

Abstract
The covalent attachment of sugars to growing glycan chains is heavily reliant on a specific family of solute transporters (SLC35), the nucleotide sugar transporters (NSTs) that connect the synthesis of activated sugars in the nucleus or cytosol, to glycosyltransferases that reside in the lumen of the endoplasmic reticulum (ER) and/or Golgi apparatus. This review provides a timely update on recent progress in the NST field, specifically we explore several NSTs of the SLC35 family whose substrate specificity and function have been poorly understood, but where recent significant progress has been made. This includes SLC35 A4, A5 and D3, as well as progress made towards understanding the association of SLC35A2 with SLC35A3 and how this relates to their potential regulation, and how the disruption to the dilysine motif in SLC35B4 causes mislocalisation, calling into question multisubstrate NSTs and their subcellular localisation and function. We also report on the recently described first crystal structure of an NST, the SLC35D2 homolog Vrg-4 from yeast. Using this crystal structure, we have generated a new model of SLC35A1, (CMP-sialic acid transporter, CST), with structural and mechanistic predictions based on all known CST-related data, and includes an overview of reported mutations that alter transport and/or substrate recognition (both de novo and site-directed). We also present a model of the CST-del177 isoform that potentially explains why the human CST isoform remains active while the hamster CST isoform is inactive, and we provide a possible alternate access mechanism that accounts for the CST being functional as either a monomer or a homodimer. Finally we provide an update on two NST crystal structures that were published subsequent to the submission and during review of this report.
Keywords: Nucleotide sugar transporters, CMP-sialic acid transporter, Golgi apparatus, Endoplasmic reticulum, SLC35
Graphical Abstract
1. Introduction
Protein glycosylation is the most prevalent and complex post-translational modification that occurs in a wide range of organisms over all three domains. The process of glycosylation covalently attaches sugars to the growing end of glycan chains on nascent proteins and/or lipids, catalysed by glycosyltransferases. The decoration of these molecules with a range and diversity of sugars play integral roles in every aspect of biology. The diversity of the proteome is further augmented through variation provided by glycosylation, including diversity arising from differences in glycosidic linkage and monosaccharide composition, to the degree of branching and chain length. In eukaryotes, sugars are activated through attachment to a nucleotide before being transported across the membrane from the nucleus or cytosol into the ER or Golgi lumen. This transport process is accomplished by a family of transmembrane proteins called nucleotide sugar transporters (NSTs) [1].
The solute carrier family of human NSTs are designated as SLC35 (as per HGNC). Table 1 highlights the predominant members of this family and their key characteristics, including specificity, localisation and UniProtKB Primary Accession Number. The SLC35 family are highly conserved type III trans-membrane proteins that are divided into 7 subfamilies (SLC35A-G). These subfamilies are then further differentiated according to the purported substrate that is being transported [2]. Most of the members of the SLC35 family have been identified over many years as facilitating the transport of a cytosolic nucleotide sugar, coupled with the antiport of the corresponding luminal nucleotide monophosphate [1,[3], [4], [5], [6]]. Several of the members of the SLC35 subfamilies have been labelled orphan transporters as their substrate specificity and physiological functions are yet to be determined. Recent research has shed some light on these orphan transporters and found that they may not be directly involved in glycosylation per se, but rather may be involved in regulating homeostasis through coupling to an NST with transport activity [[7], [8], [9]].
Table 1.
Selected members of the SLC35 nucleotide sugar transporter family.
| Gene name and aliases | Protein name and aliases | Substrate(s) | Sub-cellular localisation | Length of protein sequence | UniProtKB Primary Accession Number |
|---|---|---|---|---|---|
| SLC35A1 | CMP-Sia transporter, (CST) | CMP-Sia | Exclusively Golgi | 337 | P78382 |
|
SLC35A2 UGALT UGT UGTL |
UDP-Galactose translocator, UGP-Gal Tr, (UGT). | UDP-Gal; UDP-GlcNAc | Golgi and/or ER | 396 | P78381 |
| SLC35A3 | UDP-N-acetylglucosamine transporter, UDP-GlcNAc transporter, Golgi UDP-GlcNAc Tr, (NGT) | UDP-GlcNAc | Predominantly Golgi | 325 | Q9Y2D2 |
| SLC35A4 | Probable UDP-sugar transporter MGC2541 | Putative UDP-Gal | Predominantly Golgi | 324 | Q96G79 |
|
SLC35A5 (ORF) UNQ164 (OFR) PRO190 |
Probable UDP-sugar transporter | Putative UDP-sugar | 424 | Q9BS91 | |
|
SLC35B1 UGTREL1 |
UDP-Galactose Transporter related protein 1, UGTrel1, hUGTrel1, HUT-1 (homolog), AXER. | ADP/ATP Exchanger | 322 | P78383 | |
|
SLC35B2 PAPST1 PSEC0149 |
Adenosine 3′-phospho 5′-phosphosulfate transporter 1, PAPS transporter 1. | PAPS | Exclusively Golgi | 432 | Q8TB61 |
|
SLC35B3 C6orf196 PAPST2 (ORF) CGI-19 |
Adenosine 3′-phospho 5′-phosphosulfate transporter 2, PAPS transporter 2. | PAPS | Exclusively Golgi | 401 | Q9H1N7 |
|
SLC35B4 YEA4 PSEC0055 |
UDP-Xyl transporter, YEA4 homolog | UDP-Xyl; UDP-GlcNAc | Golgi and/or ER (Dependent on C-terminal tagging) and expression system | 331 | Q969S0 |
|
SLC35C1 FUCT1 |
GDP-Fucose transporter 1 (GFT) | GDP-Fuc | Predominantly Golgi | 364 | Q96A29 |
|
SLC35C2 OVCOV1 C20orf5 (ORF) CGI-15 |
Ovarian cancer-overexpressed gene 1 protein | Putative GDP-Fuc transporter | Golgi and/or ER | 365 | Q9NQQ7 |
|
SLC35D1 KIAA0260 UGTREL7 |
UDP-Glucuronic acid/UDP-N-acetylgalactosamine dual transporter, UGTrel7. | UDP-GlcA; UDP-GalNAc | Exclusively ER | 355 | Q9NTN3 |
|
SLC35D2 HFRC UGTREL8 |
UDP-GlcNAc/UDP-Glc/GDP-Man transporter, HFRC1, SQV7L, UGTrel8 | UDP-GlcNAc; UDP-Glc; GDP-Man (not humans) | Exclusively Golgi | 337 | Q76EJ3 |
|
SLC35D3 FRCL1 |
Fringe connection-like protein 1 | Substrate unknown | 416 | Q5M8T2 | |
|
SLC35F2 FLJ13018 |
SLC35F2 | Substrate unknown | Possibly outer cell membrane | 374 | Q8IXU6 |
Each SLC35 gene name should be italics, and have only been bolded to distinguish them from their know aliases which are shown below as required. These aliases should also remain italics.
As more research comes to light, the association of two or more transporters to form a complex seems to be much more prevalent. A number of studies delving into the members of the SLC35A2-5 family suggests they are all functionally linked, whether in glycosylation itself or in a regulatory manner [10]. There are further indications that they may also form multiprotein complexes with glycosyltransferases (Mgats) [11]. This review will highlight a range of previously designated SLC35 orphan transporters that have now been associated with either transport or regulatory roles directly and indirectly linked to glycosylation, including glycosaminoglycans (GAGS) biosynthesis [12]. Interestingly research on SLC35B4 specifically has suggested that the localisation and function of many of these SLC35 transporters be re-evaluated as experimental disruption to their C-terminal region can cause mislocalisation, especially in yeast expression systems [13]. Recently the crystal structure of the yeast Vrg-4 transporter (homologous to SLC35D2) has become available [14]. This important breakthrough has enabled us to build a template-generated model of SLC35A1 (CMP-sialic acid transporter (CST)), one of the best characterised transporters in the family, which has allowed us to propose a structural basis for CST transport including the rationale for several CST mutations that cause congenital disorders of glycosylation.
2. SLC35A2 UDP-Galactose Transporter (UGT) and SLC35A3 UDP-N-Acetylglucosamine Transporter (NGT)
Both the UGT and NGT will be discussed together as there is significant evidence that these two transporters are functionally linked. Since the SLC35A2 gene is located on the X-chromosome, most patients with related CDG's are female, though Ng et al. [15] reported two patients who were somatic mosaic for the deleterious variants. Hence it is likely that the presence of a functional allele is required for survival [16]. The molecular basis of one of these new SLC35A2-CDGs was recently shown to be a Gly266Val mutation. Although the patient was found to be heterozygous, a loss of activity resulted from random X-inactivation of the competent allele. Its functional relevance was confirmed by complementation of UGT-deficient MDCK-RCAr and CHO-Lec8 cells with the wild-type UGT but not by the mutant transporter [17].
Previously known, and newly discovered variants in this UGT are comprehensively covered in a review by Yates et al. [16]. In addition to these, a further five somatic variants of SLC35A2 have been found to be associated with intractable neocortical epilepsy when observed in the brain [18,19]. This area of research is progressing steadily with a further nine novel disease causing SLC35A2 variants being recently described [20]. This brings the current total of known subjects of this rare disorder to 62 individuals [21]. It is important to note the severity of truncated glycans that inactivation of SLC35A2, and the resulting abolition of UDP-Gal transport into the Golgi Apparatus [[22], [23], [24]], effects N- and O-linked glycans, GAGs and glycosphingolipids. Ng et al., [21] comprehensively investigated 30 individual SLC35A2-CDG patients with profound neurological and developmental impairments, most with epilepsy and/or skeletal abnormalities. Unexpectedly, the majority of these patients did not display the common biomarker used to diagnose SLC35A2-CDG, a carbohydrate-deficient serum protein, transferrin. Separation of serum transferrin is the standard screening test for CDG where the glycans are either truncated or non-existant [17]. The inaccuracy of this diagnostic assay led to the inception of a new biochemical assay to assess SLC35A2-dependent UDP-Gal transport activity in primary fibroblasts [21]. It was shown that transport activity was directly correlated to the ratio of wild-type to mutant alleles in fibroblasts [21]. The lack of this traditional biomarker in SLC35A2-CDG patients may be explained by normalisation of transferrin glycosylation after supplementary intake of dietary Gal [17].
Novel compound heterozygous mutations in SLC35A3 (missense and frame-shift variants) (Arg25Cys and Leu300Gln respectively) were identified from the parents of two non-consanguineous siblings with severe epileptic encephalopathy with skeletal abnormalities. The missense Arg25Cys is in a highly conserved region that flanks the first trans-membrane domain (TMD) of the protein. With this residue substitution, using software for cysteine state and disulfide bond partner prediction (DiANNA), a novel disulphide bond between Cys25 and Cys183 that could cause the protein to misfold was predicted. As expected, the del/ins variant caused a frameshift that resulted in a premature stop codon [25].
Although CDG patients usually display a wide-range of systemic symptoms, patients with an SLC35A2 mutation are rare, and generally have disorders related to the central nervous system (CNS) [26]. These disorders are generally X-linked epileptic encephalopathies [18] but also, as stated previously, include skeletal abnormalities and congenital cardiac disease [16,21]. Interestingly SLC35A3 mutations also display CNS-related symptoms such as epilepsy and autism [25,27] but also musculoskeletal abnormalities in humans, which for this transporter was previously only shown in cattle [25,28].
Previously it was proposed that the function of NGT and UGT in galactosylation might be coupled [29], and results from a further study supported this hypothesis. The construction of six novel chimeric proteins were analysed in UGT-deficient mammalian cell lines, and showed that the UGT and NGT may replace the function played by each other when appropriate regions of these transporters are preserved. An important exception to this is the UGT short N-terminal region comprised of 35 amino acid residues that is crucial for galactosylation of N-glycans [10].
Subsequently, evidence has been provided that complex formation between NSTs and glycosyltransferases may be more common than previously suggested. Research focussed on inherently-linked mediators of N-linked protein glycosylation; UGT, NGT and acetylglucosaminyltranferases (Mgats) has shown that UGT and NGT are also in close proximity to Mgats, further suggesting that all three may form multiprotein complexes mediating N-glycan biosynthesis. Both splice variants of UGT (UGT1 localised to the Golgi and UGT2 dual localised to ER and Golgi) are involved in these complexes though an important observation was that only Mgat4B was in close proximity to UGT2 [11].
A recent study investigating the complex-formation within the Golgi of the NSTs SLC35A2, A3 and A4 using in vivo Bimolecular Fluorescence Complementation (BiFC) FRET interaction screens [30] identified novel ternary complexes between Mgats and SLC35A2, A3 and A4. These complexes were found to self-assemble into multi-enzyme/multi-transporter complexes, and helped facilitate synthesis of complex N-glycans in the medial-Golgi. It is still unclear how these sub-complex assemblies function in N-glycan processing, but it was significant that multi-enzyme-transporter complexes can exist in live cells, potentially coupling the sugar supplied by the transporter directly to the transferase in the same complex [30].
3. SLC35A4 and SLC35A5 Transporters
Until now, SLC35A4 has been described as a putative UDP-sugar (possibly Gal) transporter but recent work has suggested its role may not be in glycosylation but have a modulatory or regulatory role related to the UGT (SLC35A2) and NGT (SLC35A3). Recent work has shown SLC35A4 does not form homodimers and only directly associates with one other transporter, the orphan SLC35A5–a putative UDP-sugar transporter [7]. SLC35A4 localises mainly to the Golgi apparatus and has been thought to contain an even number of TMDs with both the N- and C- termini facing the cytosol. However, recent in silico topology predictions suggest an uneven number of spans resulting in the N-terminus of SLC35A4 directed towards the cytosol and not the Golgi lumen [7]. As previously reported, the transporters SLC35A2 and SLC35A3 are now considered to form a functional complex [11,29]. Keeping this in mind, in situ proximity ligation assays on SLC35A4 have placed it 10–40 nm proximal to the subcellular location of these complexes. Although SLC35A4 knock-outs showed no significant glycosylation changes the mutation did influence the distribution/relocalisation of these SLC35A2/SLC35A3 complexes. Whereas overexpression of SLC35A4 negatively affects interaction between the two proteins that form the SL35A2/SLC35A3 complex, but only where the SLC35A2 protein is the Golgi localised splice variant (UGT1). Further to this, SLC35A2-deficient cells cannot co-express SLC35A3 and SLC35A4 [7].
Later in this review we will discuss SLC35B4 C-terminal tagging that affects subcellular localization of NSTs with C-terminal dilysine motifs that are considered ER retention/retrieval signals. We also previously mentioned that Golgi-resident NSTs, such as the first splice variant of SLC35A2 or SLC35A4, can be tagged either at the N- or C-terminus without affecting their subcellular localization. However, SLC35A5 is not the same in this regard, being the only member of the SLC35 family that contains possible UDP-sugar binding regions amongst several diacidic motifs exposed in the cytosolically located C-terminus tail. As well as potentially being the first mammalian NST to engage in the transport of three different UDP-sugars (UDP-GlcA, UDP-GalNAc, and UDP-GlcNAc) it is also suggested that the SLC35A5 may be a superior regulatory protein that coordinates the action of all other SLC35A subfamily members [8].
4. SLC35C1: GDP-Fucose Transporter (GFT)
A detailed understanding of the GFT has been hampered by the absence of an appropriate mutant cell line lacking GFT activity, which has been instrumental in progressing the NST field. However, using the codon-optimised SLC35C1 successful expression of human GFT in the cytoplasmic membrane of E. coli has now been achieved. This expression system, which is based on that used by us to express the CST in E. coli [31], generated a functional GFT that was confirmed by affinity and kinetic data mined from inverted membrane vesicles [32].
A recent study demonstrated that the GFT and its corresponding fucosyltranferase (Fut9) are key regulators of ricin toxicity in humans [33]. The plant toxin, ricin, has the potential to be used as a powerful biological weapon due to its extreme toxicity, ease of access and lack of an antidote [34]. It was established that fucosylation is crucial for ricin toxicity through its inhibitory effects of LewisX, with genetic ablation of the human SLC35C1 conferring resistance to ricin [33].
Interestingly, the upregulation of GFT in the mammary glands has been shown to play an essential role in the metabolism of milk glycoconjugates in mammals. Expression of the GFT, and two sialyltransferases, are upregulated during lactating stages of water buffalo compared to non-lactating stages [35].
5. SLC35B1: ADP/ATP Exchanger
SLC35B1 is commonly referred to as the UDP-galactose transporter-related protein 1 (UGTrel1), although there are several splice-variants (UniProtKB-P78383). HUT-1 is the homolog of UDP-Gal transporter in Caenorhabditis elegans, where it is involved in the maintenance of ER homeostasis and essential for larval development [36].
Recently, SLC35B1 has been identified as having a critical role in ATP/ADP exchange in the ER, and as such, the protein is referred to as AXER (ATP/ADP exchanger in the ER membrane) [37]. These findings provide an explanation as to why AXER orthologs in Saccharomyces cerevisiae (HUT1), Schizosaccharomyces pombe (HUT1), and Caenorhabditis elegans (HUT-1) have been found to play a more general role in ER homeostasis as would normally be expected for an NST. SLC35B2 has been shown to have the highest mRNA expression in the skeletal muscle [38], which would correspond to the expected function of an ADP/ATP exchanger due to high energy requirements in these tissues.
6. SLC35D3–Substrate/s Unknown
Sosicka et al. [7] were not alone in suggesting certain members of SLC35 family had modulatory roles instead of direct glycosylation roles. Just as proposed with the orphan SLC35A4, Wei et al. [39] reported that the role of SLC35D3 in brain neurons is to enhance the formation of a particular complex to induce autophagy. It is in this role as a regulator that SLC35D3 increases tissue-specific autophagic activity required for the survival of subsets of dopaminergic neurons. Furthermore, the authors did not indicate a direct glycosylation role for this protein. Zhang et al. [40] had previously investigated the role of SLC35D3 in dopaminergic neurons. They determined that 2 recessive mutations (K404X and insL201) in the SLC35D3 gene resulted in subcellular mislocalisation of SLC35D3 resulting in impaired dopamine signalling in striatal neurons. This suggests that SLC35D3 may be a candidate gene for patients with Metabolic Syndrome (MetS), which is involved in metabolic control in the CNS by regulating dopamine signalling. The presence of SLC35D3 in non-neuronal tissues is only known so far in platelets, hence SLC35D3 has been determined as specifically regulating the contents of dense granules [41]. Platelet dense granules consist exclusively of low-molecular-weight components such as serotonin, adenine nucleotides, pyrophosphate, calcium, and polyphosphate [42,43] that assist in blood clotting when released from activated platelets [41]. Therefore, SLC35D3 has also been implicated in other recessive metabolic disorders, including Hermansky-Pudlak syndrome (HPS) and Chediak-Higashi syndrome (CHS) [44].
7. SLC35B4: UDP-Xyl/UDP-GlcNAc Transporter
It has previously been shown that human SLC35B4 overexpressed in CHO-K1 cells localised to the Golgi apparatus [45]. In contrast, another study showed that overexpressed SLC35B4 in Madin-Darby canine kidney wild-type (MDCK-wt) and MCDK mutant cells localised exclusively in the ER [46], while homologous proteins from other species have been found in the ER and epithelium [47,48]. These contradictory data prompted further investigation to clarify the correct subcellular localisation of the human SLC35B4 [13]. Using SLC35B4-deficient HepG2 cells and indirect immunofluorescent staining, Bazan et al. [13] found that a dilysine motif at position 329 was crucial for ER localisation in human cells. This led them to agree with Ishikawa et al. [48] who observed that deletion of the KKVE C-terminal region in the D. melanogaster homolog Efr resulted in a mislocalisation of the protein mainly to the Golgi apparatus.
There are two splice variants of SLC35B4 with the longer variant containing a C-terminal dilysine motif (KDSKKN) that is slightly different to the classic ER localisation motifs [[49], [50], [51]]. The most important results of the Bazan et al. [13] study was that the position of any attached fusion tag to recombinant SLC35B4 strongly affects the subcellular localisation of the protein. This calls into question several studies that have incorporated fusion-tagging that may have contributed to incorrect SLC35B4 sub-cellular localisation including important work by Ashikov et al. [45] and Mkhikian et al. [52]. Significantly, this change of localisation for SLC35B4 also throws doubt on the previously predicted biological significance of this NST, and it has been suggested that the substrate specificity of a number of NSTs, particularly those exhibiting multi-substrate specificity (reviewed in [53]) determined using yeast heterologous system, should now be re-assessed [13].
8. SLC35D2: GDP-Mannose/UDP-Glc/UDP-GlcNac Transporter
The GDP-mannose transporter (GMT) from Leishmania donovani and Saccharomyces cerevisiae has been identified and characterised [54,55], with its biological importance and background covered in our previous review Hadley et al. [53]. In human's the NST encoded by hfrc1 was first characterised as being a multi-substrate specific NST, though unlike the fungal and protozoan equivalent hfrc1 when expressed in mammalian cells was only able to transport UDP-GlcNAc and UDP-Glc and not GDP-Man [56]. Advancement in the structural knowledge of SLC35D2 has increased significantly due to elucidation of the crystal structure of the yeast GDP-Man transporter Vrg-4 (Vanadate resistance glycosylation protein 4) by Parker and Newstead [14]. The crystal structure of Vrg-4 was determined to a resolution of 3.2 Å confirming a topology of 10-transmembrane α-helices connected by short loops with N and C termini located on the cytoplasmic side of the Golgi membrane [14]. This topology is similar to those proposed for the majority of the other NSTs in the SLC35 family. Functional studies on proteoliposome reconstituted Vrg-4 demonstrated that is a strict antiporter requiring either GDP-Man or GMP for transport to occur. The potential binding site for the sugar and phosphate was suggested by the positively charged central cavity. This central binding site was also able to discriminate between mono-, di- and triphosphates with transport rates being faster when the proteoliposome was preloaded with GDP-Man as opposed to the slow rate of GMP, suggesting an asymmetrical transport mechanism. Parker & Newstead [14] also observed that the nucleotide sugar binds in an extended conformation with both the nucleotide and the sugar complexed within separate binding pockets, thus confirming ligand binding studies suggested previously using STD NMR [57].
A x-ray crystal snapshot of Vrg-4 shows a state that is closed to the cytoplasm but open to the lumen of the Golgi apparatus. The rationale for transport suggested by the x-ray crystal structure showed that TMD6-TMD7 are packaged against TMD8-TMD9 sealing a large polar cavity from the cytoplasm whereas TMD1-TMD2 and TMD3-TMD4 are opened outwards. A cytoplasmic-facing model was generated to investigate the structural changes involved in transport. This model showed that alternating between the Golgi- and cytoplasmic-facing states results from TMD1 and TMD3 packing together and TMD6 and TMD8 moving apart, thus opening the binding site to the cytoplasm. The specific positive charge on TMD4 was shown to be crucial for activity with an alanine substitution resulting in a severe reduction in activity. Even with conservative substitutions of Lys118Arg, activity was still reduced by 50%.
Importantly, the coupling between substrate binding and conformational rearrangement is imperative in all transporters. In Vrg-4, the bound GDP-Man spans the binding site facilitating substrate-induced reorientation. By modelling GMP into the nucleotide-binding pocket on TMD7, they found the phosphate was in close proximity to the Lys118 of TMD4. This was given as a possible explanation for the necessity of a positive charge at this position. The ribose also makes similar interactions with TMD1 and TMD8 helping to coordinate movement. This ribose interaction was also proposed as essential for the CST based on STD NMR experiments [58].
The crystal structure of the Vrg-4 suggests a general alternating-access mechanism, where binding of the nucleotide sugar in the two specificity pockets while in the cytoplasmic-facing state, triggers reorganisation that results in the formation of the Golgi open state. Dissociation of the nucleotide sugar would vacate the binding site for the nucleotide monophosphate to bind, triggering reorientation back to the cytoplasmic state. The differing transport rates between GDP-Man and GMP stated previously was attributed to this asymmetrical transport mechanism as the nucleotide sugar participates in more interactions with the transporter than the nucleotide monophosphate. Due to this, GDP-Man may then be more effective in reorientating the transporter giving rise to the preference of transporting GDP-Man over GMP into the Golgi when both substrates are present in the cytoplasm. Interestingly, the oligomeric state of the functional Vrg-4 still remains unclear. Vrg-4 crystallised as eight molecules in the P1 unit cell of which four adopt a dimer arrangement with two lipids (monoolein) bound at the dimer interface, and four arrange as monomeric molecules with no lipid-mediated dimerization [14]. Even though there is a possibility that the functional Vrg-4 exists as a dimer, the transport mechanism is explained as a function of the monomer; the functional role of a dimer was not addressed.
9. SLC35A1: CMP-Sialic Acid Transporter (CST)
The CST remains one of the best characterised transporters of the SLC35 family, with research on its role in CDG's featuring prominently over the last few years. Until recently, the proposed topology of the CST has been based upon epitope-tagging, site-directed mutagenesis and hydrophobicity plots [59]. Since this initial proposal, the topology has been refined by many as highlighted in our previous review [53] but it still falls short of suggesting a potential structure and transport mechanism. As a high-resolution x-ray crystal structure for any human NST is yet to be published, the revised topologies have been our basis for attempting to interpret CST mechanism. Fortunately, we have now been able to take advantage of the 3D crystal structure of the GMT homolog Vrg-4 and use it as a template for an in silico-generated protein prediction homology model. Based on what we now know, this model has allowed us to make further predictions regarding the structure, transport mechanism and potentially the oligomeric functional state of the CST. Insights from our model may help better understand the structural reason for the reduced or abolished transport activity and/or lack of substrate specificity due to specific mutations.
We originally generated a model of the CST (CST homology-1SLAC1) based on the template of the bacterial SLAC1 anion channel (PDB 3M73) (Supplementary Fig. S1). After the release of the Vrg-4 crystal structure, we progressed our model of the CST using the SPARKS-X structural modelling tool [60] using the GMT homolog x-ray structure as a template, followed by further refinement using the MODELLER package [61] to relax the knotted loop formed between Leu70 and Pro85 by default. The output structure was visualised using YASARA (Bioinformatics 30,2981—2982, Version 18.4.24) and is our current structure represented in Fig. 1 (CST homology-2Vrg-4). Our predicted structural model gives insight into the rationale behind previously reported CST loss of function (reviewed in [53]). We examined these structural and functional limitations in light of the refined template-based model generated by SPARKS-X and the Vrg-4 crystal structure [14]. The structural and mechanistic predictions in this review are based on all CST-related data currently published and include an overview of all mutations that altered transport and/or substrate recognition (both de novo and site-directed).
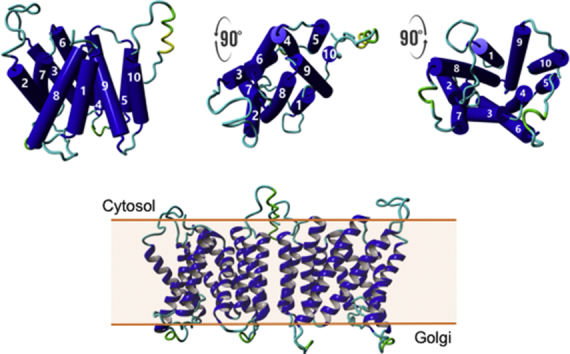
Fig. 1.
Refined CST model (CST homology-2Vrg-4) using the MODELLER package to relax the knotted loop formed between Leu70 and Pro85 by default. The initial model was generated using the SPARKS-X structural modelling tool based on the Vrg-4 template. This current model shows the TMD7 α-helix has a noticeable bend visible in both the ribbon (A) and TMD topology diagram (B). The model structure rotated forward 90° showing the binding site closed to cytosol (C), and rotated back 90° showing the binding site open to Golgi (D).
Our original SPARKS-X model CST homology-1SLAC1 (Supplementary Fig. S1A) split TMD7 into two shorter α-helices that we refer to as TMD7a and TMD7b (the TMD7 N-terminal half and the TMD7-C-terminal half, respectively). However, our current CST model, CST homology-2Vrg-4, shows TMD7 as a single α-helix but with a noticeable bend (Fig. 1 and Supplementary Fig. S1C). All modelled structures presented here are based on this current MODELLER-refined Vrg-4 template-based version (CST homology-2Vrg-4) though we will still refer to the N- and C– terminal of the TMD7 α-helix, as TMD7a and 7b.
9.1. Role of Glycine Pairs
Twenty years ago, Eckhardt et al. [62] observed that a single missense mutation Gly189Glu was directly responsible for the inactivation of CST transport activity. Further investigation convinced them that it was the size of the amino acid rather than the charge itself that was responsible for the inactivity. Ten years later, Lim et al. [63] agreed that increased steric hindrance associated with the exchange of Gly189 for a larger amino acid was directly responsible for the reduction in CST activity, and then further proposed that not just Gly189, but multiple glycine pairs may contribute to a glycine channel through which CMP-Sia could be translocated.
As previously reported [53], the suggestion of a Gly-rich hydrophilic channel or pore contradicts the hypothesis that the CST is a simple solute carrier that operates in an antiporter mechanism. We suggested that the two hypotheses could be reconciled by a ligand-binding event promoting a conformational change allowing the formation of this Gly-rich hydrophilic channel. Based on our current CST homology-2Vrg-4 model, this putative channel does not look likely if the protein is active as a monomer, though the glycines may play an integral role if the functional CST exists as a homodimer. While the Vrg-4 template is available as a crystallographic dimer, both subunits are found in the Golgi-open conformation which is not representative of the proposed antiporter mechanism. In order to observe the dimeric interface of the CST in a more biologically relevant conformation, we generated a dimeric model (CST homology-dimerTPT) based on the occluded conformation of the triose-phosphate/phosphate translocator (TPT) in complex with 3-phosphoglycerate (PDB 5y79) [64] (Fig. 2). Residues predicted to be disordered by SPOT-Disorder 2 [65] (Supplementary Fig. S2) have been excluded from the visualisations to improve clarity. The Parker & Newstead [14] structure shows the dimerization of Vrg-4 is lipid-mediated at the dimer interface, which could also be the case with the CST.
Fig. 2.

The potential CST occluded homodimer (CST homology-dimerTPT) is schematically based on the crystallographic dimer of the triose-phosphate/phosphate translocator in complex with 3-phosphoglycerate (PDB 5y79). Residues predicted as disordered have been deleted for clarity (Supplementary Fig. S2). Glycine pairs shown are Gly153 & Gly154 (green) in TM5; Gly177 & 179 (red) in TM6; Gly189 & Gly192 (magenta) in TM6; and Gly256 & Gly257 (yellow) in TM8. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 2 highlights that the position of the 4 glycine pairs in our CST model do not face in towards the putative active site, but instead, congregate on the outside of the same face. It seems most likely that they contribute to a “helix packing effect”, where two helices converge in a helix-helix cross [66]. This allows for one α-helix to subtly shift past another whilst also acting as a molecular notch to retain α-helices interactions when changing alternate-access conformation. Either way, Lim et al. [63] and others have shown that these glycine residues are essential for function and they are also highly conserved in diverse NST homologs.
With respect specifically to Gly189, our model places this residue medially in TMD6, adjacent to the predicted bound ligand. A feasible role for this residue, rather than contributing to a putative glycine channel, is that it facilitates van der Waals interactions between the ligand and the α-helical backbone in the active site. Hence a residue with a sidechain in this position would either sterically hinder the ligand or weaken ligand binding, ultimately affecting CST activity. This same phenomenon is seen in rhodopsin that also has several key transmembrane glycine residues. Gly121 specifically forms part of the retinal binding pocket and is thought to form a cavity for the methyl group of retinal [[66], [67], [68]]. Substitutions of larger hydrophobic residues for Gly121 led to steric hinderance [67] in a similar way as that observed for Gly189 in the CST [63].
9.2. The Importance of Tyr214
Not only does the Parker & Newstead [14] structure of Vrg-4 give us a basic understanding of the overall topology of the CST, it also highlights crucial differences. One of these is a modification in the nucleotide binding pocket from that of the conserved FYNN-motif seen in the Vrg-4 to the homologous motif in the CST of NIQM-motif with Gln212 replacing the asparagine. Parker & Newstead [14] propose that the extension of the glutamine side chain over the asparagine may be to help facilitate the recognition of cytosine, which is smaller than guanine. Although this ligand orientation works well with the Vrg-4 structure, it contradicts the CST ligand binding and orientation studies completed by Maggioni et al. [69]. STD-NMR spectroscopy experiments using site-directed mutants of CST have shown that Tyr214 is important in nucleotide sugar recognition predominantly by coordinating the Sia, not the cytosine moiety as proposed by Parker & Newstead [14]. In our CST homology-2Vrg-4 model, the Tyr214 faces the active site (Fig. 3), whereas the Parker & Newstead x-ray crystal structure reveals that Tyr214 is facing the membrane, and attributes its function as anchoring Gln212 to stabilise its interaction with the cytosine. Both these arguments cannot hold true simultaneously.
Fig. 3.
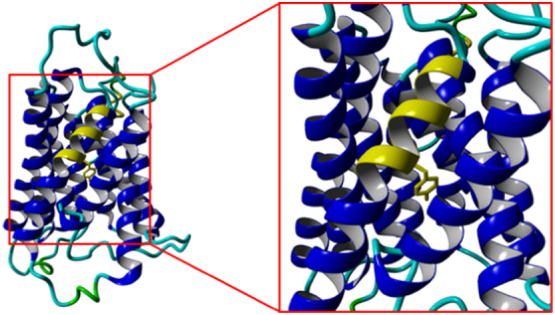
The stretch of amino acids between Val208-Gly217 (CST-TMD7a highlighted in yellow) is well documented as being essential for CST activity. In our CST homology-2Vrg-4 model the Tyr214 faces the active site, permitting direct interaction with CMP-Sia. The Parker and Newstead structure has Tyr214 facing the membrane and attributes Tyr214 function as anchoring Gln212 to stabilise its interaction with the cytosine. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The importance of the CST Tyr214 is well-described (see references in review [53]). Takeshima-Futagami et al. [70] indicated that the Val208-Gly217 stretch (CST-TMD7a highlighted in yellow in Fig. 3 and Supplementary Fig. S1) was essential for CMP-Neu5Ac transport activity. The authors also specifically identified Tyr214 and Ser216 as being critical for substrate recognition. Maggioni et al. [69] have further verified that the hydroxyl side-chains of one, or both, of these amino acids made specific interactions (hydrogen bonds) with the Sia moiety, not the cytosine of the substrate. It should be noted that Takeshima-Futagami et al. [70] indicate that Tyr214CST was more important than Ser216CST in the recognition of CMP-Sia.
Sialyl motifs include a recognisable stretch of residues that reside in the substrate binding sites and are common in sialyltransferases [71]. Takeshima-Futagami et al. [70] however found no similar pattern of Sialyl motifs in the CST. Datta et al. [71] proposed that the hydroxy group of Ser320 participated in the binding of the cytidine moiety of CMP-Sia in the sialyltransferases ST6Gal I. Interestingly, Ser216 and Tyr214 must not contribute in the same way in the CST because the chimeric constructs analysed by Takeshima-Futagami et al. [70], featuring the UGT and chimeric UGT/CST, lacked these two equivalent residues, and were still shown to interact with CMP. This is also demonstrated by the fact that UDP-Gal and UMP were released from microsomal vesicles in exchange for CMP [72].
9.3. Substrate-Induced Reorientation with TMD7a as the Potential Conformational Trigger
TMD7 seems to be a key player in the structure and function of the CST as highlighted by many researchers. As previously explained, our original SPARKS-X model split TMD7 into two α-helices joined by a short loop (Supplementary Fig. S1). Although our CST homology-2Vrg-4 model does not structurally split TMD7 into two separate α-helices, it seems to be functionally split into C– and N-terminal ends. TMD7a is folded towards the middle of the transporter and appears to block access to the active site from the cytosolic side (Fig. 3 and Supplementary Fig. S1). The two residues (Tyr214 and Ser216) shown to be critical for substrate recognition of the Sia portion of the ligand are located in this domain and give credence to a possible mechanistic trigger. In the case of a functional monomeric CST when our model is open to the cytosolic side, the substrate CMP-Sia could enter and bind to the active site. Part of this binding looks to include interactions between the Sia moiety and the hydroxyl side-chain of the Tyr214 and/or Ser216, which could result in movement of the TMD7a helix inwards, partially blocking access to the cytosolic side. Perhaps this inwards movement or tension on TMD7a triggers the conformational change required to alternatively open the transporter's active site to the Golgi lumen. Once this occurs and the CMP-Sia is released, CMP may then be able to bind, but without the interaction of Sia, the ligand would not interact with the TMD7a's Tyr214. This inaction could allow a more energetically favourable relaxation of TMD7a, hence triggering a reversion to the cytosolic-facing active site; the relaxed, or more energetically favourable state.
Perhaps this CMP-Sia binding-induced reorientation can be better explained if the functional state of the CST is dimeric. In this example of a potential substrate-induced reorientation and alternate access dimer model (Fig. 4), CMP-Sia binds in an extended form entering the CST from the cytosolic-side (with the nucleotide and sugar housed within separate binding pockets [14,58]), while CMP correspondingly binds on the Golgi-facing side of the homodimer (Fig. 4A). The binding of CMP-Sia induces a substrate-recognised reorientation of the CST, allowing CMP-Sia to be released in the Golgi lumen. In doing this, the CMP is given alternate access to be released in the cytosol (Fig. 4B). In the next step, CMP-Sia again binds from the cytosolic side of the CST, whilst CMP is bound on the Golgi-lumen side (Fig. 4C). The binding of CMP-Sia again induces a substrate-recognised reorientation of the CST to give access for the CMP-Sia to be released in the Golgi lumen and the CMP is correspondingly released in the cytosol (Fig. 4D). As mentioned by Parker & Newstead [14], binding of the extended nucleotide sugar must be the rate-limiting step of this process as there are more connections and interactions at play between the transporter and the substrate during binding. This extended binding time by the larger and more complex of the two antiported molecules (CMP-Sia) not only allows the release of CMP-Sia on the Golgi lumen but also allows extra time for CMP to take its place, binding in the active binding site to be antiported back to the cytosol.
Fig. 4.
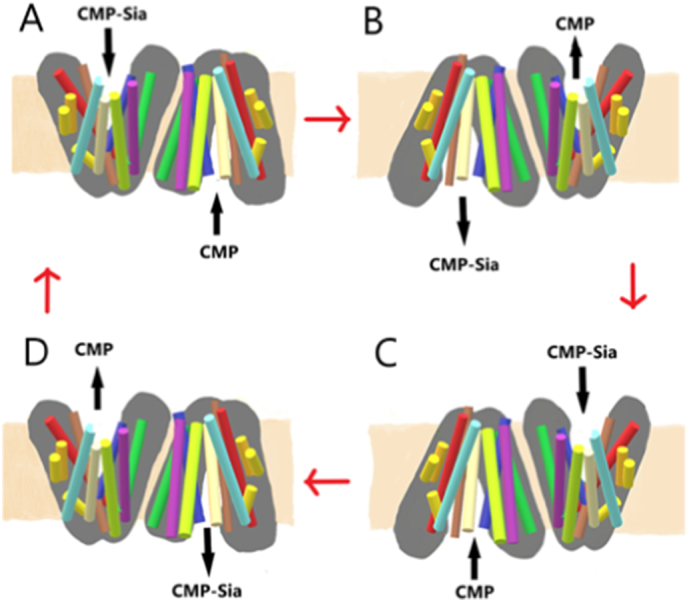
Potential substrate-induced reorientation and alternate access dimeric model. (A). CMP-Sia binds to the CST binding site in an extended form entering from the cytosolic side, while CMP binds from the Golgi-facing side of the dimeric CST. (B) The binding of CMP-Sia induces a reorientation of the CST to allow CMP-Sia to be released in the Golgi lumen while CMP is released into the cytosol. (C) CMP-Sia again binds from the cytosolic side of the CST, whilst CMP is bound on the Golgi-lumen side. (D) The binding of CMP-Sia again induces a reorientation of the CST to allow CMP-Sia to be released into the Golgi lumen and the CMP is released into the cytosol.
STD NMR spectroscopy analyses of the Tyr214Ala CST in complex with CMP-Neu5Ac shows complete loss of the STD NMR signal intensities of the Sia moiety even though significant binding of CMP still remains [58]. If Tyr214 is the conformational trigger, then Tyr214Ala would effectively lock the CST in the cytosolic-open conformation, rendering it inactive (Fig. 4, steps A and C). This inactivation has previously been shown [69,70] and the importance of Ser216 and Tyr214 is not only inferred experimentally but also highlighted by its high evolutionary conservation in the CST.
As alluded to previously, Ser216 and Tyr214 are even more interesting when considering their context in the hUGT/hCST chimera [70]. Both Tyr214Gly and Ser216Phe individual mutations showed loss of CST activity, but retained UGT activity. It is vitally important to note that the UGT and CST can recognise both UMP and CMP as the antiport substrate [72]. This again supports the hypothesis that the lack of interaction between the Sia moiety and TMD7a allows the relaxation of the transporter to the more favourable energetic state of facing the cytosol if the CST is a functional monomer. If dimeric, the binding of UMP/CMP may be of much less significance than the binding of the corresponding nucleotide sugar. Further, the chimeric UGT/CST showed that the CMP-Sia recognising motif is somewhere in TMD7 as the UGT with CST-derived TMD7, is capable of recognising, and competent in transporting, CMP-Sia. Whereas hUGT recognition of UDP-Gal is much more complicated and is proposed to be recognised in spatially dispersed regions. In the UGT some of the contact points, including those on TMD1 and TMD8 are critical, whereas either the subset on TMD2, TMD3, and TMD7, or those on TMD9 and TMD10 may be dispensable provided the others are available [72]. As we have previously reported [53], the necessity of Tyr214 was confirmed through experiments utilising the CST-inactive hUGT/hCST chimera G1-B, where Gly239UGT and Phe241UGT were separately mutated to the corresponding amino acid residues in the CST sequence, Tyr (Tyr214CST) and Ser (Ser216CST), respectively. This showed conclusively that addition of just the Tyr in this position conferred the ability to transport CMP-Sia in addition to UDP-Gal [70].
9.4. SLC35A1 Congenital Disorders of Glycosylation
SLC35A1 deficiency has been referred to as congenital disorder of N-linked glycosylation IIf (CDG-IIf), with the new nomenclature being SLC35A1-CDG [73]. Abnormal protein glycosylation is implicated in numerous genetic and acquired diseases ranging from diabetes, cancer, and inflammatory disease to neurodegenerative and neuromuscular disease [74]. Over 125 genetic disorders have been attributed to many glycosylation pathways [75], but a distinct difference should be made between the disorders of the pathways compared to direct mutations of the NST itself. Insights from studying these diseases in human medicine not only allows identification of therapeutic targets but gives us further understanding of the physiological and pathological results of these mutations in humans. For further information, a recent comprehensive review of CDG's across all glycosylation pathways has been published by Ng & Freeze [76]. Our review will briefly focus on the Congenital Disorders of Glycosylation (CDG's) that result directly from mutations in the SLC35A1 (CST) and the mechanistic and structural implications of these mutations in our predicted model.
The four individual cases presenting with rare CST-linked CDG that we review possess different mutations. Case 1 deals with two inactive alleles whereas Case 2, Case 3, Case 4 are point mutations. The discovery of active and inactive isoforms through Case 1 has led to extensive inspection of SLC35A1 splice variants especially the human del177 isoform. The structural anomalies in the transporter from all four cases are discussed here, and references for in-depth detail on the physiological aspects of the patients provided.
Case 1
del177 Isoform
A novel SLC35A1-CDG reported by Martinez-Duncker et al. [77] was characterised by macrothrombocytopenia, neutropenia and complete lack of the sialyl-Lex antigen on polymorphonuclear cells caused by inactive SLC35A1 alleles. This inactivation of the CST is more complicated than the point mutations identified in Case 2, Case 3, Case 4 as it involves multiple deletions, and is further complicated by completely different mutations in each contributing allele. In particular a double microdeletion was discovered in the paternal allele that induced a frameshift mutation resulting in a premature stop codon. Whereas the maternal allele had a 130-bp deletion that covered 66% of exon 6, again resulting in a premature stop codon. Interestingly, the maternal mutation involved a homozygous insertion of a 4 bp CACT that created a new U2 spliceosomal snRNA site. This is extremely important as it is in direct competition with the normal U2 snRNA site and can therefore induce a range of different splice variations from wild-type to complete skipping of exon 6 and several active and inactive variations in-between. This CACT insert was shown to have a gene frequency in the screened population of 32%, but unfortunately in this case, the splice-varied maternal allele was the deleterious 130 isoform, and so coupled with the paternal inactive allele, induced the disease.
Several years later, Salinas-Marin et al. [78] further evaluated a range of these isoforms, with their data initially seeming difficult to reconcile from a structure/function perspective. One of the functional splice variants of the human CST, known as the del177 isoform corresponds to deletion of the entire exon 6, which in hamsters, renders the CST inactive. Surprisingly, the del177 isoform in humans is functional. It was postulated that the non-functionality of the hamster del177 isoform in contrast to the functional human del177 isoform could reside in the 23 amino acid difference in sequence between the human and the hamster transporters. Their proposed topology of the active human del177 translated to a protein with 8 TMDs as a result of the loss of amino acids 192–251, which includes loop 6, the entire TMD7, and loop 7. This would fuse the remaining amino acids of TMD6 and TMD8 into a new 31 amino acid TMD6 in the del177 isoform. This active human del177 isoform (Fig. 5 highlighted in yellow) seems to defy previous findings as it excludes the amino acids shown to be essential for CST activity (see Hadley et al. [53]) including Tyr214, Ser216 and 236KGFF239 (4th luminal loop).
Fig. 5.

Multiple sequence alignment of del177 isoform from hamster, mouse and human. The deleted region in the del177 isoforms is highlighted in yellow, with previously proposed critical residues in boxed in red. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
There are two conundrums here that need to be considered. Firstly, how is the human del177 isoform still functional sans-TMD7? Secondly, why is the hamster del177 isoform inactive given it only differs from the hCST by 23 different amino acids? The answer to both these questions as posed by Salinas-Marin et al. [78] might lie in one or more of these 23 amino acids replacing the critical function of Y214 and 236KGFF239 (4th luminal loop). In an attempt to answer these questions, we generated a human del177 model using SPARKS-X and found that Tyr207 occupies the same space as the essential Tyr214 in the CST-WT (Fig. 6 A and B). We suggest that this analogous residue is why the human del177 isoform remains functional. We also generated a hamster del177 model using the same methodology and found that the Tyr207 sidechain in this model is not oriented towards the active site (Fig. 6C). This difference in the Tyr207 orientation may singularly be responsible for the inactive hamster CST del177 isoform compared to the functional human CST del177 isoform.
Fig. 6.

Structural alignment of the human CST-del177 (blue) and hamster del177 (yellow) (A). The human CST-del177 with the Tyr207 sidechain (highlighted in red) facing in towards the active site (B). The hamster del177 with the Tyr207 sidechain (highlighted in green) rotated away from the active site (C). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
A further observation resulting from our CST homology-2Vrg-4 model was the number of predicted TMDs in both the CST del177 isoforms, human and hamster. Using SPARKS-X to generate an alignment with the original Vrg-4 template, the CST-del177 human and hamster still translates into the typical 10 TMDs that defines the CST-wt as opposed to the 8 TMD's suggested by Salinas-Marin et al. [78]. The predicted 8-TMD model contains a long 31-residue α-helix whereas our 10-TMD model consists of a similar topology to the wild-type but with TMD7 and 9 α-helices being much shorter. TMD7 has 8 residues (Leu199-Lys206) and TMD9 has 10 residues (Leu223-Leu231). Although these may seem unusually small, the minimum length of an α-helix required to produce an α-helix-like CD spectrum is calculated to be two to three turns (seven to eleven residues) so both are within feasible limits [79].
Case 2
Q101H (Gln101His) TMD3.
The Gln101His mutation reported by Mohamed et al. [80] occurs within TMD3. This residue is critical as it is highly conserved across all species even down to Drosophila melanogaster. Gln101His still allows the CST to remain stable but reduces the activity of the transporter by 50%. Interestingly, our CST homology-2Vrg-4 model shows the Gln sidechain faces inwards, directly into the purported active site (Supplementary Fig. S3). In this position, one could assume the charged His would probably have a more deleterious impact on ligand binding rather than any hindering of the stability of the protein in the membrane.
Case 3
Thr156Arg and Glu196Lys.
Recent novel CST mutations have been discovered that decrease the overall transport rate resulting in a patient presenting with encephalopathy [81] . Although the neurological symptoms are quite similar between Case 2, Case 3, they are still distinguishable from each other. These mutations, which have not been previously identified, are rare compound heterozygous mutations Thr156Arg and Glu196Lys.
Ng et al. [81] postulated that as Thr156Arg localised to the Golgi lumen side of TMD-5, and Glu196Lys was at the cytosolic side of TMD-6, these mutations might disrupt interactions that orient the transporter within the membrane. They based their hypothesis on the assumption that NSTs can form hetero or homodimers with other transporters. Sosicka et al. [7] also claim confirmation of homomer formation by the CST, however no data was provided confirming this. Sosicka et al. [7] also stated that “many nucleotide sugar transporters form homomers...” so therefore “…one may assume that homooligomerization is an intrinsic feature of NSTs”. Our CST homology-2Vrg-4 model could support both a monomeric and dimeric antiporter mechanism, but the functional oligomeric state for the functional NST is yet to be confirmed. Regarding the Tyr156Arg and Glu196Lys variations, our model shows these mutations occur on the same face of the transporter but at opposite ends. As such if they were not impacting dimerization, they could possibly interact and disrupt both external and internal membrane interfaces, restricting the open/closed conformations (Supplementary Fig. S4). Further credence to this is given when looking at Thr156Arg in conjunction with Gly153Ile and Gly154Ile mutants discussed in Case 4 below.
Case 4
Ser147Pro.
Two offspring, both homozygous for a missense mutation Ser147Pro in SLC35A1 [82], were found with heterozygous consanguineous parents. This mutation resulted in macrothrombocytopenia and other related symptoms that were already reported in case 2 (Gln101His). Ser147 is located in TMD5 and the insertion of a proline into an α-helix would likely create instability of the protein. Ser147 would not seem to be involved directly in ligand binding (TMD5 is distant to the active site) so disruption to protein stability is probable. Interestingly, Ser147 is very close to the Thr156Arg mutation of Case 3 in TMD5. As expressed above, our CST homology-2Vrg-4 model shows TMD5 is distant to the active site (Fig. 1B–D). The fact that these mutations both reduce activity suggests hindering of the conformational change. To give further support to this idea, other residues in this region Gly153Ile and Gly154Ile also show clear reductions in activity [63]. In conclusion it appears that any disruption to this region reflects in reduced activity. Either TMD5 must be pivotal in an alternative access conformation role or perhaps this is evidence of disruption to a potential dimer?
10. Update of Recently Published NST Crystal Structures
During the review process of this manuscript new structural information regarding a CST homolog from the plant Zea mays [83], and the mouse CST [84] became available. It should be noted that the Zea mays NST reported by Nji et al. [83] shares only 28% homology with the human CST, and plants do not express Sia [85] (a family of 9-carbon α-keto acids that include 5-N-acetylneuraminic acid (Neu5Ac), 5-N-glycolylneuraminic acid (Neu5Gc) or deaminoneuraminic acid (KDN) [86]). Therefore it is highly improbable that this NST transports CMP-Sia in vivo, instead it is more than likely that it transports CMP-KDO (KDO is an 8-carbon keto-deoxy acid that is found in prokaryotes and plants but not humans [87]). Nevertheless, the structures reported by Nji et al. [83] as well as Ahuja & Whorton [84] confirmed the overall topology of our mammalian CST model. For example, in agreement with our CST model, TM5 and TM10 in the Zea mays CST homolog appears to form a dimerization domain that acts as a scaffold, anchoring the transporter whilst rearrangement of the transport domain bundles occur. Furthermore, as now confirmed by Nji et al., [83] this arrangement is similar to that predicted for the structural homolog TPT from which we generated our dimeric model CST homology-dimerTPT (Fig. 2) that was based on its occluded conformation in complex with 3-phosphoglycerate (PDB 5y79) [64]. In addition, Ahuja & Whorton [84] describes TMD 5, 9 and 10 in the mouse CST structure as moving independently of the rest of the protein. Considering that TMD's 5 and 10 are distant to the active site, it is again possible that these regions may be playing a stabilising or structural role whilst the transport domain/s undergo rearrangement enabling substrate translocation.
Nji et al. [83] and Ahuja & Whorton [84] also concur with the analysis presented in this review that there is no similarity between the Sia binding site in the CST and the Man binding site in the GDP-Man bound Vrg4 structure [14], although the exact structural elements specifically involved in substrate recognition and binding that triggers transport in our opinion remains to be fully resolved. We propose that the alternative access mechanism is triggered by the binding of CMP-Sia, particularly the interaction of Sia and Tyr214, whereas Nji et al. [83] and Ahuja & Whorton [84] suggests that instead, binding of CMP governs the initiation of translocation with the Sia moiety chaperoned into the inner cavity of the transporter. This possibility does not preclude the alternative access mechanism we propose in Fig. 4 where instead of the trigger being specific binding interaction with Sia, the trigger is potentially binding to CMP, leading to the conformational changed detailed by Ahuja & Whorton [84].
However, we still need to reconcile the importance of Tyr214 in CMP-Sia binding and its role in CST function. We and others, using site-directed mutagenesis and chimeric studies, employing different transport detection/analysis methods (STD NMR spectroscopy [69] and complementation analysis in Lec2 and Lec8 cells [70]), provide strong evidence that TMD7 and Tyr214 in particular, are involved in Sia binding. Similarly deletion and/or mutation of Tyr214 leads to dramatic alteration of substrate specificity and complete loss of activity in the UGT/CST chimera [70]. Despite the fact that the new NST x-ray crystal structures (mouse CST [84] and plant CST homolog [83]), previous functional data [69] [70], and the CST homology-2Vrg-4 model generated in this report all agree that Tyr214 interacts with the CST substrates and facilitates transport, which verifies our del177 model suggesting that del177-Tyr207 replaces the function of the mCST-Tyr214, the mechanistic importance of Tyr214 suggested by these studies is not reflected in the mouse CST structure Ahuja & Whorton [84]. We believe that the exact role of Tyr214 in intiating/triggering the conformational change that permits transport of CMP/CMP-Sia by the CST requires further investigation and clarification.
11. Conclusion and Future Perspective
There has been significant progress and discovery in the NST field over the past 5 years. Many of the orphan transporters have now been postulated as regulatory proteins coupled to other NSTs rather than being directly involved in glycosylation themselves. In addition, transporters that were thought to be single substrate-specific are now being uncovered as possible multi-substrate transporters. In the absence of a published x-ray crystal structure of any mammalian NST at the time of submission and subsequent review process, we used a template-based in silico-generated model to help shed light on the structural aspects of the best studied NST, the CMP-sialic acid transporter, specifically relating to the CST mechanism of action. We have used our model to structurally and mechanistically rationalise currently published CST-related functional data and mutations that altered transport and/or substrate recognition. Importantly, our model and the associated mechanistic and functional insights are supported by the recently published NST x-ray crystal structures [83,84], and as such can now be used to further assess NST structure function relationships.
Declarations of Competing Interest
None.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2019.08.002.
Appendix A. Supplementary Data
Supplementary material
References
- 1.Caffaro C.E., Hirschberg C.B. Nucleotide sugar transporters of the Golgi apparatus: from basic science to diseases. Acc Chem Res. 2006;39:805–812. doi: 10.1021/ar0400239. [DOI] [PubMed] [Google Scholar]
- 2.Fredriksson R., Nordstrom K.J., Stephansson O., Hagglund M.G., Schioth H.B. The solute carrier (SLC) complement of the human genome: phylogenetic classification reveals four major families. FEBS Lett. 2008;582:3811–3816. doi: 10.1016/j.febslet.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 3.Abeijon C., Mandon E.C., Hirschberg C.B. Transporters of nucleotide sugars, nucleotide sulfate and ATP in the Golgi apparatus. Trends Biochem Sci. 1997;22:203–207. doi: 10.1016/s0968-0004(97)01053-0. [DOI] [PubMed] [Google Scholar]
- 4.Capasso J.M., Hirschberg C.B. Mechanisms of glycosylation and sulfation in the Golgi apparatus: evidence for nucleotide sugar/nucleoside monophosphate and nucleotide sulfate/nucleoside monophosphate antiports in the Golgi apparatus membrane. Proc Natl Acad Sci U S A. 1984;81:7051–7055. doi: 10.1073/pnas.81.22.7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milla M.E., Clairmont C.A., Hirschberg C.B. Reconstitution into proteoliposomes and partial purification of the Golgi apparatus membrane UDP-galactose, UDP-xylose, and UDP-glucuronic acid transport activities. J Biol Chem. 1992;267:103–107. [PubMed] [Google Scholar]
- 6.Tiralongo J., Ashikov A., Routier F., Eckhardt M., Bakker H. Functional expression of the CMP-sialic acid transporter in Escherichia coli and its identification as a simple mobile carrier. Glycobiology. 2006;16:73–81. doi: 10.1093/glycob/cwj029. [DOI] [PubMed] [Google Scholar]
- 7.Sosicka P., Maszczak-Seneczko D., Bazan B., Shauchuk Y., Kaczmarek B. An insight into the orphan nucleotide sugar transporter SLC35A4. Biochim Biophys Acta (BBA) Mol Cell Res. 2017;1864:825–838. doi: 10.1016/j.bbamcr.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 8.Sosicka P., Bazan B., Maszczak-Seneczko D., Shauchuk Y., Olczak T. SLC35A5 Protein-A Golgi complex member with putative nucleotide sugar transport activity. Int J Mol Sci. 2019;20:276. doi: 10.3390/ijms20020276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He J., Jin Y., Zhou M., Li X., Chen W. Solute carrier family 35 member F2 is indispensable for papillary thyroid carcinoma progression through activation of transforming growth factor-β type I receptor/apoptosis signal-regulating kinase 1/mitogen-activated protein kinase signaling axis. Cancer Sci. 2017;109:642–655. doi: 10.1111/cas.13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sosicka P., Jakimowicz P., Olczak T., Olczak M. Short N-terminal region of UDP-galactose transporter (SLC35A2) is crucial for galactosylation of N-glycans. Biochem Biophys Res Commun. 2014;454:486–492. doi: 10.1016/j.bbrc.2014.10.098. [DOI] [PubMed] [Google Scholar]
- 11.Maszczak-Seneczko D., Sosicka P., Kaczmarek B., Majkowski M., Luzarowski M. UDP-galactose (SLC35A2) and UDP-N-acetylglucosamine (SLC35A3) transporters form glycosylation-related complexes with Mannoside Acetylglucosaminyltransferases (Mgats) J Biol Chem. 2015;290:15475–15486. doi: 10.1074/jbc.M115.636670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sasisekharan R., Raman R., Prabhakar V. Glycomics approach to structure-function relationships of glycosaminoglycans. Annu Rev Biomed Eng. 2006;8:181–231. doi: 10.1146/annurev.bioeng.8.061505.095745. [DOI] [PubMed] [Google Scholar]
- 13.Bazan B., Wiktor M., Maszczak-Seneczko D., Olczak T., Kaczmarek B. Lysine at position 329 within a C-terminal dilysine motif is crucial for the ER localization of human SLC35B4. PLoS ONE. 2018;13 doi: 10.1371/journal.pone.0207521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parker J.L., Newstead S. Structural basis of nucleotide sugar transport across the Golgi membrane. Nature. 2017;551:521–524. doi: 10.1038/nature24464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ng B.G., Buckingham K.J., Raymond K., Kircher M., Turner E.H. Mosaicism of the UDP-galactose transporter SLC35A2 causes a congenital disorder of glycosylation. Am J Hum Genet. 2013;92:632–636. doi: 10.1016/j.ajhg.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yates T.M., Suri M., Desurkar A., Lesca G., Wallgren-Pettersson C. SLC35A2-related congenital disorder of glycosylation: defining the phenotype. Eur J Paediatr Neurol. 2018;22:1095–1102. doi: 10.1016/j.ejpn.2018.08.002. [DOI] [PubMed] [Google Scholar]
- 17.Dorre K., Olczak M., Wada Y., Sosicka P., Gruneberg M. A new case of UDP-galactose transporter deficiency (SLC35A2-CDG): molecular basis, clinical phenotype, and therapeutic approach. J Inherit Metab Dis. 2015;38:931–940. doi: 10.1007/s10545-015-9828-6. [DOI] [PubMed] [Google Scholar]
- 18.Winawer M.R., Griffin N.G., Samanamud J., Baugh E.H., Rathakrishnan D. Somatic SLC35A2 variants in the brain are associated with intractable neocortical epilepsy. Ann Neurol. 2018;83:1133–1146. doi: 10.1002/ana.25243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sim N.S., Seo Y., Lim J.S., Kim W.K., Son H. Brain somatic mutations in <em>SLC35A2</em> cause intractable epilepsy with aberrant N-glycosylation. Neurol Genet. 2018;4 doi: 10.1212/NXG.0000000000000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vals M.-A., Ashikov A., Ilves P., Loorits D., Zeng Q. Clinical, neuroradiological and biochemical features of SLC35A2-CDG patients. J Inherit Metab Dis. 2019;42:553–564. doi: 10.1002/jimd.12055. [0] [DOI] [PubMed] [Google Scholar]
- 21.Ng B.G., Sosicka P., Agadi S., Almannai M., Bacino C.A. SLC35A2-CDG: Functional characterization, expanded molecular, clinical, and biochemical phenotypes of 30 unreported individuals. Hum Mutat. 2019;40:908–925. doi: 10.1002/humu.23731. [0] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brandli A.W., Hansson G.C., Rodriguez-Boulan E., Simons K. A polarized epithelial cell mutant deficient in translocation of UDP-galactose into the Golgi complex. J Biol Chem. 1988;263:16283–16290. [PubMed] [Google Scholar]
- 23.Ishida N., Yoshioka S., Iida M., Sudo K., Miura N. Indispensability of transmembrane domains of Golgi UDP-galactose transporter as revealed by analysis of genetic defects in UDP-galactose transporter-deficient murine had-1 mutant cell lines and construction of deletion mutants. J Biochem. 1999;126:1107–1117. doi: 10.1093/oxfordjournals.jbchem.a022556. [DOI] [PubMed] [Google Scholar]
- 24.Oelmann S., Stanley P., Gerardy-Schahn R. Point mutations identified in Lec8 Chinese hamster ovary glycosylation mutants that inactivate both the UDP-galactose and CMP-sialic acid transporters. J Biol Chem. 2001;276:26291–26300. doi: 10.1074/jbc.M011124200. [DOI] [PubMed] [Google Scholar]
- 25.Marini C., Hardies K., Pisano T., May P., Weckhuysen S. Recessive mutations in SLC35A3 cause early onset epileptic encephalopathy with skeletal defects. Am J Med Genet A. 2017;173:1119–1123. doi: 10.1002/ajmg.a.38112. [DOI] [PubMed] [Google Scholar]
- 26.Kimizu T., Takahashi Y., Oboshi T., Horino A., Koike T. A case of early onset epileptic encephalopathy with de novo mutation in SLC35A2: clinical features and treatment for epilepsy. Brain Dev. 2017;39:256–260. doi: 10.1016/j.braindev.2016.09.009. [DOI] [PubMed] [Google Scholar]
- 27.Edvardson S., Ashikov A., Jalas C., Sturiale L., Shaag A. Mutations in SLC35A3 cause autism spectrum disorder, epilepsy and arthrogryposis. J Med Genet. 2013;50:733–739. doi: 10.1136/jmedgenet-2013-101753. [DOI] [PubMed] [Google Scholar]
- 28.Thomsen B., Horn P., Panitz F., Bendixen E., Petersen A.H. A missense mutation in the bovine SLC35A3 gene, encoding a UDP-N-acetylglucosamine transporter, causes complex vertebral malformation. Genome Res. 2006;16:97–105. doi: 10.1101/gr.3690506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maszczak-Seneczko D., Sosicka P., Majkowski M., Olczak T., Olczak M. UDP-N-acetylglucosamine transporter and UDP-galactose transporter form heterologous complexes in the Golgi membrane. FEBS Lett. 2012;586:4082–4087. doi: 10.1016/j.febslet.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 30.Khoder-Agha F., Sosicka P., Escriva Conde M., Hassinen A., Glumoff T. N-acetylglucosaminyltransferases and nucleotide sugar transporters form multi-enzyme-multi-transporter assemblies in golgi membranes in vivo. Cell Mol Life Sci. 2019;76:1821–1832. doi: 10.1007/s00018-019-03032-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maggioni A., von Itzstein M., Gerardy-Schahn R., Tiralongo J. Targeting the expression of functional murine CMP-sialic acid transporter to the E. coli inner membrane. Biochem Biophys Res Commun. 2007;362:779–784. doi: 10.1016/j.bbrc.2007.08.070. [DOI] [PubMed] [Google Scholar]
- 32.Förster-Fromme K., Schneider S., Sprenger G.A., Albermann C. Functional expression of a human GDP-l-fucose transporter in Escherichia coli. Biotechnol Lett. 2017;39:219–226. doi: 10.1007/s10529-016-2233-x. [DOI] [PubMed] [Google Scholar]
- 33.Taubenschmid J., Stadlmann J., Jost M., Klokk T.I., Rillahan C.D. A vital sugar code for ricin toxicity. Cell Res. 2017;27:1351. doi: 10.1038/cr.2017.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bigalke H., Rummel A. Medical aspects of toxin weapons. Toxicology. 2005;214:210–220. doi: 10.1016/j.tox.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 35.Song S., Ou-Yang Y., Huo J., Zhang Y., Yu C. Molecular cloning, sequence characterization, and tissue expression analysis of three water buffalo (Bubalus bubalis;) genes – ST6GAL1, ST8SIA4, SLC35C1. Arch Anim Breeding. 2016;59:363–372. [Google Scholar]
- 36.Dejima K., Murata D., Mizuguchi S., Nomura K.H., Gengyo-Ando K. The ortholog of human solute carrier family 35 member B1 (UDP-galactose transporter-related protein 1) is involved in maintenance of ER homeostasis and essential for larval development in Caenorhabditis elegans. FASEB J. 2009;23:2215–2225. doi: 10.1096/fj.08-123737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klein M.-C., Zimmermann K., Schorr S., Landini M., Klemens P.A.W. AXER is an ATP/ADP exchanger in the membrane of the endoplasmic reticulum. Nat Commun. 2018;9:3489. doi: 10.1038/s41467-018-06003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nishimura M., Suzuki S., Satoh T., Naito S. Tissue-specific mRNA expression profiles of human solute carrier 35 transporters. Drug Metab Pharmacokinet. 2009;24:91–99. doi: 10.2133/dmpk.24.91. [DOI] [PubMed] [Google Scholar]
- 39.Wei Z.-B., Yuan Y.-F., Jaouen F., Ma M.-S., Hao C.-J. SLC35D3 increases autophagic activity in midbrain dopaminergic neurons by enhancing BECN1-ATG14-PIK3C3 complex formation. Autophagy. 2016;12:1168–1179. doi: 10.1080/15548627.2016.1179402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Z., Hao C.-J., Li C.-G., Zang D.-J., Zhao J. Mutation of SLC35D3 causes metabolic syndrome by impairing dopamine signaling in striatal D1 neurons. PLoS Genet. 2014;10:e1004124. doi: 10.1371/journal.pgen.1004124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chintala S., Tan J., Gautam R., Rusiniak M.E., Guo X. The Slc35d3 gene, encoding an orphan nucleotide sugar transporter, regulates platelet-dense granules. Blood. 2007;109:1533–1540. doi: 10.1182/blood-2006-08-040196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rendu F., Brohard-Bohn B. The platelet release reaction: granules' constituents, secretion and functions. Platelets. 2001;12:261–273. doi: 10.1080/09537100120068170. [DOI] [PubMed] [Google Scholar]
- 43.McNicol A., Israels S.J. Platelet dense granules: structure, function and implications for haemostasis. Thromb Res. 1999;95:1–18. doi: 10.1016/s0049-3848(99)00015-8. [DOI] [PubMed] [Google Scholar]
- 44.Shotelersuk V., Dell'Angelica E.C., Hartnell L., Bonifacino J.S., Gahl W.A. A new variant of Hermansky-Pudlak syndrome due to mutations in a gene responsible for vesicle formation. Am J Med. 2000;108:423–427. doi: 10.1016/s0002-9343(99)00436-2. [DOI] [PubMed] [Google Scholar]
- 45.Ashikov A., Routier F., Fuhlrott J., Helmus Y., Wild M. The human solute carrier gene SLC35B4 encodes a bifunctional nucleotide sugar transporter with specificity for UDP-xylose and UDP-N-acetylglucosamine. J Biol Chem. 2005;280:27230–27235. doi: 10.1074/jbc.M504783200. [DOI] [PubMed] [Google Scholar]
- 46.Maszczak-Seneczko D., Olczak T., Olczak M. Subcellular localization of UDP-GlcNAc, UDP-Gal and SLC35B4 transporters. Acta Biochim Pol. 2011;58 [PubMed] [Google Scholar]
- 47.Roy S.K., Chiba Y., Takeuchi M., Jigami Y. Characterization of yeast Yea4p, a uridine diphosphate-N-acetylglucosamine transporter localized in the endoplasmic reticulum and required for chitin synthesis. J Biol Chem. 2000;275:13580–13587. doi: 10.1074/jbc.275.18.13580. [DOI] [PubMed] [Google Scholar]
- 48.Ishikawa H.O., Ayukawa T., Nakayama M., Higashi S., Kamiyama S. Two pathways for importing GDP-fucose into the endoplasmic reticulum lumen function redundantly in the O-fucosylation of Notch in Drosophila. J Biol Chem. 2010;285:4122–4129. doi: 10.1074/jbc.M109.016964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Itin C., Kappeler F., Linstedt A.D., Hauri H.-P. A novel endocytosis signal related to the KKXX ER-retrieval signal. EMBO J. 1995;14:2250–2256. doi: 10.1002/j.1460-2075.1995.tb07219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andersson H., Kappeler F., Hauri H.-P. Protein targeting to endoplasmic reticulum by dilysine signals involves direct retention in addition to retrieval. J Biol Chem. 1999;274:15080–15084. doi: 10.1074/jbc.274.21.15080. [DOI] [PubMed] [Google Scholar]
- 51.Benghezal M., Wasteneys G.O., Jones D.A. The C-terminal dilysine motif confers endoplasmic reticulum localization to type I membrane proteins in plants. Plant Cell. 2000;12:1179. doi: 10.1105/tpc.12.7.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mkhikian H., Mortales C.-L., Zhou R.W., Khachikyan K., Wu G. Golgi self-correction generates bioequivalent glycans to preserve cellular homeostasis. Elife. 2016;5 doi: 10.7554/eLife.14814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hadley B., Maggioni A., Ashikov A., Day C.J., Haselhorst T. Structure and function of nucleotide sugar transporters: current progress. Comput Struct Biotechnol J. 2014;10:23–32. doi: 10.1016/j.csbj.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma D., Russell D.G., Beverley S.M., Turco S.J. Golgi GDP-mannose uptake requires Leishmania LPG2. A member of a eukaryotic family of putative nucleotide-sugar transporters. J Biol Chem. 1997;272:3799–3805. [PubMed] [Google Scholar]
- 55.Dean N., Zhang Y.B., Poster J.B. The VRG4 gene is required for GDP-mannose transport into the lumen of the Golgi in the yeast, Saccharomyces cerevisiae. J Biol Chem. 1997;272:31908–31914. doi: 10.1074/jbc.272.50.31908. [DOI] [PubMed] [Google Scholar]
- 56.Suda T., Kamiyama S., Suzuki M., Kikuchi N., Nakayama K. Molecular cloning and characterization of a human multisubstrate specific nucleotide-sugar transporter homologous to Drosophila fringe connection. J Biol Chem. 2004;279:26469–26474. doi: 10.1074/jbc.M311353200. [DOI] [PubMed] [Google Scholar]
- 57.Maggioni A., Meier J., Routier F., Haselhorst T., Tiralongo J. Direct investigation of the Aspergillus GDP-mannose transporter by STD NMR spectroscopy. Chembiochem. 2011;12:2421–2425. doi: 10.1002/cbic.201100483. [DOI] [PubMed] [Google Scholar]
- 58.Maggioni A., von Itzstein M., Tiralongo J., Haselhorst T. Detection of ligand binding to nucleotide sugar transporters by STD NMR spectroscopy. Chembiochem. 2008;9:2784–2786. doi: 10.1002/cbic.200800526. [DOI] [PubMed] [Google Scholar]
- 59.Eckhardt M., Gotza B., Gerardy-Schahn R. Membrane topology of the mammalian CMP-sialic acid transporter. J Biol Chem. 1999;274:8779–8787. doi: 10.1074/jbc.274.13.8779. [DOI] [PubMed] [Google Scholar]
- 60.Yang Y., Faraggi E., Zhao H., Zhou Y. Improving protein fold recognition and template-based modeling by employing probabilistic-based matching between predicted one-dimensional structural properties of query and corresponding native properties of templates. Bioinformatics. 2011;27:2076–2082. doi: 10.1093/bioinformatics/btr350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Webb B., Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Bioinformatics. 2016;54:5.6.1–5.6.37. doi: 10.1002/cpbi.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eckhardt M., Gotza B., Gerardy-Schahn R. Mutants of the CMP-sialic acid transporter causing the Lec2 phenotype. J Biol Chem. 1998;273:20189–20195. doi: 10.1074/jbc.273.32.20189. [DOI] [PubMed] [Google Scholar]
- 63.Lim S.F., Lee M.M., Zhang P., Song Z. The Golgi CMP-sialic acid transporter: a new CHO mutant provides functional insights. Glycobiology. 2008;18:851–860. doi: 10.1093/glycob/cwn080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee Y., Nishizawa T., Takemoto M., Kumazaki K., Yamashita K. Structure of the triose-phosphate/phosphate translocator reveals the basis of substrate specificity. Nat Plants. 2017;3:825–832. doi: 10.1038/s41477-017-0022-8. [DOI] [PubMed] [Google Scholar]
- 65.Paliwal K., Hanson J., Zhou Y., Yang Y. Improving protein disorder prediction by deep bidirectional long short-term memory recurrent neural networks. Bioinformatics. 2016;33:685–692. doi: 10.1093/bioinformatics/btw678. [DOI] [PubMed] [Google Scholar]
- 66.Javadpour M.M., Eilers M., Groesbeek M., Smith S.O. Helix packing in polytopic membrane proteins: role of glycine in transmembrane helix association. Biophys J. 1999;77:1609–1618. doi: 10.1016/S0006-3495(99)77009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Han M., Lin S.W., Smith S.O., Sakmar T.P. The effects of amino acid replacements of glycine 121 on transmembrane helix 3 of rhodopsin. J Biol Chem. 1996;271:32330–32336. doi: 10.1074/jbc.271.50.32330. [DOI] [PubMed] [Google Scholar]
- 68.Han M., Groesbeek M., Sakmar T.P., Smith S.O. The C9 methyl group of retinal interacts with glycine-121 in rhodopsin. Proc Natl Acad Sci U S A. 1997;94:13442–13447. doi: 10.1073/pnas.94.25.13442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maggioni A., von Itzstein M., Rodriguez Guzman I.B., Ashikov A., Stephens A.S. Characterisation of CMP-sialic acid transporter substrate recognition. Chembiochem. 2013;14:1936–1942. doi: 10.1002/cbic.201300298. [DOI] [PubMed] [Google Scholar]
- 70.Takeshima-Futagami T., Sakaguchi M., Uehara E., Aoki K., Ishida N. Amino acid residues important for CMP-sialic acid recognition by the CMP-sialic acid transporter: analysis of the substrate specificity of UDP-galactose/CMP-sialic acid transporter chimeras. Glycobiology. 2012;22:1731–1740. doi: 10.1093/glycob/cws116. [DOI] [PubMed] [Google Scholar]
- 71.Datta A.K., Chammas R., Paulson J.C. Conserved cysteines in the sialyltransferase sialylmotifs form an essential disulfide bond. J Biol Chem. 2001;276:15200–15207. doi: 10.1074/jbc.M010542200. [DOI] [PubMed] [Google Scholar]
- 72.Aoki K., Ishida N., Kawakita M. Substrate recognition by nucleotide sugar transporters-further characterization of substrate recognition regions by analyses of UDP-galactose/CMP-sialic acid transporter chimeras and biochemical analysis of the substrate specificity of parental and chimeric transporters. J Biol Chem. 2003;278:22887–22893. doi: 10.1074/jbc.M302620200. [DOI] [PubMed] [Google Scholar]
- 73.Jaeken J., Hennet T., Matthijs G., Freeze H.H. CDG nomenclature: time for a change! Biochim Biophys Acta. 2009;1792:825–826. doi: 10.1016/j.bbadis.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lefeber D.J. Protein-specific glycoprofiling for patient diagnostics. Clin Chem. 2016;62:9–11. doi: 10.1373/clinchem.2015.248518. [DOI] [PubMed] [Google Scholar]
- 75.Freeze Hudson H., Chong Jessica X., Bamshad Michael J., Ng Bobby G. Solving glycosylation disorders: fundamental approaches reveal complicated pathways. Am J Hum Genet. 2014;94:161–175. doi: 10.1016/j.ajhg.2013.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ng B.G., Freeze H.H. Perspectives on glycosylation and its congenital disorders. Trends Genet. 2018;34:466–476. doi: 10.1016/j.tig.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martinez-Duncker I., Dupre T., Piller V., Piller F., Candelier J.J. Genetic complementation reveals a novel human congenital disorder of glycosylation of type II, due to inactivation of the Golgi CMP-sialic acid transporter. Blood. 2005;105:2671–2676. doi: 10.1182/blood-2004-09-3509. [DOI] [PubMed] [Google Scholar]
- 78.Salinas-Marin R., Mollicone R., Martinez-Duncker I. A functional splice variant of the human Golgi CMP-sialic acid transporter. Glycoconj J. 2016;33:897–906. doi: 10.1007/s10719-016-9697-8. [DOI] [PubMed] [Google Scholar]
- 79.Manning M.C., Illangasekare M., Woody R.W. Circular dichroism studies of distorted α-helices, twisted β-sheets, and β-turns. Biophys Chem. 1988;31:77–86. doi: 10.1016/0301-4622(88)80011-5. [DOI] [PubMed] [Google Scholar]
- 80.Mohamed M., Ashikov A., Guillard M., Robben J.H., Schmidt S. Intellectual disability and bleeding diathesis due to deficient CMP—sialic acid transport. Neurology. 2013;81:681–687. doi: 10.1212/WNL.0b013e3182a08f53. [DOI] [PubMed] [Google Scholar]
- 81.Ng B.G., Asteggiano C.G., Kircher M., Buckingham K.J., Raymond K. Encephalopathy caused by novel mutations in the CMP-sialic acid transporter, SLC35A1. Am J Med Genet A. 2017;173:2906–2911. doi: 10.1002/ajmg.a.38412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kauskot A., Pascreau T., Adam F., Bruneel A., Reperant C. A mutation in the gene coding for the sialic acid transporter SLC35A1 is required for platelet life span but not proplatelet formation. Haematologica. 2018;103:e613. doi: 10.3324/haematol.2018.198028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nji E., Gulati A., Qureshi A.A., Coincon M., Drew D. Structural basis for the delivery of activated sialic acid into Golgi for sialyation. bioRxiv. 2019:580449. doi: 10.1038/s41594-019-0225-y. [DOI] [PubMed] [Google Scholar]
- 84.Ahuja S., Whorton M.R. Structural basis for mammalian nucleotide sugar transport. Elife. 2019;8 doi: 10.7554/eLife.45221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zeleny R., Kolarich D., Strasser R., Altmann F. Sialic acid concentrations in plants are in the range of inadvertent contamination. Planta. 2006;224:222–227. doi: 10.1007/s00425-005-0206-8. [DOI] [PubMed] [Google Scholar]
- 86.Tiralongo J. Sialobiology: structure, biosynthesis and function. sialic acid glycoconjugates in health and disease. Bentham Science Publishers; Sharjah, UAE: 2013. Introduction to sialic acid structure, occurrence, biosynthesis and function; pp. 3–32. [Google Scholar]
- 87.Münster-Kühnel A.K., Tiralongo J., Krapp S., Weinhold B., Ritz-Sedlacek V. Structure and function of vertebrate CMP–sialic acid synthetases. Glycobiology. 2004;14:43R–51R. doi: 10.1093/glycob/cwh113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material