

Abstract
Amnestic mild cognitive impairment (aMCI) is a transitional stage between normal aging and Alzheimer disease (AD), and is associated with an increased risk of AD. Many studies have shown that apolipoprotein E epsilon 4 (APOE ε4) genotype is a major genetic predictor of AD progression, especially in patients with aMCI. However, the application of APOE genotyping in the diagnosis of MCI progressing to AD is limited by its low sensitivity and specificity, which often leads to high false-positive rate. The aim of this study was to evaluate serum brain-derived neurotrophic factor (BDNF) and hippocampal volume as predictors of aMCI to AD transition in APOE ε4 genotype patients.
A total of 178 subjects were diagnosed with aMCI. The patients with aMCI that progressed to AD within 2 years were included in the MCI-AD group (n = 86), those maintaining an aMCI diagnosis after 2 years were placed in the MCI-MCI group (n = 92), and neurologically healthy age-matched individuals were set as controls (n = 90). APOE genotypes were determined. Blood samples from all subjects were drawn at baseline, 12 months, and 24 months for serum BNDF assessments. Hippocampal delineations were monitored by magnetic resonance imaging.
Compared to control group, aMCI-AD patients (the patients with aMCI that progressed to AD within 2 years) exhibited worse performance on cognitive and neuropsychological batteries. Meanwhile, we found that aMCI-AD patients were associated with abnormally low serum BDNF level and greater hippocampal volume loss than MCI-MCI patients (patients maintaining an aMCI diagnosis after 2 years). Moreover, patients with aMCI who were carriers of APOE ε4 showed a notable decrease in serum BDNF and a significant reduction in hippocampal volume, especially in those who progressed to AD.
The present study demonstrates that aMCI that evolves into AD in patients with the APOE ε4 genotype may be predicted by hippocampal volume and serum BDNF.
Keywords: Alzheimer disease, amnestic mild cognitive impairment, apolipoprotein E epsilon 4, brain-derived neurotrophic factor, hippocampal volume
1. Introduction
Alzheimer disease (AD) is a progressive neurodegenerative disease associated with loss of memory and cognitive decline.[1] The earliest clinical feature of individuals with incipient AD is usually mild impairment of episodic memory, many of these patients fulfill the criteria for mild cognitive impairment (MCI), but are still not demented. Mounting evidences have shown that MCI is a transitional stage between normal aging and dementia, and is associated with increased risk of AD.[1] A previous research has shown that the rate of progression to clinically diagnosable AD is 10% to 15% per year among persons who meet the criteria for amnestic MCI (aMCI), in contrast with a rate of 1% to 2% per year among healthy elderly individuals.[2] aMCI refers to the subtype that has a primary memory component, either alone or in conjunction with other cognitive-domain impairments, but of insufficient severity to constitute dementia.[2] Increasing evidences have shown that the prevalence of apolipoprotein E epsilon 4 (APOE ε4) is substantially higher in patients with aMCI than in control individuals, and the presence of ≥1 APOE ε4 alleles is associated with a more rapid rate of progression.[2] Importantly, the presence of APOE ε4 is associated with increased risk of progression from MCI to AD-type dementia.[3] However, the use of APOE genotyping in the diagnosis of aMCI that evolves into AD is limited because of its low sensitivity and specificity.[4] These findings suggest that APOE ε4 has considerable deleterious effects on memory performance and might be used to predict disease progression in combination with AD biomarkers and neuroimaging approaches.
In recent years, a great deal of interest has been generated concerning the use of novel biomarkers in the early or even preclinical detection of AD.[5] These excitements have been boosted showing that upcoming disease-modifying drugs against AD seem most effective if treatment is initiated before the neuronal loss has become too widespread.[6] Several studies showed that patients with AD or MCI who were APOE ε4 carriers exhibit greater medial temporal lobe atrophy, particularly in the hippocampal area.[7] Structural magnetic resonance imaging (MRI) studies also found that, compared with noncarriers, APOE ε4 carriers have accelerated age-related loss in cortical thickness and hippocampal volume that are tightly coupled to decline in cognitive performance.[8] However, whether human ApoE isoform status affects hippocampal volume in aMCI patients that converts to AD remains to be well-established. In the last years, there were growing evidences for an involvement of neurotrophins such as brain-derived neurotrophic factor (BDNF) in the pathogenesis of AD.[9] It has been proved that BDNF played an important role in neuronal survival, differentiation, and synaptic plasticity in the central nervous system, particularly in brain regions susceptible to degeneration in AD.[10] Reduced BDNF concentration in hippocampal and cortical neurons may contribute to degeneration and may occur during early stages of AD. In addition, the decrease of BDNF serum levels in AD may reflect a lack of trophic support and thus contribute to progressive degeneration in disease. However, more studies are needed to determine the diagnostic capabilities of those biomarkers, especially in terms of diagnosing early AD.[11] Thus, this study aimed to evaluate the relationship between hippocampal volume and serum BDNF as predictors of aMCI that converts to AD in patients with the APOE ε4 genotype.
2. Materials and methods
2.1. Participants
A total of 1697 outpatients who complained of memory impairment from 3 centers in China from June 2012 to July 2016 were screened. At baseline assessment, 178 subjects were diagnosed with aMCI (64 with single-domain aMCI and 114 multiple-domain aMCI). Single-domain aMCI and multiple-domain aMCI are two main clinical subtypes of aMCI. The single-domain aMCI indicates memory is the only domain impaired, and multiple-domain aMCI (aMCI-MD) occurs when besides the memory deficit, at least another cognitive domain is impaired (eg, executive function, language, or visuospatial skills).[12] The patients with aMCI that progressed to AD within 2 years were placed in the MCI-AD group (n = 86, 46 males, mean age = 69.2 ± 5.1), those maintaining an aMCI diagnosis after 2 years were placed in the MCI-MCI group (n = 92, 48 males, mean age = 72.8 ± 6.2), and 90 neurologically healthy age-matched individuals were included as controls (n = 90, 44 males, mean age = 74.2 ± 5.2). Written informed consents were obtained from all participants, and this study was approved by the ethics committees of the Harrison International Peace Hospital, China.
2.2. Clinical and neuropsychological assessments
All participants received a multidisciplinary clinical evaluation through the Department of Neurology, Harrison International Peace Hospital. These evaluations included detailed medical history, physical, psychiatric, and neurological examinations. All clinical investigations were performed by clinicians with expertise in diagnosing aMCI and dementia. The criteria of aMCI described by Petersen were used.[13] Patients who received a diagnosis of AD were required to meet the DSM-IIIR criteria of dementia and the criteria of probable AD defined by the National Institute of Neurological and Communicative Disorders-Stroke/Alzheimer's Disease and Related Disorders Association (NINCDS-ADRDA).[14] Of the 1697 outpatients who complained of memory impairment, eligibility assessments were conducted following below inclusion and exclusion criteria. The inclusion criteria were as follows: the absence of memory complaints or any other cognitive symptoms; objective memory impairment when performing cognitive tests of the assessment battery that was not severe enough to reach dementia diagnosis; the preservation of general cognitive functioning; preserved or minimal impairments in activities of daily living; and no demented. The inclusion criteria for controls were a healthy, age-matched control group determined to have no memory pathologies and evidence of cognitive impairment. Subjects with severe psychiatric disorders were excluded.
Secondary measures were assessed in all subjects at baseline, including the scores of the Mini Mental Status Examination (MMSE), the Clinical Dementia Rating (CDR) sum of boxes (the sum of individual CDR domain scores), the Activities of Daily Living Scale (ADL), the Global Deterioration Scale, and Geriatric Depression Scale. Follow-up assessments were performed for all patients and controls in which the same clinical and neuropsychological protocols were administered.
2.3. APOE genotyping
Genomic DNA was isolated from the whole blood of each subject, and APOE genotypes were determined according to a standard protocol using the oligonucleotides and reaction conditions described by Hixson and Vernier.[15] Briefly, amplification products were digested with HhaI (Fermentas, Ottawa, ON, Canada), subjected to electrophoresis on a 4% agarose gel, stained with ethidium bromide and analyzed with a ChampGel 6000 (Sagecreation, Beijing, China). Ten percent of genotypes were repeated as a quality check, with complete concordance.
2.4. BDNF analysis
Blood samples from all subjects were drawn in the morning at baseline and at the 12- and 24-month assessments. Peripheral venous blood (maximum 4 mL) was sampled into anticoagulant-free tubes (serum) between 8:00 and 8:30 am to take in account a possible circadian rhythm. Tubes were immediately immersed in melting ice. To minimize the source of platelets, serum was centrifuged within 30 minutes after gaining and stored at −80°C until further analysis. The serum BDNF levels were measured using a high-sensitivity quantitative enzyme immunoassay (ELISA) (R&D Systems, Minneapolis, MN).
2.5. Magnetic resonance imaging
All subjects were imaged with 1.5-T General Electric Signa device according to a standardized imaging protocol. The protocol began with a unit as a series of 124 1.5-mm coronal brain slices using a 3D fast spoiled gradient recalled echo with the following parameters: TR: minimum, TE: minimum, 15 degree flip angle, voxel size = 0.96 × 0.96 × 1.5 mm. The images were hand-processed to remove scalp, skull, and meninges. The hippocampal delineation procedure was adapted from existing protocols. Using the software BrainImage Java (Stanford University), one experienced blinded rater traced the exterior boundaries of each hippocampus.
2.6. Statistical analysis
We performed Pearson χ2 test to assess differences in the distributions of gender and APOE ε4 frequencies among the different groups. Nonparametric Kruskal-Wallis one-way analysis of variance (ANOVA) was performed followed by the Mann–Whitney U test to assess the normality of the distribution for each continuous variable. If data were normally distributed, parametric statistical tests were conducted for all analyses. For all quantitative data, results are expressed as the mean (±SD). ANOVA was used to assess the mean differences in the sociodemographic data, clinical variables, and scores on the cognitive and neuropsychological tests among the different diagnostic groups. In addition, we carried out Bonferroni analyses for multiple comparisons to address mean differences in the scores of the cognitive and neuropsychological tests among each group. All statistical analyses were performed using SPSS v17.0 for Windows (SPSS Inc., Chicago, IL), and α levels were set at 0.05.
3. Results
3.1. Baseline demographic and clinical characteristics of all subjects
Table 1 shows the sociodemographic data, APOE ε4 frequency and the scores on the cognitive and neuropsychological assessments in the 3 groups at baseline. We screened a total of 1697 outpatients consecutively recruited from 3 centers, and 268 completed the baseline assessment. A total of 86 patients with aMCI progressed to AD, with 92 maintaining an aMCI diagnosis after 2 years.
Table 1.
Baseline demographic and clinical characteristics of all subjects.
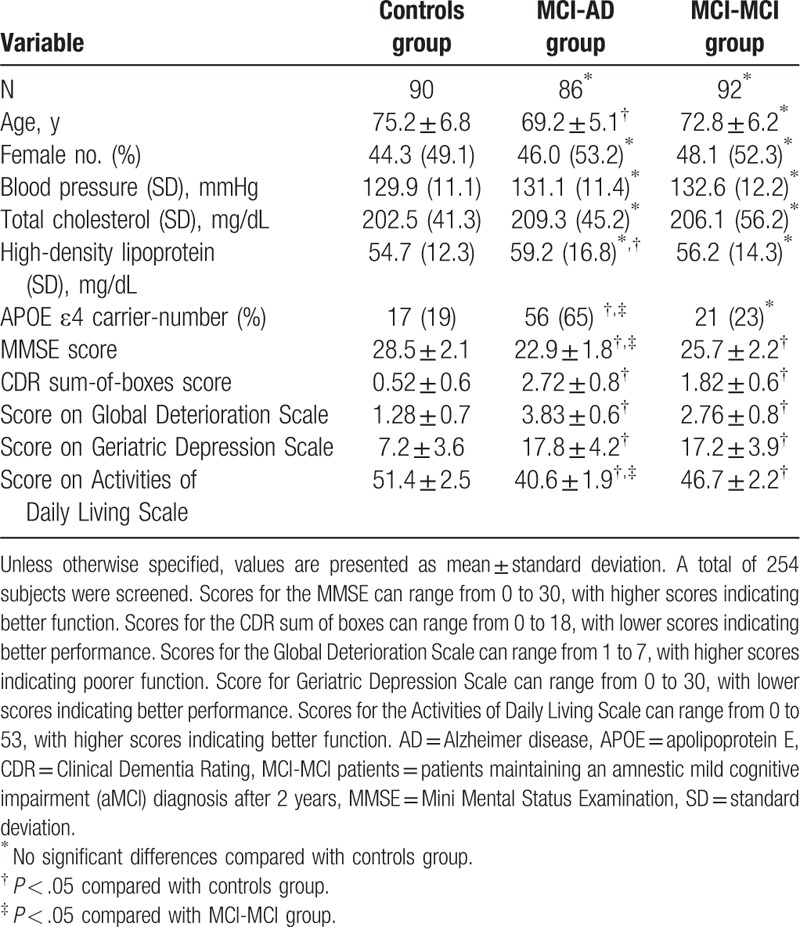
At baseline, patients in MCI-AD group (patients with aMCI that progressed to AD within 2 years) were significantly younger than that in control group (P < .05). No difference was observed between MCI-AD and MCI-MCI (patients maintaining an aMCI diagnosis after 2 years) group in terms of age and sex. Percentage of patients possessing APOE ε4 allele was significantly higher in MCI-AD group than that in MCI-MCI (P < .05) and controls (P < .05), indicating that possession of the APOE ε4 allele was a major predictor of progression to AD.
Next, we compared the scores on the cognitive and neuropsychological assessments among the 3 analyzed groups. We observed that scores for the MMSE, CDR sum-of-boxes, global deterioration scale, geriatric depression scale, activities of daily living scale were also higher in both MCI-AD and MCI-MCI groups, compared with controls (P < .05). However, only scores of MMSE and activities of daily living scale in MCI-AD group were lower than that in MCI-MCI group.
3.2. Analysis of the expression levels of serum BDNF and hippocampal volume in all subjects
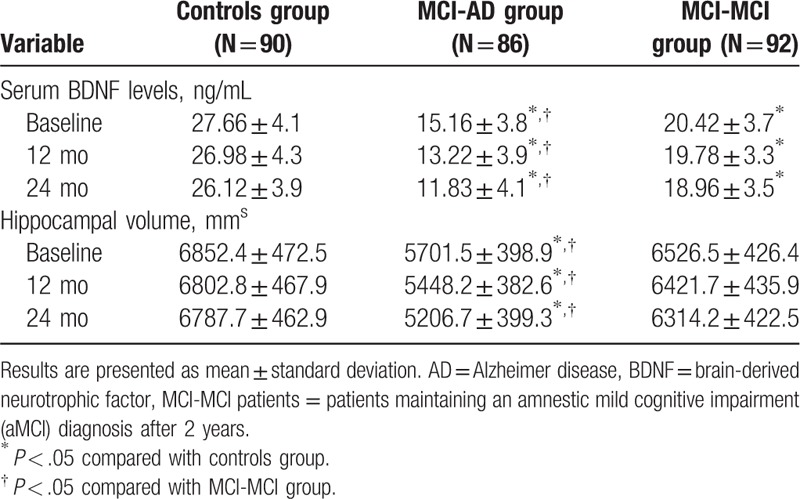
We examined the expression levels of serum BDNF and hippocampal volume in all subjects at 3 time points: baseline, 12 months later, and 24 months later (Table 2). Our data showed that there were statistically significant decreases in the serum BDNF levels in both the MCI-AD and MCI-MCI patients compared to the normal controls (P < .05) at all the 3 time points. Of note, the serum BDNF levels in the MCI-AD patients were notably decreased compared with those in the MCI-MCI (P < .05) at the 3 time points. Meanwhile, we found that the hippocampal volume of the patients in the MCI-AD group were significantly smaller when compared with those in MCI-MCI group (P < .05) and the control group (P < .05) at baseline, 12 months later, and 24 months later. These results indicate that aMCI patients with an abnormally low serum BDNF and greater hippocampal volume loss have a higher risk of progressing to AD.
Table 2.
The expression levels of serum BDNF and hippocampal volume in all subjects.
3.3. Analysis of serum BDNF and hippocampal volume in subjects harboring APOE ε4
Furthermore, we compared the differences in the expression levels of serum BDNF and hippocampal volume at baseline in subjects harboring APOE ε4 (Fig. 1). A total of 17 (17/90, 18.9%) control individuals harbored APOE ε4 compared with 56 in the MCI-AD (56/86, 65.1%) group and 21 (21/92, 22.8%) in the MCI-MCI group. We found that there were significant decreases in serum BDNF levels in both the MCI-AD and MCI-MCI patients possessing the APOE ε4 genotype compared to control group (P < .01, P < .01). Notably, the APOE ε4 carriers in the MCI-AD and MCI-MCI groups exhibited a marked decrease in serum BDNF levels compared to APOE ε4 noncarriers (P < .05, P < .05, Fig. 1A). Similarly, hippocampal volume in the APOE ε4 carriers of both the MCI-AD and MCI-MCI groups were evidently reduced than those of control groups (P < .01, P < .05). MCI-AD and MCI-MCI patients possessing the APOE ε4 had lower hippocampal volume than those without APOE ε4 (P < .05, P < .05, Fig. 1B). Collectively, patients with aMCI who were carriers of APOE ε4 showed a notable decrease in serum BDNF and a significant reduction in hippocampal volume, especially in those who progressed to AD.
Figure 1.
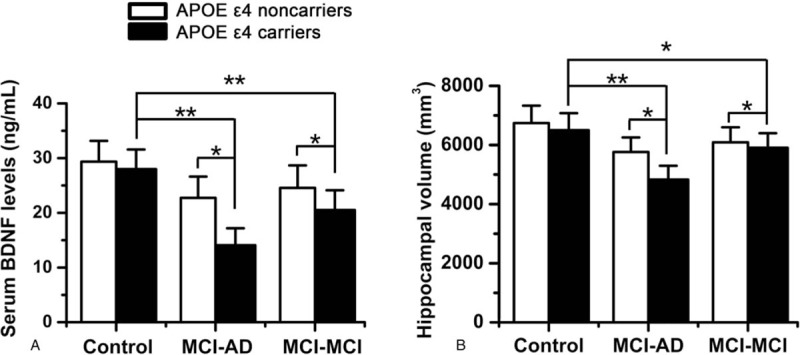
The expression levels of serum BDNF level and hippocampal volume in subjects who harbor APOE ε4. (A) The expression levels of serum BDNF level at baseline. (B) The hippocampal volume at baseline. The data are shown as the means (±SD). ∗P < .05, ∗∗P < .01. AD = Alzheimer disease, APOE = apolipoprotein E, BDNF = brain-derived neurotrophic factor, MCI-MCI patients = patients maintaining an amnestic mild cognitive impairment (aMCI) diagnosis after 2 years.
4. Discussion
The aim of the present study was to evaluate hippocampal volume and serum BDNF as predictors of conversion from MCI to AD in patients with the APOE ε4 genotype. Consistent with previous studies, possession of the APOE ε4 allele is a major predictor of progression to AD, especially in patients with aMCI.[1] Recent studies have proven that patients with aMCI who harbor APOE ε4 exhibit distinct cognitive profiles, which seem to resemble those of patients in the early stages of AD.[7] Our demographic and clinical profiles showed that patients with aMCI that evolved into AD experienced worse performances on cognitive and neuropsychological batteries than control group, and 65.1% of MCI-AD patients carried APOE ε4, supporting the role APOE ε4 carrier status in developing dementia.[16] This finding is consistent with the results from a longitudinal study that emphasized conversion to dementia was driven strongly by a sharp decline in functional ability.[17] However, whether progressive serum BDNF and hippocampal volume reductions are related in the conversion process in patients with aMCI who harbor APOE ε4, are not yet available.
A previous study has highlighted that reduced hippocampal volume, an early neural correlate of dementia, is commonly observed in patients with MCI.[18] It has been proved that patients with AD or MCI who are APOE ε4 carriers exhibits greater medial temporal lobe atrophy, particularly in the hippocampal area.[7] Structural MRI studies also found that, compared with noncarriers, APOE ε4 carriers have accelerated age-related loss in cortical thickness and hippocampal volume that are tightly coupled to decline in cognitive performance.[8] Numerous studies suggest that APOE4 carriers demonstrate increased vulnerability to developing AD, which is manifested through neurodegeneration.[7] However, it is unclear whether neurodegenerative and resultant clinical trajectories are accelerated in aMCI patients with the APOE ε4 genotype, leading to a faster conversion to dementia stages than those who are not APOE ε4 carriers. In the present study, we observed that patients with aMCI that progressed to AD exhibited a marked decrease in hippocampal volume compared to the controls in the baseline assessment. Furthermore, the APOE ε4 carriers in the MCI-AD group exhibited a marked decrease in hippocampal volume compared to the MCI-MCI group and the controls. A possible hypothesis is that the APOE ε4 peptide may elicit an increase in intracellular calcium levels and subsequent death of embryonic hippocampal neurons.
It had been proved that strong correlations were found between neuron number and both hippocampal volume and brain volume, demonstrating that volume and neuron content were related in normal subjects and that the relationship was maintained in AD.[19] An alternative theory was that the effect of the APOE ε4 allele on reduced hippocampal volume might be a result of the inability of the brain to compensate for this combined assault.[20] Smaller hippocampal volume and the APOE ε4 allele may act synergistically in a greater disruption of neurological functioning, which may in turn lower an individual's cognitive reserve capacity, the individual's ability to successfully contend with increasing pathology.[21] Collectively, these findings support the suggestion that hippocampal atrophy in aMCI is a result of neuron loss, and may be aggravated in the presence of APOE ε4.
Furthermore, hippocampal neurogenesis is known for its role in the plasticity of cognitive functions.[22] It provides a substrate for dynamic and flexible aspects of learning and memory. Dysfunctional neurogenesis resulting from early disease manifestations could, therefore, exacerbate neuronal vulnerability to AD and contribute to memory impairment.[23] A previous study has proved that APOE is required for maintenance of the neural stem or progenitor cell pool in the adult dentate gyrus region of the hippocampus.[24] In APOE-TR mice, ApoE ε4 inhibits hippocampal neurogenesis by impairing maturation of hilar γ-aminobutyric acid-containing interneurons, which contributes to learning and memory deficits. These results demonstrate an important pathological role of ApoE ε4 in impairment of neurogenesis, which might contribute to AD pathogenesis. Moreover, during the course of adult neurogenesis, presence of the neurotrophic factors, including BDNF, is crucial for the survival and differentiation of neuronal populations. In addition, BDNF signaling is important for hippocampal synaptic plasticity and learning. BDNF is expressed throughout the brain, especially in the prefrontal cortex and the hippocampus.[25] It has been reported that the deposition of beta-amyloid protein is involved in the synthesis of BDNF and signal transduction, resulting in a blockage of, synaptic function, and accelerated neuronal degeneration.[10] Thus, in view of neuroprotective effects of BDNF in general, the demonstrated decrease of BDNF in AD may contribute to the development of this neurodegenerative disease because of a lack of neurotrophic support.[9] In agreement with these above studies, our data showed that serum BDNF levels were significantly decreased in aMCI patients compared to normal individuals. Moreover, serum BDNF levels in APOE ε4 carriers were significantly decreased compared to noncarriers in both the MCI-AD and MCI-MCI groups, whereas the MCI-AD patients showed a notable decrease in serum BDNF levels. Together, these findings indicated that there might be a synergistic effect of reduced serum BDNF levels and hippocampal volume in aMCI patients that promoted earlier conversion to AD, particularly in those who were APOE ε4 carriers.
The present study suggested that APOE ε4 genotype influenced the conversion of patients from MCI to AD. It has been previously reported that AD was closely correlated with vascular risk factors, and early detection and treatment of vascular risk factors may prevent or at least postpone the evolution of the disease.[26,27] Our study added evidences on the field of AD disease and indicated that AD represented a multifactorial disease where either genetic or acquired conditions interact. In future, it would be interesting to investigate whether this interaction is simply additive or even synergistic, and the mechanism underlying this interaction to influence the AD pathophysiology, to provide evidence to controlled AD.
There were several limitations in this study. First, the enrolled participant number in the study was limited, and it is necessary to validate our findings in a larger population. Second, this is a single-center study which can only reflect the pattern of aMCI patients in this geographical region, and studies in multiple cohorts are needed. Third, we only investigated the function of serum BDNF and hippocampal volume for aMCI-AD progression, and studies focusing on other biomarkers are warranted.
In summary, our study provided evidence that there was a significant decrease in serum BDNF and notable reduction in hippocampal volume in aMCI patients that progressed to AD, especially in those who harbored the APOE ε4 allele, suggesting that combination of these diagnostic strategies may increase the predictive power to identify AD patients at the prodromal stages of the disease.
Author contributions
Conceptualization: Lei Zheng.
Data curation: Yan Fang, Naiyi Du, Yali Duo.
Formal analysis: Yan Fang, Naiyi Du, Longyan Xing, Yali Duo.
Investigation: Longyan Xing.
Methodology: Yan Fang, Naiyi Du, Lei Zheng.
Project administration: Yan Fang, Yali Duo.
Resources: Longyan Xing.
Supervision: Yan Fang, Lei Zheng.
Writing – original draft: Lei Zheng.
Writing – review & editing: Lei Zheng.
Footnotes
Abbreviations: AD = Alzheimer disease, aMCI = amnestic mild cognitive impairment, aMCI-AD patients = the patients with amnestic mild cognitive impairment (aMCI) that progressed to Alzheimer's disease (AD) within 2 years, APOE = apolipoprotein E, BDNF = brain-derived neurotrophic factor, CSF = cerebrospinal fluid, ELISA = enzyme-linked immunosorbent assay, GDS = the Geriatric depression scale, MCI = mild cognitive impairment, MCI-MCI patients = patients maintaining an amnestic mild cognitive impairment (aMCI) diagnosis after 2 years.
The authors report no conflicts of interest.
This work was supported by the Hebei Medical Science Research Program (20130345) of the Health Department of Hebei Province.
References
- [1].Liu CC, Liu CC, Kanekiyo T, et al. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 2013;9:106–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Petersen RC, Thomas RG, Grundman M, et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med 2005;352:2379–88. [DOI] [PubMed] [Google Scholar]
- [3].Houlden H, Crook R, Backhovens H, et al. ApoE genotype is a risk factor in nonpresenilin early-onset Alzheimer's disease families. Am J Med Genet 1998;81:117–21. [DOI] [PubMed] [Google Scholar]
- [4].Saunders AM, Hulette O, Welsh-Bohmer KA, et al. Specificity, sensitivity, and predictive value of apolipoprotein-E genotyping for sporadic Alzheimer's disease. Lancet 1996;348:90–3. [DOI] [PubMed] [Google Scholar]
- [5].Desai AK, Grossberg GT. Diagnosis and treatment of Alzheimer's disease. Neurology 2005;6412 suppl 3:S34–39. [DOI] [PubMed] [Google Scholar]
- [6].DeKosky ST, Marek K. Looking backward to move forward: early detection of neurodegenerative disorders. Science 2003;302:830–4. [DOI] [PubMed] [Google Scholar]
- [7].Farlow MR, He Y, Tekin S, et al. Impact of APOE in mild cognitive impairment. Neurology 2004;63:1898–901. [DOI] [PubMed] [Google Scholar]
- [8].Espeseth T, Westlye LT, Fjell AM, et al. Accelerated age-related cortical thinning in healthy carriers of apolipoprotein E epsilon 4. Neurobiol Aging 2008;29:329–40. [DOI] [PubMed] [Google Scholar]
- [9].Laske C, Stransky E, Leyhe T, et al. BDNF serum and CSF concentrations in Alzheimer's disease, normal pressure hydrocephalus and healthy controls. J Psychiatr Res 2007;41:387–94. [DOI] [PubMed] [Google Scholar]
- [10].Mattson MP, Maudsley S, Martin B. BDNF and 5-HT: a dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci 2004;27:589–94. [DOI] [PubMed] [Google Scholar]
- [11].Blennow K, Hampel H, Weiner M, et al. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol 2010;6:131–44. [DOI] [PubMed] [Google Scholar]
- [12].Brambati SM, Belleville S, Kergoat MJ, et al. Single- and multiple-domain amnestic mild cognitive impairment: two sides of the same coin? Dement Geriatr Cogn Disord 2009;28:541–9. [DOI] [PubMed] [Google Scholar]
- [13].Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med 2004;256:183–94. [DOI] [PubMed] [Google Scholar]
- [14].Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:270–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res 1990;31:545–8. [PubMed] [Google Scholar]
- [16].Ramakers IH, Visser PJ, Aalten P, et al. The association between APOE genotype and memory dysfunction in subjects with mild cognitive impairment is related to age and Alzheimer pathology. Dement Geriatr Cogn Disord 2008;26:101–8. [DOI] [PubMed] [Google Scholar]
- [17].Gomar JJ, Bobes-Bascaran MT, Conejero-Goldberg C, et al. Utility of combinations of biomarkers, cognitive markers, and risk factors to predict conversion from mild cognitive impairment to Alzheimer disease in patients in the Alzheimer's disease neuroimaging initiative. Arch Gen Psychiatry 2011;68:961–9. [DOI] [PubMed] [Google Scholar]
- [18].Chung JK, Plitman E, Nakajima S, et al. Depressive symptoms and small hippocampal volume accelerate the progression to dementia from mild cognitive impairment. J Alzheimers Dis 2016;49:743–54. [DOI] [PubMed] [Google Scholar]
- [19].Kril JJ, Hodges J, Halliday G. Relationship between hippocampal volume and CA1 neuron loss in brains of humans with and without Alzheimer's disease. Neurosci Lett 2004;361:9–12. [DOI] [PubMed] [Google Scholar]
- [20].Bu G. Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat Rev Neurosci 2009;10:333–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Stern Y. What is cognitive reserve? Theory and research application of the reserve concept. J Int Neuropsychol Soc 2002;8:448–60. [PubMed] [Google Scholar]
- [22].Aimone JB, Li Y, Lee SW, et al. Regulation and function of adult neurogenesis: from genes to cognition. Physiol Rev 2014;94:991–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mu Y, Gage FH. Adult hippocampal neurogenesis and its role in Alzheimer's disease. Mol Neurodegener 2011;6:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yang CP, Gilley JA, Zhang G, et al. ApoE is required for maintenance of the dentate gyrus neural progenitor pool. Development 2011;138:4351–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Driscoll I, Martin B, An Y, et al. Plasma BDNF is associated with age-related white matter atrophy but not with cognitive function in older, non-demented adults. PLoS One 2012;7:e35217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lattanzi S, Brigo F, Vernieri F, et al. Visit-to-visit variability in blood pressure and Alzheimer's disease. J Clin Hypertens (Greenwich) 2018;20:918–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Xiang J. Carotid atherosclerosis promotes the progression of Alzheimer's disease: a three-year prospective study. Exp Ther Med 2017;14:1321–6. [DOI] [PMC free article] [PubMed] [Google Scholar]