

Abstract
In the United States, the evolving landscape of state-legal marijuana use for recreational and/or medical purposes has given rise to flourishing markets for marijuana and derivative products. The popularity of these products highlights the relative absence of safety, pharmacokinetic, and drug interaction data for marijuana and its constituents, most notably the cannabinoids. This review articulates current issues surrounding marijuana terminology, taxonomy, and dosing; summarizes cannabinoid pharmacology and pharmacokinetics; and assesses the drug interaction risks associated with co-consuming marijuana with conventional medications. Existing pharmacokinetic data are currently insufficient to fully characterize potential drug interactions precipitated by marijuana constituents. As such, increasing awareness amongst researchers, clinicians, and federal agencies regarding the need to conduct well-designed in vitro and clinical studies is imperative. Mechanisms that help researchers navigate the legal and regulatory barriers to conducting these studies would promote rigorous evaluation of potential marijuana-drug interactions and inform health care providers and consumers about the possible risks of co-consuming marijuana products with conventional medications.
Keywords: cannabinoid, drug interaction, marijuana, natural product, pharmacokinetics
1. Introduction
Marijuana, or cannabis, is the most commonly consumed scheduled or illicit substance worldwide (Degenhardt et al., 2013; Vergara et al., 2017). State restrictions on access to marijuana and derivative products in the United States have been relaxing since 1996, fostering the appearance of state-legal dispensaries and an increasingly diverse market of commercially available products (Jikomes and Zoorob, 2018; Maxwell and Mendelson, 2016; Vergara et al., 2017). Patterns of consumer use are similarly in flux. In 2013, there were an estimated 181.8 million recreational users aged 15 to 64 years (WHO, 2016). By 2016, the percentage of individuals aged 12 years or older who were current marijuana users was higher than that from 2002 to 2015. This increase was due largely to increased consumption by young adults aged 26 years or older, whose use has increased more than that of young adults aged 18 to 25 years.
Given these changes, the absence of definitive data regarding constituent composition, pharmacokinetics, safety, and efficacy of marijuana products is increasingly conspicuous. These gaps in scientific knowledge about marijuana products are compounded by the lack of definitive information regarding the risk of co-consuming marijuana products with conventional drugs (both prescription and over-the-counter). The shifting legal landscape affords an unprecedented opportunity to study the drug interaction liability of marijuana amidst a wealth of information regarding constituents, consumption patterns, and safety data from both prescription and illicit synthetic derivatives.
As with most natural products, evaluating the drug interaction risk of marijuana and constituents (e.g., cannabinoids) is difficult due to the complex phytochemistry of the plant and the abundance of derivative products on the commercial market. However, marijuana is unique among natural products in that its constituent composition has been a subject of forensic examination for decades. Additionally, synthetic cannabinoids have been approved for prescription use by regulatory agencies in multiple countries. To facilitate increasing research on the pharmacokinetics and drug interaction risk of marijuana products, this review summarizes cannabinoid constituents, pharmacology, and pharmacokinetics in humans, as well as probable marijuana-drug interactions precipitated by concomitant consumption of marijuana products with conventional drugs.
2. Marijuana products and consumption
According to United States law, the Schedule I substance Cannabis sativa (C. sativa) encompasses all non-fibrous components of any marijuana strain (21 USC 802). Pragmatically, the botanical taxonomy of marijuana is far from standardized. Cannabis formally refers to a genus of flowering, dioecious plants from which a variety of psychoactive products are prepared; the term also may be used informally to refer to derivative products (WHO, 2016). Accordingly, both Cannabis and cannabis, as well as the common name of the plant (marijuana) are used throughout this review.
Although speciation of Cannabis is not well understood, probable taxonomic divisions include C. sativa, C. indica, C. ruderalis, and perhaps C. afghanica; in United States markets, most strains are varieties of C. sativa and C. indica (Dufresnes et al., 2017; Hillig, 2005; Small, 2015; Welling et al., 2016). In addition, “drug” strains cultivated for medicinal or recreational use tend to exhibit greater genetic variation than fibrous Cannabis (hemp) strains, probably because Cannabis is dioecious, and female plants are preferentially cultivated for their buds (Dufresnes et al., 2017; ElSohly et al., 2017).
Members of the genus Cannabis produce resin containing a host of phytocannabinoids, some with pharmacological utility. Primarily produced by epidermal trichomes and resin glands, which intersperse with the flowering buds of Cannabis, the bulk of the resin is concentrated in the floral clusters of female plants (de Pasquale et al., 1974). Psychoactive and therapeutic effects in humans are primarily a function of the relative content of two dibenzopyran cannabinoids produced by Cannabis varieties: [(−)Δ9-trans-(6aR, 10aR)-tetrahydrocannabinol] (THC), the primary psychoactive constituent, and cannabidiol (CBD), the primary non-psychoactive constituent (Figure 1A) (Grotenhermen, 2016). THC and CBD are the major cannabinoids in 11 C. sativa and C. indica strains (Fischedick et al., 2010). As such, cannabinoid studies in humans have focused largely on THC and CBD, and to some extent on cannabinol (CBN), an aromatized monoterpenoid derivative of THC present in marijuana smoke condensate (ElSohly and ElSohly, 2007). Although there is extreme inter-strain variability in THC:CBD ratios, in general, C. indica dominant strains, which are short and wide-leafed, tend to produce more CBD relative to THC; the taller and more narrow-leafed C. sativa dominant strains tend to produce more THC relative to CBD (Dufresnes et al., 2017; Hillig and Mahlberg, 2004).
Figure 1: Structures of THC, CBD, CBN, and their major metabolites.
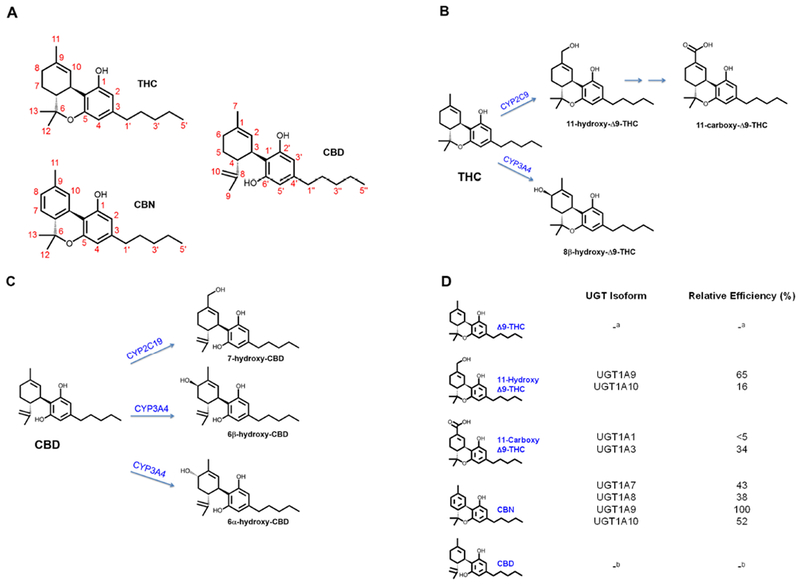
(A) Structures of THC, CBD, and CBN. (B) CYP2C9 catalyzes formation of the primary active THC metabolite, 11-OH-THC, which is subsequently oxidized to the inactive THC-COOH, whereas CYP3A4 catalyzes formation of a second primary, but inactive metabolite, 8β-OH-THC. (C) Primary metabolites of CBD are formed by CYP2C19 (7-OH-CBD) and CYP3A4 (6β-OH-CBD and 6α-OH-CBD). (D) Relative efficiency of glucuronide conjugation of THC and its metabolites, CBN, and CBD by various UGT enzymes, calculated as a percent of UGT1A9 Vmax/Km (Mazur et al., 2009).
a not determined due to lack of UGT activity against THC.
b not determined due to very low UGT activity toward CBD.
Routes of administration for marijuana and derivative preparations (e.g., wax, oils, and resins) include inhalation (smoking), oral consumption, or topical application. The increasing popularity of vaporizing instruments has produced a market for water-soluble emulsions or solutions of cannabinoids for inhalation. Oral forms (edibles) are often prepared by solubilizing constituents of the whole plant in lipids, incorporating into food products, and cooking to de-carboxylate cannabinoid constituents.
Dosing recommendations for marijuana remain nascent, albeit some states have defined a standard dose of THC per serving of edible product (e.g., 10 mg in Washington, California, and Colorado; 5 mg in Oregon). Precise dosing is complicated by inaccurate labeling, a problem inherent to the natural product market (Ekor, 2014; Raynor et al., 2011). For example, among marijuana products purchased online, 26% contained less CBD than indicated on the label (Bonn-Miller et al., 2017). Cannabinoid content of marijuana also has increased substantially over the last two decades such that the THC concentration of confiscated marijuana reportedly increased by 103% between 1998 and 2008 (ElSohly et al., 2016; Niesink et al., 2015). Recommended medical dosing with whole plant products is to “start low, go slow, and stay low,” titrating slowly over approximately two-week increments (MacCallum and Russo, 2018). Prescription cannabinoids in the United States are labeled with low initial doses for twice-daily administration (e.g., 2.5 mg dronabinol, 1 mg nabilone, 2.5 mg CBD) (Valeant Pharmaceuticals International, 2006; Greenwich Biosciences, Inc., 2018; AbbVie, Inc., 2017).
Adverse events reportedly ensuing from medical marijuana consumption appear to pertain primarily to THC content; thus, medical users are advised to consume THC with CBD to attenuate THC-associated anxiety and tachycardia and to limit the total daily dose-equivalent of THC to 30 mg/day or less. Higher doses of CBD likely are safe (MacCallum and Russo, 2018). In general, a joint contains approximately one-half gram of marijuana; however, differences in strain and cannabinoid composition preclude accurate estimation of the dose consumed after smoking one joint.
3. Major constituents
“Cannabinoid” originally referred to numerous structurally related C21 hydrocarbon compounds isolated from C. sativa; however, after THC was identified in 1964 and subsequently identified as the primary psychoactive constituent in marijuana, the term evolved to encompass compounds with pharmacological effects and target receptors similar to those of THC (Ford et al., 2017; Gaoni and Mechoulam, 1964; Stout and Cimino, 2014). As such, cannabinoids include structurally diverse compounds produced by plants (phytocannabinoids) (Hanus et al., 2016), humans (endocannabinoids) (Martin et al., 1999), or synthetic chemistry (synthetic cannabinoids) (Di Marzo and Petrocellis, 2006; Ford et al., 2017).
The phytocannabinoids in marijuana compose ten major sub-types (Elsohly and Slade, 2005). However, in total, the marijuana plant probably contains some 500 chemical constituents (ElSohly et al., 2016; ElSohly et al., 2017; Ross, 1995; WHO, 2016). Most of the known Cannabis constituents were identified between 1960 and 1990; more recently discovered constituents were isolated from “high potency” strains containing >20% THC by dry weight (Radwan et al., 2015). Different laboratories report varying cannabinoid content in popular commercial strains, with these between-lab differences persisting even after controlling for plausible confounds (Jikomes and Zoorob, 2018).
Synthetic cannabinoids are used as both prescription medications (e.g., dronabinol, nabilone) and drugs of abuse (e.g., “K2”, “Spice”). Other synthetic cannabinoids include the HU series (developed at the Hebrew University), the CP series (developed by Pfizer Inc.), and the JWH series (developed by JW Huffman) (Castaneto et al., 2014; Tait et al., 2016). Initially developed as pharmacological probes of the endogenous cannabinoid system, these synthetic cannabinoids may also be used as drugs of abuse. Synthetic cannabinoids not approved for human consumption have higher affinity for cannabinoid receptors and stronger pharmacologic effects than THC (Tait et al., 2016). As such, adverse effects are considered to be more intense than those observed with THC (Le Boisselier, et al., 2017).
4. Pharmacology
Marijuana has psychotropic effects, including both psychological stimulation and sedation, as well as somatic effects, including analgesia, antinociception, and orexigenia (Grotenhermen, 2003; Kumar et al., 2001). At high doses, marijuana induces anxiety, tachycardia, and hypertension (Grotenhermen, 2003). Indications for prescription marijuana products include chemotherapy-associated nausea and vomiting (dronabinol and nabilone), AIDS-associated anorexia (dronabinol), and rare epilepsy syndromes (CBD) (Valeant Pharmaceuticals International, 2006; Greenwich Biosciences, Inc., 2018; AbbVie, Inc., 2017). Although cannabinoids have been studied for an extremely wide range of other indications, CBD in particular is increasingly under investigation for indications that benefit from central nervous system cannabinoid receptor activation, including generalized anxiety, obsessive compulsive disorder, panic disorder, and psychosis (Blessing et al., 2015; van Amsterdam et al., 2018).
The complex endocannabinoid system underlying these effects, including endocannabinoid production mechanisms, cannabinoid receptors, and cannabinoid-degrading enzymes, is a phylogenetically ancient system that appears to be conserved in all vertebrates (Elphick and Egertova, 2005). The major endocannabinoids in humans are derivatives of arachidonic acid: N-arachidonylethanolamine, or anandamide; 2-arachidonylglycerol; 2-arachidonylglyceryl ether, or noladin ether; and N-arachidonyl-dopamine (Di Marzo, 2009; Grotenhermen, 2004; Kendall and Yudowski, 2016; Pertwee, 2002).
The mechanism of action of phyto-, endo-, and synthetic cannabinoids generally involves activation of two G-protein coupled cannabinoid receptors, CB1 and CB2, which were cloned in the early 1990s (Kumar et al., 2001; Pertwee, 1997). CB1 is expressed in both the central and peripheral nervous systems, whereas CB2 is expressed mainly in the peripheral nervous system, especially in spleen and thymus, and may contribute to the immunomodulatory actions of cannabinoids (Grotenhermen, 2004; Grotenhermen and Muller-Vahl, 2012; Kumar et al., 2001; Schatz et al., 1997). Activation of cannabinoid receptors inhibits production of adenyl cyclase and may modulate calcium and potassium channels (Grotenhermen, 2004; Kumar et al., 2001; Szabo and Schlicker, 2005) (Figure 2).
Figure 2: Effects of cannabinoid ligands on the central nervous system and peripheral nervous system.
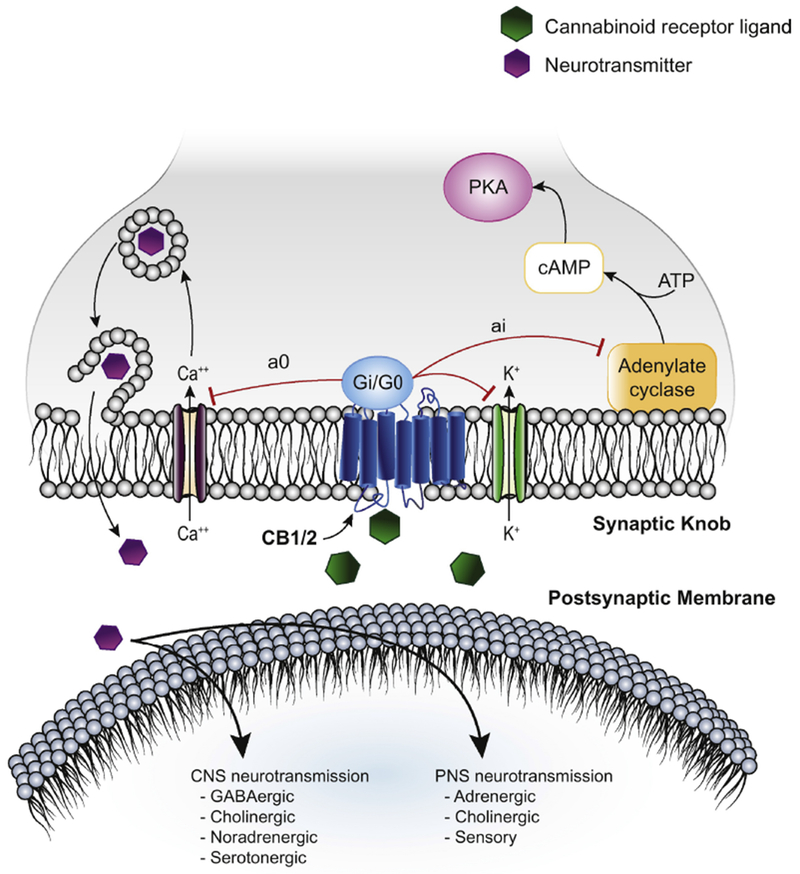
Activation of the cannabinoid 1 receptor (CB1) and cannabinoid 2 receptor (CB2) by cannabinoid ligands inhibits production of adenylate cyclase via activation of a coupled Gi/G0 G-protein subunit. CB1/2 activation also promotes exocytosis of a variety of neurotransmitters, which act broadly on the central and peripheral nervous system (CNS and PNS), perhaps by modulation of calcium and potassium transport.
The affinities of phyto- and endocannabinoids for CB1 and CB2 have been elucidated using competitive binding assays that measure displacement of the synthetic cannabinoid radioligand, [3H]-CP-55,940, or surrogate measures such as inhibition of cAMP production downstream of cannabinoid receptor activation (Felder et al., 1995; McPartland et al., 2007; Navarro et al., 2018; Showalter et al., 1996). Early experiments with CB1-overexpressing Chinese hamster ovary cell membranes and CB2-overexpressing mouse pituitary cells showed more potent displacement of [3H]-CP-55,940 by phytocannabinoids compared to endocannabinoids (Felder et al., 1995). For example, THC was a stronger displacer than anandamide at CB1 by an order of magnitude (Ki, 53 versus >500 nM), and THC and CBN were stronger displacers than anandamide and adrenyl-ethanolamide at CB2 (Ki, <300 versus >2000 nM) (Felder et al., 1995). THC (100 nM) also inhibited forskolin-stimulated cAMP production by ≥50% compared to control in Chinese hamster ovary cells overexpressing CB1 and CB2 (Rinaldi-Carmona et al., 1996). A more recent study reported that cannibigerol displaced [3H]-CP-55,940 at concentrations >1000 nM (Navarro et al., 2018). A partial agonist of CB2, cannabigerol (100 nM) reduced the effect of synthetic CB1/CB2 agonists on cAMP production in cells over-expressing both CB1 and CB2 (Navarro et al., 2018).
These observations have implications about the mechanism of action of phytocannabinoids. The relatively potent inhibition of radioligand binding to CB1 and CB2 in these in vitro systems suggests that the phytocannabinoids exhibit higher affinity for human CB1 and CB2 compared to some endocannabinoids. Although THC is generally believed to be an agonist of both CB1 and CB2, with effects and affinities similar to those of the endocannabinoids anandamide and 2-arachidonoylglycerol, THC has been reported to have pharmacologic effects akin to cannabinoid receptor antagonists. Other investigators have reported THC to be a CB2 antagonist. These conflicting observations are discussed in detail elsewhere (Pertwee, 2008).
The aforementioned inhibition data indicate preferential binding of cannabinoids to cannabinoid receptors. For example, THC displaced [3H]-CP-55,940 from CB2 by a greater extent (1.4 times) than from CB1, and CBN displaced [3H]-CP-55,940 from CB1 by a greater extent (3.8 times) than from CB2 (Felder et al., 1995). CBD was reported to bind weakly to CB1 and CB2 (Ki, >10 μM and 1.8 μM, respectively), whereas a synthesized enantiomer, (+)-CBD, bound more strongly to CB1 and CB2 (Ki, 17 nM and 211 nM, respectively) (Hanus et al., 2005). These data suggest that CBD does not exert its pharmacological effects via cannabinoid receptors (Hanus et al., 2005). More recently, CBD has been proposed to exert its pharmacological effects via binding to 5-HT (serotonin) receptors (Martinez-Pinilla et al., 2017).
5. Pharmacokinetics
The pharmacokinetics of primary phytocannabinoids have been reviewed extensively (Agurell et al., 1986; Grant et al., 2017; Grotenhermen, 2003; Huestis, 2007; Lucas et al., 2018; Stout and Cimino, 2014). The following section summarizes the pharmacokinetic properties of THC and CBD, and to some extent CBN, as they relate to the risk of marijuana-drug interactions.
5.1. Absorption
The rate of cannabinoid absorption into the systemic circulation varies with route of administration. When inhaled (i.e., smoked), systemic absorption of THC, CBD, and CBN is rapid, with maximum plasma concentrations (Cmax) achieved within 0.02-0.5 hours (1.2-30 minutes) (Table 1 and Supplementary Table 2; note that Table 1 summarizes data from the Supplementary Tables). After oral consumption (i.e., as a prescription drug or in food or oil), the Cmax of THC, CBD, and CBN is achieved within 1-3.4 hours (Supplementary Table 3). Like the rate of absorption, the extent of absorption of cannabinoids into the systemic circulation - as assessed by Cmax and area under the plasma concentration versus time curve (AUC) - varies substantially with route of administration (Figure 3, Figure 4). Compared to oral administration, mean dose-normalized Cmax for THC, CBD, and CBN was 3-35 times higher, and the AUC for THC was two times higher, after smoking. Cmax and AUC for THC, CBD, and CBN were similar between oral administration and use of an oral mucosal spray (Table 1).
Table 1.
Dose-normalized Cmax and AUC of key cannabinoids and metabolites in humans after various routes of administration of cannabinoids.
| Cannabinoid | Route of
Administration |
|||
|---|---|---|---|---|
| Intravenous | Inhalation | Oral | Oral mucosal | |
| Dose range (mg) | ||||
| THC | 0.5-5.0 | 8.9-70 | 2.5-90 | 5.4-22 |
| CBD | 20 | 2.0-19 | 1.5-800 | 5.0-20 |
| CBN | 20 | 1.6-19 | 3.3-40 | - |
| Cmax/Dose (nM/mg) | ||||
| THC | 130 [90.0-200] | 9.9 [7.0-14.1] | 1.50 [1.32-1.71] | 1.47 [0.63-3.39] |
| 11-OH-THCa | 4.7 [2.7-8.1] | 0.65 [0.40-1.05] | 1.10 [0.90-1.34] | 1.48 [1.05-2.10] |
| COOH-THCa | 23.4 [15.5-35.4] | 3.73 [2.31-6.00] | 13.3 [9.74-18.0] | 22.7-58.1c |
| CBD | 109b | 18.2b | 0.60 [0.31-1.51] | 0.50 [0.20-1.3] |
| CBN | 67.4b | 20.6b | 0.58b | - |
| AUC/Dose (min/L) | ||||
| THC | 1.5 [0.90-2.4] | 0.14 [0.10-0.21] | 0.07 [0.05-0.10] | 0.11 [0.03-0.34] |
| 11-OH-THCa | 0.15-0.50c | 0.03 [0.02-0.05] | 0.14 [0.12-0.16] | 0.16 [0.11-0.22] |
| COOH-THCa | - | 0.33 [0.20-0.55] | 3.7 [2.2-6.3] | 3.4-9.6c |
| CBD | 0.83b | 0.25b | 0.06-0.11c | 0.03 [0.02-0.07] |
| CBN | 0.76b | 0.28b | - | - |
Values denote geometric means and 95% confidence intervals or range of data presented in the Supplementary Tables. Dashes indicate data were not available.
Normalized to administered dose of THC
Data from one available study
Data depict range from two studies
Figure 3: Effect of route of administration on dose-normalized Cmax of THC, 11-OH-THC, COOH-THC, CBD, and CBN.

Dose-normalized Cmax after administration of parent cannabinoid (THC, CBD, or CBN). 11-OH-THC and COOH-THC were measured after administration of THC via various routes; metabolite Cmax was normalized to the administered THC dose. Symbols represent mean values from individual studies (see Supplementary Table). Horizontal and vertical lines represent geometric means and corresponding 95% confidence intervals, respectively. Data were not available for CBN administered via oral mucosal spray.
Figure 4: Effect of route of administration on dose-normalized AUC of THC, 11-OH-THC, COOH-THC, CBD, and CBN.

Dose-normalized AUC after administration of parent cannabinoid (THC, CBD, or CBN). 11-OH-THC and COOH-THC were measured after administration of THC via various routes; metabolite AUC was normalized to the administered THC dose. Symbols represent mean values from individual studies (see Supplementary Table). Horizontal and vertical lines represent geometric means and corresponding 95% confidence intervals, respectively. Data were not available for COOH-THC following intravenous administration of THC nor for CBN administered orally and via oral mucosal spray.s
Assuming linear pharmacokinetics, the estimated absolute bioavailability of THC after inhalation, oral administration, or administration by oral mucosal spray is low (5-7%), suggesting substantial pre-systemic (first-pass) metabolism and/or incomplete absorption into the systemic circulation (Table 1). Incomplete absorption may be the primary driver of the low bioavailability of THC from marijuana smoke, as 23-30% of THC is lost to pyrolysis, and another 40-50% is lost to non-inhaled smoke, leaving 20-37% of the initial THC amount available for pulmonary absorption (Perez-Reyes, 1990). Similarly, the absolute bioavailability of CBD after inhalation is ~30%, whereas that after oral administration (7-13%) or by oral mucosal spray (4%) is much lower (Table 1). These data suggest substantial first-pass metabolism and/or incomplete absorption of CBD after extravascular administration.
5.2. Distribution
Cannabinoids sequester in fatty tissues due to their high lipophilicity (logP for THC, CBD, and CBN is 6.97, 5.79, and 4.38, respectively) (Levitt, 2007; Thomas et al., 1990). Animal studies show the highest accumulation in adipose, liver, and lung tissue (Kreuz and Axelrod, 1973). THC is highly protein bound, primarily to lipoproteins, with a fraction unbound in plasma of less than 5% (Klausner et al., 1975). Human blood-to-plasma ratios range from 0.39-0.63 for THC, 0.55-0.59 for the acid metabolite of THC (COOH-THC), and 0.60 for the primary metabolite of THC, 11-OH-THC (Karschner et al., 2012). These data indicate that THC and these metabolites do not partition appreciably into erythrocytes.
5.3. Metabolism
Cytochrome P450 (CYP) 2C9 is the major enzyme that catalyzes the oxidation of THC to the primary metabolite, 11-OH-THC, which is also psychoactive; 11-OH-THC is oxidized further to the inactive COOH-THC (Figure 1B) (Bland et al., 2005; Bornheim et al., 1992). Compared to healthy human participants homozygous for the CYP2C9 reference allele (CYP2C9*1), participants homozygous for the reduced functional allele (CYP2C9*3) showed a three-fold higher median THC plasma AUC when administered a single oral dose of THC (15 mg); the corresponding median COOH-THC AUC was 70% lower in CYP2C9*3/*3 carriers compared to CYP2C9*1/*1 carriers (Sachse-Seeboth et al., 2009). CYP3A4 is primarily responsible for catalyzing the oxidation of THC to the second primary but inactive metabolite, 8β-OH-THC (Figure 1B) (Bornheim et al., 1992; Watanabe et al., 2007), which is stereoselectively preferred over 8α-OH-THC. Although other phytocannabinoids and metabolites have other therapeutic effects, such as analgesia, they do not produce appreciable psychoactive effects in humans; thus, THC and 11-OH-THC are generally considered the psychoactive constituents in marijuana (Awasthi et al., 2018; Sharma et al., 2012).
CBD is oxidized to form hydroxylated metabolites (Figure 1C). Side-chain oxidation forms seven other metabolites (Jiang et al., 2011). Formation of 7-OH-CBD is catalyzed primarily by CYP2C19 based on metabolite profiling with recombinant enzymes, a high correlation between CBD 7-hydroxylation and (S)-4’-mephenytoin hydroxylation in phenotyped human liver microsomes, and inhibition of this pathway in human liver microsomes by omeprazole (CYP2C19 inhibitor) but not sulfaphenazole (CYP2C9 inhibitor) (Jiang et al., 2011). CYP3A4 generates other minor metabolites, including several with alcohols on the pentyl side chain of CBD (Figure 1C) (Jiang et al., 2011). One case report showed that CBD is metabolized to more than 30 urinary metabolites (Harvey and Mechoulam, 1990). Notably, numbering systems are a source of confusion in the marijuana literature, as the metabolically labile allylic sites at C-11 and C-8 in Δ9-THC are equivalent to C-7 and C-6 in CBD. For example, in older literature, Δ9-THC is sometimes referred to as Δ1-THC (using the monoterpenoid numbering system).
Dose-normalized Cmax and AUC of the THC metabolites 11-OH-THC and COOH-THC after administration of THC varies with route of administration (Figure 3, 4 and Table 1). After inhalation, mean dose-normalized Cmax of THC was higher than that of 11-OH-THC and COOH-THC, whereas the AUC of COOH-THC was higher than that of 11-OH-THC and THC; both the Cmax and AUC of COOH-THC was higher than those of 11-OH-THC. After administration of THC orally and as an oral mucosal spray, mean dose-normalized Cmax and AUC of COOH-THC were higher than those of THC and 11-OH-THC (Table 1). These observations may be due to a relatively slower clearance and/or smaller volume of distribution of COOH-THC compared to that of THC and 11-OH-THC.
Of the major phytocannabinoids, only CBN is glucuronidated (Mazur et al., 2009). CBN glucuronidation is catalyzed by the UDP-glucuronosyltransferase (UGT) 1A subfamily, with Km values ranging from 3.4 μM (UGT1A9) to 59 μM (UGT1A10) (Mazur et al., 2009). 11-OH-THC is glucuronidated by UGT1A9 and UGT1A10, with Km values of 7.3 and 72 μM, respectively. COOH-THC is glucuronidated by UGT1A1 and UGT1A3, with Km values of 170 and 68 μM, respectively.
5.4. Elimination
Plasma concentration-time profiles for THC and CBD are qualitatively similar and can be described by two- or three-compartment pharmacokinetics (Heuberger et al., 2015; Hunt and Jones, 1980; Wall et al., 1983). The initial half-life after inhalation ranges from 1.4-12.8 minutes, whereas the terminal half-life has been estimated at 20-30 hours (Heuberger et al., 2015; Ohlsson et al., 1986). Assuming an average weight of 70 kg, mean THC plasma clearance after intravenous administration was 9.9 ml/min/kg and ranged from 2.8-14 ml/min/kg, indicating a low to medium hepatic extraction ratio (Grotenhermen, 2003). Mean THC plasma clearance was reported to be higher in men than in women (14 ± 4.8 vs. 8.1 ± 5.2 ml/min/kg) (Naef et al., 2004), suggesting a potential sex-related difference in THC clearance. In other reports, mean plasma clearance of CBD and CBN after intravenous administration to small groups of young men (n < 9) was comparable to or higher than that of THC (12-20 and 16-22 vs. 11-17 ml/min/kg, respectively), assuming an average weight of 70 kg (Ohlsson et al., 1982; Ohlsson et al., 1986; Johansson et al., 1987). Additional studies involving larger numbers of subjects are needed to confirm these observations.
Urinary excretion of THC and 11-OH-THC persists for up to 3 days after a single dose of THC and for more than 3 weeks in chronic marijuana smokers (Johansson et al., 1989; Leighty et al., 1976; Lowe et al., 2009; Skopp and Potsch, 2008). Thus, differences in study design produce wide variations in measurements for excretion of THC and metabolites. Although some studies report 20% urinary excretion of THC and metabolites, these results are likely to be time-dependent, as only 0.22% of an 8-mg THC dose was excreted in urine as THC and metabolites during the 0-8 hour collection interval after consumption via smoking (Brenneisen et al., 2010; Hunt and Jones, 1980; Wall et al., 1983).
THC and CBD, along with their alcohol and acid metabolites, can in principal be converted to their ether and/or acyl glucuronides by conjugation with glucuronic acid; such metabolites have been detected in human plasma and urine (Figure 1D) (Desrosiers et al., 2014). After intravenous administration of [3H]-labeled THC, 20% of the total radioactive dose was excreted into urine as conjugated metabolites, and 80% was excreted into the feces. Of the THC excreted into the feces, 20% represented unconjugated 11-OH-THC, and 28% represented COOH-THC (Wall et al., 1983). Less than 5% of THC was excreted unchanged in the feces. Unlike THC, a large proportion of CBD was excreted unchanged into the feces after intravenous administration (Ohlsson et al., 1986). The large fraction of 11-OH-THC and CBD excreted into the feces suggests transport of these compounds into the bile by canalicular efflux transporters [e.g., P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), and/or multi-drug resistance associated protein 2 (MRP2)].
5.5. Pharmacogenomics
The majority of studies evaluating the pharmacogenomics of cannabinoids have focused on those that modulate the risk of dependency (Hryhorowicz et al., 2018). Because cannabinoids are substrates for P-gp (Bonhomme-Faivre et al., 2008; Brzozowska et al., 2016; Spiro et al., 2012; Zhu et al., 2006) and BCRP (Brzozowska et al., 2016; Spiro et al., 2012), polymorphisms in either gene that encodes these proteins (ABCB1 and ABCG2, respectively) could influence cannabinoid disposition and the likelihood of marijuana-drug interactions mediated by either transporter. As described earlier (section 5.3), among the enzymes that catalyze THC metabolism in humans (primarily CYP2C9, CYP2C19, and CYP3A4), the CYP2C9*2 and CYP2C9*3 polymorphisms may lead to reduced formation of 11-OH-THC (Sachse-Seeboth et al., 2009).
6. Drug interactions
6.1. In vitro and animal studies
6.1.1. Interactions with cytochrome P450s
Cannabinoids exhibit multifaceted interactions with the CYPs (Alsherbiny and Li, 2018). For example, THC induced human CYP1A1 mRNA expression in a murine hepatoma cell line (Hepa-1) expressing an inducible CYP1A1 gene, possibly via interactions with the aryl hydrocarbon receptor (Roth et al., 2001). However, THC inhibited CYP1A/1B1 activity (7-ethoxyresorufin O-deethylation) in human liver microsomes (Table 2). THC has been reported to inhibit the activity of other CYPs in human liver microsomes and recombinant systems, with Ki values ranging from 1.3 μM (CYP2C9) to 29 μM (CYP2A6) (Table 2, Table 3). THC also has shown time-dependent inhibition of CYP1A1 and CYP2A6 activity in recombinant systems with similar efficiency (kinact/KI, ~0.02 min−1μM−1) (Table 4).
Table 2.
Inhibition of cytochrome P450 activities in human liver microsomes by cannabinoids.
| Enzyme(s) | Substrate | Reaction | Cannabinoid (μM) | Reference | |||||
|---|---|---|---|---|---|---|---|---|---|
| THC | CBD | CBN | |||||||
| Ki | IC50 | Ki | IC50 | Ki | IC50 | ||||
| CYP1A/1B1 | 7-Ethoxyresorufin | O-deethylation | 4.7 | - | 1.8 | - | 0.081 | - | (Yamaori et al., 2010) |
| CYP2D6 | Dextromethorphan | O-demethylation | - | 23 | 2.4 | - | - | 25 | (Yamaori et al., 2011) |
| CYP2C9 | (S)-Warfarin | 7-hydroxylation | 1.5 | - | 5.6 | - | 0.9 | - | (Yamaori et al., 2012) |
| CYP2C9 | Diclofenac | 4’-hydroxylation | 1.3 | - | 9.9 | - | 1.3 | - | |
| CYP3A4/5 | Diltiazem | N-demethylation | - | - | 6.1 | - | - | - | (Yamaori et al., 2011) |
Dashes indicate data were not available.
Table 3.
Inhibition of cytochrome P450 activities in recombinant enzymes or transfected cell systems by cannabinoids.
| Enzyme | Substrate | Reaction | Cannabinoid (μM) | Reference | |||||
|---|---|---|---|---|---|---|---|---|---|
| THC | CBD | CBN | |||||||
| Ki | IC50 | Ki | IC50 | Ki | IC50 | ||||
| CYP1A1 | 7-Ethoxyresorufin | O-deethylation | 4.8 | - | 0.16 | - | 0.54 | - | (Yamaori et al., 2010) |
| CYP1A2 | 7.5 | - | 2.7 | - | 0.08 | - | |||
| CYP1B1 | 2.5 | - | 3.6 | - | 0.15 | - | |||
| CYP2A6 | Coumarin | 7-hydroxylation | 29 | - | 55 | - | 40 | - | (Yamaori et al., 2011) |
| CYP2B6 | 7-Benzoxyresorufin | O-debenzylation | 2.8 | - | 0.69 | - | 2.6 | - | |
| CYP2D6 | Dextromethorphan | O-demethylation | - | 17 | 2.7 | - | - | 12 | (Yamaori et al., 2011) |
| CYP2D6 | AMMC | O-demethylation | - | 21 | 1.2 | - | - | 24 | |
| CYP2C9 | (S)-Warfarin Diclofenac | 7-hydroxylation 4’-hydroxylation |
1.4 1.4 |
- | 1.0 2.3 |
- | 0.9 1.3 |
- | (Yamaori et al., 2012) |
| CYP2C19 | (S)-Mephenytoin | 4’-hydroxylation | 1.93 | - | 0.79 | - | - | - | (Jiang et al., 2013) |
| CYP3A4 | Diltiazem | N-demethylation | - | >50 | 1.0 | - | - | >50 | (Yamaori et al., 2011) |
| CYP3A5 | - | 36 | 0.2 | - | - | >50 | |||
| CYP3A7 | - | 30 | 12 | - | - | 24 | |||
Table adapted from (Stout and Cimino, 2014; Zendulka et al., 2016). Dashes indicate data were not available. AMMC, 3-[2-(N,N-diethyl-N-methylammonium)ethyl]-7-methoxy-4-methylcoumarin iodide.
Table 4.
Time-dependent inhibition of recombinant cytochrome P450 activity by cannabinoids.
| Enzyme | Substrate | Reaction | THC | CBDa | CBN | Reference | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KI (μM) | kinact (min−1) | kinact/KI (min−1μM−1) | KI (μM) | kinact (min−1) | kinact/KI (min−1μM−1) | KI (μM) | kinact (min−1) | kinact/KI (min−1μM−1) | ||||
| CYP1A1 | 7-Ethoxyresorufin | O-deethylation | 1.2 | 0.023 | 0.019 | 5.9 | 0.316 | 0.054 | 0.09 | 0.019 | 0.21 | (Yamaori et al., 2010) |
| CYP1A2 | - | - | - | 0.59 | 0.109 | 0.19 | - | - | - | |||
| CYP1B1 | - | - | - | 3.1 | 0.027 | 0.0087 | - | - | - | |||
| CYP2A6 | Coumarin | 7-hydroxylation | 0.86 | 0.017 | .020 | - | - | - | 1.01 | 0.0091 | 0.0090 | (Yamaori et al., 2011) |
Also showed time-dependent inhibition of 7-ethoxyresorufin O-deethylation in human liver microsomes (kinact, 0.152 min−1; KI, 2.1 μM). Dashes indicate data were not available.
Like THC, CBD has been reported to inhibit the activity of many CYPs, showing the highest potency towards 7-ethoxyresofurin O-deethylation by both human liver microsomes (Table 2) and recombinant CYP1A1 (Table 3). CBD also showed time-dependent inhibition of 7-ethoxyresorufin O-deethylation by recombinant CYP1 enzymes, with highest efficiency toward CYP1A2 (kinact/KI, 0.18 versus ≤0.05 min−1μM−1, respectively) (Table 4). CBN was the most efficient time-dependent inhibitor of 7-ethoxyresorufin O-deethylation by recombinant CYP1A1 (kinact/KI, 0.21 min−1μM−1) (Table 4). The resorcinol moiety of 7-ethoxyresorufin has been implicated in the generation of a reactive intermediate (Yamaori et al., 2014). Pre-incubation of human liver microsomes with CBD (64 μM) decreased formation of CYP3A-mediated THC and cyclosporine metabolites, but not CYP2C9-mediated THC metabolites (Jaeger et al., 1996). Additional studies are needed to assess the time-dependent inhibition kinetics of these cannabinoids towards CYP3A and other CYPs.
In vitro assays used to date to characterize the inhibition kinetics of THC, CBD, and CBN carry a major limitation. Based on the absence of data on non-specific binding of the cannabinoids, many published investigations of CYP-mediated cannabinoid-drug interactions did not account for experimental factors such as poor aqueous solubility of cannabinoids and extensive non-specific binding of cannabinoids to assay materials (e.g., microsomal proteins, glassware, and plasticware). Thus, reported Ki, or IC50 values may be overestimated, leading to underestimation of the true interaction potential. These inaccuracies were addressed via in vitro-to-in vivo extrapolation using the predicted concentrations of THC and CBD in the intestine, as well as unbound plasma concentrations in the portal venous and systemic circulation, after administration of representative medical doses by various routes (Bergamaschi et al., 2011; Lile et al., 2013) (Tables 5, 6). Unbound plasma concentrations were estimated using a conservative unbound fraction (fu,p) of 0.03, based on plasma protein binding of THC (Garrett and Hunt, 1974; Klausner et al., 1975; Widman et al., 1974). Because values for non-specific binding of cannabinoids to assay materials were not available, fu,p was used as a surrogate to correct the reported Ki, or IC50 values.
Table 5.
Predicted cytochrome P450-mediated clinical drug interaction potential of THC administered orally or by inhalation.
| Enzyme | IC50 or Ki corrected for binding (μM)b | Oral Dose (40 mg) | Inhaled Dose (34 mg) | Reference | ||
|---|---|---|---|---|---|---|
| AUCRgut | AUCRhep | AUCRsys | AUCRsys | |||
| CYP1A1 | 0.14 | N/A | 1.15 | 1.01 | 1.11 | (Yamaori et al, 2010) |
| CYP1A2a | 0.23 | N/A | 1.10 | 1.01 | 1.07 | (Yamaori et al, 2010) |
| CYP1B1 | 0.074 | N/A | 1.30 | 1.03 | 1.21 | (Yamaori et al, 2010) |
| CYP2A6 | 0.87 | N/A | 1.03 | 1.00 | 1.02 | (Yamaori et al, 2011) |
| CYP2B6 | 0.084 | N/A | 1.26 | 1.02 | 1.18 | (Yamaori et al, 2011) |
| CYP2D6 | 0.51 | N/A | 1.04 | 1.00 | 1.03 | (Yamaori et al, 2011) |
| CYP2C9a | 0.041 | N/A | 1.53 | 1.05 | 1.37 | (Yamaori et al, 2012) |
| CYP2C19a | 0.057 | N/A | 1.38 | 1.03 | 1.27 | (Jiang et al., 2013) |
| CYP3A4 | >1.5 | >340 | 1.01 | 1.00 | 1.00-1.01 | (Yamaori et al, 2011) |
| CYP3A5 | 1.1 | 480 | 1.02 | 1.00 | 1.01 | (Yamaori et al, 2011) |
Clinical studies suggest the potential for marijuana-drug interactions.
Published Ki or IC50 values (Table 3) corrected for non-specific binding to assay materials using fu,p = 0.03 (based on plasma protein binding of THC) as a surrogate (Garrett and Hunt, 1974; Klausneret al., 1975; Widman et al., 1974).
Predicted potential for drug interactions mediated by inhibition of cytochromes P450 by THC consumed orally or inhaled. Predicted AUC ratio (AUCR) for intestine (AUCRgut) ≥ 11, liver (AUCRhep) ≥ 1.25, and systemic circulation (AUCRsys) ≥ 1.02 indicate strong presystemic intestinal, presystemic hepatic, or systemic drug interaction potential, respectively. Values in shaded cells exceed the cut-off values recommended by the FDA (FDA, 2017).
- AUCRgut = 1+(Ig/binding corrected Ki or IC50), where intestinal luminal concentration (Ig) was calculated as dose/250 mL (507,200 nM)
- AUCRhep = 1+(Ihep,u/Ki or IC50), where Ihep,u was calculated as 22 nM , where Cmax= 64 nM (Lile et al., 2013); Fa = 1 (estimated based on FDA guidance), ka = 0.02 min−1 (estimated using Phoenix WinNonlin and confirmed by (Heuberger et al., 2015)), QheP (liver blood flow) = 1500 ml/min (assumption), RB (blood:plasma ratio) = 0.4 (Schwilke et al., 2009)
- AUCRsys = 1+(Isys/Ki or IC50), where Isys = Cmax,u = 1.91 nM (orally) (Lile et al., 2013) or 15.5 nM (inhalation) (Huestis et al., 1992)
Table 6.
Predicted cytochrome P450-mediated clinical drug interaction potential of CBD administered orally or by inhalation.
| Enzyme | IC50 or Ki corrected for binding (μM)b | Oral Dose (1500 mg) | Inhaled Dose (20 mg) | Reference | ||
|---|---|---|---|---|---|---|
| AUCRgut | AUCRhep | AUCRsys | AUCRsys | |||
| CYP1A1 | 0.0047 | N/A | 176 | 1.35 | 3.21 | (Yamaori et al, 2010) |
| CYP1A2a | 0.081 | N/A | 11 | 1.02 | 1.13 | (Yamaori et al, 2010) |
| CYP1B1 | 0.11 | N/A | 8.5 | 1.01 | 1.09 | (Yamaori et al, 2010) |
| CYP2A6 | 1.65 | N/A | 1.5 | 1.00 | 1.01 | (Yamaori et al, 2011) |
| CYP2B6 | 0.021 | N/A | 40 | 1.08 | 1.49 | (Yamaori et al, 2011) |
| CYP2D6 | 0.081 | N/A | 11 | 1.02 | 1.13 | (Yamaori et al, 2011) |
| CYP2C9a | 0.069 | N/A | 13 | 1.02 | 1.15 | (Yamaori et al, 2012) |
| CYP2C19a | 0.024 | N/A | 35 | 1.07 | 1.43 | (Jiang et al., 2013) |
| CYP3A4 | 0.030 | 6.3 × 105 | 28 | 1.05 | 1.34 | (Yamaori et al, 2011) |
| CYP3A5 | 0.0059 | 3.2 × 106 | 140 | 1.28 | 2.76 | (Yamaori et al, 2011) |
Clinical studies suggest the potential for marijuana-drug interactions.
Published Ki or IC50 values (Table 3) corrected for non-specific binding to assay materials using fu,p=0.03 (based on plasma protein binding of THC) as a surrogate (Garrett and Hunt, 1974; Klausner et al., 1975; Widman et al., 1974).
Predicted potential for drug interactions mediated by inhibition of cytochromes P450 by CBD consumed orally or inhaled. Predicted AUC ratio (AUCR) for the intestine (AUCRgut) ≥ 11, liver (AUCRhep) ≥ 1.25, and systemic circulation (AUCRsys) ≥ 1.02 indicate strong presystemic intestinal, presystemic hepatic, or systemic dug interaction potential, respectively. Values in shaded cells exceed the cutoff values recommended by the FDA (FDA, 2017).
Ig (intestinal luminal concentrations) = Dose/250 mL (1.90 × 107 nM), Ihep,u = 815 nM, and Isys,u = 1.62 nM (oral) or 10.3 nM (inhalation).
- AUCRgut = 1+(Ig/binding corrected Ki or IC50)
- AUCRhep = 1+(Ihep,u/Ki or IC50), where , where Cmax = 54 nM (Bergamaschi et al., 2011); Fa= 1 (estimated based on FDA guidance), ka = 0.02 min−1 (estimated using Phoenix WinNonlin and confirmed by (Heuberger et al., 2015)), Qhep (liver blood flow) = 1500 ml/min (assumption), RB (blood:plasma ratio) = 0.4 (Heuberger et al., 2015; Schwilke et al., 2009)
- AUCRsys = 1+(Isys/Ki or IC50), where Isys = Cmax,u = 1.62 nM (THC, oral) (Fusar-Poli et al., 2009) and 10.3 nM (THC, inhalation) (Ohlsson et al., 1986)
These predictions generally indicated a high potential for first-pass marijuana-drug interactions precipitated by THC and CBD after oral administration due to inhibition of CYPs expressed in the intestine and liver. CBD showed a higher potential than THC to precipitate systemic drug interactions after oral administration or inhalation. In summary, orally administered THC (40 mg) was predicted to inhibit intestinal CYP3A4 and CYP3A5, as well as hepatic CYP1B1, CYP2B6, CYP2C9, and CYP2C19, during first passage through the intestine and liver; systemic interactions also were predicted for these four hepatic CYPs (Table 5). Orally administered CBD (1500 mg) was predicted to inhibit all of the evaluated CYPs during first pass and to inhibit all but CYP1B1 and CYP2A6 systemically (Table 6). Inhaled THC (34 mg) was predicted to precipitate systemic interactions by inhibiting all evaluated CYPs except CYP3A4 and CYP3A5, whereas inhaled CBD (20 mg) was predicted to precipitate systemic interactions by inhibiting all evaluated CYPs except CYP2A6. Well-designed clinical studies are needed to test the accuracy of these predictions.
6.1.2. Interactions with UDP-Glucuronosyltransferases (UGTs)
CBD was reported to inhibit ethanol glucuronidation, catalyzed largely by UGT1A9 and UGT2B7, in human liver microsomes, with a Ki, of 3.1 mg/L (67 μM) (Al Saabi et al., 2013). In contrast, CBN at 5, 10, and 15 mg/ml (16, 32, and 48 μM) increased ethanol glucuronidation in a concentration-dependent manner, by approximately 2.5-to 6-fold relative to control. CBD and CBN (15 mg/ml) were further reported to inhibit ethanol glucuronidation by recombinant UGT1A9, by ~35-50% relative to control (Al Saabi et al., 2013). CBD also inhibited ethanol glucuronidation by recombinant UGT2B7, by 70%, whereas CBN increased activity by 4.2-fold. All of these effects required supraphysiological concentrations of CBD and CBN, suggesting UGT-mediated cannabinoid-ethanol interactions are unlikely to occur in vivo. However, the effects of cannabinoids on other UGT substrates, e.g., some opioids and benzodiazepines, remain to be determined, both in vitro and in vivo.
6.1.3. Interactions with transporters
Cannabinoids are both substrates and inhibitors of drug transporters (Table 7). Studies in mice showed THC to be a substrate for P-gp (Abcbl) in the gut and for both P-gp and Bcrp (Abcg2) at the blood-brain barrier. Abcb1a/b and Abcg2 knockout mice showed up to a 4.2- and 6.1-fold increase in the brain/blood THC ratio, respectively, compared to wild-type mice. Similarly, P-gp deficient CF1 mice demonstrated a 2.2-fold higher systemic AUC of THC compared to wild-type mice (Bonhomme-Faivre et al., 2008). Unlike THC, CBD was shown not to be a substrate for P-gp and Bcrp, as evidenced by lack of differences in the brain/blood CBD ratios between Abcb1a/b and Abcg2 knockout mice and wild-type mice (Brzozowska et al., 2016).
Table 7.
Cannabinoid interactions with drug transporters.
| Cannabinoids as substrates for efflux transporters | |||||
|---|---|---|---|---|---|
| Transporter | Cannabinoid | Dose or Concentration | Experimental system(s) | Key observations | Reference |
| P-gp | THC | 25 mg/kg (oral) | Mice | 2× higher AUC in CF1 (P-gp-deficient) mice compared to wild type mice | (Bonhomme-Faivre et al., 2008) |
| THC | 3 mg/kg (ip) | Mice | 1.5-4.2× higher brain/blood THC ratio in Abcb1a/b(−/−)(−/−) mice compared to wild type mice | (Spiro et al., 2012) | |
| CBD | 10 mg/kg (sc) | Mice | Brain/blood CBD ratio in Abcb1a/b(−/−) mice was not different from that in wild type mice | (Brzozowska et al., 2016) | |
| CBN | 1-100 μM | Human P-gp membranes | P-gp ATPase activity was stimulated, with CIint = 0.7 min−1 × 10−3 | (Zhu et al., 2006) | |
| Bcrp | THC | 3 mg/kg (ip) | Mice | 1.9-6.1× higher brain/blood THC ratio in Abcg2(−/−) mice compared to wild type mice | (Spiro et al., 2012) |
| CBD | 10 mg/kg (sc) | Brain/blood CBD ratio was similar between Abcg2(−/−) and wild type mice | (Brzozowska et al., 2016) | ||
| Cannabinoids as inhibitors of efflux transporters | |||||
| Transporter | Cannabinoid | Probe Substrate | Experimental system(s) | Key observations | Reference |
| P-gp | THC, CBD, CBN | Rhodamine 123 | Human CEM/VLB100 cells | No increase in rhodamine 123 accumulation in the presence of cannabinoid (10 μM) after a 1-hour exposure | (Holland et al., 2006) |
| CBD | Verapamil, Rhodamine 123 | Human P-gp membranes; LLC-PK1/MDRI cells | IC50 = 39.6 μM for
verapamil-stimulated P-gp ATPase activity in human P-gp
membranes IC50= 8.44 ± 0.58 μM for rhodamine 123 accumulation in LLC-PK1/MDRI cells |
(Zhu et al., 2006) | |
| THC, CBN | Verapamil | Human P-gp membranes | No inhibition of P-gp ATPase activity | (Zhu et al., 2006) | |
| THC | Risperidone, Clozapine | Mice | Decreased brain accumulation of risperidone (~25-50%) and 9-OH-risperidone (~33-63%) in mice treated with 1 mg/kg THC ip for 14 days followed by a 14-day wash-out. Repeated THC exposure did not impact clozapine (a non P-gp substrate) neurobehavioral effects. | (Brzozowska et al., 2017) | |
| BCRP/Bcrp | THC, CBD, CBN | Sulfasalazine, Mitoxantrone | Human BCRP membranes; Murine MEF3.8/Bcrp1 cells | IC50 values for
sulfasalazine-stimulated ATPase activity ranged from 4.4-7.3
μM. Mitoxantrone toxicity (IC50) in MEF3.8/Bcrp1 cells was significantly reduced to 2 μM (THC) and 5 μM (CBD and CBN). |
(Holland et al., 2007) |
| CBD | Mitoxantrone, Glyburide | Human CEM/VLB100 cells, BeWo cells, Jar cells; Human perfused placenta | Significant increase in mitoxantrone
accumulation following exposure to 10 μM CBD in BeWo, Jar, and
MCF7/P-gp cell lines. CBD (15 μM) increased glyburide fetal-to-maternal ratio 1.4-fold in human perfused placenta. |
(Feinshtein et al., 2013) | |
| MRP1 | CBD, CBN, THC | Fluo3, Vincristine | 2008/MRP1 cells | IC50 for Fluo3 accumulation: CBD
= 128 μM, CBN = 145 μM, THC = 161
μM IC50 for vincristine accumulation: CBD = 38 μM, CBN = 30.9 μM, THC=107 μM |
(Holland et al., 2008) |
| Effects of cannabinoids on efflux transporter expression | |||||
| Transporter | Cannabinoid | Dose or Concentration | Experimental system(s) | Key observations | Reference |
| P-gp | CBD, THC | 0.1, 1, 10 μM | Human CEM/VLB100 cells | P-gp expression increased by 50% after a 72-hour exposure. | (Holland et al., 2006) |
| CBD, THC | 10 μM | Human CEM/VLB100 cells | MDR1 mRNA expression increased by 2 and 2.5 fold, respectively, at a 4-hour, but not 8- or 48-hour, incubation. | (Arnold et al., 2012) | |
| CBD | 15 μ | BeWo cells, Jar cells, MCF7/P-gp cells | A 72-hour exposure resulted in ~60% decrease in P-gp expression in BeWo and Jar cell lines but a ~2.5 fold increase in P-gp expression in MCF7/P-gp cell line. | (Feinshtein et al., 2013) | |
| THC | 1 mg/kg THC ip for 14 days followed by 14 day wash-out | Mice | Increased expression of P-gp in the ventrolateral septum, nucleus accumbens core, and the paraventricular nucleus of the thalamus | (Brzozowska et al., 2017) | |
| BCRP | THC, CBD, CBN | 0.4, 4, 5 μM | WiDr human colon cells, MEF3.8/BCRP cells | No change in ABCG2 (BCRP) expression | (Holland et al., 2007) |
| CBD | 15 uM | BeWo cells, Jar cells | A 72-hour exposure resulted in ~2 fold increase of BCRP expression in BeWo and Jar cell lines. | (Feinshtein et al., 2013) | |
THC, CBD, and CBN (10 μM) had no inhibitory effects on P-gp activity (rhodamine 123 accumulation) relative to vehicle in the human T lymphoblastoid leukemia cell line CEM/VLB100 after a 1-hour exposure (Holland et al., 2006) (Table 7). A subsequent study by the same investigators showed that, relative to vehicle-treated cells, a 4-hour (but not an 8- or 48-hour) exposure to THC or CBD (10 μM) led to an increase in MDR1 (ABCB1) mRNA expression by 2-2.5 fold and a decrease in rhodamine 123 accumulation (Arnold et al., 2012). However, a 72-hour exposure to either cannabinoid decreased P-gp expression by up to 50% in a dose-dependent manner; the decrease in protein expression was accompanied by an increase in rhodamine 123 accumulation. Mice treated with THC (1 mg/kg) daily for 14 days showed an increase in P-gp localization in brain microvessels compared to control mice, which led to a 25-50% decrease in brain accumulation of the P-gp substrate risperidone (Brzozowska et al., 2017). These in vitro-in vivo incongruities with THC may be due to species differences in P-gp regulation and/or differences in transport properties between substrates.
THC, CBD, and CBN increased intracellular accumulation of the BCRP/Bcrp probe substrate mitoxantrone in the mouse MEF3.8/Bcrp1 cell line and inhibited basal and substrate (sulfasalazine)-stimulated ATPase activity in human BCRP-containing membranes (Holland et al., 2007) (Table 7), indicating inhibition of BCRP/Bcrp activity by these cannabinoids. Likewise, compared to vehicle, CBD (15 μM) increased the fetal-to-maternal ratio of the BCRP substrate glyburide by 1.4-fold in a perfused human placenta model, indicating increased placental transport of glyburide via BCRP inhibition (Feinshtein et al., 2013). The same study showed, compared to vehicle-treated cells, CBD to inhibit mitoxantrone uptake in the human placenta cell line BeWo, with IC50 values ranging from 20-40 μM. Further studies in BeWo cells following a 72-hour exposure to CBD (15 μM) demonstrated an increase in BCRP expression by 2-fold and a decrease in P-gp expression by >50% (Feinshtein et al., 2013). Lastly, THC, CBD, and CBN increased the intracellular accumulation of the MRP1 substrates Fluo3 and vincristine in human 2008/MRP1/ABCC ovarian carcinoma cells, with IC50S ranging from 30-110 μM (Holland et al., 2008).
The above observations suggest that cannabinoids can inhibit BCRP and MRP1 activity, decrease protein expression of P-gp, and increase protein expression of BCRP. However, the concentrations used in these experiments greatly exceeded systemic concentrations likely to be observed in humans, suggesting systemic transporter-mediated drug interactions with cannabinoids are unlikely. However, transporter-mediated interactions may be possible during first-pass through the intestine and/or liver upon oral administration. The effects of cannabinoids on other efflux transporters, as well as uptake transporters, have not been reported.
6.2. Human clinical drug interactions
Despite the widespread use and availability of marijuana products, substantive deficiencies remain regarding the potential risks for marijuana or cannabinoids to precipitate interactions with conventional drugs. Well-designed clinical studies are particularly sparse, and the few published studies to date involved CYPs as potential targets. For example, oral administration of CBD (Epidiolex®) (5-25 mg/kg/day) to patients with refractory epilepsy (aged 4-19 years) receiving the anti-seizure drug, clobazam, led to a profound increase (500 ± 300%) in mean (± SD) plasma concentrations of the active metabolite N-desmethylclobazam compared to baseline (absence of CBD) (Geffrey et al., 2015). After dosage adjustment of clobazam, due to reports of sedation,N-desmethylclobazam concentrations remained higher (by two-to sixfold) at eight weeks compared to baseline. This pharmacokinetic interaction was attributed to CBD-mediated inhibition of CYP2C19, which catalyzes the metabolism of N-desmethylclobazam (Gaston and Szaflarski, 2018; Geffrey et al., 2015).
Older studies substantiate the likelihood of pharmacokinetic interactions between cannabinoids and CYP substrates. A study published in 1978 showed the mean clearance of theophylline, a CYP1A2 substrate, to be approximately 40% higher in both habitual smokers of marijuana cigarettes and of tobacco cigarettes compared to non-smoking control subjects (~73 versus 52 ml/kg/h) (Jusko et al., 1978). These observations suggested that marijuana smoking may induce CYP1A2, similar to tobacco smoking. A study published in 1980 involving healthy male marijuana users administered oral CBD (600 mg/day for 5-12 days) with a single oral dose of hexobarbital (500 mg) exhibited a 36% reduction in apparent oral clearance and a 51% increase in the AUC of hexobarbital (Benowitz et al., 1980), a substrate for CYP2C19 (Knodell et al., 1988). These data suggested inhibition of CYP2C19 by CBD, consistent with the more recent observations with N-desmethylclobazam (Geffrey et al., 2015).
Other clinical studies failed to show an effect of oral THC on the pharmacokinetics of drugs administered intravenously, specifically cyclophosphamide (CYP2B6/CYP2C9/CYP3A substrate), docetaxel (CYP1B1/CYP2B6/CYP3A substrate), doxorubicin (carbonyl reductase 1/P-gp substrate), and irinotecan (CYP3A/UGT1A1 substrate) (Bertholee et al., 2016; Ekhart et al., 2008; Kosel et al., 2002; Lal et al., 2010; Riggs et al., 1981; Roy et al., 1999; Solas et al., 2007). Consumption of 200 ml of marijuana tea containing 18% THC and 0.8% CBD (amount of each cannabinoid not specified) for 15 days also showed no effect on the pharmacokinetics of intravenous irinotecan and docetaxel (Engels et al., 2007). Likewise, THC-containing cigarettes and oral THC (as dronabinol) had no effect on the pharmacokinetics of oral indinavir (CYP3A/P-gp substrate) and nelfinavir (CYP2C19/CYP3A/P-gp substrate). As nelfinavir is a substrate for CYP2C19, the lack of an interaction contrasts with the aforementioned studies involving N-desmethylclobazam and hexobarbital; however, the contribution of CYP2C19 to nelfinavir clearance is inconclusive (Damle et al., 2009; Hirt et al., 2008; Kattel et al., 2015).
One case report described a probable interaction between smoked marijuana and warfarin (Yamreudeewong et al., 2009). In brief, a 56-year-old white man who had been taking warfarin for 11 years since undergoing surgery for mechanical heart valve replacement was admitted to the hospital due to upper gastrointestinal bleeding. Upon admission, his International Normalized Ratio (INR) was supratherapeutic (10.4). He was administered oral vitamin K, and his INR decreased to 1.8 the next day. He was discharged seven days after admission. Fifteen days later, he was readmitted due to a constant nosebleed and increased bruising, with an INR of 11.6. After treatment, he was discharged with an INR of 1.14. The patient mentioned smoking marijuana more frequently throughout these two hospitalizations due to depression. After counseling by the pharmacist about a potential marijuana-warfarin interaction, he stopped smoking marijuana. During the nine months when he did not smoke marijuana, his INR ranged from 1.08 to 4.4, with no major bleeding complications. Potential mechanisms of this probable interaction, which was based on the Horn Drug Interaction Probability Scale, included inhibition of CYP2C9-mediated warfarin metabolism and, to a lesser extent, displacement of warfarin from plasma protein binding sites by marijuana constituents.
Epidiolex® was recently approved by the United States Food and Drug Administration for treatment of seizures associated with Lennox-Gastaut syndrome or Dravet syndrome in patients aged two years and older (Greenwich Biosciences, 2018). The labeling states a risk of drug interactions with substrates for CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, UGT1A9, and UGT2B7 (Greenwich Biosciences, 2018). With the exception of CYP2C19 and possibly CYP2C9, these potential risks are based on in vitro predictions. Accordingly, opportunities exist to confirm or refute these predictions via controlled clinical studies.
7. Conclusions
The risk of marijuana-drug interactions precipitated by cannabinoids remains challenging to study due to the Schedule I classification of marijuana and marijuana products, the diversity of marijuana strains and derivative products on the commercial market, and the variety of routes of administration. However, the sheer magnitude of forensic and academic literature on marijuana, its constituents, and its drug interaction potential collectively presents a useful primer for designing and executing future studies on this critical, understudied public health issue.
Results from the few available clinical studies indicate a risk for marijuana-drug interaction mediated via inhibition of hepatic CYP2C19 by THC and CBD. Additionally, one case report suggested a risk for an interaction mediated via inhibition of hepatic CYP2C9 by marijuana constituents. Finally, in vitro-in vivo extrapolation suggested a risk for interactions mediated via inhibition of intestinal CYP3A and hepatic CYP1B1, CYP2B6, CYP2C9, and CYP2C19 by THC and CBD and of hepatic CYP1A1, CYP1A2, CYP2A6, CYP2D6, and CYP3A by CBD (Table 5, 6). To date, in vitro and clinical studies evaluating marijuana-drug interactions have focused predominately on the CYPs as interaction targets. Effects of marijuana and its constituents on other drug metabolizing enzymes (e.g., the UGTs) remain less characterized. Regarding drug transporters, in vitro and animal studies have thus far focused exclusively on efflux transporters, specifically P-gp, BCRP, and MRP1. The effects of cannabinoids on other efflux, as well as uptake, transporters remain a critical knowledge gap, particularly as the lipophilic cannabinoids tend to accumulate in tissues upon chronic marijuana use.
Another understudied problem is that of potential marijuana-drug interactions precipitated in lung tissue via inhalation of marijuana. For example, THC and other constituents in marijuana smoke, such as polycyclic aromatic hydrocarbons, which activate the aryl hydrocarbon receptor, may induce lung CYP1A1 (Roth et al., 2001). This potential interaction would be particularly critical for pregnant women, in whom placental CYP1A activity can be altered by tobacco smoke (Stejskalova and Pavek, 2011). Such alterations may have implications for fetal growth and development.
In addition to the many gaps in scientific knowledge regarding marijuana-drug interactions, researchers face multiple legal and regulatory hurdles that preclude clinical, even in vitro, evaluation in a timely manner. Although THC and CBD are available as prescription drugs, the doses used typically are much less than the THC and CBD content in commercial products consumed for medical and recreational purposes. The widespread availability of these products highlights the urgent need for well-designed in vitro and clinical studies to investigate potential marijuana-drug interactions. This public health need should prompt the development of policies regarding marijuana research, with the end goal of informing health care providers and consumers about the safety of consuming marijuana products concomitantly with conventional medications.
Supplementary Material
Acknowledgments
All authors were supported in part by the National Institutes of Health National Center for Complementary and Integrative Health grant U54 AT008909. MFP an EJJ were supported in part by the National Institute of General Medical Sciences grant R01 GM077482. JDU and GP-V were supported in part by the National Institute on Drug Abuse grant P01 DA032507. JSM was supported in part by the National Institute of General Medical Sciences grant R01 GM129863 and National Cancer Institute grant R01 CA182963.
Abbreviations
- AUC
area under the plasma concentration-time curve
- CBD
cannabidiol
- CB1
cannabinoid receptor 1
- CB2
cannabinoid receptor 2
- CBN
cannabinol
- Cmax
maximum plasma concentration
- CYP
cytochrome P450
- IC50
half-maximal inhibitory concentration
- Ki
inhibitory constant
- THC
(−)Δ9-trans-(6aR, 10aR)-tetrahydrocannabinol
- Δ8-THC
(–)-Δ8-trans-tetrahydrocannabinol
- UGT
uridine 5′-diphospho-glucuronosyltransferase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
The authors declare no conflicts of interest.
References
- Agurell S, Halldin M, Lindgren JE, Ohlsson A, Widman M, Gillespie H, et al. (1986). Pharmacokinetics and metabolism of delta 1-tetrahydrocannabinol and other cannabinoids with emphasis on man. Pharmacol Rev, 38, 21–43. [PubMed] [Google Scholar]
- Al Saabi A, Allorge D, Sauvage FL, Tournel G, Gaulier JM, Marquet P, et al. (2013). Involvement of UDP-glucuronosyltransferases UGT1A9 and UGT2B7 in ethanol glucuronidation, and interactions with common drugs of abuse. Drug Metab Dispos, 41, 568–574. [DOI] [PubMed] [Google Scholar]
- Alsherbiny MA, & Li CG (2018). Medicinal Cannabis-Potential Drug Interactions. Medicines (Basel), 6, pii: E3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold JC, Hone P, Holland ML, & Allen JD (2012). CB2 and TRPV1 receptors mediate cannabinoid actions on MDR1 expression in multidrug resistant cells. Pharmacol Rep, 64, 751–757. [DOI] [PubMed] [Google Scholar]
- Awasthi R, An G, Donovan MD, & Boles Ponto LL (2018). Relating Observed Psychoactive Effects to the Plasma Concentrations of Delta-9-Tetrahydrocannabinol and Its Active Metabolite: An Effect-Compartment Modeling Approach. J Pharm Sci, 107, 745–755. [DOI] [PubMed] [Google Scholar]
- Benowitz NL, Nguyen TL, Jones RT, Herning RI, & Bachman J (1980). Metabolic and psychophysiologic studies of cannabidiol-hexobarbital interaction. Clin Pharmacol Ther, 28, 115–120. [DOI] [PubMed] [Google Scholar]
- Bergamaschi MM, Queiroz RH, Zuardi AW, & Crippa JA (2011). Safety and side effects of cannabidiol, a Cannabis sativa constituent. Curr Drug Saf, 6, 237–249. [DOI] [PubMed] [Google Scholar]
- Bertholee D, Maring JG, & van Kuilenburg AB (2016). Genotypes Affecting the Pharmacokinetics of Anticancer Drugs. Clin Pharmacokinet, 56, 317–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bland TM, Haining RL, Tracy TS, & Callery PS (2005). CYP2C-catalyzed delta9-tetrahydrocannabinol metabolism: kinetics, pharmacogenetics and interaction with phenytoin. Biochem Pharmacol, 70, 1096–1103. [DOI] [PubMed] [Google Scholar]
- Blessing EM, Steenkamp MM, Manzanares J, & Marmar CR (2015). Cannabidiol as a Potential Treatment for Anxiety Disorders. Neurotherapeutics, 12, 825–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhomme-Faivre L, Benyamina A, Reynaud M, Farinotti R, & Abbara C (2008). Disposition of Delta tetrahydrocannabinol in CF1 mice deficient in mdr1a P-glycoprotein. Addict Biol, 13, 295–300. [DOI] [PubMed] [Google Scholar]
- Bonn-Miller MO, Loflin MJE, Thomas BF, Marcu JP, Hyke T, & Vandrey R (2017). Labeling Accuracy of Cannabidiol Extracts Sold Online. JAMA, 318, 1708–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornheim LM, Lasker JM, & Raucy JL (1992). Human hepatic microsomal metabolism of delta 1-tetrahydrocannabinol. Drug Metab Dispos, 20, 241–246. [PubMed] [Google Scholar]
- Brenneisen R, Meyer P, Chtioui H, Saugy M, & Kamber M (2010). Plasma and urine profiles of Delta9-tetrahydrocannabinol and its metabolites 11-hydroxy-Delta9-tetrahydrocannabinol and 11-nor-9-carboxy-Delta9-tetrahydrocannabinol after cannabis smoking by male volunteers to estimate recent consumption by athletes. Anal Bioanal Chem, 396, 2493–2502. [DOI] [PubMed] [Google Scholar]
- Brzozowska N, Li KM, Wang XS, Booth J, Stuart J, McGregor IS, et al. (2016). ABC transporters P-gp and Bcrp do not limit the brain uptake of the novel antipsychotic and anticonvulsant drug cannabidiol in mice. PeerJ, 4, e2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzozowska NI, de Tonnerre EJ, Li KM, Wang XS, Boucher AA, Callaghan PD, et al. (2017). The Differential Binding of Antipsychotic Drugs to the ABC Transporter P-Glycoprotein Predicts Cannabinoid-Antipsychotic Drug Interactions. Neuropsychopharmacology, 42, 2222–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaneto MS, Gorelick DA, Desrosiers NA, Hartman RL, Pirard S, & Huestis MA (2014). Synthetic cannabinoids: epidemiology, pharmacodynamics, and clinical implications. Drug Alcohol Depend, 144, 12–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damle BD, Uderman H, Biswas P, Crownover P, Lin C, & Glue P (2009). Influence of CYP2C19 polymorphism on the pharmacokinetics of nelfinavir and its active metabolite. Br J Clin Pharmacol, 68, 682–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pasquale A, Tumino G, & de Pasquale RC (1974). Micromorphology of the epidermic surfaces of female plants of Cannabis sativa L. Bull Narc, 26, 27–40. [PubMed] [Google Scholar]
- Degenhardt L, Whiteford HA, Ferrari AJ, Baxter AJ, Charlson FJ, Hall WD, et al. (2013). Global burden of disease attributable to illicit drug use and dependence: findings from the Global Burden of Disease Study 2010. Lancet, 382, 1564–1574. [DOI] [PubMed] [Google Scholar]
- Desrosiers NA, Himes SK, Scheidweiler KB, Concheiro-Guisan M, Gorelick DA, & Huestis MA (2014). Phase I and II cannabinoid disposition in blood and plasma of occasional and frequent smokers following controlled smoked cannabis. Clin Chem, 60, 631–643. [DOI] [PubMed] [Google Scholar]
- Di Marzo V (2009). The endocannabinoid system: its general strategy of action, tools for its pharmacological manipulation and potential therapeutic exploitation. Pharmacol Res, 60, 77–84. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, & Petrocellis LD (2006). Plant, synthetic, and endogenous cannabinoids in medicine. Annu Rev Med, 57, 553–574. [DOI] [PubMed] [Google Scholar]
- Dufresnes C, Jan C, Bienert F, Goudet J, & Fumagalli L (2017). Broad-Scale Genetic Diversity of Cannabis for Forensic Applications. PLoS One, 12, e0170522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekhart C, Doodeman VD, Rodenhuis S, Smits PH, Beijnen JH, & Huitema AD (2008). Influence of polymorphisms of drug metabolizing enzymes (CYP2B6, CYP2C9, CYP2C19, CYP3A4, CYP3A5, GSTA1, GSTP1, ALDH1A1 and ALDH3A1) on the pharmacokinetics of cyclophosphamide and 4-hydroxycyclophosphamide. Pharmacogenet Genomics, 18, 515–523. [DOI] [PubMed] [Google Scholar]
- Ekor M (2014). The growing use of herbal medicines: issues relating to adverse reactions and challenges in monitoring safety. Front Pharmacol, 4, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elphick MR, & Egertova M (2005). The phylogenetic distribution and evolutionary origins of endocannabinoid signalling. Handb Exp Pharmacol, 283–297. [DOI] [PubMed] [Google Scholar]
- ElSohly HN, & ElSohly MA (2007). Marijuana and the cannabinoids (pp. 67–96): Humana Press Inc. [Google Scholar]
- ElSohly MA, Mehmedic Z, Foster S, Gon C, Chandra S, & Church JC (2016). Changes in Cannabis Potency Over the Last 2 Decades (1995–2014): Analysis of Current Data in the United States. Biol Psychiatry, 79, 613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ElSohly MA, Radwan MM, Gul W, Chandra S, & Galal A (2017). Phytochemistry of Cannabis sativa L. Prog Chem Org Nat Prod, 103, 1–36. [DOI] [PubMed] [Google Scholar]
- Elsohly MA, & Slade D (2005). Chemical constituents of marijuana: the complex mixture of natural cannabinoids. Life Sci, 78, 539–548. [DOI] [PubMed] [Google Scholar]
- Engels FK, de Jong FA, Sparreboom A, Mathot RA, Loos WJ, Kitzen JJ, et al. (2007). Medicinal cannabis does not influence the clinical pharmacokinetics of irinotecan and docetaxel. Oncologist, 12, 291–300. [DOI] [PubMed] [Google Scholar]
- FDA. (2017). Clinical drug interaction studies - study design, data analysis, and clinical implications (guidance for industry). Food and Drug Administration Center for Drug Evaluation and Research (CDER). Document number UCM292362. URL: https://www.fda.gov/downloads/drugs/guidances/ucm292362.pdf Access date 1/15/2019.
- FDA. (2017). In vitro metabolism- and transporter-mediated drug-rug interaction studies (guidance for industry). In: Food and Drug Administration Center for Drug Evaluation and Research (CDER). Document number UCM581965. URL: https://www.fda.gov/downloads/Drugs/Guidances/UCM581965.pdf Access date 1/15/19.
- Feinshtein V, Erez O, Ben-Zvi Z, Erez N, Eshkoli T, Sheizaf B, et al. (2013). Cannabidiol changes P-gp and BCRP expression in trophoblast cell lines. PeerJ, 1, e153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinshtein V, Erez O, Ben-Zvi Z, Eshkoli T, Sheizaf B, Sheiner E, et al. (2013). Cannabidiol enhances xenobiotic permeability through the human placental barrier by direct inhibition of breast cancer resistance protein: an ex vivo study. Am J Obstet Gynecol, 209, 573.e571–573.e515. [DOI] [PubMed] [Google Scholar]
- Feinshtein V, Erez O, Ben-Zvi Z, Eshkoli T, Sheizaf B, Sheiner E, et al. (2013). Cannabidiol enhances xenobiotic permeability through the human placental barrier by direct inhibition of breast cancer resistance protein: an ex vivo study. Am J Obstet Gynecol, 209, 573 e571–573 e515. [DOI] [PubMed] [Google Scholar]
- Felder CC, Joyce KE, Briley EM, Mansouri J, Mackie K, Blond O, et al. (1995). Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol, 48, 443–450. [PubMed] [Google Scholar]
- Fischedick JT, Hazekamp A, Erkelens T, Choi YH, & Verpoorte R (2010). Metabolic fingerprinting of cannabis sativa L., cannabinoids and terpenoids for chemotaxonomic and drug standardization purposes. Phytochemistry, 71, 2058–2073. [DOI] [PubMed] [Google Scholar]
- Ford BM, Tai S, Fantegrossi WE, & Prather PL (2017). Synthetic Pot: Not Your Grandfather’s Marijuana. Trends Pharmacol Sci, 38, 257–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusar-Poli P, Crippa JA, Bhattacharyya S, Borgwardt SJ, Allen P, Martin-Santos R, et al. (2009). Distinct effects of {delta}9-tetrahydrocannabinol and cannabidiol on neural activation during emotional processing. Arch Gen Psychiatry, 66, 95–105. [DOI] [PubMed] [Google Scholar]
- Gaoni Y, & Mechoulam R (1964). Isolation, Structure, and Partial Synthesis of an Active Constituent of Hashish. JACS, 86, 1646–1647. [Google Scholar]
- Garrett ER, & Hunt CA (1974). Physiochemical properties, solubility, and protein binding of delta9-tetrahydrocannabinol. J Pharm Sci, 63, 1056–1064. [DOI] [PubMed] [Google Scholar]
- Gaston TE, & Szaflarski JP (2018). Cannabis for the Treatment of Epilepsy: an Update. Curr Neurol Neurosci Rep, 18, 73. [DOI] [PubMed] [Google Scholar]
- Geffrey AL, Pollack SF, Bruno PL, & Thiele EA (2015). Drug-drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia, 56, 1246–1251. [DOI] [PubMed] [Google Scholar]
- Grant KS, Petroff R, Isoherranen N, Stella N, & Burbacher TM (2017). Cannabis Use during Pregnancy: Pharmacokinetics and Effects on Child Development. Pharmacol Ther, 182, 133–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwich Biosciences, I. (2018). EPIDIOLEX® (cannabidiol) oral solution, CX [package insert]. Greenwich Biosciences, Inc, Carlsbad, CA. [Google Scholar]
- Grotenhermen F (2003). Pharmacokinetics and pharmacodynamics of cannabinoids. Clin Pharmacokinet, 42, 327–360. [DOI] [PubMed] [Google Scholar]
- Grotenhermen F (2004). Clinical Pharmacodynamics of Cannabinoids. Journal of Cannabis Therapeutics, 4. [Google Scholar]
- Grotenhermen F (2016). Medicinal uses of marijuana and cannabinoids. Crit Rev in Plant Sci, 35, 378–405. [Google Scholar]
- Grotenhermen F, & Muller-Vahl K (2012). The therapeutic potential of cannabis and cannabinoids. Dtsch Arztebl Int, 109, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanus LO, Meyer SM, Munoz E, Taglialatela-Scafati O, & Appendino G (2016). Phytocannabinoids: a unified critical inventory. Nat Prod Rep, 33, 1357–1392. [DOI] [PubMed] [Google Scholar]
- Hanus LO, Tchilibon S, Ponde DE, Breuer A, Fride E, & Mechoulam R (2005). Enantiomeric cannabidiol derivatives: synthesis and binding to cannabinoid receptors. Org Biomol Chem, 3, 1116–1123. [DOI] [PubMed] [Google Scholar]
- Harvey DJ, & Mechoulam R (1990). Metabolites of cannabidiol identified in human urine. Xenobiotica, 20, 303–320. [DOI] [PubMed] [Google Scholar]
- Heuberger JA, Guan Z, Oyetayo OO, Klumpers L, Morrison PD, Beumer TL, et al. (2015). Population pharmacokinetic model of THC integrates oral, intravenous, and pulmonary dosing and characterizes short- and long-term pharmacokinetics. Clin Pharmacokinet, 54, 209–219. [DOI] [PubMed] [Google Scholar]
- Hillig K (2005). Genetic evidence for speciation in Cannabis (Cannabaceae). Genet Resourc Crop Evol, 52, 161–180. [Google Scholar]
- Hillig KW, & Mahlberg PG (2004). A chemotaxonomic analysis of cannabinoid variation in Cannabis (Cannabaceae). Am J Bot, 91, 966–975. [DOI] [PubMed] [Google Scholar]
- Hirt D, Mentre F, Tran A, Rey E, Auleley S, Salmon D, et al. (2008). Effect of CYP2C19 polymorphism on nelfinavir to M8 biotransformation in HIV patients. Br J Clin Pharmacol, 65, 548–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland ML, Allen JD, & Arnold JC (2008). Interaction of plant cannabinoids with the multidrug transporter ABCC1 (MRP1). Eur J Pharmacol, 591, 128–131. [DOI] [PubMed] [Google Scholar]
- Holland ML, Lau DT, Allen JD, & Arnold JC (2007). The multidrug transporter ABCG2 (BCRP) is inhibited by plant-derived cannabinoids. Br J Pharmacol, 152, 815–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland ML, Panetta JA, Hoskins JM, Bebawy M, Roufogalis BD, Allen JD, et al. (2006). The effects of cannabinoids on P-glycoprotein transport and expression in multidrug resistant cells. Biochem Pharmacol, 71, 1146–1154. [DOI] [PubMed] [Google Scholar]
- Hryhorowicz S, Walczak M, Zakerska-Banaszak O, Slomski R, & Skrzypczak-Zielinska M (2018). Pharmacogenetics of Cannabinoids. Eur J Drug Metab Pharmacokinet, 43, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huestis MA (2007). Human cannabinoid pharmacokinetics. Chem Biodivers, 4, 1770–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huestis MA, Henningfield JE, & Cone EJ (1992). Blood cannabinoids. I. Absorption of THC and formation of 11-OH-THC and THCCOOH during and after smoking marijuana. J Anal Toxicol, 16, 276–282. [DOI] [PubMed] [Google Scholar]
- Hunt CA, & Jones RT (1980). Tolerance and disposition of tetrahydrocannabinol in man. J Pharmacol Exp Ther, 215, 35–44. [PubMed] [Google Scholar]
- Jaeger W, Benet LZ, & Bornheim LM (1996). Inhibition of cyclosporine and tetrahydrocannabinol metabolism by cannabidiol in mouse and human microsomes. Xenobiotica, 26, 275–284. [DOI] [PubMed] [Google Scholar]
- Jiang R, Yamaori S, Okamoto Y, Yamamoto I, & Watanabe K (2013). Cannabidiol is a potent inhibitor of the catalytic activity of cytochrome P450 2C19. Drug Metab Pharmacokinet, 28, 332–338. [DOI] [PubMed] [Google Scholar]
- Jiang R, Yamaori S, Takeda S, Yamamoto I, & Watanabe K (2011). Identification of cytochrome P450 enzymes responsible for metabolism of cannabidiol by human liver microsomes. Life Sci, 89, 165–170. [DOI] [PubMed] [Google Scholar]
- Jikomes N, & Zoorob M (2018). The Cannabinoid Content of Legal Cannabis in Washington State Varies Systematically Across Testing Facilities and Popular Consumer Products. Sci Rep, 8, 4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson E, Halldin MM, Agurell S, Hollister LE, & Gillespie HK (1989). Terminal elimination plasma half-life of delta 1-tetrahydrocannabinol (delta 1-THC) in heavy users of marijuana. Eur J Clin Pharmacol, 37, 273–277. [DOI] [PubMed] [Google Scholar]
- Johansson E, Ohlsson A, Lindgren JE, Agurell S, Gillespie H, & Hollister LE (1987). Single-dose kinetics of deuterium-labelled cannabinol in man after intravenous administration and smoking. Biomed Environ Mass Spectrom, 14, 495–499. [DOI] [PubMed] [Google Scholar]
- Jusko WJ, Schentag JJ, Clark JH, Gardner M, & Yurchak AM (1978). Enhanced biotransformation of theophylline in marihuana and tobacco smokers. Clin Pharmacol Ther, 24, 405–410. [PubMed] [Google Scholar]
- Karschner EL, Schwope DM, Schwilke EW, Goodwin RS, Kelly DL, Gorelick DA, et al. (2012). Predictive model accuracy in estimating last Delta9-tetrahydrocannabinol (THC) intake from plasma and whole blood cannabinoid concentrations in chronic, daily cannabis smokers administered subchronic oral THC. Drug Alcohol Depend, 125, 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kattel K, Evande R, Tan C, Mondal G, Grem JL, & Mahato RI (2015). Impact of CYP2C19 polymorphism on the pharmacokinetics of nelfinavir in patients with pancreatic cancer. Br J Clin Pharmacol, 80, 267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall DA, & Yudowski GA (2016). Cannabinoid Receptors in the Central Nervous System: Their Signaling and Roles in Disease. Front Cell Neurosci, 10, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausner HA, Wilcox HG, & Dingell JV (1975). The use of zonal ultracentrifugation in the investigation of the binding of delta9-tetrahydrocannabinol by plasma lipoproteins. Drug Metab Dispos, 3, 314–319. [PubMed] [Google Scholar]
- Knodell RG, Dubey RK, Wilkinson GR, & Guengerich FP (1988). Oxidative metabolism of hexobarbital in human liver: relationship to polymorphic S-mephenytoin 4-hydroxylation. J Pharmacol Exp Ther, 245, 845–849. [PubMed] [Google Scholar]
- Kosel BW, Aweeka FT, Benowitz NL, Shade SB, Hilton JF, Lizak PS, et al. (2002). The effects of cannabinoids on the pharmacokinetics of indinavir and nelfinavir. AIDS, 16, 543–550. [DOI] [PubMed] [Google Scholar]
- Kreuz DS, & Axelrod J (1973). Delta-9-tetrahydrocannabinol: localization in body fat. Science, 179, 391–393. [DOI] [PubMed] [Google Scholar]
- Kumar RN, Chambers WA, & Pertwee RG (2001). Pharmacological actions and therapeutic uses of cannabis and cannabinoids. Anaesthesia, 56, 1059–1068. [DOI] [PubMed] [Google Scholar]
- Lal S, Mahajan A, Chen WN, & Chowbay B (2010). Pharmacogenetics of target genes across doxorubicin disposition pathway: a review. Curr Drug Metab, 11, 115–128. [DOI] [PubMed] [Google Scholar]
- Leighty EG, Fentiman AF Jr., & Foltz RL (1976). Long-retained metabolites of delta9-and delta8-tetrahydrocannabinols identified as novel fatty acid conjugates. Res Commun Chem Pathol Pharmacol, 14, 13–28. [PubMed] [Google Scholar]
- Levitt DG (2007). Heterogeneity of human adipose blood flow. BMC Clin Pharmacol, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lile JA, Kelly TH, Charnigo RJ, Stinchcomb AL, & Hays LR (2013). Pharmacokinetic and pharmacodynamic profile of supratherapeutic oral doses of Delta(9) -THC in cannabis users. J Clin Pharmacol, 53, 680–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe RH, Abraham TT, Darwin WD, Herning R, Cadet JL, & Huestis MA (2009). Extended urinary Delta9-tetrahydrocannabinol excretion in chronic cannabis users precludes use as a biomarker of new drug exposure. Drug Alcohol Depend, 105, 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas CJ, Galettis P, & Schneider J (2018). The Pharmacokinetics and the Pharmacodynamics of Cannabinoids. Br J Clin Pharmacol. 84, 2477–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCallum CA, & Russo EB (2018). Practical considerations in medical cannabis administration and dosing. Eur J Intern Med, 49, 12–19. [DOI] [PubMed] [Google Scholar]
- Martin BR, Mechoulam R, & Razdan RK (1999). Discovery and characterization of endogenous cannabinoids. Life Sci, 65, 573–595. [DOI] [PubMed] [Google Scholar]
- Martinez-Pinilla E, Varani K, Reyes-Resina I, Angelats E, Vincenzi F, Ferreiro-Vera C, et al. (2017). Binding and Signaling Studies Disclose a Potential Allosteric Site for Cannabidiol in Cannabinoid CB2 Receptors. Front Pharmacol, 8, 744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell JC, & Mendelson B (2016). What Do We Know Now About the Impact of the Laws Related to Marijuana? J Addict Med, 10, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur A, Lichti CF, Prather PL, Zielinska AK, Bratton SM, Gallus-Zawada A, et al. (2009). Characterization of human hepatic and extrahepatic UDP-glucuronosyltransferase enzymes involved in the metabolism of classic cannabinoids. Drug Metab Dispos, 37, 1496–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPartland JM, Glass M, & Pertwee RG (2007). Meta-analysis of cannabinoid ligand binding affinity and receptor distribution: interspecies differences. Br J Pharmacol, 152, 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naef M, Russmann S, Petersen-Felix S, & Brenneisen R (2004). Development and pharmacokinetic characterization of pulmonal and intravenous delta-9-tetrahydrocannabinol (THC) in humans. J Pharm Sci, 93, 1176–1184. [DOI] [PubMed] [Google Scholar]
- Navarro G, Varani K, Reyes-Resina I, Sanchez de Medina V, Rivas-Santisteban R, Sanchez-Carnerero Callado C, et al. (2018). Cannabigerol Action at Cannabinoid CB1 and CB2 Receptors and at CB1-CB2 Heteroreceptor Complexes. Front Pharmacol, 9, 632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesink RJ, Rigter S, Koeter MW, & Brunt TM (2015). Potency trends of Delta9-tetrahydrocannabinol, cannabidiol and cannabinol in cannabis in the Netherlands: 2005–15. Addiction, 110, 1941–1950. [DOI] [PubMed] [Google Scholar]
- Ohlsson A, Lindgren JE, Andersson S, Agurell S, Gillespie H, & Hollister LE (1986). Single-dose kinetics of deuterium-labelled cannabidiol in man after smoking and intravenous administration. Biomed Environ Mass Spectrom, 13, 77–83. [DOI] [PubMed] [Google Scholar]
- Ohlsson A, Lindgren JE, Wahlen A, Agurell S, Hollister LE, Gillespie HK (1982). Single dose kinetics of deuterium labelled delta 1-tetrahydrocannabinol in heavy and light cannabis users. Biomed Mass Spectrom, 9(1), 6–10. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes M (1990). Marijuana smoking: factors that influence the bioavailability of tetrahydrocannabinol. NIDA Res Monogr, 99, 42–62. [PubMed] [Google Scholar]
- Pertwee RG (1997). Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol Ther, 74, 129–180. [DOI] [PubMed] [Google Scholar]
- Pertwee RG (2008). The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids:delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol, 153, 199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Ross RA (2002). Cannabinoid receptors and their ligands. Prostaglandins Leukot Essent Fatty Acids, 66, 101–121. [DOI] [PubMed] [Google Scholar]
- Radwan MM, ElSohly MA, El-Alfy AT, Ahmed SA, Slade D, Husni AS, et al. (2015). Isolation and Pharmacological Evaluation of Minor Cannabinoids from High-Potency Cannabis sativa. J Nat Prod, 78, 1271–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynor DK, Dickinson R, Knapp P, Long AF, & Nicolson DJ (2011). Buyer beware? Does the information provided with herbal products available over the counter enable safe use? BMC Med, 9, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs CE Jr., Egorin MJ, Fuks JZ, Schnaper N, Duffey P, Colvin OM, et al. (1981). Initial observations on the effects of delta 9-tetrahydrocannabinol on the plasma pharmacokinetics of cyclophosphamide and doxorubicin. J Clin Pharmacol, 21, 90S–98S. [DOI] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Calandra B, Shire D, Bouaboula M, Oustric D, Barth F, et al. (1996). Characterization of two cloned human CB1 cannabinoid receptor isoforms. J Pharmacol Exp Ther, 278, 871–878. [PubMed] [Google Scholar]
- Ross SA, ElSohly MA (1995). Constituents of Cannabis sativa L. XXVIII. A review of the natural constituents: 1980–1994. Zagazig Journal of Pharmaceutical Science, 4, 1–10. [Google Scholar]
- Roth MD, Marques-Magallanes JA, Yuan M, Sun W, Tashkin DP, & Hankinson O (2001). Induction and regulation of the carcinogen-metabolizing enzyme CYP1A1 by marijuana smoke and delta (9)-tetrahydrocannabinol. Am J Respir Cell Mol Biol, 24, 339–344. [DOI] [PubMed] [Google Scholar]
- Roy P, Yu LJ, Crespi CL, & Waxman DJ (1999). Development of a substrate-activity based approach to identify the major human liver P-450 catalysts of cyclophosphamide and ifosfamide activation based on cDNA-expressed activities and liver microsomal P450 profiles. Drug Metab Dispos, 27, 655–666. [PubMed] [Google Scholar]
- Sachse-Seeboth C, Pfeil J, Sehrt D, Meineke I, Tzvetkov M, Bruns E, et al. (2009). Interindividual variation in the pharmacokinetics of Delta9-tetrahydrocannabinol as related to genetic polymorphisms in CYP2C9. Clin Pharmacol Ther, 85, 273–276. [DOI] [PubMed] [Google Scholar]
- Schatz AR, Lee M, Condie RB, Pulaski JT, & Kaminski NE (1997). Cannabinoid receptors CB1 and CB2: a characterization of expression and adenylate cyclase modulation within the immune system. Toxicol Appl Pharmacol, 142, 278–287. [DOI] [PubMed] [Google Scholar]
- Schwilke EW, Karschner EL, Lowe RH, Gordon AM, Cadet JL, Herning RI, et al. (2009). Intra- and intersubject whole blood/plasma cannabinoid ratios determined by 2-dimensional, electron impact GC-MS with cryofocusing. Clin Chem, 55, 11881195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Murthy P, & Bharath MM (2012). Chemistry, metabolism, and toxicology of cannabis: clinical implications. Iran J Psychiatry, 7, 149–156. [PMC free article] [PubMed] [Google Scholar]
- Showalter VM, Compton DR, Martin BR, & Abood ME (1996). Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (CB2): identification of cannabinoid receptor subtype selective ligands. J Pharmacol Exp Ther, 278, 989–999. [PubMed] [Google Scholar]
- Skopp G, & Potsch L (2008). Cannabinoid concentrations in spot serum samples 24–48 hours after discontinuation of cannabis smoking. J Anal Toxicol, 32, 160–164. [DOI] [PubMed] [Google Scholar]
- Small E (2015). Evolution and classification of Cannabis sativa (Marijuana, Hemp) in relation to human utilization. Bot Rev, 81, 189–294. [Google Scholar]
- Solas C, Simon N, Drogoul MP, Quaranta S, Frixon-Marin V, Bourgarel-Rey V, et al. (2007). Minimal effect of MDR1 and CYP3A5 genetic polymorphisms on the pharmacokinetics of indinavir in HIV-infected patients. Br J Clin Pharmacol, 64, 353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiro AS, Wong A, Boucher AA, & Arnold JC (2012). Enhanced brain disposition and effects of Delta9-tetrahydrocannabinol in P-glycoprotein and breast cancer resistance protein knockout mice. PLoS One, 7, e35937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stejskalova L, & Pavek P (2011). The function of cytochrome P450 1A1 enzyme (CYP1A1) and aryl hydrocarbon receptor (AhR) in the placenta. Curr Pharm Biotechnol, 12, 715–730. [DOI] [PubMed] [Google Scholar]
- Stout SM, & Cimino NM (2014). Exogenous cannabinoids as substrates, inhibitors, and inducers of human drug metabolizing enzymes: a systematic review. Drug Metab Rev, 46, 86–95. [DOI] [PubMed] [Google Scholar]
- Szabo B, & Schlicker E (2005). Effects of cannabinoids on neurotransmission. Handb Exp Pharmacol, 327–365. [DOI] [PubMed] [Google Scholar]
- Tait RJ, Caldicott D, Mountain D, Hill SL, & Lenton S (2016). A systematic review of adverse events arising from the use of synthetic cannabinoids and their associated treatment. Clin Toxicol (Phila), 54, 1–13. [DOI] [PubMed] [Google Scholar]
- Thomas BF, Compton DR, & Martin BR (1990). Characterization of the lipophilicity of natural and synthetic analogs of delta 9-tetrahydrocannabinol and its relationship to pharmacological potency. J Pharmacol Exp Ther, 255, 624–630. [PubMed] [Google Scholar]
- van Amsterdam J, Vervloet J, de Weert G, Buwalda VJA, Goudriaan AE, & van den Brink W (2018). Acceptance of pharmaceutical cannabis substitution by cannabis using patients with schizophrenia. Harm Reduct J, 15, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara D, Bidwell LC, Gaudino R, Torres A, Du G, Ruthenburg TC, et al. (2017). Compromised External Validity: Federally Produced Cannabis Does Not Reflect Legal Markets. Sci Rep, 7, 46528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall ME, Sadler BM, Brine D, Taylor H, & Perez-Reyes M (1983). Metabolism, disposition, and kinetics of delta-9-tetrahydrocannabinol in men and women. Clin Pharmacol Ther, 34, 352–363. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Yamaori S, Funahashi T, Kimura T, & Yamamoto I (2007). Cytochrome P450 enzymes involved in the metabolism of tetrahydrocannabinols and cannabinol by human hepatic microsomes. Life Sci, 80, 1415–1419. [DOI] [PubMed] [Google Scholar]
- Welling M, Shapter T, Rose T, Liu L, Stanger R, & King G (2016). A belated green revolution for Cannabis: virtual genetic resources for fast-track cultivar development. Front Plant Sci, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. (2016). The health and social effects of nonmedical cannabis use. Geneva, Switzerland: World Health Organization. [Google Scholar]
- Widman M, Agurell S, Ehrnebo M, & Jones G (1974). Binding of (+)- and (minus)-delta-1-tetrahydrocannabinols and (minus)-7-hydroxy-delta-1-tetrahydrocannabinol to blood cells and plasma proteins in man. J Pharm Pharmacol, 26, 914–916. [DOI] [PubMed] [Google Scholar]
- Yamaori S, Ebisawa J, Okushima Y, Yamamoto I, & Watanabe K (2011). Potent inhibition of human cytochrome P450 3A isoforms by cannabidiol: role of phenolic hydroxyl groups in the resorcinol moiety. Life Sci, 88, 730–736. [DOI] [PubMed] [Google Scholar]
- Yamaori S, Koeda K, Kushihara M, Hada Y, Yamamoto I, & Watanabe K (2012). Comparison in the in vitro inhibitory effects of major phytocannabinoids and polycyclic aromatic hydrocarbons contained in marijuana smoke on cytochrome P450 2C9 activity. Drug Metab Pharmacokinet, 27, 294–300. [DOI] [PubMed] [Google Scholar]
- Yamaori S, Kushihara M, Yamamoto I, & Watanabe K (2010). Characterization of major phytocannabinoids, cannabidiol and cannabinol, as isoform-selective and potent inhibitors of human CYP1 enzymes. Biochem Pharmacol, 79, 1691–1698. [DOI] [PubMed] [Google Scholar]
- Yamaori S, Maeda C, Yamamoto I, & Watanabe K (2011). Differential inhibition of human cytochrome P450 2A6 and 2B6 by major phytocannabinoids. Forensic Toxicology, 29, 117–124. [Google Scholar]
- Yamaori S, Okamoto Y, Yamamoto I, & Watanabe K (2011). Cannabidiol, a major phytocannabinoid, as a potent atypical inhibitor for CYP2D6. Drug Metab Dispos, 39, 2049–2056. [DOI] [PubMed] [Google Scholar]
- Yamaori S, Okushima Y, Yamamoto I, & Watanabe K (2014). Characterization of the structural determinants required for potent mechanism-based inhibition of human cytochrome P450 1A1 by cannabidiol. Chem Biol Interact, 215, 62–68. [DOI] [PubMed] [Google Scholar]
- Yamreudeewong W, Wong ΗK, Brausch LM, & Pulley KR (2009). Probable interaction between warfarin and marijuana smoking. Ann Pharmacother, 43, 1347–1353. [DOI] [PubMed] [Google Scholar]
- Zendulka O, Dovrtelova G, Noskova K, Turjap M, Sulcova A, Hanus L, et al. (2016). Cannabinoids and Cytochrome P450 Interactions. CurrDrug Metab, 17, 206–226. [DOI] [PubMed] [Google Scholar]
- Zhu HJ, Wang JS, Markowitz JS, Donovan JL, Gibson BB, Gefroh HA, et al. (2006). Characterization of P-glycoprotein inhibition by major cannabinoids from marijuana. J Pharmacol Exp Ther, 317, 850–857. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.