through mitochondrial biogenesis (MB), whose involvement in podocyte injury was recently shown27,28, may stimulate podocyte recovery from injury.
Studies have shown that MB is an adaptive response to acute injuries and is necessary to meet the increased metabolic and energy demands during the process of organ recovery29. In a mouse model of acute kidney injury, Jesinkey et al.,30, reported that treatment with the potent, long-acting and specific β2-AR agonist formoterol promoted MB and recovery of renal function when administered after injury. Our recent analysis of mRNA profiling of injury-induced genes and pathways in podocytes revealed several genes that participate in MB. In this study, we show that MB induced by the β2-AR agonist formoterol accelerates recovery of podocytes from injury and restores lost glomerular filtration function in mice models of podocytopathy.
RESULTS
Understanding the events that contribute to podocyte recovery from injury are key to developing new therapies. Since mitochondrial dysfunction was noted in various injury models (figure S1A–F and figure S2A) and several reports now show that MB plays an integral part in cellular recovery30,31, we hypothesized that recovery of podocytes from injury can be mediated through increased MB.
MB is induced during podocyte recovery from injury.
To test whether MB is induced during recovery, mitochondrial copy number, a marker of MB, was evaluated in immortalized cultured human podocytes32,33 that were injured with PS (protamine sulphate) or NTS (nephrotoxic serum) and were subsequently assessed for recovery in serum supplemented media as described earlier33,34. Injury induced changes to podocyte actin cytoskeleton and localization of Neph1 were largely restored (Figure 1A–C and figure S2B&C). Importantly, mtDNA copy number showed enhancement during the recovery phase (Figure 1D). Furthermore, qPCR analysis showed an increase in mitochondrial components (Figure 1E). Collectively, these results suggest that MB participates in the recovery of podocytes from injury.
Figure 1: MB is induced during recovery of podocytes from injury:

(A) Podocytes treated with PS showed actin cytoskeleton (Green) disorganization with accumulation of actin stress fibers at the cell periphery, and reduced Neph1 (Red) at cell-cell junctions. Recovery induced with supplementation of serum (serum induced recovery) restored actin cytoskeleton organization and localization of Neph1 at the cell junctions. Scale bar 20μm. (B) The quantitative analysis showed significant increase in the number of healthy podocytes (>40%) during recovery. (C) The quantitative assessment of Neph1 localization showed significant relocalization of Neph1 (~40%) at the cell membrane during recovery. (D) Analysis of mtDNA copy number showed significant enhancement during recovery of podocytes from injuries with NTS or PS. (E) Upregulation of β2-AR2 and various other mitochondrial components as evaluated by qPCR. All experiments were performed at least in triplicates. Data are presented in mean±SEM and p-values were calculated using a 2-tailed t-test. *P≤0.05, **P≤0.01, control vs. injury; aP≤0.05, PS vs. PS+recovery; bP≤0.05 NTS vs. NTS+recovery.
Formoterol enhanced MB in podocytes.
We further hypothesized that pharmacological-induced increase of MB may contribute to enhanced recovery of podocytes from injury. To test this hypothesis, we first examined MB using the maximal oxygen consumption rate (OCR) in podocytes. FCCP (carbonyl cyanide-p-trifluoromethoxyphenylhydrazone), is known to induce maximal OCR through the ETC and is a marker of MB35. FCCP increased OCR in differentiated cultured podocytes (p<0.05) compared to the basal OCR (Figure 2A). To determine if maximal OCR levels can be further elevated, differentiated podocytes were treated with formoterol (30 nM), a potent inducer of MB30,36, for 24 h and FCCP-induced OCR was determined. Formoterol further increased the maximum OCR, when compared to vehicle controls (Figure 2B), suggesting that formoterol induced MB in podocytes. To confirm formoterol-induced MB in podocytes, RNA was isolated from podocytes treated with formoterol (30 nM) for 24 h and qPCR (quantitative PCR) was performed to analyze various genes involved in mitochondrial function. Expression of PGC-1α, ATPase-6, mitochondrially encoded cytochrome b (MT-CYTB), and cytochrome c oxidase I (CO-I) were all increased (Figure 2C). Overall, these results provide evidence of pharmacological induction of MB by formoterol in podocytes.
Figure 2: Formoterol induced MB in podocytes.

(A & B) FCCP uncoupled maximum OCR measurements were made in cultured human podocytes treated with 30 nM formoterol for 24 h and showed significantly enhanced mitochondrial oxygen consumption. *P≤0.05, 2-tailed t-test. (C) qPCR analysis of podocytes treated with either control vehicle or 30nM formoterol showed relative fold change in the expression of PGC-1a, ATPase 6, CYTB, FN1 and CO1 genes that are involved in MB. All experiments were performed at least in triplicates. Data are presented in mean±SEM and p-values were calculated using a 2-tailed t-test. *P≤0.05, **P≤0.01, ***p≤001 vs. control. (D) Rat glomeruli were stained with β2-AR (Green) and Synaptopodin (Red) antibodies (arrows mark the presence of β2-AR in podocytes). Scale bar 10μm (E) Cultured human podocytes immuno-stained with phalloidin (Green) and β2-AR (Red) showed membrane and nuclear staining of β2-AR. Scale bar 20μm (F) Western blot analysis of β2-AR expression in podocyte cell lysate. (G) β2-AR knockdown (β2-AR-KD) podocytes were generated using five different sets of shRNA and the qPCR analysis showed maximal (~80%) knockdown using shRNA # 83, which was subsequently used for all experiments. (H) mtDNA copy number analysis showed induction of mtDNA by formoterol, whereas, β2-AR-KD blunted formoterol-induced increase in mtDNA copy number. Data are presented in mean±SEM. *P<0.05, control-KD+vehicle vs control-KD+formoterol; #p<0.05, control-KD+vehicle vs β2-AR-KD+ vehicle.
β2-adrenergic receptor (β2-AR) knockdown attenuated MB in cultured podocytes.
To determine if formoterol mediates its affect through β2-AR, we first evaluated expression of β2-AR in glomeruli and human podocytes. Immunostaining analysis showed that β2-AR was expressed in podocytes and its localization at the membrane was noted (Figure 2D–E), which is consistent with previous reports37,38. Interestingly, nuclear staining for β2-AR was also observed (Figure 2F), which has not been reported in other cell types. The presence of β2-AR was further confirmed using western blot analysis of podocyte cell lysates (Figure 2F).
Next, we decreased β2-AR in human podocytes using specific shRNA (Figure 2G). mtDNA copy number analysis in β2-AR knockdown podocytes showed decreased mtDNA and that the loss of β2-AR attenuated formoterol induced increase in mtDNA copy number (Figure 2H). Finally, we determined if the pharmacological β2-AR antagonist ICI 118,551 blocked formoterol-induced PGC-1α expression36. The β2-AR inhibitor blocked the formoterol-induced increase in PGC-1α expression (Figure S2D). Collectively, these results show that formoterol mediates its effect through β2-AR.
Formoterol restored mitochondrial damage and enhanced recovery of podocytes from injury.
We hypothesized formoterol-induced MB would lead to enhanced recovery of podocytes after injury. Therefore, we first evaluated the effect of formoterol on mitochondrial morphology, which is indicative of mitochondrial damage23, in podocytes. Using Mito Tracker staining, elongated/filamentous mitochondria were observed under basal conditions and were fragmented and granular in response to injury PS treatment (Figure 3A). Treatment with formoterol largely restored the filamentous structure of mitochondria. Quantitative analysis confirmed ~70% increase in elongated mitochondria upon formoterol treatment, when compared to vehicle treated injured podocytes (Figure 3B). To further support these observations OCR and ATP levels were estimated, which showed an increased OCR and ATP production in formoterol-treated injured podocytes (Figure 3C–E).
Figure 3: Formoterol restored injury-induced changes to mitochondrial morphology, OCR and ATP:

(A) MitoTracker staining was used to assess mitochondrial morphology in cultured human podocytes. Elongated mitochondria were observed in healthy untreated podocytes (arrows), but shortened fragmented mitochondria were noted in podocytes injured by PS. Treatment with formoterol restored elongated morphology to a larger extent. Scale bar 10μm. (B) The quantitative analysis showed more than 70% recovery of mitochondrial morphology upon formoterol treatment of PS-injured cells. (C–D) Formoterol treatment along with PS-injury significantly enhanced the basal as well as uncoupled OCR in cultured podocytes. *P≤0.05 vs. untreated control (2-tailed t-test). (E) ATP measurements showed significant enhancement of ATP production in PS+formoterol treated cells. *P≤0.05 vs. untreated control.
We determined whether this restoration of mitochondrial morphology and increase in MB by formoterol translates to enhanced recovery of podocytes from injury. Two in-vitro podocyte injury models, nephrotoxic serum (NTS) and protamine sulphate (PS) that induce cytoskeletal damages that are characterized by a gradual increase in stress fibers39,40 moving to cell periphery as the injury progresses and mislocalization of slit-diaphragm protein Neph1 were employed39,41,42. Similar changes to cell morphology were noted when cultured podocytes were treated with either NTS or PS (Figure 4). After 8h following establishment of injury, the injured cells were treated with formoterol (30 nM) and recovery of actin cytoskeleton organization and the restoration of Neph1 at cell membrane/junctions was analyzed (Figure 4A–B). The quantitative assessment revealed that formoterol treated NTS and PS injured podocytes resulted in increased recovery of ~27% and ~40% respectively in actin cytoskeleton organization, when compared to vehicle control (Figure 4B&E). The results further showed nearly 100% recovery of Neph1 at the membrane following formoterol treatment (Figure 4C&F) of NTS and PS injured podocytes. Collectively, these results provide evidence for a protective or reparative role of formoterol in enhancing the recovery of podocytes from injury.
Figure 4: Formoterol treatment enhanced recovery of podocytes from PS and NTS induced injuries:

(A) Podocytes treated with NTS show damaged actin cytoskeleton (Green) with accumulation of actin stress fibers at cell periphery, and loss of Neph1 (Red) at the cell-cell junctions. In contrast, significant recovery of actin cytoskeletal organization was noted in formoterol treated cells along with restoration of Neph1 staining at cell-cell junctions (arrows). Scale bar 20μm. (B) The quantitative analysis showed ~27% increase in the number of healthy podocytes with a concomitant decrease in NTS injured podocytes upon formoterol treatment, while the vehicle treated podocytes recovered minimally. (C) Quantitation further showed complete relocalization of Neph1 at cell membrane upon formoterol treatment of NTS injured podocytes. Data are presented in mean±SEM, One-way ANOVA, ###P≤0.0001 vs. untreated control; ***P≤0.0001 NTS+vehicle vs. NTS+formoterol. (D) PS injured podocytes showed damaged actin cytoskeleton (Green) with accumulation of actin stress fibers at cell periphery, and loss of Neph1 (Red) at the cell-cell junctions. Recovery of actin cytoskeletal organization was noted in formoterol treated cells with restoration of Neph1 staining at the cell junctions (arrows). Scale bar 20μm. (E) The quantitative analysis of actin cytoskeleton reorganization showed significant increase in the number of healthy podocytes (~40%), with a concomitant decrease in PS injured podocytes upon treatment with formoterol, whereas the recovery in vehicle treated podocytes was minimal. (F) The quantitative assessment of Neph1 relocalization at podocyte cell membrane shows, complete relocalization of Neph1 at cell membrane in formoterol treated PS injured podocytes. Data are presented in mean±SEM, One-way ANOVA, ###P≤0.0001 vs. untreated control; *** P≤0.0001 PS+vehicle vs. PS+formoterol.
Formoterol restored glomerular filtration in acute and chronic models of glomerular injury.
To determine if formoterol induces a similar protective effect under in vivo conditions, we tested the effect of formoterol in two different glomerular injury models, NTS and adriamycin (ADR). A schematic diagram depicting experimental design is shown in 2Figure 5A & B. Urine samples from each mouse were collected and analyzed by SDS-PAGE and ELISA to determine the extent of albuminuria. Please note that in both the models formoterol was administered only after the proteinuria was established (4h for NTS and one week for ADR, Figures 5C&D respectively)..
Figure 5: Formoterol accelerated glomerular recovery from acute and chronic models of glomerular injury:

The schematic of experimental plan to evaluate in-vivo significance of formoterol using NTS (A) and ADR (B) induced glomerular injury models. (C) Urine samples were analyzed by SDS-PAGE and Coomassie blue staining and showed significant reduction in albuminuria starting at day3, which extended to day 7 in NTS+formoterol treated mice group, whereas minimal reduction was noted in the control NTS+vehicle treated mice group. (D) Similar urine analysis showed significant reduction in albuminuria at day 10 post formoterol treatment in ADR+formoterol treated mice, whereas minimal reduction was noted in the control ADR+vehicle mice. (E) Measurement of urine albumin/creatinine ratios by ELISA showed initiation of albuminuria at 4h post NTS injection, which further increased at days 1 and 2 in both groups. The ratios dropped to preinjection levels in NTS+formoterol treated mice group but remained significantly elevated in control mice group at days 5–7. 2-tailed t-test. n=5 mice per group. Data are presented in means±SEM. *P≤0.01, **P≤0.001 vs. control. (F) Measurements of urine albumin/creatinine ratios were made at 1 week post ADR-injection. The ratios remained significantly elevated in both the groups at day 5, but significant reduction was noted in ADR+formoterol mice at day 10 post formoterol treatment. 2-tailed t-test (*p<0.05, ADR+vehicle vs ADR+formoterol). n=5 mice per group. Data are presented as mean±SEM. In-vivo results were reproduced in three independent experiments with n=5, each group, each experiments.
Albuminuria occurred at 4h and one week in NTS and ADR models respectively, with no differences in albuminuria observed between control and formoterol treated groups (Figure 5C–F). Interestingly, marked reduction in albuminuria was noted in the NTS+formoterol and ADR+formoterol treated groups starting at days 4 and 5 respectively (Figure 5C–F). Collectively, these findings suggest that formoterol treatment promoted recovery from injury-induced albuminuria. Sera from these mice did not show any changes in creatinine levels (Figure S3A).
Formoterol restored injury-induced glomerular damage.
To evaluate histological changes, kidney sections from all experimental and control mice (sacrificed at 7 days post NTS injection), were stained with hematoxylin and eosin (H&E), periodic acid-Schiff (PAS) and Masson’s Trichrome staining (Figure 6A and figure S3B–C). Quantitative analyses of histological features were performed in a blinded fashion, which revealed focal atrophy and dilatation of tubules, with round cell interstitial infiltration in control NTS+vehicle treated groups (Figure 6B); in contrast, these changes were notably reduced in the NTS+formoterol treated group (Figure 6B). Additionally, the sections from NTS+vehicle mice group showed significant tubular dilatation with PAS positive casts, which were reduced in the kidneys of NTS+formoterol treated mice (Figure 6B). Further histological data analysis confirmed reduction of fibrosis, glomerular sclerosis and notably reduced loss of brush borders, in NTS+formoterol treated mice (Figure 6B). Estimated glomerular damage (specified by glomerular scoring of 25%, 25–50% and more than 50% damage) showed that treatment of injured mice with formoterol reduced glomerular damage among all evaluated categories (Figure 6C). Ultrastructural analysis using transmission electron microscopy further revealed reduced podocyte foot process damage in NTS+formoterol treated mice, when compared to the control NTS+vehicle treated mice (Figure 6D). To further understand the effect of formoterol treatment on renal outcome in the context of a chronic kidney disease, we extended the NTS-induced glomerular injury model from 7 to 14 days. The urine analysis including SDS-PAGE and UACR from NTS+formoterol treated mice showed significant reduction in albuminuria after 7 days, which persisted till the 14th day (Figure S4A–C). Similar histological observations were noted in this NTS extended model, where sections were analyzed 14-days post-injury with and without formoterol treatment and showed significant recovery of renal pathology in NTS+formoterol treated mice (Figure S4D).
Figure 6: Formoterol restored injury-induced glomerular damage.

(A) Histological analysis of mice (sacrificed at day 7 post NTS injection) kidney sections shows that formoterol treatment (NTS+formoterol) reduced focal atrophy, proteinaceous tubular caste and tubular dilation as compared to control (vehicle) mice. Scale bar 50μm. (B) Major histological features were individually scored, and the comparative analysis showed reduction in loss of brush borders, degeneration of proximal tubules and reduced proteinaceous casts, tubular dilation and fibrosis in NTS+formoterol treated mice. 2-tailed t-test, ***P≤0.0001 NTS+vehicle vs. NTS+formoterol. (C) Glomerular scores demonstrating varying levels of glomerulosclerosis in control and NTS+formoterol treated mice are presented and show increased number of normal glomeruli in NTS+formoterol treated mice, along with reduction in sclerotic glomeruli. 2-tailed t-test, **P≤0.001, ***P≤0.0001 NTS+vehicle vs. NTS+formoterol. n=5 mice each group (both kidneys were scored) (D) Representative images from the TEM analysis show significant damage to podocyte foot processes in control NTS+vehicle treated mice, whereas, normal foot processes were present in the NTS+formoterol treated mice. Scale bar 2μm. (E) Histological analysis of mice (sacrificed at day 10 post formoterol injection) kidney sections showed that formoterol treatment (ADR+formoterol) reduced ADR-induced glomerular sclerosis, focal atrophy, proteinaceous tubular caste and tubular dilation. Scale bar 50μm. (F) Glomerulosclerosis severity score was calculated and was significantly reduced for ADR+formoterol mice. (**P≤0.001, NTS+vehicle vs. NTS+formoterol). n=5 mice each group.
Similar observations were noted in the ADR model where glomerulosclerosis ranging from mild segmental sclerosis to severe global sclerosis, numerous proteinaceous casts, and an interstitial hypercellularity with higher manifestation was noted in ADR+vehicle treated mice; in comparison these changes were reduced in the ADR+formoterol mice (Figure 6E, figure S3D). Accordingly, the glomerulosclerosis severity scores derived from these images (indicative of glomerular damage) were higher for ADR+vehicle treated mice than the ADR+formoterol mice (Figure 6F).
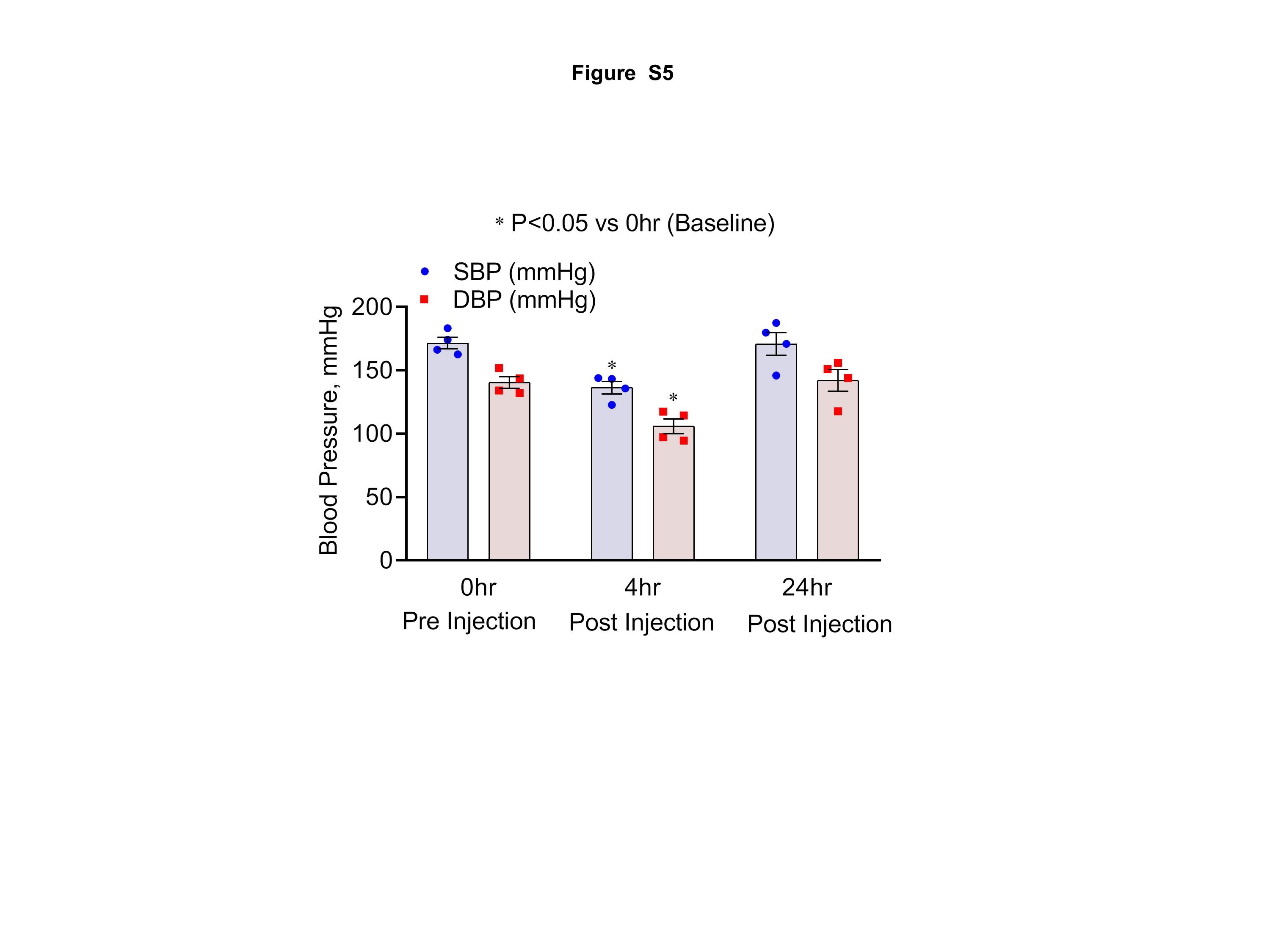
To determine if formoterol treatment affected blood pressure in these mice, blood pressure measurements were performed over a period of 24h following formoterol treatment. The results showed, that while there was a marginal reduction in blood pressure at 4h, it returned to baseline within 24h (Figure S5).
Formoterol treatment restored injury-induced mislocalization of Neph1.
Our previous studies have shown that Neph1 mis-localizes in response to injury41,43. Importantly, re-localization of Neph1 to the membrane serves as a marker of recovery from injury as previously demonstrated41,42. To determine if formoterol-induced recovery restored Neph1 localization, kidney sections from NTS+vehicle and NTS+formoterol treated mice were immunostained with Neph1 and synaptopodin antibodies (Figure 7A). While NTS induced mislocalization of Neph1, formoterol treatment restored the original distribution of Neph1 and increased its colocalization with synaptopodin (Figure 7A). Pearson’s correlation coefficient (Rr) analysis further supported increased colocalization of Neph1 and synaptopodin in the glomeruli of NTS+formoterol treated mice (Figure 7B). Thus, formoterol-induced recovery involves restoring membrane localization of slit diaphragm protein Neph1, which contributes to the structural integrity of podocytes.
Figure 7: Formoterol treatment restored Neph1 localization at the podocyte cell membrane and reduced cellular apoptosis:

(A) Mice kidney sections were immunostained with Neph1 (Green) and Synaptopodin (Red) antibodies and DAPI (Blue). NTS induced mislocalization of Neph1 was largely restored by formoterol treatment, where increased Neph1 localization at the podocytes membrane and colocalization with synaptopodin was visible. (B) The Pearson’s correlation coefficient (Rr) analysis showed increased colocalization of Neph1 and Synaptopodin in NTS+formoterol treated mice. Data are presented in mean±SEM. One-way ANOVA, ***P≤0.001 NTS+vehicle vs. NTS+formoterol. (C & D) Apoptosis was measured in the kidney sections using TUNEL assay. Significant amounts of TUNEL positive (Green) nuclei (blue DAPI) were present in NTS+vehicle treated control mice and positive control, whereas they were largely absent in the NTS+formoterol treated mice (white arrows). Data are presented in mean±SEM. One-way ANOVA, ###P≤0.001 Control vs NTS+vehicle; ***P≤0.001 NTS+vehicle vs. NTS+formoterol.
Formoterol treatment prevented injury-induced cell death and podocyte loss in mice glomeruli:
The progression of a glomerular disease involves podocyte effacement, loss of slit diaphragm and cell death44–47. It has been reported that animal models of FSGS exhibit podocyte apoptosis that may lead to “podocytopenia” and consequently glomerulosclerosis48–51. To evaluate cell death in our models, kidney sections from NTS+vehicle and NTS+formoterol treated mice were analyzed using the TUNEL assay. While a significant number of TUNEL positive nuclei were observed in the glomeruli of NTS+vehicle treated mice and positive controls, they were largely absent in the NTS+formoterol treated mice (Figure 7C). Quantitative analysis further confirmed reduction in TUNEL positive nuclei in the glomeruli of NTS+formoterol treated mice (Figure 7D). Finally, WT1 staining was performed to examine podocyte loss, which showed that in comparison to control, WT1 positive cells were reduced in the glomeruli of NTS+vehicle mice, and treatment with formoterol restored WT1 positive cells to a larger extent (Figure S6).
Formoterol treatment induced MB in mice.
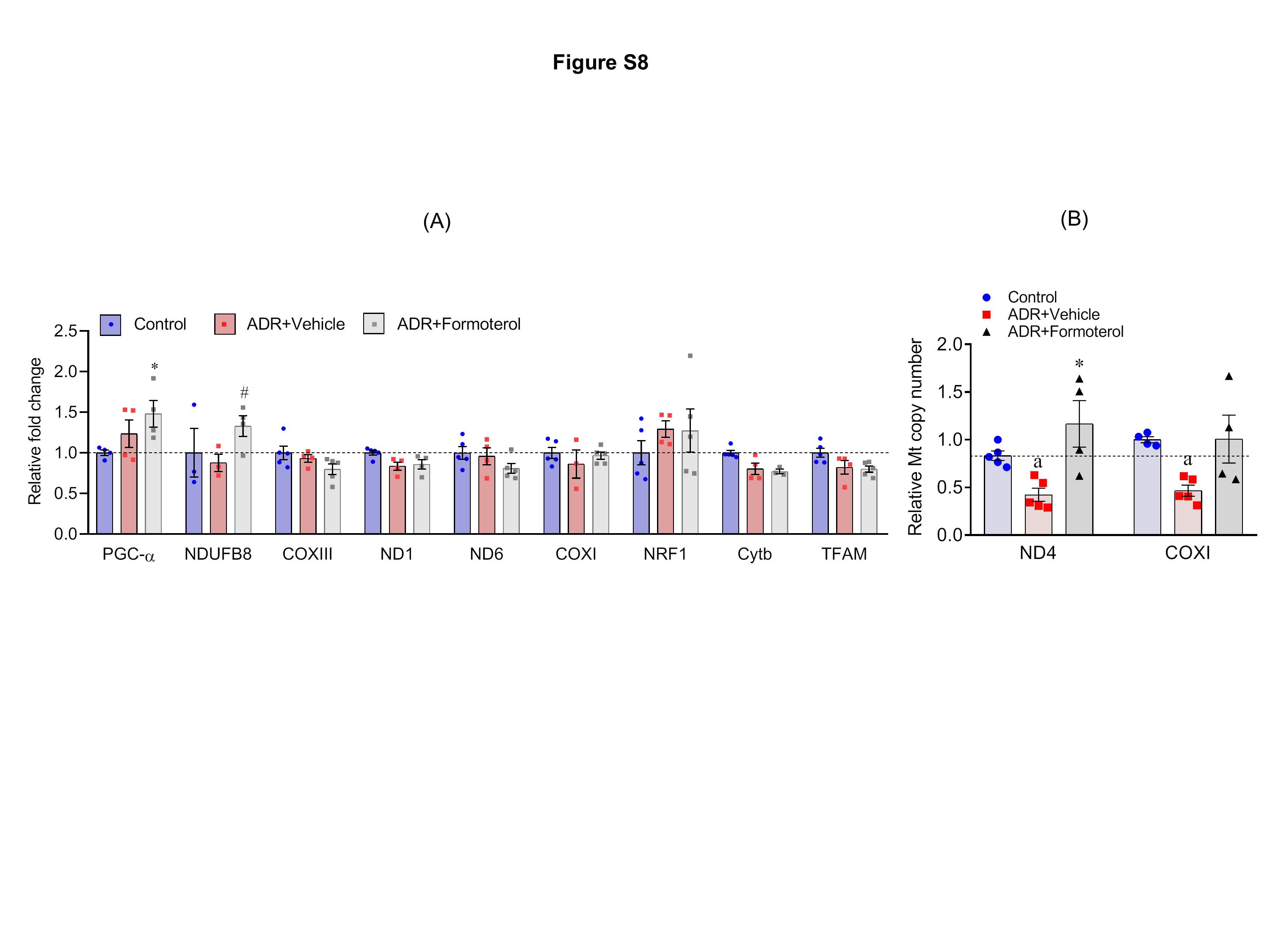
Formoterol mediated its effect through β2-AR (Figure 2H), and to further highlight the mechanism through which formoterol stimulated glomerular recovery, we evaluated the effect of formoterol on MB in the NTS model. Independent evaluations were performed from the kidney and glomerular lysates derived from the vehicle and formoterol treated mice (n=5). These lysates were subjected to immunoblot analysis using PGC-1α, OXPHOS and GAPDH antibodies. Results showed significant induction of PGC-1α protein in the kidney and glomerular lysate of NTS+formoterol treated mice (Figure 8A–C, figure S7A–C). The immunoblot and quantitative analysis further showed that mitochondrial protein complexes I and V, the complex III protein (markers of mitochondrial dysfunction), which were reduced in response to injury by NTS (Figure 8A & C), were upregulated in NTS+formoterol-treated mice (Figure 8A–C, figure S7A–C). qPCR analysis of mitochondrial genes also showed that NTS-induced injury decreased the expression of PGC-1α and ND6 genes (Figure 8D), whereas formoterol treatment resulted in a upregulation of PGC-1α, NDUFB8, COXIII and NRF1 genes (Figure 8D). Similar induction for PGC-1α was noted in ADR+formoterol treated mice (Figure S8A).
Figure 8: Formoterol treatment induced the expression of mitochondrial proteins:

(A) Markers of MB in mice kidney lysates were evaluated by western blotting using PGC-1α, OXPHOS (cocktail antibodies of mitochondrial ETC complex I to V) and GAPDH antibodies. (B–C) Densitometric analysis of immunoblots showed increased expression of PGC-1α (One-way ANOVA, *P≤0.05 NTS+vehicle vs. NTS+formoterol; ##P≤0.01 vehicle vs NTS+formoterol) and OXPHOS (mitochondrial complex I, III and V) proteins in NTS+formoterol treated mice (One-way ANOVA, **P≤0.01, ***P≤0.001 NTS+vehicle vs. NTS+formoterol; ##P≤0.01 vehicle vs NTS+formoterol). Data are presented in mean±SEM. (D) The qPCR analysis showed that formoterol treatment upregulated the expression of PGC-α, NDUFB8, ND6, COXIII and NRF1 genes that are involved in MB. Data are presented in mean±SEM. One-way ANOVA, **P≤0.05, **P≤0.01, ***P≤0.001 NTS+vehicle vs. NTS+formoterol; aaP≤0.01 control vs NTS+vehicle; #P≤0.05, ###P≤0.05 control vs NTS+formoterol. (E) mtDNA copy number analysis of kidney samples from NTS injured mice showed that mtDNA copy number significantly increased (~2fold) upon formoterol treatment. *P≤0.05 control vs NTS+formoterol.
To further evaluate MB, DNA from the kidneys (Figure 8F) and glomeruli (Figure S7E & S8B) of control (vehicle only), NTS+vehicle, NTS+formoterol and ADR+formoterol treated mice were subjected to mtDNA copy number analysis using ND4 and COX1 primers. Results showed that in comparison to control and vehicle treated mice, there was an increase in ND4 and COX1 expression in NTS+formoterol and ADR+formoterol treated mice, which is consistent with the role of formoterol as a MB inducer (Figure 8E, figure S7E & S8B).
Formoterol treatment enhanced the expression of PGC-1α in mice glomeruli and podocytes.
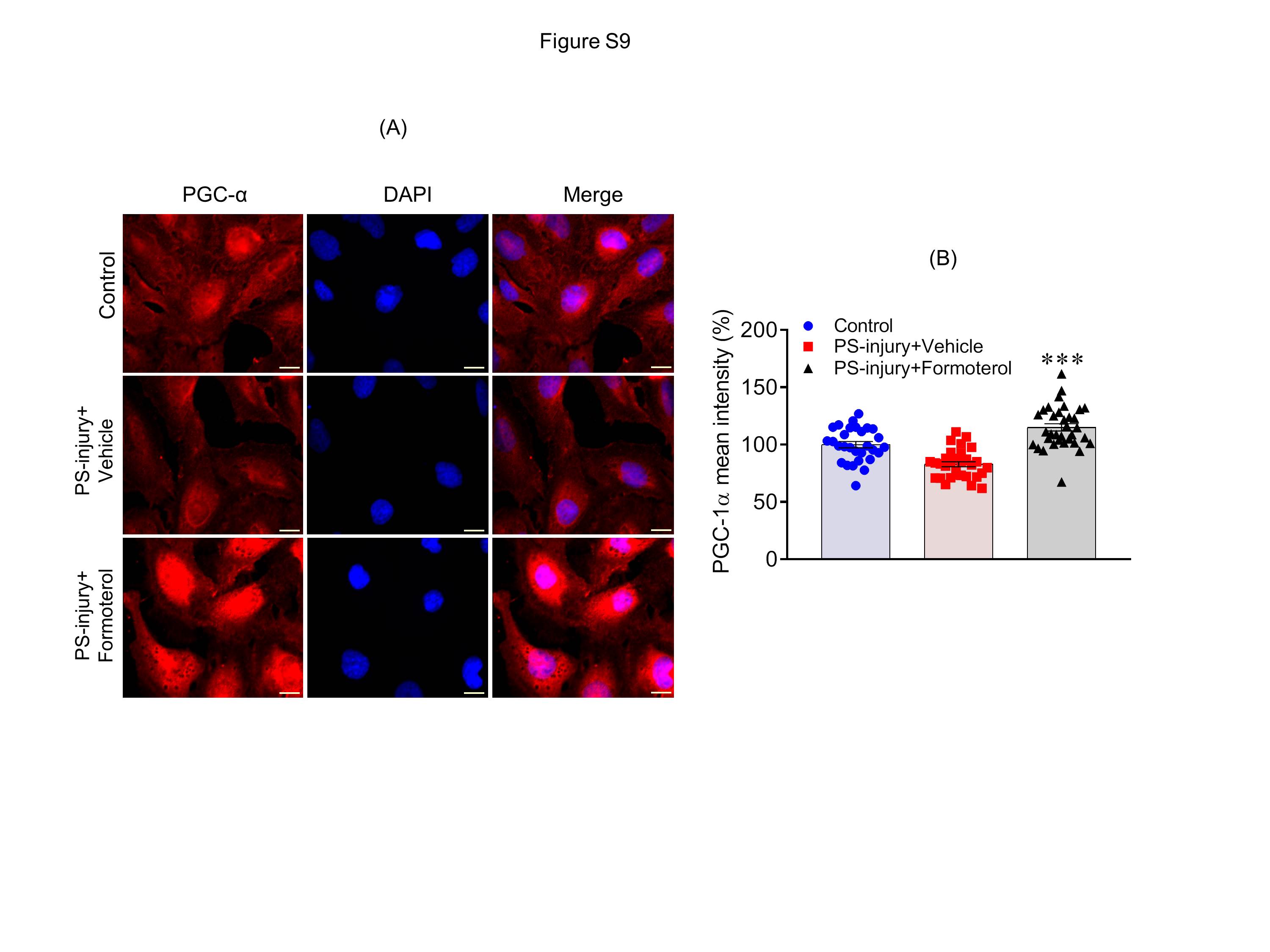
Although increased expression of PGC-1α was observed in kidney lysates from formoterol-treated mice, we wanted to evaluate PGC-1α levels in glomeruli and podocytes, especially since podocyte dysfunction is the primary outcome in NTS and ADR induced injuries52,53. Kidney sections from controls, NTS+formoterol and ADR+formoterol mice were immunostained with PGC-1α antibody and analyzed by confocal microscopy. The results showed increased PGC-1α staining in the formoterol-treated mice, which overlapped with the podocyte marker synaptopodin (Figure 9A&C); in contrast, no upregulation of PGC-1α in the glomeruli of control or NTS/ADR treated mice was noted (Figure 9A&C). Quantitation of PGC-1α fluorescence intensity further showed increased expression of PGC-1α in the glomeruli of NTS+formoterol treated mice (Figure 9B&D and S9). Similarly, increased expression of PGC-1α was noted in cultured podocytes that were treated with formoterol (Figure S10).
Figure 9: Formoterol treatment induced the expression of mitochondrial genes in mice glomeruli:

(A) Immunostaining analysis of kidney sections using PGC-1α (Green) and Synaptopodin (Red) antibodies and DAPI (Blue) showed increased PGC-1α staining in the NTS+formoterol treated mice. (B) The quantitative analysis of mean pixel intensity showed increased PGC-1α expression in the glomeruli of NTS+formoterol treated mice. Data are presented in mean±SEM. One-way ANOVA, ***P≤0.001 NTS+vehicle vs. NTS+formoterol. (C) Immunostaining with PGC-1α (Green) and Synaptopodin (Red) antibodies and DAPI (Blue) showed increased PGC-1α staining in the glomeruli of ADR+formoterol treated mice. (D) The quantitative analysis of mean pixel intensity showed increased PGC-1α expression in the glomeruli of ADR+formoterol treated mice. Data are presented in mean±SEM. One-way ANOVA, ***P≤0.001 NTS+vehicle vs. NTS+formoterol.
DISCUSSION
Podocytes are the primary target in majority of glomerular diseases9,54,55 and their dysfunction leads to ESRD55,56. The pharmacological treatment options for podocytes are severely limited and are focused primarily on prevention and survival of podocytes from injury5,9,57. In this study, we sought to discover a pharmacological approach directed towards accelerating podocytes recovery in mouse models of glomerular injury. Interestingly, several studies show that podocytes have propensity to recover from injury54,55,58; however, little is known about the underlying mechanisms involved in the recovery process57. In this study, we demonstrate that podocyte MB can be pharmacologically targeted using an FDA approved β2-AR2 agonist, formoterol, to enhance recovery from injury, thus presenting β2-AR2 as a novel therapeutic target in podocytes.
MB is an adaptive response to maintain high-energy demands and metabolic homeostasis following injury59. It is also characterized as a process that increases mitochondrial copy number and ATP output, and can occur under basal conditions59. Therefore, MB has been recognized as a potential therapeutic target to treat mitochondrial dysfunction that is commonly seen in gastrointestinal, respiratory, neurological, renal and numerous other disorders30,60–62. The initial evidence for the induction of MB in podocytes came from our mRNA profiling of PAN-induced injury of podocytes, where an increase in PGC-1α was observed. Importantly, formoterol treatment further enhanced the expression of this protein, which stimulates MB through induction of various mitochondrially-encoded genes36,59. Since our previously published work showed involvement of formoterol-induced MB in recovery from ischemiareperfusion (I/R) injury30, we hypothesized that induction of MB may accelerate podocyte recovery after injury. MB is commonly evaluated through induction of various markers of ETC and consistent with other studies30,63, our qPCR and immunoblot results showed that podocyte injury down regulated these markers. Importantly, these markers were upregulated upon treatment of podocytes with formoterol, suggesting a critical role for MB in podocyte injury. We further report that maximal OCR, a marker of MB30, was also induced by formoterol35 in differentiated cultured human podocytes (Fig. 2B). Thus, in addition to MB, formoterol increased mitochondrial function in podocytes, which further supports the ability of podocytes to respond to formoterol treatment.
Injury to podocytes is commonly assessed through actin cytoskeleton disorganization and mis-localization of slit diaphragm proteins such as Nephrin and Neph139,41,42. Previous studies, where podocytes were therapeutically targeted were primarily focused on preventing injury-induced damage to podocyte actin cytoskeleton42,64, which provides preemptive value in treating podocytopathies, but is clinically less significant. In contrast, formoterol treatment was administered in-vitro and in-vivo systems after the injury was established, which is of high clinical significance. Our results show that formoterol treatment was restored podocyte actin cytoskeletal organization and relocalization of Neph1 to the membrane, NTS and PS in-vitro injury models. Further support for therapeutic significance of formoterol was obtained from in vivo studies, where the effect of formoterol was tested in an acute NTS injury model that targets podocytes leading to progressive glomerulonephritis65 renal dysfunction and proteinuria65,66. When administered following establishment of injury at 4h, formoterol treatment resulted in early signs of recovery at 72h post injury, and by days 5–7, significant attenuation of proteinuria was noted. Importantly, all histological injury hallmarks, including focal atrophy, dilatation of the tubules, and round cell interstitial infiltrations were decreased in formoterol treated mice kidneys. In addition, the numbers of sclerotic glomeruli were significantly reduced, with a concomitant increase in normal glomeruli (Figure 6A). Although similar results were noted when the effect of formoterol was tested on ADR, a chronic model of glomerular injury, recovery in this model was less as compared to the NTS injury model. Such differences could be attributed to distinct injury mechanisms in these models that in addition to MB may contribute towards disease pathogenesis. However, it was interesting that the protective effects were noted in both models, suggesting that MB does participate in recovery from injury in these models. We also believe that testing additional models will further assist in establishing the therapeutic potential of formoterol in treating glomerular diseases and broaden the clinical scope of this β2-AR agonist.
Our results also highlight a mechanism for formoterol-mediated recovery, where formoterol mediated its affect through β2-AR and immunoblot and qPCR analysis showed induction of PGC-α, NDUFB8, COXIII and NRF1 and complex I, III and V proteins of the ETC that are involved in MB (Figure 8A–D). Notably, glomerular staining revealed that formoterol-induced recovery also increased the expression of PGC-1α in mouse podocytes. PGC-1α is a transcriptional coactivator and the master regulator of mitochondrial energy metabolism that interacts with other transcriptional factors to regulate MB27,30,67. It is regulated by nuclear receptors of the PPAR family, and PPARα and γ agonists have been shown to induce anti-albuminuric effects in clinical trials68. Experimental evidence also suggests that PPAR agonists may directly improve podocytes response to injury and exert cytoprotective effects68. In contrast to previous findings69, a recent study showed that increased expression of PGC1α alters mitochondrial properties and induces proliferation and dedifferentiation of podocytes leading to glomerulopathy27. Collectively, these studies highlight the importance of a physiologically balanced and regulated expression of PGC1α that is critical for the recovery of podocytes from injury. Therefore, moderately elevated levels of PGC1α may participate in podocytes recovery from injury; a concept that is more likely supported from this study. Unlike formoterol, it has been shown that the short-acting β2 agonist salbutamol-mediated activation of β2-AR attenuated monocyte activation, pro-inflammatory and pro-fibrotic responses in diabetic kidney disease through the inhibition of NF-kB and β-arrestin2 signaling70. This further suggests that additional molecular events and signaling pathways may participate in formoterol-mediated activation of β2-AR, which needs further investigation.
Although studies focused on understanding disease mechanisms has resulted in significant progress in our understanding of pathophysiology of glomerular diseases such as FSGS, the treatment options for this disease remain limited and include immunosuppressive drugs as frontline therapy57. Unfortunately, many patients do not respond to immune-based therapies and drugs such as cyclosporine54 and rituximab58, which although target podocytes offer limited success71. The ability of formoterol to induce rapid recovery of damaged podocytes, presents a novel therapeutic alternative that could benefit FSGS patients. While this conclusion is based on limited glomerular injury models, testing various other injury models will determine widespread application of this therapeutic approach in treating other glomerular diseases. In conclusion, we present a viable therapeutic approach, where targeting MB by formoterol after the establishment of injury, induces rapid podocyte recovery and restored glomerular filtration function.
METHODS
Podocytes Cell Culture:
Human podocytes were cultured in RPMI 1640-based medium supplemented with 10% fetal bovine serum (FBS) (Invitrogen), 2 g/liter of sodium bicarbonate (NaHCO3), insulin-transferrin-selenium (Sigma-Aldrich), and 200 units/ml penicillin and streptomycin (Roche Applied Science) as described previously32,33,41. The podocyte cells were grown on collagen-coated culture dishes at 33°C and 5% CO 2, and were differentiated by thermo switching to 37°C as described previously32,72,73. Podocyte injuries were developed using PAN (100μg/ml) and Adriamycin (0.25μg/ml) for 48 hours, 5% NTS for 16hours and 600μg/ml protamine sulphate for 8hours in serum free media34,41. To initiate recovery, the injury medium was removed and serum-mediated recovery was initiated by the addition of medium containing serum (0.2%) as described earlier33. In a similar fashion, the drug-induced recovery was initiated by addition of formoterol or vehicle in serum free medium for the indicated times.
RNA-Seq:
Differentiated human podocytes were treated with PAN (100μg/ml) for 48h. The cells were lyzed and RNA isolation was performed using the Qiagen RNA isolation kit. All experiments were performed in triplicates. RNA samples were submitted for RNA sequencing and bioinformatics analysis to either Novogene or the profiling and Bioinformatics Shared Resource core facility of the Medical University of South Carolina (MUSC). RNA sequencing was performed using an Illumina HiScanSQ as described previously74. Briefly, RNA integrity was verified on an Agilent 2200 Tape Station (Agilent Technologies). Total RNA (100–200ng) was used to prepare RNA-Seq libraries using the TruSeq RNA Sample Prep kit as per the manufacturer instructions (Illumina, San Diego, CA). Bioinformatics analysis was performed at the MUSC bioinformatics core facility as described previously74,75. Data was processed using the Trimmomatic program, and the sequences were confirmed using FastQC and aligned with human genome build HG19 using Tophat (Bowtie2). Differential expression analysis was performed using the DESeq2 R package. Differential Expressing Genes (DEGs) p-values were adjusted using the Benjamini and Hochberg’s approach and adjusted P-value <0.05 found by DESeq2 were assigned as differentially expressed (GEO Accession # GSE124622). The SABiosciences website was used to identify genes involved in mitochondrial energy metabolism and generate DEGs list, which was used to construct the heat-maps.
Oxygen Consumption Rate (OCR) measurement:
The OCR measurements were performed using a Seahorse Bioscience XF-96 instrument as described previously35. Differentiated human podocytes were plated and treated with vehicle controls (DMSO 0.1%), blank controls, and formoterol (30 nM). The XF-96 protocol consists of five measurements of basal OCR (1 measurement/1.5 min), injection of p-trifluoromethoxyphenylhydrazone (FCCP) (0.5 μM), and three measurements of uncoupled OCR (1 measurement/1.5 min). The consumption rates were calculated from continuous average slope of O2 partitioning among plastic, atmosphere, and cellular uptake76.
Indirect immunofluorescence microscopy:
Podocytes were grown on coverslips and injury was induced by PS (500ug/ml) or 5% NTS for 8h and 12h respectively. The recovery was initiated by addition of 30nM formoterol or control vehicle. Podocytes were fixed with 4% paraformaldehyde (in 1× PBS), followed by permeabilization with 0.1% SDS. Staining of podocytes was performed with Neph1 antibody, phalloidin and DAPI as described previously41. Fluorescence microscopy was performed with Leica confocal microscope and images were collected under constant parameters. Representative images from a minimum of three experiments are presented in the figures 3. Estimation of mean pixel intensity was analyzed using Image-J software as described previously33. Statistical analysis was performed using GraphPad Prism software (version 7.01); oneway ANOVA was performed, and the p-values were adjusted using Tukey’s multiple comparisons test. Podocyte injuries induced by PS and NTS were categorized into three groups, healthy, moderately injured and severely injured, based on the cell morphology as shown in figure S1A. Similarly, mitochondrial morphology was analyzed using MitoTracker staining performed as per the manufacturer protocol (MitoTracker FM, Invitrogen # M7514).
β2-AR knockdown (KD) in human podocytes:
β2-AR KD was performed using shRNA Lentivirus obtained from sigma. Five sets of Lentiviral particles were used as described, TRCN0000008083, TRCN0000008084, TRCN0000008086, TRCN0000356690 and TRCN0000356689. Stable human podocytes with β2-AR KD were obtained through puromycin selection.
Mouse models of glomerular injury:
Ten weeks old mice (C57BL/6N genetic background) used in our experiments were obtained from Charles River Laboratory. Each experimental and control group consisted of five mice. A pilot experiment was performed to select an optimal NTS (Probetex INC, PTX-001) dose. Based on this, 80μl NTS was identified as the minimum amount of NTS required to induce consistent proteinuria in C57BL/6N mouse strain. NTS (80μl) was injected retro-orbitally as described previously43. Urine samples from individual mice were collected at preinjection, 4h and at every 24h post NTS injection for seven days. Control vehicle or formoterol (1 mg/kg body weight) was administered (intraperitoneally) 4h post NTS injection, when a significant amount of proteinuria was noted. Mice receives a repeated dose of formoterol every 24h for 6 days. Detailed experimental plans for drug administration and urine collection are provided in the schematic diagram presented in Figure 5A. Urine samples were spun at 4000xg for 5 minutes and then frozen at −80°C for subsequent analysis. Similarly, Adriamycin (ADR)-induced chronic model of glomerular injury was tested in BALB/c mice obtained from Jackson’s laboratory. 7mg/kg ADR (doxorubicin hydrochloride) was injected in these mice as described previously73,77. Experimental design, urine collection and formoterol dosing (2mg/kg body weight) plan are presented in schematic figure (Figure 5B).
Urine Analysis:
2.5μl of each urine sample was diluted 5-fold with water and mixed with 2X sample buffer and analyzed by 10% SDS-PAGE followed by CB staining. Urine albumin/creatinine ratios were obtained using an enzyme-linked immunosorbent assay (ELISA) Albuwell kit (Exocell) and creatinine companion kit (Exocell), whereas, serum creatinine was measured using the QuantiChrom creatinine assay kit (DICT-500), and the results were analyzed by an unpaired one-tailed t test (GraphPad Prism 7) as described previously43,78.
Blood pressure measurement:
Systolic and diastolic blood pressure were measured by tail cuff method using the CODA system (Kent Scientific Torrington, CT)79. Tail-cuff blood pressure data are from an average of 10–15 measurements for each experimental animal.
Histological and ultra-structural Analysis:
Mouse kidneys were isolated following perfusion of mice with Hanks buffered salt solution (HBSS) for 3 min; the kidneys were excised, decapsulated, transected, and then fixed for 12h in 4% paraformaldehyde, rinsed, and stored in 70% ethanol and submitted to the MUSC Histology Core for paraffin embedding and sectioning. Five-micrometer sections were made from blocks, deparaffinized using xylene-ethanol (EtOH) and processed for hematoxylin and eosin (H&E), periodic acid-Schiff (PAS) and Masson’s Trichrome staining. Representative images were collected on an inverted Zeiss Axiovert-200-M confocal microscope at the Cell & Molecular Imaging facility of MUSC. Histological analysis and scoring was performed by Dr. Judit Megyesi. Mice kidney samples were fixed in 2% glutaraldehyde and 2% paraformaldehyde, stored overnight and submitted to the electron microscopy core facility at MUSC for transmission electron microscopy (TEM).
Isolation of mice glomeruli:
Mice glomeruli were isolated using magnetic beads-based procedure as described previously66. Total RNA was isolated from mice glomeruli and was evaluated by qPCR using specific primers. Protein and DNA were isolated from these mice glomeruli for western blotting and mtDNA estimation.
Immunoblotting:
Mice kidneys were lysed in RIPA buffer using a beads beater and tissue lysate was collected following centrifugation. Protein estimation was performed using BCA procedure and 40 μg proteins were loaded in each well of a 7.5–10% SDS-PAGE gel. Western blotting was performed using primary antibodies against PGC-α, GAPDH and OX-PHOS (cocktail of complex I-V) (Invitrogen). Images were collected and densitometric analysis was performed using the LI-CORE imaging station.
Quantitative real-time PCR:
Total RNA was isolated from human podocytes and mouse kidney by Qiagen RNA isolation kit and cDNA was prepared using ThermoScript RT-PCR Systems (Invitrogen, Carlsbad, CA) as per the manufacturer protocol. Real-time q-PCR was performed using SsoAdvanced, Universal SYBR Green Supermix according to manufacturer instructions. The qPCR from human podocytes was performed using specific qPCR primers for genes PGC-α, ATPase 6, CYTB, ND1, NRF1, FN1, CO1 and ND6. Mouse kidney qPCR was performed using specific qPCR primers for PGC-α, NDUFB8, COXIII, ND1, ND6 COXI, NRFI, Cytb1 and TFAM genes as described previously36,80,81. RPS13 primers were used for normalization. Primer sequences for all genes used in qPCR studies are listed in the Table SI.
Immunohistochemistry (IHC):
Mouse kidneys were fixed, paraffin embedded and sectioned, and processed for Immunohistochemistry as described previously82. Kidney sections were Immunostained using specific primary antibodies for PGC-1α, Neph1 and Synaptopodin, followed by Alexa Fluor-labeled secondary antibodies and Mounted with DAPI containing mounting medium. Images were collected using an SP5 Leica confocal microscope fitted with 60X oil objective. All acquisition parameters were kept constant throughout the imaging of various samples. Mean pixel intensity estimation and colocalization analysis using Pearson’s correlation coefficient was performed by Image-J software as described previously33. Statistical analysis included one-way ANOVA as described above.
Measurement of mitochondrial DNA (mtDNA):
Real-time PCR was used to determine relative quantities of mtDNA in human podocytes, mice glomeruli and mouse kidney tissues. Genomic DNA was extracted using DNeasy Blood and Tissue kit (QIAGEN # 69504). PCR products were amplified using 10–25 ng of cellular DNA as described previously36,83. mtDNA estimation from mice kidney and glomeruli was measured using the NADH dehydrogenase subunit 4 (ND4) and COXI gene primers. The nuclear DNA and apoB gene were used for normalization36,83. The mtDNA estimation was made using D-loop region and was normalized against the b-actin gene to calculate relative mtDNA copy number36,83. Primer details for mtDNA estimation are provided in table S2
TUNEL assay:
Apoptosis was measured using the terminal transferase-dUTP-nick-end labeling (TUNEL) assay as per manufacturer’s protocol. Briefly, Kidney sections were permeabilized with 0.5% Triton X-100 for 10 min, washed with 1× PBS (twice) and then incubated with TUNEL reaction mixture for 60 min at 37°C in a humidified dark chamber. Kidney sections were subsequently washed with 1×PBS (X3), mounted with DAPI, and analyzed using a fluorescence microscope as described previously47.
Animal Study approval:
All animal studies were approved under the protocol number # IACUC-2018–00360 by the MUSC IACUC (Institutional Animal Care and Use Committee) and were conducted as per the NIH guidelines for Care and Use of Laboratory Animals.
Statistical Analyses:
Each data set is presented as mean±SEM. The unpaired 2-tailed t-test or one-way ANOVA was performed using the GraphPad Prism 7 software. A p value of ≤ 0.05 was considered as statistically significant.
Supplementary Material
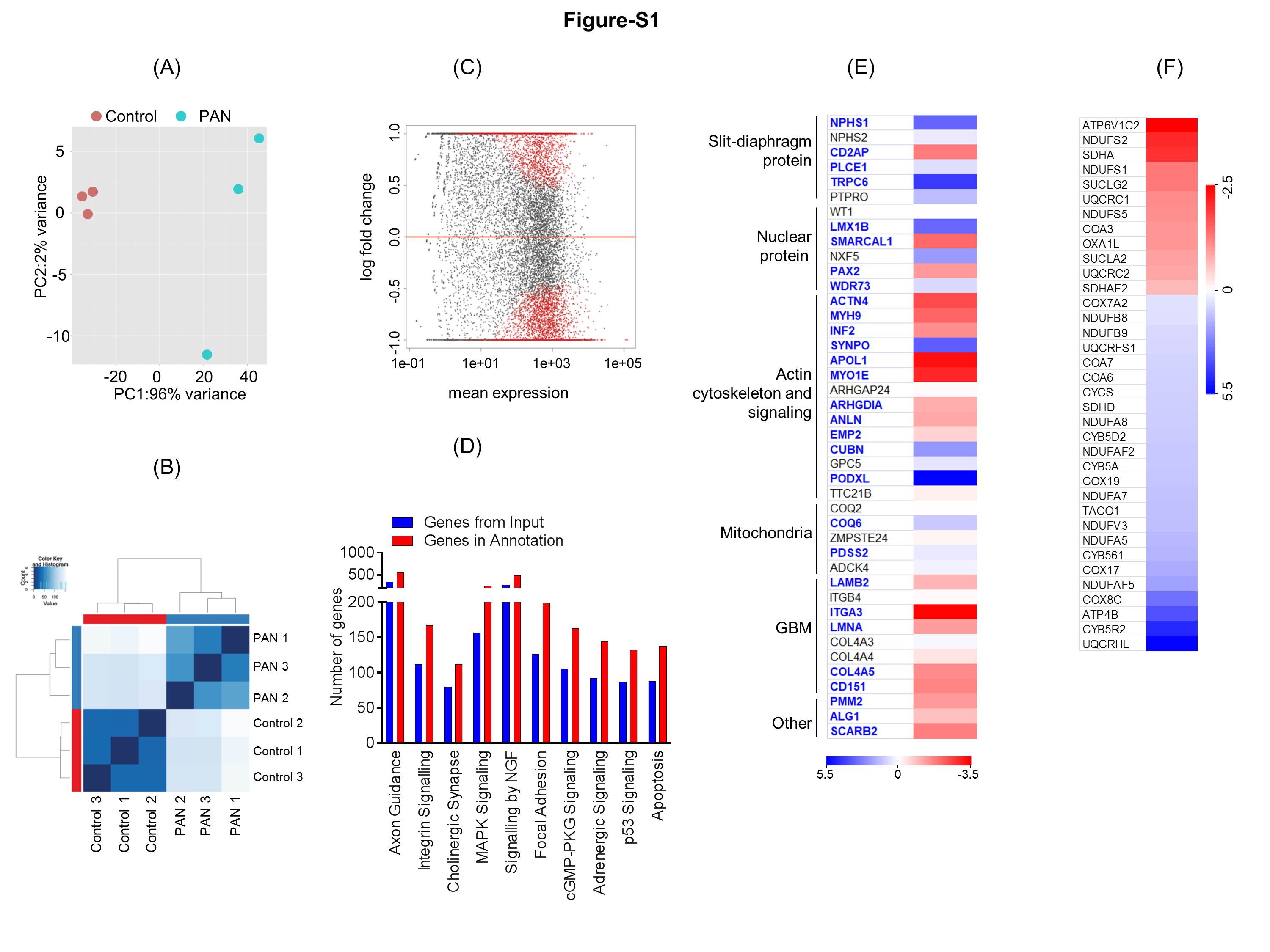
Figure S1: (A) Principal component analysis of RNA-seq gene expression data from biological replicates of control (vehicle treated) and PAN treated human podocytes (n=3). (B) MA plot of log2 fold changes in gene expression versus means of normalized counts showed DEGs between control and PAN treated human podocytes. DEGs with adjusted p-value <0.05 are represented by red dots. (C) Sample-to-sample distances are demonstrated. The heatmap shows Euclidean distances between the two samples as calculated from regularized log transformation. This distance matrix provides an overview of similarities and dissimilarities between the samples and reveals that the experimental samples fall into two distinct groups: control and PAN treated. (D) Gene Ontology (GO) analysis for the enrichment of major biological pathways was analyzed and plotted based on the statistical significance. Top ten enriched pathways (Padj < 0.05) were plotted. (E) Heat map was plotted from the RNA-Seq data against genes known to associate with nephrotic syndrome and were found to be significantly altered upon PAN injury in podocytes. Letters in blue and bold denote statistical significance (Padj < 0.05). (F) Heatmap of the RNA-seq data collected from PAN injured podocytes was plotted for mitochondrial electron transport chain DEGs (mitochondrial complex I to V).
{kind=link}
Figure S7: Formoterol treatment induced the expression of mitochondrial genes in mice glomeruli: (A) Markers of MB in isolated mice glomeruli were evaluated by western blotting using PGC-1α, OXPHOS (cocktail antibodies of mitochondrial ETC complex I to V) and GAPDH antibodies. (B–C) Densitometric analysis of immunoblots showed increased expression of PGC-1α and OXPHOS (mitochondrial complex I, II, III and IV) proteins in NTS+formoterol treated mice. One-way ANOVA, *P≤0.05, ***P≤0.001 NTS+vehicle vs. NTS+formoterol; #P≤0.05, ##P≤0.01, ###P≤0.001 vehicle vs NTS+formoterol. Mitochondrial complex I and II were significantly reduced upon NTS injury (NTS+vehicle Vs control). bP≤0.05, bbP≤0.00, NTS+vehicle vs. control vehicle. Data are presented as mean±SEM. (D) qPCR analysis further showed formoterol treatment upregulated the expression of PGC-α, NDUFB8, ND1, ND6, COXI, COXIII, CYTB, NRF1 and TFAM genes in NTS+formoterol treated mice. Data are presented in mean±SEM. One-way ANOVA, *P≤0.05, **P≤0.01, ***P≤0.001 NTS+vehicle vs. NTS+formoterol; aP≤0.01 control vs NTS+vehicle; #P≤0.05, ##P≤0.01 ###P≤0.001 control vs NTS+formoterol. (E) Analysis of mtDNA copy number from isolated glomeruli showed significant reduction in NTS+vehicle mice, but significant recovery was noted in NTS+formoterol mice (One way ANOVA, bbP≤0.01, control vs NTS+vehicle; *P≤0.05, NTS+vehicle vs NTS+formoterol, #P≤0.05, control vs NTS+formoterol). Data are presented as mean±SEM.
{kind=link}
Figure S8: Formoterol treatment induced expression of mitochondrial genes and mtDNA copy number in glomeruli of ADR-injured mice: (A) qPCR analysis of isolated glomeruli showed significant upregulation of PGC-α and NDUFB8 in ADR+formoterol treated mice. Data are presented in mean±SEM. One-way ANOVA, *P≤0.05 control vs. ADR+formoterol; #P≤0.05, ADR+vehicle vs ADR+formoterol). (B) The mtDNA copy number analysis showed that ADR treatment significantly reduced mtDNA in the mice glomeruli (ADR+vehicle) but addition of formoterol (ADR+formoterol) restored lost mtDNA. One way ANOVA, aP≤0.05, control vs NTS+vehicle; *P≤0.05, ADR+vehicle vs ADR+formoterol. Data are presented in mean±SEM.
{kind=link}
Figure S9: Formoterol treatment induced the expression of mitochondrial genes in mice glomeruli: (A) Immunostaining analysis of kidney sections using PGC-1α (Green) and Synaptopodin (Red) antibodies and DAPI (Blue) showed increased PGC-1α staining in the NTS+formoterol treated mice.
{kind=link}
Figure S10: Formoterol treatment induced expression of PGC-1α in cultured podocytes. (A) Human cultured podocytes treated with PS-injury and supplemented with vehicles or formoterol were immunostained with PGC-1α (Red) and DAPI (blue). Scale Bar = 20μm (B) Quantitative analysis showed significant upregulation of PGC-1α expression in formoterol treated podocytes. Data are presented in mean±SEM. One-way ANOVA, ***P≤0.0001 PS-injury+vehicle vs. PS-injury+formoterol
{kind=link}
Table S1: Sequences of qPCR primers for mice and human
Table S2: Sequences of primers used for analyzing mtDNA copy number in mice and human.
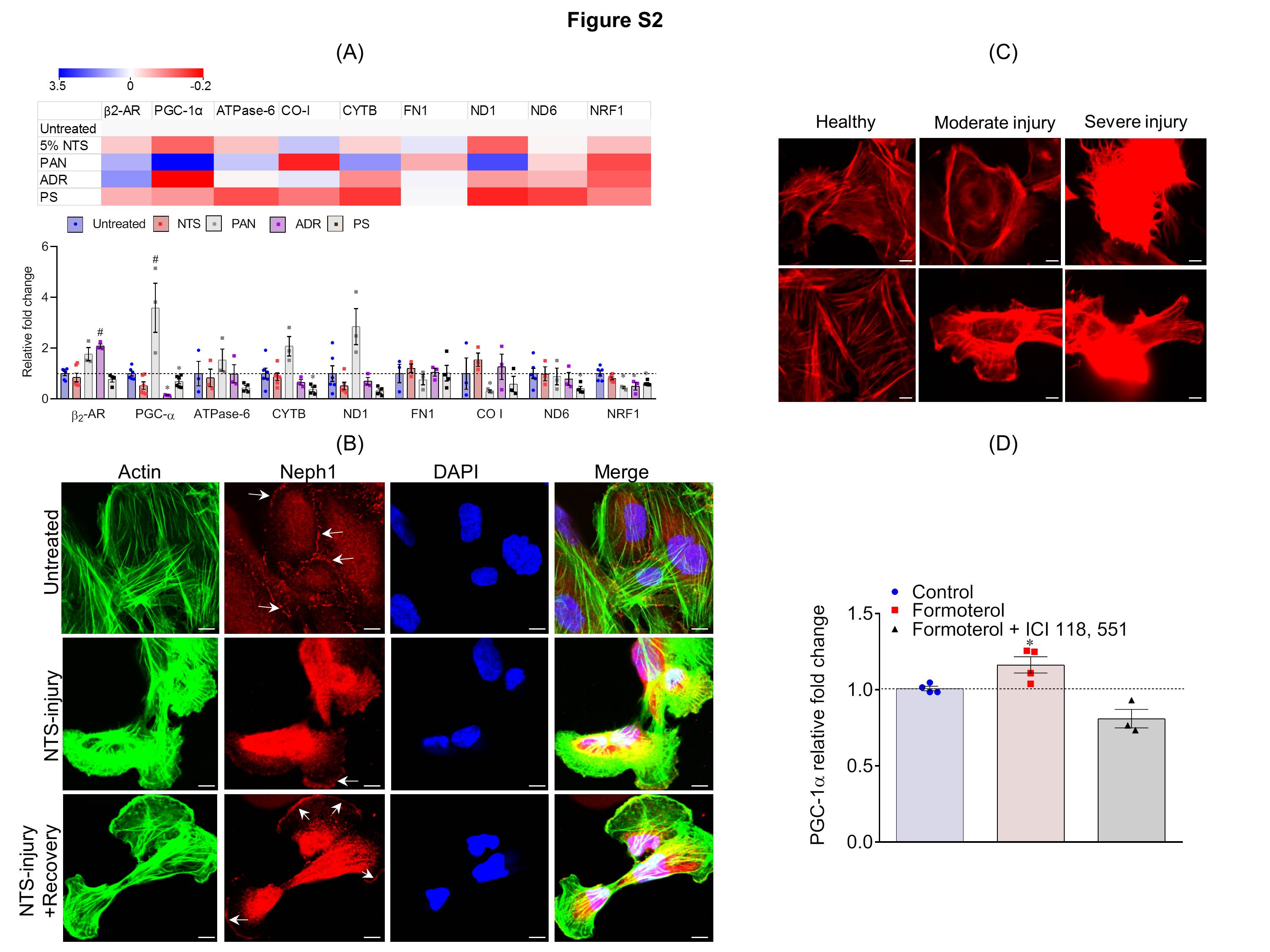
Figure S2: (A) qPCR analysis of the genes involved in mitochondrial function were analyzed in podocytes treated with various injury reagents including PAN, ADR, NTS (5%) and PS. Heatmap (upper panel) and bar graph (lower panel) were plotted that show differential expression of genes involved in mitochondrial dysfunction. Data are presented in mean±SEM. #P<0.05, control vs. Injury (up-regulated); *P<0.05, control vs. Injury (down-regulated). (B). Injury in cultured podocytes was defined at three levels, severe, moderate and healthy, based on the actin cytoskeleton morphology. Scale bar 20μm (C) Podocytes treated with NTS and immuno-stained with phalloidin (Green) showed accumulation of actin stress fibers at podocyte cells periphery and loss of Neph1 (Red) from podocyte membrane. Replacement with serum led to the restoration of actin cytoskeleton organization and Neph1 localization at the cell membrane. Scale bar 20μm. (D) Quantitative assessment of PGC-1α expression by qPCR shows that ICI-118,551 (β2-adrenergic receptor agonists) treatment attenuated the formoterol-induced increase of PGC-1α expression. *P<0.05, control vs. formoterol.
{kind=link}
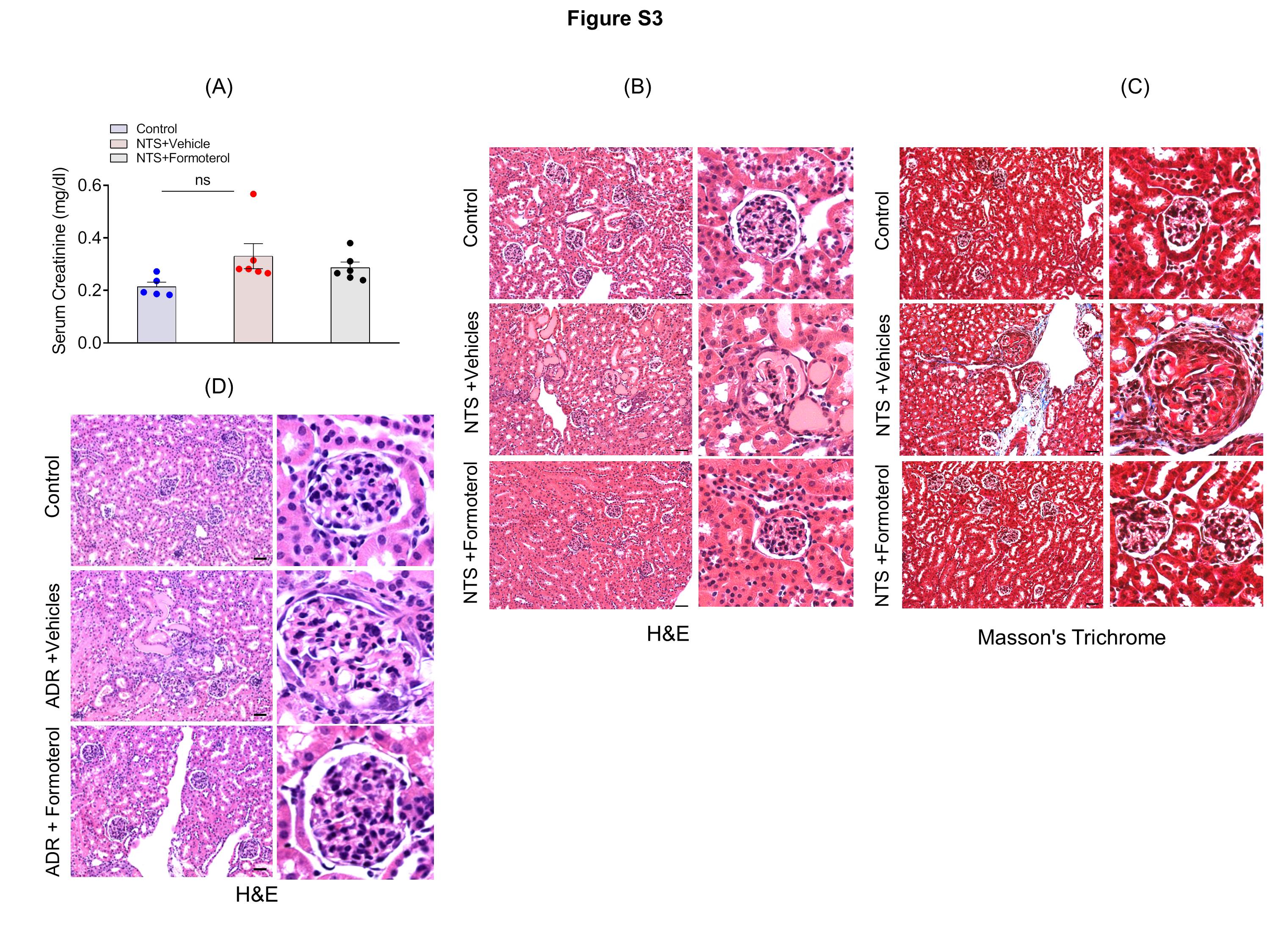
Figure S3: (A) Serum creatinine analysis among control, NTS+vehicle and NTS+formoterol group did not show any significant changes in the level (p>0.05). (B–C) Mice kidney sections were stained with hematoxylin and eosin (H&E) and Masson’s Trichrome for histological analysis. NTS+formoterol treated mice showed reduced focal atrophy, proteinaceous tubular caste and tubular dilation when compared to NTS+vehicle treated control mice. Scale bar 50μm. (D) H&E staining of mice kidney sections showed increased in ADR-induced glomerulosclerosis, and proteinaceous tubular caste (ADR+vehicle), which was significantly reduced upon formoterol treatment (ADR+formoterol). Scale bar 50μm.
{kind=link}
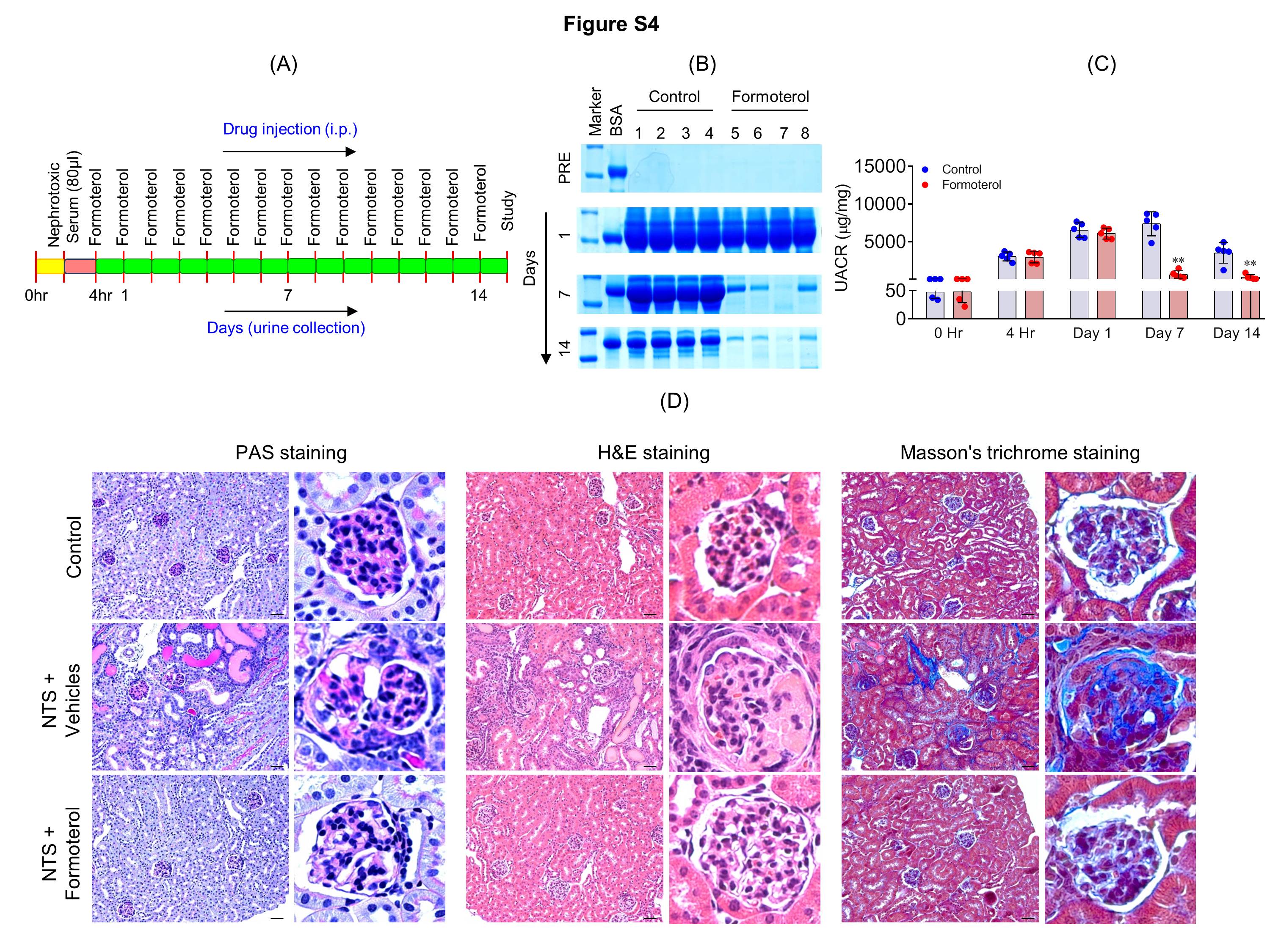
Figure S4: Formoterol treatment enhanced glomerular recovery in the extended model of NTS-induced glomerular injury. (A) The schematic of experimental plan for the extended model of NTS-injury. (B) Urine samples were analyzed by SDS-PAGE and Coomassie blue staining and showed significant reduction in albuminuria starting at day7, which persisted till day 14 in the NTS+formoterol treated mice group. (C) Measurement of urine albumin/creatinine ratios by ELISA showed initiation of albuminuria at 4h post NTS injection, which further increased at day 1 in both the groups. The ratios dropped significantly in NTS+formoterol treated mice group at days 7 and 14. 2-tailed t-test. n=5 mice per group. Data are presented in means±SEM. **P≤0.001 vs. control. (D) Histological analysis of mice (sacrificed at day 14 post NTS injection) kidney sections stained with PAS, H&E and Masson’s trichrome show reduced focal atrophy, proteinaceous tubular caste, tubular dilation and fibrosis in NTS+formoterol treated mice. Scale bar 50μm.
{kind=link}
Figure S5: Baseline Blood pressure measurements were made at 0, 4 and 24 hours after formoterol treatment. Figure shows summary of both systolic and diastolic blood pressure during each time point. Blood pressure decreased at 4 hours (about 20–25%), and returned to baseline at 24 hours of treatment.
{kind=link}
Figure S6: (A) The podocyte loss in mice sections was evaluated by immunostaining with WT1 antibody (Red) and DAPI (Blue). The WT1 positive cells were significant reduced in the glomeruli of NTS+vehicle mice, and treatment with formoterol restored WT1 positive cells to a larger extent. (B) Quantitative analysis showed increased number of WT1 positive cells in NTS+formoterol treated mice when compared to NTS+vehicle. Data are presented in mean±SEM. One-way ANOVA, ###P≤0.001 NTS+vehicle vs. control; **P≤0.01 NTS+vehicle vs. NTS+formoterol.
{kind=link}
TRANSLATIONAL STATEMENT.
The incidences of glomerular diseases such as FSGS are growing at a rapid pace, and are leading causes of nephrotic syndrome resulting in ESRD (End Stage Renal Failure) worldwide. Podocytes are the primary target in these glomerular diseases, and there are limited therapeutic agents available to treat podocytopathies. In this study, we report mitochondrial dysfunction as one of the critical events that participates in podocyte injury and we further determine that therapeutically targeting mitochondrial function prevents podocyte loss and function. We identified β2-adrenergic receptor agonist formoterol as the lead therapeutic compound with ability to induce mitochondrial biogenesis leading to podocyte recovery from injury. Using various in-vitro and in-vivo models of podocyte injury we demonstrate that formoterol enhanced the recovery of actin cytoskeleton reorganization post injury in cultured podocytes and prevented progression of proteinuria and preserved renal pathology in acute and chronic mouse models of glomerulopathy.
ACKNOWLEDGEMENTS
National Institutes of Health, NIDDK, Grant RO1 2R01DK087956-06A1 and R56 DK116887-01A1 to D.N. are duly acknowledged. NIH Grant GM084147 and the Biomedical Laboratory Research and Development Program of the Department of Veterans Affairs BX-000851 to R.G.S are duly acknowledged. Carl Gottschalk award to S.-H. K and Ben J. Lipps Research Fellowship to A.K.S from American Society of Nephrology are also acknowledged. We thank Babita Kumari for technical assistance in the laboratory.
Sources of support: This work was supported in whole or in part by the NIH Grants 2R01DK087956-06A1 & R56 DK116887-01A1 to D.N. and NIH Grant GM084147 (R.G.S) and the Biomedical Laboratory Research and Development Program of the Department of Veterans Affairs BX-000851 (R.G.S). The authors also thank the American Society of Nephrology for the Carl W Gottschalk Scholar Grant to S.H.K. and Ben J. Lipps Research Fellowship to A.K.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary information is available at Kidney International’s website: www.kidney-international.org.
REFERENCES
- 1.Reiser J and Altintas MM, Podocytes. F1000Res. 5 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haraldsson B, Nystrom J, and Deen WM, Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev. 88:451–487, 2008 [DOI] [PubMed] [Google Scholar]
- 3.Brinkkoetter PT, Ising C, and Benzing T, The role of the podocyte in albumin filtration. Nat Rev Nephrol. 9:328–336, 2013 [DOI] [PubMed] [Google Scholar]
- 4.Kitiyakara C, Kopp JB, and Eggers P, Trends in the epidemiology of focal segmental glomerulosclerosis. Semin Nephrol. 23:172–182, 2003 [DOI] [PubMed] [Google Scholar]
- 5.Assady S, Wanner N, Skorecki KL, and Huber TB, New Insights into Podocyte Biology in Glomerular Health and Disease. J Am Soc Nephrol. 28:1707–1715, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maisonneuve P, Agodoa L, Gellert R, Stewart JH, Buccianti G, Lowenfels AB, et al. , Distribution of primary renal diseases leading to end-stage renal failure in the United States, Europe, and Australia/New Zealand: results from an international comparative study. Am J Kidney Dis. 35:157–165, 2000 [DOI] [PubMed] [Google Scholar]
- 7.Rosenberg AZ and Kopp JB, Focal Segmental Glomerulosclerosis. Clin J Am Soc Nephrol. 12:502–517, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ranganathan S, Pathology of Podocytopathies Causing Nephrotic Syndrome in Children. Front Pediatr. 4:32, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muller-Deile J and Schiffer M, Podocytes from the diagnostic and therapeutic point of view. Pflugers Arch. 469:1007–1015, 2017 [DOI] [PubMed] [Google Scholar]
- 10.Korbet SM, Treatment of primary FSGS in adults. J Am Soc Nephrol. 23:1769–1776, 2012 [DOI] [PubMed] [Google Scholar]
- 11.Deegens JK and Wetzels JF, Immunosuppressive treatment of focal segmental glomerulosclerosis: lessons from a randomized controlled trial. Kidney Int. 80:798–801, 2011 [DOI] [PubMed] [Google Scholar]
- 12.Kveder R, Therapy-resistant focal and segmental glomerulosclerosis. Nephrol Dial Transplant. 18 Suppl 5:v34–37, 2003 [DOI] [PubMed] [Google Scholar]
- 13.Carney EF, Glomerular disease: autophagy failure and mitochondrial dysfunction in FSGS. Nat Rev Nephrol. 11:66, 2015 [DOI] [PubMed] [Google Scholar]
- 14.Hagiwara M, Yamagata K, Capaldi RA, and Koyama A, Mitochondrial dysfunction in focal segmental glomerulosclerosis of puromycin aminonucleoside nephrosis. Kidney Int. 69:1146–1152, 2006 [DOI] [PubMed] [Google Scholar]
- 15.Zhao M, Yuan Y, Bai M, Ding G, Jia Z, Huang S, et al. , PGC-1alpha overexpression protects against aldosterone-induced podocyte depletion: role of mitochondria. Oncotarget. 7:12150–12162, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weinberg JM, Venkatachalam MA, Roeser NF, Saikumar P, Dong Z, Senter RA, et al. , Anaerobic and aerobic pathways for salvage of proximal tubules from hypoxia-induced mitochondrial injury. Am J Physiol Renal Physiol. 279:F927–943, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feldkamp T, Kribben A, Roeser NF, Senter RA, and Weinberg JM, Accumulation of nonesterified fatty acids causes the sustained energetic deficit in kidney proximal tubules after hypoxia-reoxygenation. Am J Physiol Renal Physiol. 290:F465–477, 2006 [DOI] [PubMed] [Google Scholar]
- 18.Goto Y, Nonaka I, and Horai S, A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 348:651–653, 1990 [DOI] [PubMed] [Google Scholar]
- 19.Quinzii C, Naini A, Salviati L, Trevisson E, Navas P, Dimauro S, et al. , A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am J Hum Genet. 78:345–349, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heeringa SF, Chernin G, Chaki M, Zhou W, Sloan AJ, Ji Z, et al. , COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J Clin Invest. 121:2013–2024, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mollet J, Giurgea I, Schlemmer D, Dallner G, Chretien D, Delahodde A, et al. , Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J Clin Invest. 117:765–772, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Godel M, Hartleben B, Herbach N, Liu S, Zschiedrich S, Lu S, et al. , Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest. 121:2197–2209, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang W, Wang Y, Long J, Wang J, Haudek SB, Overbeek P, et al. , Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 15:186–200, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartleben B, Godel M, Meyer-Schwesinger C, Liu S, Ulrich T, Kobler S, et al. , Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. 120:1084–1096, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benoit G, Machuca E, and Antignac C, Hereditary nephrotic syndrome: a systematic approach for genetic testing and a review of associated podocyte gene mutations. Pediatr Nephrol. 25:1621–1632, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamagata K, Muro K, Usui J, Hagiwara M, Kai H, Arakawa Y, et al. , Mitochondrial DNA mutations in focal segmental glomerulosclerosis lesions. J Am Soc Nephrol. 13:1816–1823, 2002 [DOI] [PubMed] [Google Scholar]
- 27.Li SY, Park J, Qiu C, Han SH, Palmer MB, Arany Z, et al. , Increasing the level of peroxisome proliferator-activated receptor gamma coactivator-1alpha in podocytes results in collapsing glomerulopathy. JCI Insight. 2 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chuang PY, Cai W, Li X, Fang L, Xu J, Yacoub R, et al. , Reduction in podocyte SIRT1 accelerates kidney injury in aging mice. Am J Physiol Renal Physiol. 313:F621–F628, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Medeiros DM, Assessing mitochondria biogenesis. Methods. 46:288–294, 2008 [DOI] [PubMed] [Google Scholar]
- 30.Jesinkey SR, Funk JA, Stallons LJ, Wills LP, Megyesi JK, Beeson CC, et al. , Formoterol restores mitochondrial and renal function after ischemia-reperfusion injury. J Am Soc Nephrol. 25:1157–1162, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaw JM and Winge DR, Shaping the mitochondrion: mitochondrial biogenesis, dynamics and dysfunction. Conference on Mitochondrial Assembly and Dynamics in Health and Disease. EMBO Rep. 10:1301–1305, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saleem MA, Ni L, Witherden I, Tryggvason K, Ruotsalainen V, Mundel P, et al. , Co-localization of nephrin, podocin, and the actin cytoskeleton: evidence for a role in podocyte foot process formation. Am J Pathol. 161:1459–1466, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wagner MC, Rhodes G, Wang E, Pruthi V, Arif E, Saleem MA, et al. , Ischemic injury to kidney induces glomerular podocyte effacement and dissociation of slit diaphragm proteins Neph1 and ZO-1. J Biol Chem. 283:35579–35589, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tian X, Kim JJ, Monkley SM, Gotoh N, Nandez R, Soda K, et al. , Podocyte-associated talin1 is critical for glomerular filtration barrier maintenance. J Clin Invest. 124:1098–1113, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beeson CC, Beeson GC, and Schnellmann RG, A high-throughput respirometric assay for mitochondrial biogenesis and toxicity. Anal Biochem. 404:75–81, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wills LP, Trager RE, Beeson GC, Lindsey CC, Peterson YK, Beeson CC, et al. , The beta2-adrenoceptor agonist formoterol stimulates mitochondrial biogenesis. J Pharmacol Exp Ther. 342:106–118, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huber TB, Gloy J, Henger A, Schollmeyer P, Greger R, Mundel P, et al. , Catecholamines modulate podocyte function. J Am Soc Nephrol. 9:335–345, 1998 [DOI] [PubMed] [Google Scholar]
- 38.Boivin V, Jahns R, Gambaryan S, Ness W, Boege F, and Lohse MJ, Immunofluorescent imaging of beta 1- and beta 2-adrenergic receptors in rat kidney. Kidney Int. 59:515–531, 2001 [DOI] [PubMed] [Google Scholar]
- 39.Zheng CX, Chen ZH, Zeng CH, Qin WS, Li LS, and Liu ZH, Triptolide protects podocytes from puromycin aminonucleoside induced injury in vivo and in vitro. Kidney Int. 74:596–612, 2008 [DOI] [PubMed] [Google Scholar]
- 40.Vega-Warner V, Ransom RF, Vincent AM, Brosius FC, and Smoyer WE, Induction of antioxidant enzymes in murine podocytes precedes injury by puromycin aminonucleoside. Kidney Int. 66:1881–1889, 2004 [DOI] [PubMed] [Google Scholar]
- 41.Arif E, Rathore YS, Kumari B, Ashish F, Wong HN, Holzman LB, et al. , Slit diaphragm protein Neph1 and its signaling: a novel therapeutic target for protection of podocytes against glomerular injury. J Biol Chem. 289:9502–9518, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mundel P, Reiser J, Zuniga Mejia Borja A, Pavenstadt H, Davidson GR, Kriz W, et al. , Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res. 236:248–258, 1997 [DOI] [PubMed] [Google Scholar]
- 43.Sagar A, Arif E, Solanki AK, Srivastava P, Janech MG, Kim SH, et al. , Targeting Neph1 and ZO-1 protein-protein interaction in podocytes prevents podocyte injury and preserves glomerular filtration function. Sci Rep. 7:12047, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang W, Liu H, Zhu S, Woodson M, Liu R, Tilton RG, et al. , Sirt6 deficiency results in progression of glomerular injury in the kidney. Aging (Albany NY). 9:1069–1083, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zoja C, Garcia PB, Rota C, Conti S, Gagliardini E, Corna D, et al. , Mesenchymal stem cell therapy promotes renal repair by limiting glomerular podocyte and progenitor cell dysfunction in adriamycin-induced nephropathy. Am J Physiol Renal Physiol. 303:F1370–1381, 2012 [DOI] [PubMed] [Google Scholar]
- 46.Pavenstadt H, Kriz W, and Kretzler M, Cell biology of the glomerular podocyte. Physiol Rev. 83:253–307, 2003 [DOI] [PubMed] [Google Scholar]
- 47.Reidy KJ, Villegas G, Teichman J, Veron D, Shen W, Jimenez J, et al. , Semaphorin3a regulates endothelial cell number and podocyte differentiation during glomerular development. Development. 136:3979–3989, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim YH, Goyal M, Kurnit D, Wharram B, Wiggins J, Holzman L, et al. , Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN-treated rat. Kidney Int. 60:957–968, 2001 [DOI] [PubMed] [Google Scholar]
- 49.Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, et al. , Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol. 16:2941–2952, 2005 [DOI] [PubMed] [Google Scholar]
- 50.Wiggins RC, The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int. 71:1205–1214, 2007 [DOI] [PubMed] [Google Scholar]
- 51.Cybulsky AV, Takano T, Papillon J, Guillemette J, Herzenberg AM, and Kennedy CR, Podocyte injury and albuminuria in mice with podocyte-specific overexpression of the Ste20-like kinase, SLK. Am J Pathol. 177:2290–2299, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee VW and Harris DC, Adriamycin nephropathy: a model of focal segmental glomerulosclerosis. Nephrology (Carlton). 16:30–38, 2011 [DOI] [PubMed] [Google Scholar]
- 53.Yang HC, Zuo Y, and Fogo AB, Models of chronic kidney disease. Drug Discov Today Dis Models. 7:13–19, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, et al. , The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. 14:931–938, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shankland SJ, Eitner F, Hudkins KL, Goodpaster T, D’Agati V, and Alpers CE, Differential expression of cyclin-dependent kinase inhibitors in human glomerular disease: role in podocyte proliferation and maturation. Kidney Int. 58:674–683, 2000 [DOI] [PubMed] [Google Scholar]
- 56.Fukuda A, Wickman LT, Venkatareddy MP, Sato Y, Chowdhury MA, Wang SQ, et al. , Angiotensin II-dependent persistent podocyte loss from destabilized glomeruli causes progression of end stage kidney disease. Kidney Int. 81:40–55, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Strassheim D, Renner B, Panzer S, Fuquay R, Kulik L, Ljubanovic D, et al. , IgM contributes to glomerular injury in FSGS. J Am Soc Nephrol. 24:393–406, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fornoni A, Sageshima J, Wei C, Merscher-Gomez S, Aguillon-Prada R, Jauregui AN, et al. , Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci Transl Med. 3:85ra46, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Funk JA, Odejinmi S, and Schnellmann RG, SRT1720 induces mitochondrial biogenesis and rescues mitochondrial function after oxidant injury in renal proximal tubule cells. J Pharmacol Exp Ther. 333:593–601, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Uittenbogaard M and Chiaramello A, Mitochondrial biogenesis: a therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Curr Pharm Des. 20:5574–5593, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bayeva M, Gheorghiade M, and Ardehali H, Mitochondria as a therapeutic target in heart failure. J Am Coll Cardiol. 61:599–610, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Suliman HB and Piantadosi CA, Mitochondrial Quality Control as a Therapeutic Target. Pharmacol Rev. 68:20–48, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Granata S, Dalla Gassa A, Tomei P, Lupo A, and Zaza G, Mitochondria: a new therapeutic target in chronic kidney disease. Nutr Metab (Lond). 12:49, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mathieson PW, The podocyte cytoskeleton in health and in disease. Clin Kidney J. 5:498–501, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meng XM, Nikolic-Paterson DJ, and Lan HY, Inflammatory processes in renal fibrosis. Nat Rev Nephrol. 10:493–503, 2014 [DOI] [PubMed] [Google Scholar]
- 66.Velez JC, Arif E, Rodgers J, Hicks MP, Arthur JM, Nihalani D, et al. , Deficiency of the Angiotensinase Aminopeptidase A Increases Susceptibility to Glomerular Injury. J Am Soc Nephrol 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Scarpulla RC, Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 88:611–638, 2008 [DOI] [PubMed] [Google Scholar]
- 68.Miglio G, Rosa AC, Rattazzi L, Grange C, Camussi G, and Fantozzi R, Protective effects of peroxisome proliferator-activated receptor agonists on human podocytes: proposed mechanisms of action. Br J Pharmacol. 167:641–653, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Han SH, Wu MY, Nam BY, Park JT, Yoo TH, Kang SW, et al. , PGC-1alpha Protects from Notch-Induced Kidney Fibrosis Development. J Am Soc Nephrol 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Noh H, Yu MR, Kim HJ, Lee JH, Park BW, Wu IH, et al. , Beta 2-adrenergic receptor agonists are novel regulators of macrophage activation in diabetic renal and cardiovascular complications. Kidney Int. 92:101–113, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Haraldsson B, A new era of podocyte-targeted therapy for proteinuric kidney disease. N Engl J Med. 369:2453–2454, 2013 [DOI] [PubMed] [Google Scholar]
- 72.Wagner MC, Barylko B, and Albanesi JP, Tissue distribution and subcellular localization of mammalian myosin I. J Cell Biol. 119:163–170, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Arif E, Wagner MC, Johnstone DB, Wong HN, George B, Pruthi PA, et al. , Motor protein Myo1c is a podocyte protein that facilitates the transport of slit diaphragm protein Neph1 to the podocyte membrane. Mol Cell Biol. 31:2134–2150, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Irish JC, Mills JN, Turner-Ivey B, Wilson RC, Guest ST, Rutkovsky A, et al. , Amplification of WHSC1L1 regulates expression and estrogen-independent activation of ERalpha in SUM-44 breast cancer cells and is associated with ERalpha over-expression in breast cancer. Mol Oncol. 10:850–865, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kappler CS, Guest ST, Irish JC, Garrett-Mayer E, Kratche Z, Wilson RC, et al. , Oncogenic signaling in amphiregulin and EGFR-expressing PTEN-null human breast cancer. Mol Oncol. 9:527–543, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gerencser AA, Neilson A, Choi SW, Edman U, Yadava N, Oh RJ, et al. , Quantitative microplate-based respirometry with correction for oxygen diffusion. Anal Chem. 81:6868–6878, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang Y, Wang YP, Tay YC, and Harris DC, Progressive adriamycin nephropathy in mice: sequence of histologic and immunohistochemical events. Kidney Int. 58:1797–1804, 2000 [DOI] [PubMed] [Google Scholar]
- 78.Arif E, Solanki AK, and Nihalani D, Adriamycin susceptibility among C57BL/6 substrains. Kidney Int. 89:721–723, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fitzgibbon WR, Dang Y, Bunni MA, Baicu CF, Zile MR, Mullick AE, et al. , Attenuation of accelerated renal cystogenesis in Pkd1 mice by renin-angiotensin system blockade. Am J Physiol Renal Physiol. 314:F210–F218, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li L, Pan R, Li R, Niemann B, Aurich AC, Chen Y, et al. , Mitochondrial biogenesis and peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) deacetylation by physical activity: intact adipocytokine signaling is required. Diabetes. 60:157–167, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang Q, Wang J, Deng F, Yan Z, Xia Y, Wang Z, et al. , TqPCR: A Touchdown qPCR Assay with Significantly Improved Detection Sensitivity and Amplification Efficiency of SYBR Green qPCR. PLoS One. 10:e0132666, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Johnstone DB, Zhang J, George B, Leon C, Gachet C, Wong H, et al. , Podocyte-specific deletion of Myh9 encoding nonmuscle myosin heavy chain 2A predisposes mice to glomerulopathy. Mol Cell Biol. 31:2162–2170, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yu M, Zhou Y, Shi Y, Ning L, Yang Y, Wei X, et al. , Reduced mitochondrial DNA copy number is correlated with tumor progression and prognosis in Chinese breast cancer patients. IUBMB Life. 59:450–457, 2007 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: (A) Principal component analysis of RNA-seq gene expression data from biological replicates of control (vehicle treated) and PAN treated human podocytes (n=3). (B) MA plot of log2 fold changes in gene expression versus means of normalized counts showed DEGs between control and PAN treated human podocytes. DEGs with adjusted p-value <0.05 are represented by red dots. (C) Sample-to-sample distances are demonstrated. The heatmap shows Euclidean distances between the two samples as calculated from regularized log transformation. This distance matrix provides an overview of similarities and dissimilarities between the samples and reveals that the experimental samples fall into two distinct groups: control and PAN treated. (D) Gene Ontology (GO) analysis for the enrichment of major biological pathways was analyzed and plotted based on the statistical significance. Top ten enriched pathways (Padj < 0.05) were plotted. (E) Heat map was plotted from the RNA-Seq data against genes known to associate with nephrotic syndrome and were found to be significantly altered upon PAN injury in podocytes. Letters in blue and bold denote statistical significance (Padj < 0.05). (F) Heatmap of the RNA-seq data collected from PAN injured podocytes was plotted for mitochondrial electron transport chain DEGs (mitochondrial complex I to V).
Figure S7: Formoterol treatment induced the expression of mitochondrial genes in mice glomeruli: (A) Markers of MB in isolated mice glomeruli were evaluated by western blotting using PGC-1α, OXPHOS (cocktail antibodies of mitochondrial ETC complex I to V) and GAPDH antibodies. (B–C) Densitometric analysis of immunoblots showed increased expression of PGC-1α and OXPHOS (mitochondrial complex I, II, III and IV) proteins in NTS+formoterol treated mice. One-way ANOVA, *P≤0.05, ***P≤0.001 NTS+vehicle vs. NTS+formoterol; #P≤0.05, ##P≤0.01, ###P≤0.001 vehicle vs NTS+formoterol. Mitochondrial complex I and II were significantly reduced upon NTS injury (NTS+vehicle Vs control). bP≤0.05, bbP≤0.00, NTS+vehicle vs. control vehicle. Data are presented as mean±SEM. (D) qPCR analysis further showed formoterol treatment upregulated the expression of PGC-α, NDUFB8, ND1, ND6, COXI, COXIII, CYTB, NRF1 and TFAM genes in NTS+formoterol treated mice. Data are presented in mean±SEM. One-way ANOVA, *P≤0.05, **P≤0.01, ***P≤0.001 NTS+vehicle vs. NTS+formoterol; aP≤0.01 control vs NTS+vehicle; #P≤0.05, ##P≤0.01 ###P≤0.001 control vs NTS+formoterol. (E) Analysis of mtDNA copy number from isolated glomeruli showed significant reduction in NTS+vehicle mice, but significant recovery was noted in NTS+formoterol mice (One way ANOVA, bbP≤0.01, control vs NTS+vehicle; *P≤0.05, NTS+vehicle vs NTS+formoterol, #P≤0.05, control vs NTS+formoterol). Data are presented as mean±SEM.
Figure S8: Formoterol treatment induced expression of mitochondrial genes and mtDNA copy number in glomeruli of ADR-injured mice: (A) qPCR analysis of isolated glomeruli showed significant upregulation of PGC-α and NDUFB8 in ADR+formoterol treated mice. Data are presented in mean±SEM. One-way ANOVA, *P≤0.05 control vs. ADR+formoterol; #P≤0.05, ADR+vehicle vs ADR+formoterol). (B) The mtDNA copy number analysis showed that ADR treatment significantly reduced mtDNA in the mice glomeruli (ADR+vehicle) but addition of formoterol (ADR+formoterol) restored lost mtDNA. One way ANOVA, aP≤0.05, control vs NTS+vehicle; *P≤0.05, ADR+vehicle vs ADR+formoterol. Data are presented in mean±SEM.
Figure S9: Formoterol treatment induced the expression of mitochondrial genes in mice glomeruli: (A) Immunostaining analysis of kidney sections using PGC-1α (Green) and Synaptopodin (Red) antibodies and DAPI (Blue) showed increased PGC-1α staining in the NTS+formoterol treated mice.
Figure S10: Formoterol treatment induced expression of PGC-1α in cultured podocytes. (A) Human cultured podocytes treated with PS-injury and supplemented with vehicles or formoterol were immunostained with PGC-1α (Red) and DAPI (blue). Scale Bar = 20μm (B) Quantitative analysis showed significant upregulation of PGC-1α expression in formoterol treated podocytes. Data are presented in mean±SEM. One-way ANOVA, ***P≤0.0001 PS-injury+vehicle vs. PS-injury+formoterol
Table S1: Sequences of qPCR primers for mice and human
Table S2: Sequences of primers used for analyzing mtDNA copy number in mice and human.
Figure S2: (A) qPCR analysis of the genes involved in mitochondrial function were analyzed in podocytes treated with various injury reagents including PAN, ADR, NTS (5%) and PS. Heatmap (upper panel) and bar graph (lower panel) were plotted that show differential expression of genes involved in mitochondrial dysfunction. Data are presented in mean±SEM. #P<0.05, control vs. Injury (up-regulated); *P<0.05, control vs. Injury (down-regulated). (B). Injury in cultured podocytes was defined at three levels, severe, moderate and healthy, based on the actin cytoskeleton morphology. Scale bar 20μm (C) Podocytes treated with NTS and immuno-stained with phalloidin (Green) showed accumulation of actin stress fibers at podocyte cells periphery and loss of Neph1 (Red) from podocyte membrane. Replacement with serum led to the restoration of actin cytoskeleton organization and Neph1 localization at the cell membrane. Scale bar 20μm. (D) Quantitative assessment of PGC-1α expression by qPCR shows that ICI-118,551 (β2-adrenergic receptor agonists) treatment attenuated the formoterol-induced increase of PGC-1α expression. *P<0.05, control vs. formoterol.
Figure S3: (A) Serum creatinine analysis among control, NTS+vehicle and NTS+formoterol group did not show any significant changes in the level (p>0.05). (B–C) Mice kidney sections were stained with hematoxylin and eosin (H&E) and Masson’s Trichrome for histological analysis. NTS+formoterol treated mice showed reduced focal atrophy, proteinaceous tubular caste and tubular dilation when compared to NTS+vehicle treated control mice. Scale bar 50μm. (D) H&E staining of mice kidney sections showed increased in ADR-induced glomerulosclerosis, and proteinaceous tubular caste (ADR+vehicle), which was significantly reduced upon formoterol treatment (ADR+formoterol). Scale bar 50μm.
Figure S4: Formoterol treatment enhanced glomerular recovery in the extended model of NTS-induced glomerular injury. (A) The schematic of experimental plan for the extended model of NTS-injury. (B) Urine samples were analyzed by SDS-PAGE and Coomassie blue staining and showed significant reduction in albuminuria starting at day7, which persisted till day 14 in the NTS+formoterol treated mice group. (C) Measurement of urine albumin/creatinine ratios by ELISA showed initiation of albuminuria at 4h post NTS injection, which further increased at day 1 in both the groups. The ratios dropped significantly in NTS+formoterol treated mice group at days 7 and 14. 2-tailed t-test. n=5 mice per group. Data are presented in means±SEM. **P≤0.001 vs. control. (D) Histological analysis of mice (sacrificed at day 14 post NTS injection) kidney sections stained with PAS, H&E and Masson’s trichrome show reduced focal atrophy, proteinaceous tubular caste, tubular dilation and fibrosis in NTS+formoterol treated mice. Scale bar 50μm.
Figure S5: Baseline Blood pressure measurements were made at 0, 4 and 24 hours after formoterol treatment. Figure shows summary of both systolic and diastolic blood pressure during each time point. Blood pressure decreased at 4 hours (about 20–25%), and returned to baseline at 24 hours of treatment.
Figure S6: (A) The podocyte loss in mice sections was evaluated by immunostaining with WT1 antibody (Red) and DAPI (Blue). The WT1 positive cells were significant reduced in the glomeruli of NTS+vehicle mice, and treatment with formoterol restored WT1 positive cells to a larger extent. (B) Quantitative analysis showed increased number of WT1 positive cells in NTS+formoterol treated mice when compared to NTS+vehicle. Data are presented in mean±SEM. One-way ANOVA, ###P≤0.001 NTS+vehicle vs. control; **P≤0.01 NTS+vehicle vs. NTS+formoterol.