

Abstract
The aim of this work was to predict the extent of Cytochrome P450 2D6 (CYP2D6)‐mediated drug–drug interactions (DDIs) in different CYP2D6 genotypes using physiologically‐based pharmacokinetic (PBPK) modeling. Following the development of a new duloxetine model and optimization of a paroxetine model, the effect of genetic polymorphisms on CYP2D6‐mediated intrinsic clearances of dextromethorphan, duloxetine, and paroxetine was estimated from rich pharmacokinetic profiles in activity score (AS)1 and AS2 subjects. We obtained good predictions for the dextromethorphan–duloxetine interaction (Ratio of predicted over observed area under the curve (AUC) ratio (R pred/obs) 1.38–1.43). Similarly, the effect of genotype was well predicted, with an increase of area under the curve ratio of 28% in AS2 subjects when compared with AS1 (observed, 33%). Despite an approximately twofold underprediction of the dextromethorphan–paroxetine interaction, an R pred/obs of 0.71 was obtained for the effect of genotype on the area under the curve ratio. Therefore, PBPK modeling can be successfully used to predict gene–drug–drug interactions (GDDIs). Based on these promising results, a workflow is suggested for the generic evaluation of GDDIs and DDIs that can be applied in other situations.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ A huge interindividual variability exists in the extent of drug–drug interactions (DDIs), which might be attributed to intrinsic factors affecting drug pharmacokinetics, among them genetic polymorphisms.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Can physiologically‐based pharmacokinetic (PBPK) modeling be used to predict the effect of cytochrome P450 (CYP) CYP2D6 genetic polymorphisms on the extent of DDIs?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ The study demonstrated the good performance of PBPK modeling to predict the effect of CYP2D6 genetic polymorphisms on DDIs using verified initial models and rich pharmacokinetic profiles from dedicated genetic trials to predict the effect of genotype on drug and substrate exposures.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ Using PBPK modeling to predict the combination of intrinsic (age, organ impairment, genetic polymorphism) and extrinsic factors might help to detect individuals at higher risk of clinically significant DDIs. Future integration of PBPK models in advanced computerized physician order entry systems may therefore help to increase the predictive values of DDI alerts, thereby increasing confidence in model‐informed precision medicine.
Drug–drug interactions (DDIs) are of particular clinical interest because of their high prevalence among polymorbid patients and their potential deleterious clinical implications, frequently leading to hospitalizations.1 In particular, cytochromes P450 (CYPs) explain a great amount of pharmacokinetic (PK) DDIs, many drugs being substrates and/or inhibitors or inducers of CYPs. Among them, CYP2D6 is of major clinical relevance as it metabolizes around 20% of marketed drugs and is characterized by a huge interindividual variability, which is mostly explained by genetic polymorphisms (>100 variant alleles have been described so far).2 Despite the development of DDI prediction tools integrated in computerized physician order entry systems, it still remains difficult to predict the clinical relevance of potential DDIs, as shown by the significant discrepancy between the high prevalence of potential DDIs and the low prevalence of actual DDIs leading to clinical harm.3 Indeed, a large interindividual variability exists in the extent of CYP‐mediated DDIs, which might be explained by intrinsic factors, such as age, sex, comorbidities, and genetic polymorphisms.4 As an example, the extent in the increase of area under the curve (AUC) for venlafaxine when coadministered with quinidine can vary by a 20‐fold factor in normal metabolizers of CYP2D6.5 Therefore, it seems crucial to identify other covariables responsible for this huge variability. This work proposes to use physiologically‐based PK (PBPK) modeling to identify individuals at a higher risk of DDIs because of genetic polymorphisms in metabolic enzymes. When the models with genetic data are optimized and fully validated, they can be implemented in computerized physician order entry systems to improve therapeutic management at an individual level. Such a model‐informed approach will be increasingly used to improve the efficacy and safety of a treatment for an individual patient.
After several decades of extensive investigations, pharmacogenetic testing is currently being evaluated for routine implementation.6 It was reported that more than 90% of the population carry at least one actionable genetic variant.7 These genetic variants can affect the safety and/or efficacy of some drugs. As an example, genetic polymorphisms of CYP2D6 have been shown to affect the clinical outcomes of drug treatments by tamoxifen, opioid analgesics (tramadol, codeine, oxycodone), and antidepressants and antipsychotics.8 However, not all genetic polymorphisms and medications are responsible for therapeutic accidents. Drugs with a narrow therapeutic window merit special attention, and the addition of other factors such as liver or kidney diseases may potentiate the impact and clinical relevance of DDIs. In addition, recent evidence suggests an impact of genetic polymorphism on the extent of DDIs.9, 10 The CYP2D6 activity score (AS) system developed by Gaedigk et al.11 is used to predict a CYP2D6 phenotype from genetic data. According to the Clinical Pharmacogenetic Implementation Consortium, ASs from 1 to 2 predict the extensive metabolizer phenotype.12 We recently highlighted the differential magnitude of DDIs between homozygous carriers of two fully functional alleles (AS2) and heterozygous carriers of one fully functional with one nonfunctional allele (AS1). Indeed, the AUC ratio of dextromethorphan with paroxetine was about twofold higher in the AS2 subjects when compared with the AS1 subjects.10
PBPK models divide the body into physiologically meaningful compartments in which drug‐dependent (e.g., physico‐chemical properties and in vitro data), physiology‐dependent (e.g., physiology, ethnicity, gender, and genetics), and trial‐dependent (e.g., dose regimen and concomitant medication) components are taken into account to predict the PK and pharmacodynamics of drugs in special populations. Recently, PBPK models have shown their ability to predict DDI scenarios involving intrinsic factors such as ethnicity or genetic polymorphisms in drugs undergoing multiple clearance pathways.13, 14
Therefore, the aim of the present work was to evaluate the ability of PBPK modeling to predict the extent of CYP2D6‐mediated DDIs in the presence of genetic polymorphisms using available rich PK data in different genotypes. This work addresses the issue of the variability in the extent of DDIs explained by genetic polymorphisms that help identify individuals at a higher risk of DDIs. To the best of our knowledge, this is the first study evaluating the usefulness of PBPK in predicting gene–drug–drug interactions (GDDIs) with specific inhibitors of CYP2D6—duloxetine, paroxetine, and fluoxetine—as well as the specific substrates dextromethorphan, tolterodine, and risperidone.
Materials and Methods
General
All simulations were performed using the PBPK modeling software Simcyp version 17 (Certara, Princeton, NJ). The models for dextromethorphan, dextrorphan, tolterodine, and fluoxetine were used as provided by the software and modified if required using available clinical data (see Data S1 ). A model for duloxetine was built based on available in vitro and clinical data. The risperidone model was taken from Vieira et al.13 All simulations used for model verification perform 10 trials of a given number of subjects.
Duloxetine model development and verification
An initial PBPK model for duloxetine was built using available physicochemical, in vitro, and clinical data as described in Table 1. First‐order absorption and full PBPK models using the Rodgers and Rowland equation15, 16 to predict distribution volume were used. CYP1A2‐mediated and CYP2D6‐mediated intrinsic clearances were calculated using the retrograde calculator provided by the software, assuming that the metabolic clearance of duloxetine was mediated by CYP1A2 and CYP2D6 only.17 A clinical study that evaluated the exposure change of duloxetine following steady‐state administration of paroxetine18 was used to predict the fraction of duloxetine clearance mediated by CYP2D6 (fmCYP2D6) from the mechanistic static equation,19 as follows:
| (1) |
Table 1.
Input parameters for duloxetine model
| Parameter | Value | Source |
|---|---|---|
| Molecular weight (g/mol) | 297.4 | Drugbank |
| LogP | 4.258 | Ref. 47 |
| pKa | 10.02 | Ref. 47 |
| B/P | 0.8655146 | Predicted |
| fu | 0.09 | Ref. 47 |
| ka (hour−1) | 0.168 | Ref. 17 |
| fa | 1 | Assumed based on Ref. 17 |
| Lag time (hour) | 2 | Ref. 48 |
| fuGut | 0.01508821 | Predicted |
| Q Gut (L/hour) | 17.84978 | Predicted |
| P eff,man (10−4 cm/second) | 8.752659 | Predicted with PSA and HBD |
| PSA (Å2) | 21.26 | Drugbank |
| HBD | 1 | Drugbank |
| V ss (L/kg) | 8.14 | Ref. 20 |
| CLintCYP2D6 (μL/minute/pmol CYP) | 13.33 | Retrograde model |
| CLintCYP1A2 (μL/minute/pmol CYP) | 3.21 | Retrograde model |
| CLadd = CLbile (L/hour) | 1.87 | Ref. 49 |
| KiCYP1A2 (μM) | 17.7 (fumic 0.379) | Ref. 17 |
| KiCYP2C9 (μM) | 7.1 (fumic 0.379) | Ref. 17 |
| KiCYP2D6 (μM) | 0.005 (fumic 1) | Parameter estimation |
B/P, blood plasma ratio; CLadd, additional clearance; CLbile, biliary clearance; CLint, intrinsic clearance; CYP, cytochrome P450; fa, fraction of drug absorbed following oral administration; fu, fraction of drug unbound in plasma; fuGut, fraction of drug unbound in enterocytes; fumic, unbound fraction in microsomal incubation; HBD, number of hydrogen bond donors; ka, first‐order absorption rate; Ki, competitive inhibition constant; P eff, man, human jejunum effective permeability; PSA, polar surface area; Q Gut, blood flow in gut; Ref., reference; V ss, volume of distribution at steady state; CYP, cytochrome P45; pKa, acid dissociation constant.
where k deg,CYP2D6,h is the degradation rate of CYP2D6 in the liver, [I]h is the unbound concentration of paroxetine in the portal vein, k inact is the maximal CYP2D6 inactivation rate of paroxetine, and K I is the time‐dependent inhibition (TDI) constant defined as the concentration of inhibitor reaching half k inact. Those values were taken from the paroxetine compound file in Simcyp version 17.
The model was verified with a set of PK and DDI studies that are described in Tables S1 and S2 . The contribution of CYP1A2 was verified using a DDI study with fluvoxamine in smokers.20 To that end, the population used was healthy volunteers in which the frequency of CYP1A2 ultrarapid metabolizers (UMs) was set to 100% (CYP1A2 abundance 94 pmol/mg of microsomal proteins21). Because experimental CYP2D6 competitive inhibition constant (KiCYP2D6) values (2–4 μM)17, 22 resulted in a null DDI prediction, an optimized competitive inhibition constant (Ki) value of 0.005 μM was input using sensitivity analysis with available clinical data with desipramine as a victim drug18 and verified with three independent DDI trials with desipramine, tolterodine, and metoprolol23, 24, 25 (Table S2 ).
Optimization of paroxetine model
The TDI parameters of K I and k inact in the paroxetine library compound file in version 17 were modified using optimized data taking into account the hepatocyte (pH 7.4)‐to‐plasma (pH 7.0) pH gradient.26 Briefly, the K I value was decreased from 0.315 to 0.067 μM, and k inact was increased from 10.2 to 11.55 hour−1.The model was verified for DDI predictions using a set of published DDI clinical trials (Table S3 ).
Top‐down estimation of CYP2D6‐mediated intrinsic clearances of dextromethorphan, duloxetine, paroxetine, and fluoxetine
Dextromethorphan and dextrorphan PK profiles in healthy volunteers with different CYP2D6 genotypes with ASs ranging from 1 to 3 were used to estimate the CYP2D6‐mediated intrinsic clearance (CLintCYP2D6) of dextromethorphan O‐demethylation.27 In this study, one subject had an AS of 0.5, 14 subjects were carriers of one fully functional with one nonfunctional allele (AS1), 9 subjects were carriers of two fully functional alleles (AS2), and one subject was a carrier of two functional alleles with one duplication (AS3). The CLintCYP2D6 value of dextromethorphan in Simcyp version 17 is 7.275 μL/minute per mg of microsomal proteins. The parameter was refined in each of the genotypes mentioned previously to obtain a best fit with the observed concentrations using 100 iterations around 10‐fold the initial value mentioned previously, a maximum likelihood objective function, and expectation maximization as the minimization method. As clinical data were obtained in‐house,27 covariates such as age, sex, weight, height, and CYP2D6 genotype were taken into account for the parameter estimation.28
Similarly, the genotype‐dependent CYP2D6‐mediated intrinsic clearance of duloxetine and paroxetine were estimated from available clinical data in AS1 and AS2 healthy volunteers10 using the initial models’ CLintCYP2D6 values as starting values for the parameter estimation. A total of 100 iterations around 10‐fold the initial value were interrogated, and a maximum likelihood objective function and expectation maximization as minimization method were used. As clinical data were obtained in‐house,10 covariates such as age, sex, weight, height, and CYP2D6 genotype were taken into account for the parameter estimation.28
The genotype‐dependent CYP2D6‐mediated intrinsic clearance of fluoxetine and tolterodine were similarly estimated from available clinical data in AS1 and AS2 patients29 using the initial library compound models’ CLintCYP2D6 values as initial values for parameter estimation, and 100 iterations around 10‐fold the initial value were interrogated, and a maximum likelihood objective function and expectation maximization as minimization method were used.
Simulations of GDDIs
DDIs studies were simulated in different genotypes using genotype‐dependent CYP2D6‐mediated clearances of substrates and inhibitors, which were estimated as described previously. Predicted AUC ratio (AUCR) were compared with observed values. Simulated trial characteristics were the same as the observed trials, as described in Table S4 . All simulations were run using the Healthy Volunteer population in Simcyp version 17, except for the tolterodine–fluoxetine trial for which the North European white population was used. For extensive metabolizers, including the AS1 and AS2 genotypes, the CYP2D6 phenotype distribution was set to 100% extensive metabolizers (CYP2D6 abundance in liver 8 pmol/mg of microsomal proteins). For poor metabolizers (PMs) and UMs, the CYP2D6 phenotype distribution was set to 100% PMs (i.e., absence of CYP2D6 expression in liver) and 100% UMs (CYP2D6 abundance in liver 16 pmol/mg of microsomal proteins), respectively.
Results
Duloxetine model development and verification
The duloxetine model was developed and validated for single‐dose and steady‐state exposure using a range of clinical trials in various populations. Based on the observed and simulated PK profiles (as presented in Figure 1), the prediction was judged satisfactory with all observed concentrations between the 5% and 95% percentiles of predicted concentrations. The contribution of CYP2D6 in the clearance of duloxetine (fmCYP2D6) was estimated to be 39% using Eq. (1) (see the Methods section). Therefore, the contribution of CYP1A2 was estimated to be 61%, which was confirmed by a predicted mean change of duloxetine exposure in the presence of fluvoxamine in male smokers of 5.49 when compared with a mean observed AUCR of 5.6.20 Duloxetine is a moderate inhibitor of CYP2D6.18 Using an optimized Ki value estimated from in vivo DDI data,18 good DDI predictions were obtained with tolterodine, metoprolol, and desipramine, as described in Table 2.
Figure 1.
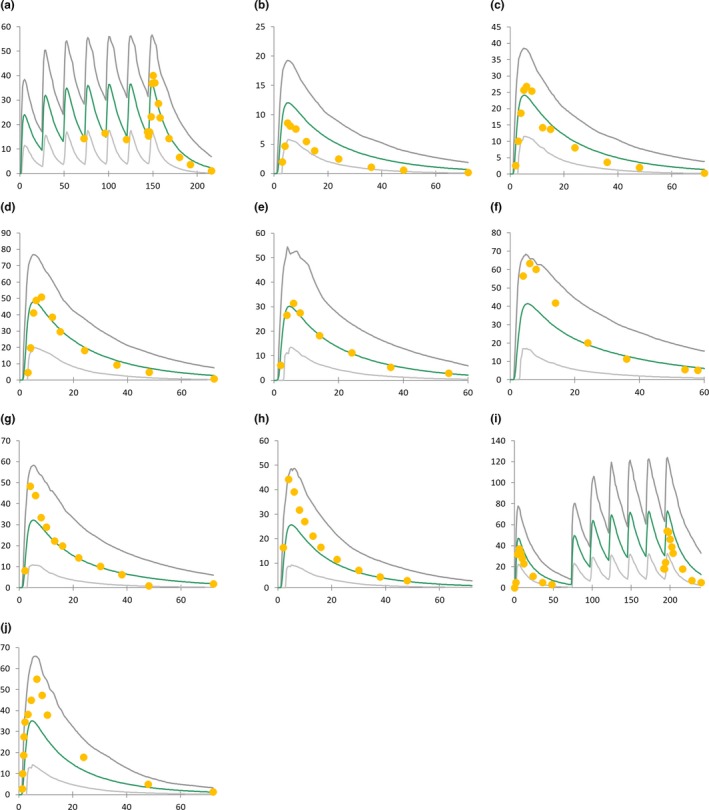
Simulated vs. observed duloxetine pharmacokinetic profiles. Green lines represent mean simulated pharmacokinetic profile, and gray lines represent the 5% and 95% percentiles of model‐predicted pharmacokinetic profiles. The x‐axis represents time after drug intake (hour), and the y‐axis represents plasma concentration (ng/mL). Observed data were taken from published trials: (a–d)42 (e, f)43 (g, h)44 (i)45 and (j)46
Table 2.
Simulated and observed drug–drug interactions with duloxetine
| Substrate | Substrate dose | Duloxetine dose | Simulated AUCRa | Observed AUCR | R pred/obs | Reference study |
|---|---|---|---|---|---|---|
| Tolterodine | 2 mg/12 hours, 9 doses | 40 mg/12 hours, 9 doses | 2.85 (2.59–2.94) | 1.71 (1.31–2.23)a | 1.67 | Ref. 16 |
| Desipramine | 50 mg SD at day 6 | 30 mg/12 hours, 20 doses | 1.75 (1.62–1.78) | 2.22 (1.95–2.51)b | 0.79 | Ref. 15 |
| Metoprolol | 100 mg SD at day 17 | 30 mg day 1 then 60 mg days 2–18 | 1.75 (1.66–1.82) | 2.80 ± 0.31c | 0.63 | Ref. 19 |
AUCR, ratio of the substrate area under the concentration‐time curve in the presence and absence of the inhibitor; R pred/obs, ratio of model‐predicted mean exposure change of substrate to observed value; SD, single dose.
Geometric mean with 95% confidence interval.
Geometric mean with 90% confidence interval.
Arithmetic mean ± standard deviation.
Paroxetine model optimization
The performance of the optimized paroxetine model was verified in terms of DDI predictions and steady‐state exposure (see Table S5 and Figure S1 ). The observed steady‐state PK profile was between 5% and 95% percentiles of predicted concentrations. The predicted change of exposure of CYP2D6 substrates (metoprolol, desipramine, imipramine) following paroxetine dosing were all within a twofold boundary of the observed AUCR and 62.5% restricted limits 75% to 125%.
Incorporation of genetic effect in CYP2D6‐mediated clearance of substrates and inhibitors
CYP2D6‐mediated clearance of victim and inhibitory compounds was modified as a function of genotype following parameter estimation using available clinical data, as described in Table 3. Using rich data from the in‐house clinical studies mentioned in the Methods section, the CYP2D6‐mediated clearance of dextromethorphan, duloxetine, and paroxetine was 2.36‐fold, 1.2‐fold, and 1.93‐fold higher, respectively, in the AS2 subjects when compared with the AS1 subjects.
Table 3.
Optimized values of CYP2D6‐mediated clearance (CLint or Vmax) of model substrates and inhibitors as a function of CYP2D6 genotype
| Compound | Pathway | Initial value | Genotype effect |
|---|---|---|---|
| Dextromethorphan | O‐demethylation | CLint = 250.85 μL/minute/mg prot |
AS1: −39% AS2: +44% AS3: +470%a |
| Tolterodine | 5‐hydroxylation | Vmax = 317 pmol/minute/mg prot |
AS0: −100%a
AS1: −74% AS2: +325% |
| Risperidone | 9‐hydroxylation | CLint = 7.55 μL/minute/pmol CYP2D6 |
PM: −100%a
EM: unchanged |
| Duloxetine | All | CLint = 13.33 μL/minute/CYP2D6 |
AS1: −31% AS2: −13% |
| Paroxetine | All | Vmax = 7.28 pmol/minute/pmol CYP2D6 |
AS1: −18% AS2: +58% |
| Fluoxetine | N‐demethylation | CLint = 52.65 μL/minute/mg prot |
AS0: −100%a
AS1: +293% AS2: +624% |
AS0, activity score 0; AS1, activity score 1; AS2, activity score 2; AS3, activity score 3; CLint, intrinsic clearance; CYP2D6, cytochrome P450 2D6; EM, extensive metabolizer; PM, poor metabolizer; prot, protein; Vmax, maximal reaction velocity.
The CLint value was increased 2.9‐fold, and CYP2D6 liver abundance increased twofold.
The CLint value was unchanged, and CYP2D6 liver abundance was defined as zero.
Simulations of GDDIs
Good predictions for GDDIs were obtained for the dextromethorphan–duloxetine interaction, with a slight overprediction (~40%) of change of substrate exposure when compared with the observed data (Table 4). Despite paroxetine model optimization with regard to TDI parameters, the DDI with dextromethorphan was still underpredicted. However, the effect of genotype was satisfactorily predicted, with R pred/obs values for the ratio of AUC increase in the AS2 subjects over the AUC increase in the AS1 subjects of 0.96 for the duloxetine‐dextromethorphan interaction and 0.71 for the paroxetine–dextromethorphan interaction.
Table 4.
Comparison of predicted and observed gene–drug–drug interaction trials
| Substrate | Inhibitor | CYP2D6 genotype | n | AUCR | AUCRx/AUCRy | Reference study | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Predicted | Observed | R pred/obs | Compared genotypes | Predicted | Observed | R pred/obs | |||||
| Dextromethorphan | Duloxetine | AS1 | 17 | 2.6 (2.5–2.7) | 1.8 (1.5–2.1) | 1.43 | x = AS2 | 1.28 | 1.33 | 0.96 | Ref. 10 |
| AS2 | 16 | 3.3 (3.2–3.4) | 2.4 (1.8–3.2) | 1.38 | y = AS1 | ||||||
| Paroxetine | AS1 | 17 | 4.7 (4.4–5.0) | 8.5 (6.7–10.8) | 0.56 | x = AS2 | 1.22 | 1.72 | 0.71 | ||
| AS2 | 16 | 5.8 (5.3–6.3) | 14.6 (10.0–21.4) | 0.40 | y = AS1 | ||||||
| Tolterodine | Fluoxetine | AS0 | 2 | 1.3 (1.3–1.40) | 1.25 | 1.06 | x = AS1 | 2.62 | 4.38 | 0.60 | Ref. 29 |
| AS1 | 4 | 3.4 (3.1–3.8) | 5.47 | 0.62 | y = AS0 | ||||||
| x = AS2 | 2.38 | 3.18 | 0.75 | ||||||||
| AS2 | 3 | 8.1 (7.4–8.9) | 17.4 | 0.47 | y = AS1 | ||||||
| Risperidone | Fluoxetine | PM | 2 | 1.4 (1.3–1.5) | 1.3 | 1.06 | x = EM | 2.12 | 3.22 | 0.66 | Ref. 50 |
| EM | 7 | 2.9 (2.7–3.1) | 4.2 | 0.70 | y = PM | ||||||
AS0, activity score 0; AS1, activity score 1; AS2, activity score 2; AUCR, ratio of the substrate area under the concentration‐time curve in the presence and absence of the inhibitor; AUCRx/AUCRy, ratio of AUCR in genotype x versus genotype y; CYP2D6, cytochrome P450 2D6; EM, extensive metabolizer; PM, poor metabolizer; R pred/obs, ratio of model‐predicted mean exposure change of substrate to observed value.
A slight underprediction of fluoxetine‐mediated DDIs was obtained with the CYP2D6 substrates tolterodine and risperidone in non‐PMs, whereas good prediction was obtained in PM subjects. For fluoxetine‐mediated DDIs, the predicted impact of genetic polymorphisms was in the twofold range of observed values with systematic underprediction (R pred/obs of 0.60 and 0.75 for AS1/AS0 and AS2/AS1, respectively).
Discussion
In this study, we used available rich PK data with different genotypes to estimate the CYP2D6‐mediated clearance of dextromethorphan, tolterodine, duloxetine, and paroxetine from beforehand validated PBPK models and subsequently performed genotype‐dependent DDI simulations to compare with clinical DDI data.
First, a PBPK model for the moderate CYP2D6 inhibitor duloxetine was developed and validated. Good predictions for single‐dose and steady‐state exposures in different populations were obtained. The optimized model predicted well CYP1A2 contribution in hepatic clearance, as verified with a DDI trial with the potent inhibitor fluvoxamine in male smokers.20 Duloxetine has been shown to increase the exposure of the CYP2D6 substrates desipramine and tolterodine up to 2.9‐fold with a 60 mg twice‐daily regimen,18, 23, 24 which could not be predicted by the bottom‐up approach in Simcyp using experimental Ki values in the range of 2–4 μM.17, 22 Instead, this value was optimized from in vivo DDI data with desipramine18 and verified with a set of independent DDI data with desipramine, metoprolol, and tolterodine.23, 24, 25 To note, the approach of using in vivo optimized rather than in vitro obtained Ki values for the modeling of DDIs was similarly performed previously with fluvoxamine and ciprofloxacine, two CYP1A2 inhibitors for which experimental Ki values resulted in the underprediction of DDIs.30, 31 The reasons for the underprediction of the DDI with duloxetine from in vitro data remains unclear, and we present herein some possible explanations. Duloxetine has been previously described as a competitive inhibitor of CYP2D6 lacking of TDI properties,32, 33 suggesting that duloxetine metabolites are not CYP2D6 inhibitors. Therefore, the underprediction of DDIs cannot be attributed to any TDI mechanism not taken into account in the model. Possible duloxetine accumulation into hepatocytes through active transport might explain the underprediction of DDI, which was not investigated in the present study. Bupropion represents another case of underprediction of CYP2D6‐mediated DDIs. Indeed, bupropion is a strong CYP2D6 inhibitor in vivo, which increases the exposure of desipramine by around fivefold.34 However, in vitro in vivo extrapolation fails to predict such DDIs from experimental data. Recently, Sager et al.35 showed that concomitant competitive inhibition and CYP2D6 downregulation by bupropion and its metabolites could explain the strong CYP2D6 inhibition observed in vivo and that taking into account the downregulation of CYP2D6 in the prediction of the DDI could improve the in vitro in vivo extrapolation of bupropion‐perpetrator DDIs. CYP2D6 has long been considered a noninducible enzyme. Therefore, CYP2D6 induction studies are not recommended by regulatory agencies and not systematically assessed in current drug development. However, recent studies have shown upregulation of CYP2D6 expression during pregnancy, which seems to be related to an enhanced HNF‐4α transactivation of the CYP2D6 promoter induced by the decreased expression of small heterodimer partner, a corepressor of various transcription factors, and increased expression of the Krüppel‐like factor 9, both occurring during pregnancy.36, 37, 38 As transcriptional regulation of CYP2D6 is not systematically investigated during drug development, we found no data regarding any regulation of CYP2D6 by duloxetine. Further investigation of the CYP2D6 downregulation by duloxetine may represent an interesting clue to explain the underprediction of DDIs between duloxetine and CYP2D6 substrates.
In preliminary simulations, the existing paroxetine PBPK model from Simcyp library version 17 resulted in the underprediction of paroxetine exposure with the two‐dose regimen used in the dextromethorphan–paroxetine DDI verification study10 and therefore in the underprediction of DDIs (data not shown), which corroborates with the observed tendency toward underprediction of DDIs between paroxetine and CYP2D6 substrates that was described previously by Marsousi et al.39 Paroxetine is both a substrate and a strong time‐dependent inhibitor of CYP2D6. Therefore, the TDI parameters K I and k inact were modified to improve the predictions of both paroxetine exposure and the DDIs. Using previously optimized TDI parameters taking into account the hepatocyte‐to‐plasma pH gradient (KI was reduced from 0.315 to 0.067 μM, and k inact was increased from 10.2 to 11.55 hour−1),26 we obtained better DDI predictions than previously reported with the Simcyp library model (Table S5 ). Moreover, the multiple dosing exposure was well recovered as shown in Figure S1 , improving confidence in the paroxetine model for the further evaluation of GDDIs. We decided to optimize the paroxetine model by modifying the TDI parameters Ki and k inact because DDI underestimation was linked with an underprediction of paroxetine exposure as well. In the model optimization part of the study, modifying the TDI parameters permitted better recovery of both the paroxetine exposure and magnitude of DDIs. Unfortunately, when simulating the in‐house data in which only two close doses of paroxetine were given, an underprediction of the DDI was observed. It is possible that the optimized paroxetine model was only working at steady state and did not capture well the paroxetine single dose. Another parameter difficult to accurately obtain is the intrahepatocyte concentration of the inhibitor that could accumulate through active transport.
At the beginning of the present study, we considered introducing genetic effect in the clearance of CYP2D6 substrates by either playing on CYP2D6 liver abundance or on CYP2D6 enzyme kinetics. In PMs, the CYP2D6 protein is absent26 and therefore the CYP2D6 abundance of PM subjects is set to zero. However, immunologic and proteomic studies failed to demonstrate a difference in the amounts of CYP2D6 between activity scores 0.5 to 2,22, 41 which seems to be mostly related to the high interindividual variability, even within a same genotype. Therefore, there was no evidence supporting a change of functional CYP2D6 abundance for non‐PM genotypes, and we rather decided to incorporate the effect of genetic polymorphisms on CYP2D6 enzyme kinetics data (intrinsic clearance (CLint) or maximal reaction velocity (Vmax)). CLint represents the ratio of Vmax over the affinity constant Km. If the affinity constant is not affected by genetic polymorphism, the effect of modifying Vmax or CLint is the same. However, it is true that using Km and Vmax provides additional information on saturation of the enzyme. The decision of playing with either Vmax of CLint was based on which parameter was informed in the initial model from the Simcyp database.
We used the full 0–8‐hour PK profiles of dextromethorphan and its metabolite dextrorphan obtained from a previous clinical trial in our group27 to estimate the CYP2D6‐mediated O‐demethylation CLint in AS1, AS2, and AS3 subjects using individual fitting. Similarly, we used the full PK profiles of duloxetine and paroxetine, both being CYP2D6 substrates as well, from another clinical trial in AS1 and AS2 subjects10 to estimate their CYP2D6‐mediated clearance. This approach allowed a good prediction of the genetic effect on the extent of DDI between dextromethorphan and paroxetine or duloxetine, although better prediction was obtained with duloxetine when compared with paroxetine. In contrast to duloxetine, whose fmCYP2D6 is around 40%, paroxetine is importantly metabolized by CYP2D6 (fmCYP2D6 > 90%). Therefore, CYP2D6 genetic polymorphisms affects the enzyme contribution in the clearance of both the victim and the inhibitor drugs, therefore adding uncertainty in both inhibitor exposure and victim drug clearance, which might explain the lower predictive performance with paroxetine.
Similar to paroxetine, we observed an underprediction of fluoxetine‐perpetrated DDIs in non‐PM subjects, which might be attributable to the CYP2D6‐mediated clearance of the inhibitor. It should be noted that fluoxetine CYP2D6‐mediated clearance in AS1 and AS2 genotypes was estimated using single fluoxetine through concentration at steady state in a limited number of individuals (only four AS1 and three AS2). It is also interesting to note that fluoxetine is a weak CYP3A4 inhibitor, which explains why the AUCR is higher than one in CYP2D6 PMs. Indeed, the contribution of CYP3A4 in tolterodine and risperidone clearance is increased in subjects lacking CYP2D6, thus resulting in significant DDIs in the presence of even weak CYP3A4 inhibitors mainly cleared by CYP2D6. The authors refer the reader to Vieira et al.13 for an insightful discussion about the impact of both comedication and genetic polymorphism on the exposure of drugs undergoing multiple clearance pathways.
Overall, both predictions and observations converged toward the following rule: the higher the activity of CYP2D6, the higher will be the extent of interaction. Indeed, the extent of DDIs is highly dependent on the contribution of the inhibited enzyme, and the higher the activity of CYP2D6 the higher is the contribution of the enzyme in the clearance of the substrate. However, it must be emphasized that numerous CYP2D6 inhibitors, including paroxetine and fluoxetine, are mainly metabolized by CYP2D6 and therefore are also more rapidly eliminated when CYP2D6 activity increases, which has an impact on the extent of DDIs. As illustrated in Figure 2, the extent of a DDI (AUCR) increases as long as the fmCYP2D6 of the substrate increases and might then diminish when the latter saturates, in which case the concentration of the inhibitor becomes a major determinant.
Figure 2.
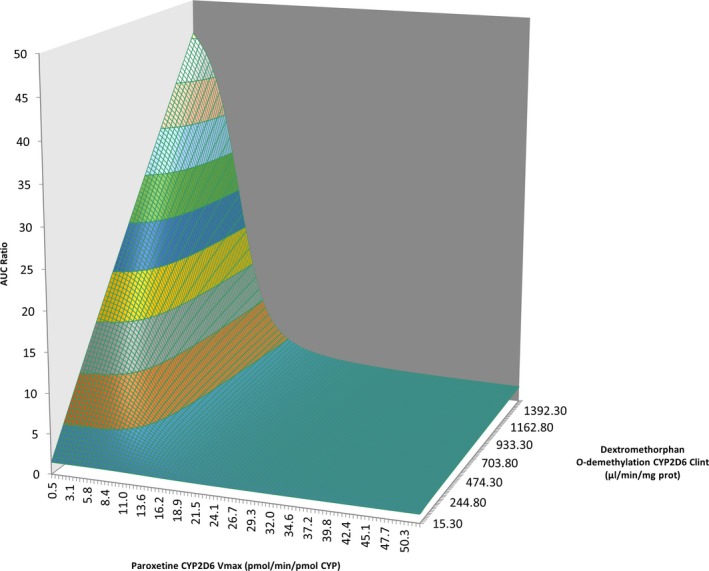
Sensitivity analysis of CYP2D6‐mediated clearance of both victim and inhibitors drugs on the extent of drug–drug interactions. AUC, area under the curve; CLint, intrinsic clearance; CYP, cytochrome P450; Vmax, maximal velocity.
Perspectives
PBPK modeling represents a promising approach to predict the combined effect of multiple intrinsic and/or extrinsic factors affecting drug PK. The extent and clinical impact of DDIs are of huge interindividual variability and poor predictability. Integrating PBPK modeling in advanced computerized physician order entry systems to predict the extent of DDIs as a function of different intrinsic factors, such as age, concomitant medications, organ impairment, and genetic polymorphisms, might help targeting individuals at high risk of clinically significant DDIs to which treatment optimization might be proposed. Previously, the PBPK approach was proposed by Patel et al.14 to predict the effect of both race and genotype on drug exposure. Similarly, Vieira et al.13 obtained good predictions with PBPK modeling for GDDIs involving drugs with multiple clearance pathways. In this article, we obtained good predictions for the dextromethorphan–duloxetine and the dextromethorphan–paroxetine DDIs in AS1 and AS2 subjects using available rich data from a dedicated GDDI clinical trial in those subjects. However, further optimization of initial PBPK models for the TDI inhibitors paroxetine and fluoxetine appears necessary based on the underestimation of DDIs observed in our study. Indeed, it is only if initial PBPK models are optimized and verified that they can further be used to predict more complex DDIs. Based on these results, we suggest a workflow as depicted in Figure 3. First, PBPK models are developed using available in vitro (bottom‐up approach) and clinical data if available (top‐down approach) and validated with a range of clinical data, including DDI studies. Available rich PK data for substrates and inhibitors from genetic studies performed during clinical development serve as a basis for the top‐down estimation of CYP2D6‐mediated clearance in different genotypes to incorporate the effect of genotype in their clearance. In vitro kinetic experiments in genotyped human liver microsomes might also represent an alternative approach, provided sufficient sample size to account for the high interindividual variability. Finally, GDDIs can be simulated to detect individuals at risk of clinically significant DDIs.
Figure 3.
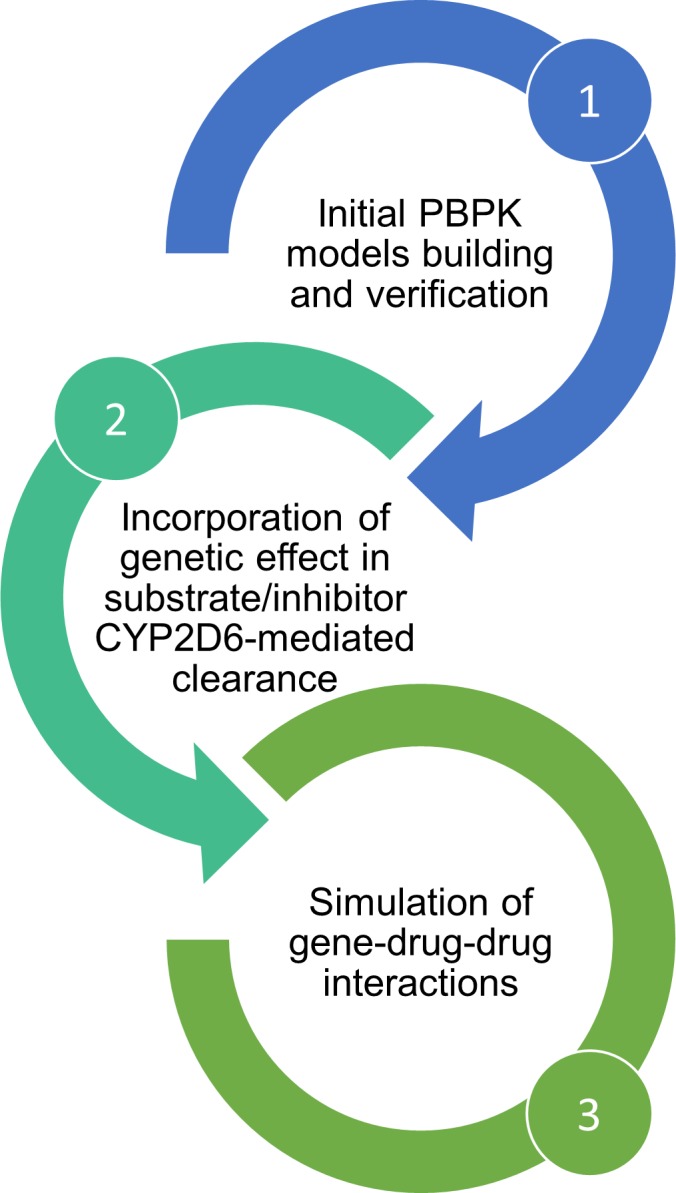
Study workflow. In a first step, physiologically‐based pharmacokinetic (PBPK) models for substrates and inhibitors were built, optimized, or used unchanged from verified library compounds in Simcyp version 17. In a second step, the Cytochrome P450 2D6 (CYP2D6) genotype‐dependent CYP2D6‐mediated clearance of substrate and inhibitors were estimated from existing in vivo data in humans. Then the third step consisted in the simulations of genotype‐dependent drug–drug interactions (DDIs) to compare with existing DDI trials.
In summary, our work demonstrates that PBPK modeling might represent a promising approach to predict the effect of genetic polymorphisms on the extent of DDIs, provided good predictive performance of the initial models and proper evaluation of the genetic effect in the exposure of substrates and inhibitors.
Funding
This work was supported by a Swiss National Science Foundation grant (SNF 320030_182361).
Conflicts of Interest
The authors declared no competing interests for this work.
Author Contributions
F.S., J.D., and Y.D. wrote the manuscript and designed the research. F.S. performed the research. F.S., J.D., and Y.D. analyzed the data.
Supporting information
Figure S1. Observed vs. simulated paroxetine steady‐state exposure. A total of 10 trials of nine healthy male CYP2D6 extensive metabolizers (age range 20–30 years) were simulated and compared with published data.
Table S1. Characteristics of simulated trials used for duloxetine PK model verification.
Table S2. Characteristics of simulated trials used for duloxetine DDI model verification.
Table S3. Characteristics of simulated trials used for modified paroxetine DDI model verification.
Table S4. Characteristics of simulated GDDI trials.
Table S5. Characteristics of simulated trials used for modified paroxetine DDI model verification.
Data S2. Supplementary material references.
Data S1. Data set: input model parameters.
Acknowledgments
The authors would like to thank Simcyp Limited, a Certara Company for the academic license for the Simcyp Population‐based Simulator and providing user support. In particular, the authors would like to thank Mian Zhang, Amaka Ezuruike, and Kim Crewe for their availability to discuss the project.
References
- 1. Marengoni, A. et al Understanding adverse drug reactions in older adults through drug‐drug interactions. Eur. J. Intern. Med. 25, 843–846 (2014). [DOI] [PubMed] [Google Scholar]
- 2. Gaedigk, A. et al The Pharmacogene Variation (PharmVar) Consortium: incorporation of the human cytochrome P450 (CYP) allele nomenclature database. Clin. Pharmacol. Ther. 103, 399–401 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zheng, W.Y. et al Drug‐drug interactions and their harmful effects in hospitalised patients: a systematic review and meta‐analysis. Eur. J. Clin. Pharmacol. 74, 15–27 (2018). [DOI] [PubMed] [Google Scholar]
- 4. Storelli, F. , Samer, C. , Reny, J.L. , Desmeules, J. & Daali, Y. Complex drug‐drug‐gene‐disease interactions involving cytochromes P450: systematic review of published case reports and clinical perspectives. Clin. Pharmacokinet. 57, 1267–1293 (2018). [DOI] [PubMed] [Google Scholar]
- 5. Lin, J.H. & Lu, A.Y. Interindividual variability in inhibition and induction of cytochrome P450 enzymes. Annu. Rev. Pharmacol. Toxicol. 41, 535–567 (2001). [DOI] [PubMed] [Google Scholar]
- 6. van der Wouden, C.H. et al Implementing pharmacogenomics in Europe: design and implementation strategy of the ubiquitous Pharmacogenomics consortium. Clin. Pharmacol. Ther. 101, 341–358 (2017). [DOI] [PubMed] [Google Scholar]
- 7. Van Driest, S.L. et al Clinically actionable genotypes among 10,000 patients with preemptive pharmacogenomic testing. Clin. Pharmacol. Ther. 95, 423–431 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Samer, C.F. , Lorenzini, K.I. , Rollason, V. , Daali, Y. & Desmeules, J.A. Applications of CYP450 testing in the clinical setting. Mol. Diagn. Ther. 17, 165–184 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bahar, M.A. , Setiawan, D. , Hak, E. & Wilffert, B. Pharmacogenetics of drug‐drug interaction and drug‐drug‐gene interaction: a systematic review on CYP2C9, CYP2C19 and CYP2D6. Pharmacogenomics 18, 701–739 (2017). [DOI] [PubMed] [Google Scholar]
- 10. Storelli, F. et al Impact of CYP2D6 functional allelic variations on phenoconversion and drug‐drug interactions. Clin. Pharmacol. Ther. 104, 148–157 (2018). [DOI] [PubMed] [Google Scholar]
- 11. Gaedigk, A. et al The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin. Pharmacol. Ther. 83, 234–242 (2008). [DOI] [PubMed] [Google Scholar]
- 12. Hicks, J.K. et al Clinical pharmacogenetics implementation consortium guideline (CPIC) for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants: 2016 update. Clin. Pharmacol. Ther. 102, 37–44 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vieira, M.D. et al PBPK model describes the effects of comedication and genetic polymorphism on systemic exposure of drugs that undergo multiple clearance pathways. Clin. Pharmacol. Ther. 95, 550–557 (2014). [DOI] [PubMed] [Google Scholar]
- 14. Patel, C. , Rathi, C. & Venkatakrishnan, K. Should race‐genotype interactions be considered in the global development of CYP2C19 substrates? A proposed framework using physiologically based pharmacokinetic modeling. J. Clin. Pharmacol. 57, 417–421 (2017). [DOI] [PubMed] [Google Scholar]
- 15. Rodgers, T. , Leahy, D. & Rowland, M. Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate‐to‐strong bases. J. Pharm. Sci. 94, 1259–1276 (2005). [DOI] [PubMed] [Google Scholar]
- 16. Rodgers, T. & Rowland, M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 95, 1238–1257 (2006). [DOI] [PubMed] [Google Scholar]
- 17. Knadler, M.P. , Lobo, E. , Chappell, J. & Bergstrom, R. Duloxetine: clinical pharmacokinetics and drug interactions. Clin. Pharmacokinet. 50, 281–294 (2011). [DOI] [PubMed] [Google Scholar]
- 18. Skinner, M.H. et al Duloxetine is both an inhibitor and a substrate of cytochrome P4502D6 in healthy volunteers. Clin. Pharmacol. Ther. 73, 170–177 (2003). [DOI] [PubMed] [Google Scholar]
- 19. US Food and Drug Administration . In Vitro Metabolism and Transporter – Mediated Drug–Drug Interaction Studies: Guidance for Industry. (US Food and Drug Administration, Silver Spring, MD, 2017) [Google Scholar]
- 20. Lobo, E.D. et al In vitro and in vivo evaluations of cytochrome P450 1A2 interactions with duloxetine. Clin. Pharmacokinet. 47, 191–202 (2008). [DOI] [PubMed] [Google Scholar]
- 21. Plowchalk, D.R. & Rowland Yeo, K. Prediction of drug clearance in a smoking population: modeling the impact of variable cigarette consumption on the induction of CYP1A2. Eur. J. Clin. Pharmacol. 68, 951–960 (2012). [DOI] [PubMed] [Google Scholar]
- 22. Storelli, F. , Desmeules, J. & Daali, Y. Genotype‐sensitive reversible and time‐dependent CYP2D6 inhibition in human liver microsomes. Basic Clin. Pharmacol. Toxicol. 124, 170–180 (2019). [DOI] [PubMed] [Google Scholar]
- 23. Hua, T.C. et al Effect of duloxetine on tolterodine pharmacokinetics in healthy volunteers. Br. J. Clin. Pharmacol. 57, 652–656 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Patroneva, A. et al An assessment of drug‐drug interactions: the effect of desvenlafaxine and duloxetine on the pharmacokinetics of the CYP2D6 probe desipramine in healthy subjects. Drug Metab. Dispos. 36, 2484–2491 (2008). [DOI] [PubMed] [Google Scholar]
- 25. Preskorn, S.H. et al Comparison of duloxetine, escitalopram, and sertraline effects on cytochrome P450 2D6 function in healthy volunteers. J. Clin. Psychopharmacol. 27, 28–34 (2007). [DOI] [PubMed] [Google Scholar]
- 26. Ming, N. , Posada, M.M. , Hall, S.D. & et Dickinson, G.G. Physiologically Based Pharmacokinetic Model Verification and Improvement for CYP2D6‐mediated Drug‐drug Interaction Prediction. Poster Session presented at the 2017 Experimental Biology Congress, Chicago, US. (2017. Apr).
- 27. Bosilkovska, M. et al Evaluation of mutual drug‐drug interaction within Geneva cocktail for cytochrome P450 phenotyping using innovative dried blood sampling method. Basic Clin. Pharmacol. Toxicol. 119, 284–290 (2016). [DOI] [PubMed] [Google Scholar]
- 28. Jamei, M. et al The Simcyp population based simulator: architecture, implementation, and quality assurance. In Silico Pharmacol. 1, 1–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brynne, N. , Svanstrom, C. , Aberg‐Wistedt, A. , Hallen, B. & Bertilsson, L. Fluoxetine inhibits the metabolism of tolterodine‐pharmacokinetic implications and proposed clinical relevance. Br. J. Clin. Pharmacol. 48, 553–563 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Karjalainen, M.J. , Neuvonen, P.J. & Backman, J.T. In vitro inhibition of CYP1A2 by model inhibitors, anti‐inflammatory analgesics and female sex steroids: predictability of in vivo interactions. Basic Clin. Pharmacol. Toxicol. 103, 157–165 (2008). [DOI] [PubMed] [Google Scholar]
- 31. Yao, C. et al Fluvoxamine‐theophylline interaction: gap between in vitro and in vivo inhibition constants toward cytochrome P4501A2. Clin. Pharmacol. Ther. 70, 415–424 (2001). [DOI] [PubMed] [Google Scholar]
- 32. Chan, C.Y. , New, L.S. , Ho, H.K. & Chan, E.C. Reversible time‐dependent inhibition of cytochrome P450 enzymes by duloxetine and inertness of its thiophene ring towards bioactivation. Toxicol. Lett. 206, 314–324 (2011). [DOI] [PubMed] [Google Scholar]
- 33. Paris, B.L. et al In vitro inhibition and induction of human liver cytochrome p450 enzymes by milnacipran. Drug Metab. Dispos. 37, 2045–2054 (2009). [DOI] [PubMed] [Google Scholar]
- 34. Jefferson, J.W. , Pradko, J.F. & Muir, K.T. Bupropion for major depressive disorder: pharmacokinetic and formulation considerations. Clin. Ther. 27, 1685–1695 (2005). [DOI] [PubMed] [Google Scholar]
- 35. Sager, J.E. et al In vitro to in vivo extrapolation of the complex drug‐drug interaction of bupropion and its metabolites with CYP2D6; simultaneous reversible inhibition and CYP2D6 downregulation. Biochem. Pharmacol. 123, 85–96 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pan, X. , Ning, M. & Jeong, H. Transcriptional regulation of CYP2D6 expression. Drug Metab. Dispos. 45, 42–48 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. He, Z.X. , Chen, X.W. , Zhou, Z.W. & Zhou, S.F. Impact of physiological, pathological and environmental factors on the expression and activity of human cytochrome P450 2D6 and implications in precision medicine. Drug Metab. Rev. 47, 470–519 (2015). [DOI] [PubMed] [Google Scholar]
- 38. Koh, K.H. et al Altered expression of small heterodimer partner governs cytochrome P450 (CYP) 2D6 induction during pregnancy in CYP2D6‐humanized mice. J. Biol. Chem. 289, 3105–3113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marsousi, N. , Desmeules, J.A. , Rudaz, S. & Daali, Y. Prediction of drug‐drug interactions using physiologically‐based pharmacokinetic models of CYP450 modulators included in Simcyp software. Biopharm. Drug Dispos. 39, 3–17 (2018). [DOI] [PubMed] [Google Scholar]
- 40. Kagimoto, M. , Heim, M. , Kagimoto, K. , Zeugin, T. & Meyer, U.A. Multiple mutations of the human cytochrome P450IID6 gene (CYP2D6) in poor metabolizers of debrisoquine. Study of the functional significance of individual mutations by expression of chimeric genes. J. Biol. Chem. 265, 17209–17214 (1990). [PubMed] [Google Scholar]
- 41. Gaedigk, A. , Dinh, J.C. , Jeong, H. , Prasad, B. & Leeder, J.S. Ten years’ experience with the CYP2D6 activity score: a perspective on future investigations to improve clinical predictions for precision therapeutics. J. Pers. Med. 8, 1–15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li, H. , Li, T. , Li, Y. & Shen, Y. Pharmacokinetics and safety of duloxetine enteric‐coated tablets in Chinese healthy volunteers: a randomized, open‐label, single‐ and multiple‐dose study. Clin. Psychopharmacol. Neurosci. 11, 28–33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lobo, E.D. et al Effects of varying degrees of renal impairment on the pharmacokinetics of duloxetine: analysis of a single‐dose phase I study and pooled steady‐state data from phase II/III trials. Clin. Pharmacokinet. 49, 311–321 (2010). [DOI] [PubMed] [Google Scholar]
- 44. Skinner, M.H. et al Effect of age on the pharmacokinetics of duloxetine in women. Br. J. Clin. Pharmacol. 57, 54–61 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tianmei, S. et al Pharmacokinetics and tolerability of duloxetine following oral administration to healthy Chinese subjects. Clin. Pharmacokinet. 46, 767–775 (2007). [DOI] [PubMed] [Google Scholar]
- 46. Roerig, J.L. et al A comparison of duloxetine plasma levels in postbariatric surgery patients versus matched nonsurgical control subjects. J. Clin. Psychopharmacol. 33, 479–484 (2013). [DOI] [PubMed] [Google Scholar]
- 47. Berezhkovskiy, L.M. Determination of drug binding to plasma proteins using competitive equilibrium binding to dextran‐coated charcoal. J. Pharmacokinet. Pharmacodyn. 33, 595–608 (2006). [DOI] [PubMed] [Google Scholar]
- 48. Cymbalta Drug Information . Eli Lilly. [cited 2018 July 2]. Available from:(http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022516lbl.pdf).
- 49. Cymbalta FDA Drug Approval Package . Bayer. [cited 2018 July 2]. Available from: (http://www.accessdata.fda.gov/drugsatfda_docs/nda/2004/021427_s000_Cymbalta.cfm)
- 50. Bondolfi, G. , et al. The effect of fluoxetine on the pharmacokinetics and safety of risperidone in psychotic patients. Pharmacopsychiatry 35, 50–56 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Observed vs. simulated paroxetine steady‐state exposure. A total of 10 trials of nine healthy male CYP2D6 extensive metabolizers (age range 20–30 years) were simulated and compared with published data.
Table S1. Characteristics of simulated trials used for duloxetine PK model verification.
Table S2. Characteristics of simulated trials used for duloxetine DDI model verification.
Table S3. Characteristics of simulated trials used for modified paroxetine DDI model verification.
Table S4. Characteristics of simulated GDDI trials.
Table S5. Characteristics of simulated trials used for modified paroxetine DDI model verification.
Data S2. Supplementary material references.
Data S1. Data set: input model parameters.