

Abstract
Rationale:
Anti-glomerular basement membrane disease (anti-GBM disease) is a rare small vessel vasculitis caused by autoantibodies directed against the glomerular and alveolar basement membranes. Anti-GBM disease is usually a monophasic illness and relapse is rare after effective treatment. This article reports a case of coexistence of recurrent anti-GBM disease and T-cell large granular lymphocytic (T-LGL) leukemia.
Patient concerns:
A 37-year-old man presented with hematuria, edema, and acute kidney injury for 2 months.
Diagnosis:
Anti-GBM disease was diagnosed by renal biopsy, in which crescentic glomerulonephritis was observed with light microscopy, strong linear immunofluorescent staining for immunoglobulin G on the GBM and positive serum anti-GBM antibody. Given this diagnosis, the patient was treated with plasmapheresis, steroids, and cyclophosphamide for 4 months. The anti-GBM antibody titer was maintained to negative level but the patient remained dialysis-dependent. One year later, the patient suffered with a relapse of anti-GBM disease, after an extensive examination, he was further diagnosed T-LGL leukemia by accident.
Interventions:
The patient received cyclosporine A therapy for T-LGL leukemia.
Outcomes:
After treatment with cyclosporine A, serum anti-GBM antibody became undetectable. During the 16 months follow-up, anti-GBM titer remained normal and abnormal T-lymphocytes in the bone marrow and peripheral blood were also decreased.
Lessons:
T-LGL leukemia is an indolent lymphoproliferative disorder that represents a monoclonal expansion of cytotoxic T cells, which has been reported to be accompanied by some autoimmune diseases. This is the first report of coincidence of T-LGL leukemia and anti-GBM disease, and suggests there are some relationships between these 2 diseases. Clinical physicians should exclude hematological tumors when faced with recurrent anti-GBM disease.
Keywords: anti-GBM disease, recurrent, T-cell large granular lymphocytic leukemia
1. Introduction
Anti-glomerular basement membrane disease (anti-GBM disease) is a rare small vessel vasculitis caused by autoantibodies directed against the noncollagenous (NC1) domain of the α3 chain of type IV collagen (α3 [IV]NC1) which is predominantly located in the glomerular and alveolar basement membranes.[1,2] Anti-GBM disease is diagnosed by a combination of positive circulating serum anti-GBM antibodies and detecting linear immunoglobulin G (IgG) deposits on the GBM on renal biopsy. Standard treatment for anti-GBM disease includes plasmapheresis which rapidly removes pathogenic autoantibody, along with cyclophosphamide (CTX) and corticosteroids, to prevent further autoantibody production. Anti-GBM disease is usually a monophasic illness and relapse is rare.[3]
T-cell large granular lymphocytic (T-LGL) leukemia is an indolent lymphoproliferative disorder that represents a monoclonal expansion of cytotoxic T cells. Patients with T-LGL leukemia usually present with neutropenia, anemia, and an increase in the number of LGLs in their peripheral blood. Most patients remain asymptomatic and are diagnosed incidentally on routine blood test.[4] It has been reported to accompany some autoimmune diseases.[5] However, to date, there has been no report of T-LGL leukemia and anti-GBM disease.
Here, we report an interesting case of recurrent anti-GBM disease with concurrent diagnosis of T-LGL leukemia.
2. Case report
A 37-year-old man initially presented with a 2-month history of hematuria and palpebral edema. He had a 30 pack-year smoking history, but was not exposed to known chemicals or solvents. He had no history of chronic diseases or drug abuse. On examination, his blood pressure was 160/110 mm Hg. Urinalysis at that time revealed hematuria (3+) and proteinuria (3+). Serum creatinine was 95 μmol/L (normal range: 50–130 μmol/L). Other investigations included a normal abdominal computed tomography (CT) and cystoscopy. He was treated with anti-hypertensive and anti-bacterial drugs for 2 weeks, but the symptom was not ameliorated. He subsequently presented to the regional hospital in October 2015 with an elevated creatinine of 273 μmol/L, low albumin of 27 g/L, urinalysis showed 5961/μL red blood cells (RBC), with a proteinuria of 5.48 g/24 h. The immunologic tests including anti-neutrophil cytoplasmic antibodies, antinuclear antibody were all negative. A renal biopsy showed crescentic glomerulonephritis and segmental membranous nephropathy, in which 60% of the glomeruli contained active cellular crescents, with strong linear GBMs staining on direct immunofluorescence that was IgG1, 2, 4 dominant; 4 glomeruli had segmental or global sclerosis (Fig. 1). Anti-GBM antibody was positive (86.99 U/L↑; ml000386, Mlbio, normal range: <20 U/L; indirect immunofluorescent assay was also positive (IIFT: kidney (monkey), Euroimmun). Then he was diagnosed anti-GBM disease and was treated with daily steroids (1 g/d × 3 d→40 mg/d) and monthly intravenous CTX (1 g).
Figure 1.
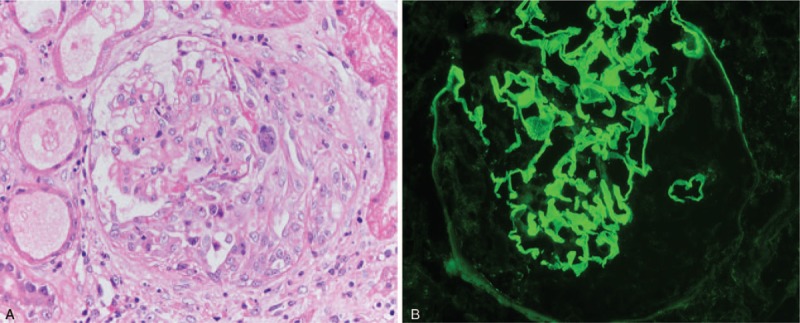
The renal pathology of the patient's renal biopsy. (A) The silver staining of the kidney biopsy showed an active cellular crescent (×400); (B) The immunofluorescence showed strong linear glomerular basement membrane staining of IgG. (×400). IgG = immunoglobulin G.
Two weeks later, without improvement of renal function, he was transferred to our hospital. Patient was found to have 6.92 g urinary protein in 24 hours, positive anti-GBM antibody, serum creatinine level of 295 μmol/L and serum albumin of 29 g/L. Cluster of differentiation (CD) antigen series were abnormal: lymphocyte population: 4.84%, CD3+: 77.49%↑, CD4+: 13.24%↓, CD8+: 59.38%↑, NK+: 4.99%↓, CD19+: 17.35%, CD20+: 17.17%, CD45: positive, CD4/CD8: 0.22↓. The total CD4+ cell counting was 0.183 × 109/L (normal range: >0.4 × 109/L). He was treated with 6 sessions of plasmapheresis. During the treatment, the patient's serum creatinine rose to 641 μmol/L, hemodialysis treatment was initiated and the patient was placed on steroid treatment and 3-month course of pulsed intravenous CTX. Anti-GBM antibody turned negative by the second month. He remained dialysis-dependent throughout the duration of his treatment.
At the fourth month, the patient presented with cough and expectoration, without hemoptysis. Pulmonary CT scan revealed bilateral upper pulmonary infection, and Asperguillus fumigatus was found in sputum culture. His total CD4+ counting was only 0.019 × 109/L which was much lower than normal. Anti-GBM antibody was negative. Pulmonary aspergillosis was considered. He was treated with voriconazole and his symptoms improved. During this period, the steroids dosage was reduced and pulsed intravenous CTX treatment was stopped while the RBC and protein in the urine decreased, the serum creatinine also decreased to 400 μmol/L. The anti-GBM antibody remained negative for nearly 1 year until the anti-GBM antibody rose to 123.39 U/L again. The serum creatinine increased to 593 μmol/L and the RBC and protein in the urine also increased. The patient had no respiratory system symptoms and the pulmonary CT was normal. There was no evidence of infection. Notably, peripheral blood CD antigen series showed that the proportion of CD3+ CD4− CD8− cells was significantly higher than normal which was 43.4%, peripheral blood lymphocyte counting was 1.99 × 109/L.
Furthermore, peripheral blood and bone marrow examination shown: classic large lymphocytes with a condensed round nuclear, abundant pale basophilic cytoplasm and small azurophilic granules were found on peripheral blood smear (Fig. 2); bone marrow smear showed a few macrolymphocytes and atypical lymphocytes (Fig. 2); peripheral blood flow cytometry found 16% abnormal T lymphocytes (CD2+, CD3+, CD5+ dim, CD7+, CD16+, CD57+, CD56+, CD4−, CD8−, TCRαβ−, TCRγδ+) (Fig. 3), the T-LGL count was 0.32 × 109/L; bone marrow flow cytometry also determined 16% abnormal T lymphocytes (CD2+, CD3+, CD7+, CD57+, CD56+, CD94+, CD5+ dim, CD16+ dim, CD4−, CD8−, TCRαβ−, TCRγδ+). Most importantly, T cell receptors (TCR) gene rearrangement indicated a rearrangement of TCRγ and TCRδ gene. Ultrasound found no lymph nodes enlargement, no splenomegaly, and no hepatomegaly. Positron emission tomography–CT found no abnormality. After a consultation, he was diagnosed CD4−/CD8− γδ T-LGL leukemia and recurrence of anti-GBM disease. He was then treated with 4 sessions of plasmapheresis and the anti-GBM antibody titer declined to 42.76 U/L. He was prescribed with cyclosporine 100 mg q12h orally.
Figure 2.
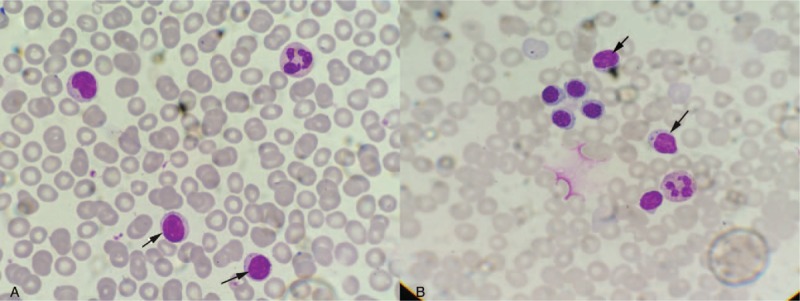
The peripheral blood and bone marrow smear. (A) Peripheral blood smear manifested classic large lymphocytes (arrow) with a condensed round nuclear, abundant pale basophilic cytoplasm, and small azurophilic granules (×1000); (B) Bone marrow smear showed a few macrolymphocytes and atypical lymphocytes (arrow) (×1000).
Figure 3.
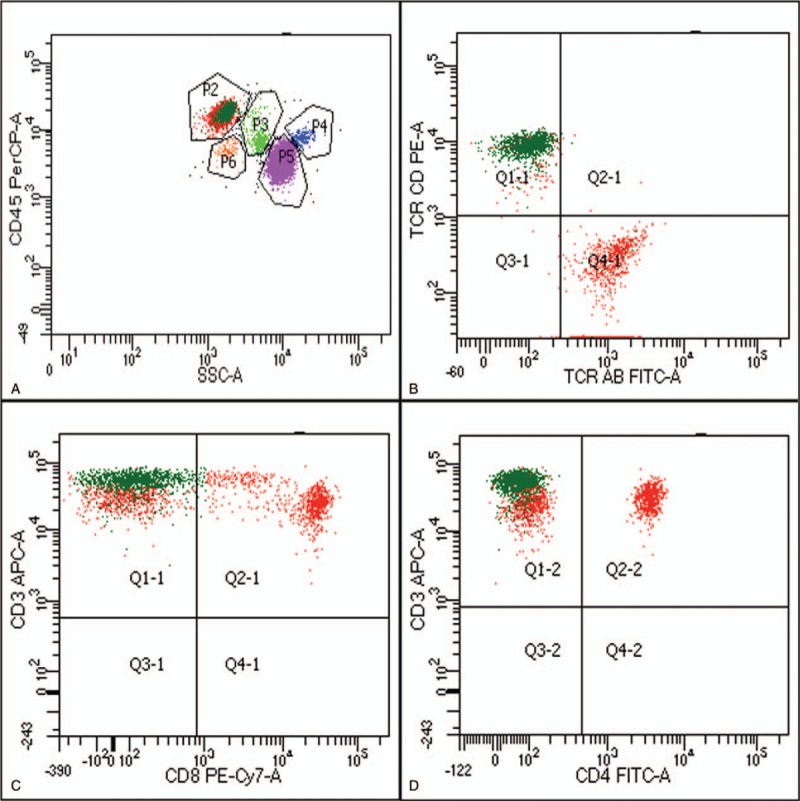
The results of peripheral blood flow cytometry. (A) CD45/SSC gating showed 40% lymphocytes (P2) and 16% CD3+ CD4− CD8− abnormal T lymphocytes (dark green dots, P3); (B) The abnormal T lymphocytes (dark green dots) were CD3+ CD8−; (C) The abnormal T lymphocytes (dark green dots) were CD3+ CD4−; (B) The abnormal T lymphocytes (dark green dots) were TCRγδ+ and TCRαβ- (P.S.TCR AB = TCRαβ, TCR CD = TCR γδ).
During the 16-month follow-up, the anti-GBM titer returned to normal and the abnormal T-lymphocytes in the bone marrow and peripheral blood were also decreased (Fig. 4). In August 2017, the peripheral blood abnormal T lymphocytes rose again and the plasma concentration of cyclosporine was not sufficient, so the dose of cyclosporine was increased to 150 mg q12h. Hence, the anti-GBM antibody stayed negative and the percent of abnormal T lymphocytes in peripheral blood had a tendency to decline.
Figure 4.
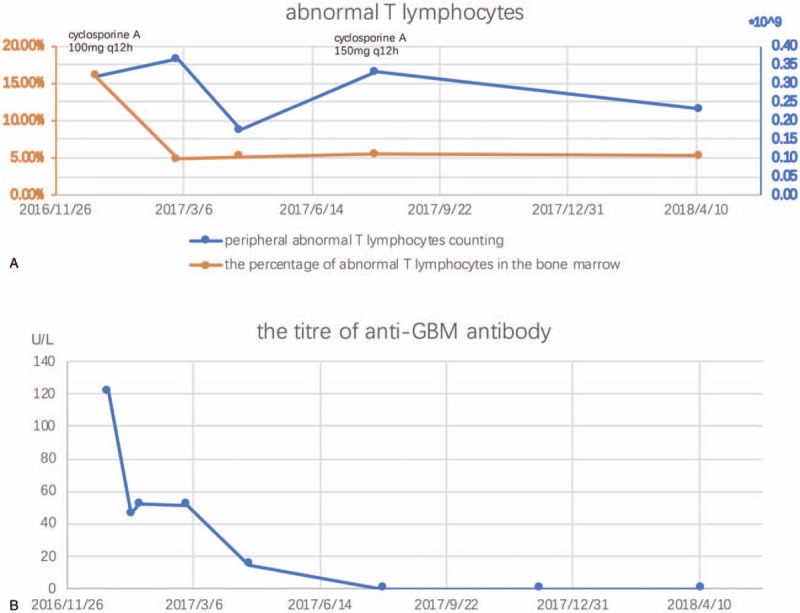
During the 16-mo follow-up, (A) The abnormal T lymphocytes in the peripheral blood and bone marrow were improved after the treatment of 100 mg cyclosporine A q12h, but in 2017.8, the peripheral abnormal T lymphocytes have a rebound and the plasma concentration of cyclosporine A was low so the dose was adjusted to 150 mg q12h and the peripheral abnormal T lymphocytes decreased again; (B) The titer of the anti-GBM antibody returned normal. GBM = glomerular basement membrane.
3. Discussion
In this article, we described a very rare case of recurrent anti-GBM disease combined with T-LGL leukemia. To our knowledge, there is no published report of anti-GBM disease accompanied with T-LGL leukemia previously.
Anti-GBM disease is usually a monophasic illness and relapse is rare, occurring in <3% of patients in the Hammersmith series.[3] Anti-GBM antibodies are usually undetectable after treatment with plasmapheresis and immunosuppressive therapy and are undetectable at 12 months in untreated disease.[6] Recurrence tends to happen after the initial symptoms have been controlled but before the antibody titer has been fully suppressed.[7] There are only a few reports of late relapses of anti-GBM disease.[7–10] This case is unusual in that it recurred following numerous negative anti-GBM titers, and in that it accompanied with coincidental T-LGL leukemia.
T-LGL leukemia represents 2% to 3% of mature lymphocytic leukemia and is defined by the World Health Organization classification as a persistent (>6 months) clonal expansion of peripheral blood T-LGL cells, usually between 2 and 20 × 109/L, without a clearly identified cause. T-LGL leukemia most frequently arise from αβ T cells (95%), with only a small subgroup (∼5%) from γδ T cells.[11] γδ T-LGL leukemia of the CD4−/CD8− subtype is rare, and data are limited in the literature.[11] γδ T cells are found mainly in mucosal surfaces and splenic red pulp and are part of the innate immune response.
Some autoimmune diseases have been reported to be associated with LGL diseases, such as rheumatoid arthritis and Felty's syndrome, scleroderma, Sjogren's syndrome, systemic lupus, polymyositis, and vasculitis.[5,12–15] Audemard et al studied a series of 11 patients displaying LGL leukemia associated with vasculitis, without anti-GBM disease.[5] Furthermore, Viny et al systematic analyzed the coexistence of T-LGL leukemia with B cell abnormalities.[16] However, the mechanism by which autoimmune phenomena develop in patients with T-LGL leukemia is not well understood. The context of immune-mediated diseases led to the hypothesis that LGL leukemia represents an exaggerated clonal immune response to a persistent antigen such as an autoantigen or viral antigen.[17]
On the other hand, Gu et al reported 3 patients with combined anti-GBM disease and Castleman disease, a lymphoproliferative disorder. They found sporadic plasma cells producing antibodies reactive with α3 (IV)NC1 in 1 patient with lymph node biopsy specimens available.[18] The large amounts of interleukin 6 produced by B cells in the germinal center of lymph nodes provide an inflammatory environment and may stimulate the proliferation of B cells and contribute to autoantibody production.
There were 3 interesting points in this case: First, the T-LGL leukemia was diagnosed incidentally on routine blood tests while the anti-GBM disease relapsed. As we mentioned previously, T-LGL leukemia is an indolent lymphoproliferative disorder. Most patients remain asymptomatic. In another word, this disease may have a long incubation period without being identified. Second, it is controversial whether the patient had T-LGL leukemia at the beginning of anti-GBM disease diagnosis. The peripheral lymphocyte population was analyzed in our hospital after the therapy of steroids and pulsed intravenous CTX. There was no significant evidence of clonal expansion of peripheral blood T-LGL cells at that time. His peripheral blood CD4+ cell counting was consistently lower than normal, which may be due to immunosuppressive therapy. But CTX is also reported to be effective in T-LGL leukemia patients. Furthermore, the therapy response was unexpected. In general, anti-GBM disease patients presenting with creatinine levels greater than 4 mg/dl, oliguria, and more than 50% crescents on renal biopsy rarely recover.[19] In this case, the patient's renal function was only borderline abnormal at the beginning of diagnosis and treatment. But aggressive therapy with plasmapheresis, corticosteroids, and CTX did not improve his renal prognosis. Gu et al reported 3 patients with combined anti-GBM disease and Castleman disease.[18] Standard treatment of methylprednisolone, CTX, and plasmapheresis did not improve their renal prognosis. All of them were treated including plasmapheresis and methylprednisolone pulse therapy, combined with COP (cyclophosphamide, vincristine, and prednisone) chemotherapy (CTX, vincristine, and prednisone)/R-CHOP (rituximab, cyclophosphamide, Adriamycin, vincristine, and prednisone) chemotherapy. The combination therapy was effective in the clearance of anti-GBM antibody and improvement of renal function. Of course, it is difficult to confirm retrospectively whether the immunosuppressive treatment covered the fact of combined T-LGL leukemia at that time. But these unusual clues reminded us to exclude underlining lymphoproliferative disorder. Third, anti-GBM antibody titer turned negative after the treatment of cyclosporine A for T-LGL leukemia. Therefore, in our case, the causal relationship between these 2 diseases appeared highly possible because therapy for T-LGL leukemia has been shown to be effective in improving both T-LGL leukemia and anti-GBM antibody simultaneously. Further investigation and researches are needed to determine the causal link between anti-GBM disease and T-LGL leukemia.
A limitation of this case is the lack of blood samples to verify the presence of T-LGL leukemia at the beginning of anti-GBM disease. Another limitation is that we did not find the pathology relationship between anti-GBM disease and T-LGL leukemia. Further follow-up and intensive study for the pathogenesis are necessary.
Author contributions
Conceptualization: Jun Xue.
Data curation: Ping Zhu, Tong Chen.
Formal analysis: Tong Chen.
Investigation: Jun Xue.
Methodology: Ping Zhu, Shaojun Liu.
Resources: Shaojun Liu, Chuanming Hao, Jun Xue.
Supervision: Chuanming Hao, Jun Xue.
Writing – original draft: Min Zhang, Nan Guan.
Writing – review and editing: Min Zhang, Chuanming Hao.
Min Zhang orcid: 0000-0002-2316-5452.
Footnotes
Abbreviations: Anti-GBM disease = anti-glomerular basement membrane disease, CTX = cyclophosphamide, T-LGL = T-cell large granular lymphocytic leukemia.
MZ and NG contributed equally to this work.
The patient has provided informed consent for publication of the case.
The authors have no conflicts of interest to disclose.
References
- [1].Saus J, Wieslander J, Langeveld JP, et al. Identification of the Goodpasture antigen as the alpha 3 (IV) chain of collagen IV. J Biol Chem 1988;263:13374–80. [PubMed] [Google Scholar]
- [2].Turner N, Mason PJ, Brown R, et al. Molecular cloning of the human Goodpasture antigen demonstrates it to be the alpha 3 chain of type IV collagen. J Clin Investig 1992;89:592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Levy JB, Turner AN, Rees AJ, et al. Long-term outcome of anti-glomerular basement membrane antibody disease treated with plasma exchange and immunosuppression. Ann Intern Med 2001;134:1033–42. [DOI] [PubMed] [Google Scholar]
- [4].Oshimi K. Clinical features, pathogenesis, and treatment of large granular lymphocyte leukemias. Intern Med 2017;56:1759–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Audemard A, Lamy T, Bareau B, et al. Vasculitis associated with large granular lymphocyte (LGL) leukemia: presentation and treatment outcomes of 11 cases. Semin Arthritis Rheum 2013;43:362–6. [DOI] [PubMed] [Google Scholar]
- [6].Bolton WK. Goodpasture's syndrome. Kidney Int 1996;50:1753–66. [DOI] [PubMed] [Google Scholar]
- [7].Levy JB, Lachmann RH, Pusey CD. Recurrent Goodpasture's disease. Am J Kidney Dis 1996;27:573–8. [DOI] [PubMed] [Google Scholar]
- [8].Trpkov K, Abdulkareem F, Jim K, et al. Recurrence of anti-GBM antibody disease twelve years after transplantation associated with de novo IgA nephropathy. Clin Nephrol 1998;49:124–8. [PubMed] [Google Scholar]
- [9].Fonck C, Loute G, Cosyns JP, et al. Recurrent fulminant anti-glomerular basement membrane nephritis at a 7-year interval. Am J Kidney Dis 1998;32:323–7. [DOI] [PubMed] [Google Scholar]
- [10].Borza DB, Chedid MF, Colon S, et al. Recurrent Goodpasture's disease secondary to a monoclonal IgA1-kappa antibody autoreactive with the alpha1/alpha2 chains of type IV collagen. Am J Kidney Dis 2005;45:397–406. [DOI] [PubMed] [Google Scholar]
- [11].Chen YH, Chadburn A, Evens AM, et al. Clinical, morphologic, immunophenotypic, and molecular cytogenetic assessment of CD4-/CD8-gammadelta T-cell large granular lymphocytic leukemia. Am J Clin Pathol 2011;136:289–99. [DOI] [PubMed] [Google Scholar]
- [12].Rose MG, Berliner N. T-cell large granular lymphocyte leukemia and related disorders. Oncologist 2004;9:247–58. [DOI] [PubMed] [Google Scholar]
- [13].Rosche B, Jacobsen M, Cepok S, et al. Myositis in a patient with large granular leukocyte leukemia. Muscle Nerve 2004;29:873–7. [DOI] [PubMed] [Google Scholar]
- [14].Ogata A, Kitano M, Fukamizu M, et al. Increased serum interleukin-18 in a patient with systemic lupus erythematosus and T-cell large granular lymphocytic leukemia. Mod Rheumatol 2004;14:267–70. [DOI] [PubMed] [Google Scholar]
- [15].Lamy T, Moignet A, Loughran TP., Jr LGL leukemia: from pathogenesis to treatment. Blood 2017;129:1082–94. [DOI] [PubMed] [Google Scholar]
- [16].Viny AD, Lichtin A, Pohlman B, et al. Chronic B-cell dyscrasias are an important clinical feature of T-LGL leukemia. Leuk Lymphoma 2008;49:932–8. [DOI] [PubMed] [Google Scholar]
- [17].Wlodarski MW, Schade AE, Maciejewski JP. T-large granular lymphocyte leukemia: current molecular concepts. Hematology 2006;11:245–56. [DOI] [PubMed] [Google Scholar]
- [18].Gu QH, Jia XY, Hu SY, et al. The clinical and immunologic features of patients with combined anti-GBM disease and Castleman disease. Am J Kidney Dis 2018;71:904–8. [DOI] [PubMed] [Google Scholar]
- [19].Greco A, Rizzo MI, De Virgilio A, et al. Goodpasture's syndrome: a clinical update. Autoimmun Rev 2015;14:246–53. [DOI] [PubMed] [Google Scholar]