

There are only a few antifungal drugs used systemically in treatment, and invasive fungal infections that are resistant to these drugs are an emerging problem in health care. In this study, we performed a high-copy-number genomic DNA (gDNA) library screening to find and characterize genes that reduce susceptibility to amphotericin B, caspofungin, and voriconazole in Saccharomyces cerevisiae.
KEYWORDS: amphotericin B, antifungal agents, caspofungin, drug resistance, genomics, multidrug resistance, voriconazole
ABSTRACT
There are only a few antifungal drugs used systemically in treatment, and invasive fungal infections that are resistant to these drugs are an emerging problem in health care. In this study, we performed a high-copy-number genomic DNA (gDNA) library screening to find and characterize genes that reduce susceptibility to amphotericin B, caspofungin, and voriconazole in Saccharomyces cerevisiae. We identified the PDR16 and PMP3 genes for amphotericin B, the RMD9 and SWH1 genes for caspofungin, and the MRS3 and TRI1 genes for voriconazole. The deletion mutants for PDR16 and PMP3 were drug susceptible, but the other mutants had no apparent susceptibility. Quantitative-PCR analyses suggested that the corresponding drugs upregulated expression of the PDR16, PMP3, SWH1, and MRS3 genes. To further characterize these genes, we also profiled the global expression patterns of the cells after treatment with the antifungals and determined the genes and paths that were up- or downregulated. We also cloned Candida albicans homologs of the PDR16, PMP3, MRS3, and TRI1 genes and expressed them in S. cerevisiae. Heterologous expression of Candida homologs also provided reduced drug susceptibility to the budding yeast cells. Our analyses suggest the involvement of new genes in antifungal drug resistance.
INTRODUCTION
Invasive fungal infections are mostly seen in patients who have been exposed to cancer chemotherapeutics or immune suppressors (1–4). There are many antifungal drugs used in local skin infections; however, the number of systemically used drugs is limited due to their side effects. Recent reports show that infectious fungi may develop resistance to new systemically used antifungals, which poses a higher risk for patients. Thus, we intended to search for drug resistance genes for amphotericin B (AmB), caspofungin (CSP), and voriconazole (VOR) by a genomic-DNA (gDNA) screening method.
Amphotericin B (5), a polyene macrolide antibiotic, was first discovered as a natural product of Streptomyces nodosus (6) and disrupts membrane permeability by binding to membrane lipids and forming pore-like structures (6). Binding of AmB to ergosterol is proposed as the main mechanism of AmB action, and the leakage of ions through the pores is considered a separate but complementary effect (7).
As an alternative to AmB, a new antifungal drug, CSP, is also used against pathogenic yeasts and molds, such as Candida spp. and Aspergillus spp. CSP is a member of the echinocandins that inhibits the synthesis of (1,3)-β-d-glucan in the cell wall (8). Noncompetitive inhibition of the (1,3)-β-d-glucan synthase (Fks1) enzyme by CSP both reduces fungal cell growth as a fungistatic effect and causes the instability to osmotic pressure that kills the fungal cells (9).
Another new antifungal is the triazole VOR, which was produced as a fluconazole derivative (10). VOR directly targets the iron atom and inhibits the cytochrome P450-dependent lanosterol 14-α-demethylase (Erg11) enzyme (11). Lack of ergosterol production disrupts fungal growth.
Identification of genes that play a role in antifungal drug susceptibility may provide further prognostic information, which may lead to improvement in the development of new agents or increase the efficacy of antifungals. In this study, we identified six genes that provide reduced susceptibility to antifungal drugs when overexpressed, and most of these genes have not been associated with any drug resistance mechanisms previously. We also cloned Candida albicans homologs of these genes, expressed them in budding yeast, and showed that they were involved in reduced drug susceptibility.
RESULTS
Screening of gDNA library and identification of drug resistance genes.
We first determined the lethal doses for AmB, CSP, and VOR under our conditions on yeast nitrogen base-2% glucose (YNB) agar medium for the Saccharomyces cerevisiae yeast strain BY4741 (12). We plated a series of serially diluted cell solutions onto agar plates containing different concentrations of each drug. The cells were not able to tolerate more than 0.4 μg/ml of AmB or 0.4 μg/ml of CSP. However, we noticed that the yeast strain that we used (BY4741) showed residual growth on high doses (100 μg/ml) of VOR (Fig. 1A, bottom). Next, we determined the MIC100 value (complete inhibition of growth) for AmB and the MIC50 values for CSP and VOR analyses in liquid YNB medium following the EUCAST microdilution procedure. AmB had a MIC100 value of 0.4 μg/ml. CSP and VOR had similar MIC50 values, 0.10 μg/ml and 0.15 μg/ml, respectively. In general, our MIC analyses and spotting assays were in agreement with each other. For screening purposes, we transferred gDNA-transformed cells onto YNB plates containing the lethal doses of the drugs. We observed four AmB colonies, two CSP colonies, and three VOR colonies showing reduced susceptibility to the corresponding drugs. Plasmids were recovered from all of these colonies and used to transform fresh wild-type (WT) cells to confirm that they were responsible for the growth in the presence of the drugs. After confirmation, plasmids were sequenced from both sites of the expression cassettes, and the genes that they harbored were identified by BLAST search using the Saccharomyces Genome Database. Genes that were present in each expression cassette are listed in Table 1.
FIG 1.

Spotting assay for AmB, CSP, and VOR resistance genes. (A) Spotting assay of wild-type cells growing on agar plates with the indicated concentrations of antifungal drugs. The cells were serially diluted, and a 5-μl aliquot was dropped for each spot. (B) Wild-type cells overexpressing PDR16 and PMP3, RMD9 and SWH1, or MRS3 and TRI1 were grown on plates containing a gradient of AmB, CSP, and VOR, respectively. Wild-type cells with an empty plasmid were used as controls (p-only). (C) Spotting assay for deletion mutants. The plates were incubated at 30°C for 3 days after inoculation and photographed.
TABLE 1.
Genomic sequences responsible for AmB, CSP, and VOR resistance
| Drug name | Colony no. | Chromosome coordinates | Covered genesa |
|---|---|---|---|
| AmB | A1, A2, A4 | Chromosome IV, bp 1009734–1019446 | DON1, YDR274C, BSC2, PMP3, MTH1, ARS430, YDR278C, part of RNH202 |
| A3 | Chromosome XIV, bp 210826–218065 | BNI4, CSL4, PDR16, part of ELA1 | |
| CSP | C1 | Chromosome I, bp 190872–196459 | YAR035C-A, SWH1 |
| C2 | Chromosome VII, bp 303197–308230 | RMD9, YGL108C, YGL109W, MLC1, ARC1 | |
| VOR | V1 | Chromosome X, bp 164101–159624 | YJL133C-A, MRS3, YJL132W |
| V2, V3 | Chromosome XIII, bp 737351–741548 | TRI1, RNH1 |
Resistance genes are shown in boldface.
Three of the AmB-resistant colonies contained the same expression cassette, which covered a 9.7-kb DNA region on chromosome (Chr) IV involving the PMP3 gene. The other resistant colony contained a 7.2-kb DNA region on Chr XIV covering the PDR16 gene. Since PDR16 is a member of the multidrug resistance family and the PMP3 gene was previously associated with AmB resistance (13, 14), we directly cloned these genes onto separate vectors to confirm that they were responsible for the reduced susceptibility that we observed. The overexpression of the PMP3 and PDR16 genes in WT cells reduced AmB susceptibility (Fig. 1B).
We also cloned candidate genes for CSP and VOR from the library cassettes and overexpressed them in WT cells to identify which ones were responsible for the drug tolerance. Overexpression of the RMD9 and SWH1 genes reduced susceptibility to CSP, and overexpression of the MRS3 or TRI1 gene reduced susceptibility to VOR (Fig. 1B).
Rmd9 plays a role in mitochondrial mRNA processing (15) and has not been associated with any drug resistance mechanisms. Swh1 is a homolog of human oxysterol binding protein and was found to be important in resistance to haloperidol, a psychiatric drug (16).
One of the genes that reduced voriconazole susceptibility, MRS3, is a member of the mitochondrial carrier family and mediates iron transport across the inner membrane (17). The other gene, TRI1, encodes a SUMOylated protein with unknown function (18). Both VOR genes have not been implicated in any drug resistance previously.
We reasoned that if the overexpression of these genes reduces drug susceptibility, then their deletion should render cells susceptible to the drugs. Therefore, we analyzed the deletion mutants for the ability to grow in the presence of the drugs and observed that absence of PDR16 and PMP3 sensitized cells to AmB, but the deletion mutants for neither CSP- nor VOR-related genes were susceptible to the drugs (Fig. 1C).
Transcriptional analyses of genes.
Our genetic-screening analyses suggested that the PMP3, PDR16, RMD9, SWH1, MRS3, and TRI1 genes reduced drug susceptibility when overexpressed from a high-copy-number plasmid. Our next question was whether these genes were transcriptionally responsive to drug treatment. Gene-based transcriptional analyses were performed using a real-time quantitative-PCR (qPCR) approach. Cells were treated with a sublethal dose of the drugs (0.2 μg/ml AmB, 0.01 μg/ml CSP, or 75 μg/ml VOR) for 2 h during the exponential growth phase. Control groups were not exposed to any drugs. Expression levels of both the PDR16 and PMP3 genes were found to be significantly upregulated by AmB (2.5-fold and 15-fold, respectively) (Fig. 2A). The voriconazole tolerance gene, MRS3, also had significant (17-fold) upregulation; however, the TRI1 gene was not activated by the drug (Fig. 2B). Treatment with CSP upregulated SWH1 expression by 1.5-fold yet downregulated RMD9 expression by 0.4-fold (Fig. 2C).
FIG 2.

Real-time qPCR analyses for resistance genes. Wild-type cells were incubated with a sublethal dose of AmB (0.2 μg/ml) (A), VOR (75 μg/ml) (B), and CSP (0.01 μg/ml) (C) for 2 h and analyzed for their transcript levels. The ACT1 gene used as an internal control. The significance of the differences was calculated by Student’s t test (P < 0.05). Each gene was analyzed in two biological samples with triplicate readings. The error bars indicate SD.
Additionally, whole-genome expression profiles of cells were monitored by microarray analyses to ascertain the cellular processes that were affected by the drugs. Genes that were upregulated or downregulated more than 2-fold were analyzed by MIPS (Munich Information Center for Protein Sequences) functional classification. The primary response of yeast cells to AmB was to activate carbohydrate and energy metabolism (see Table S1 in the supplemental material). In contrast, the amino acid metabolism and ion transport genes were inhibited (see Table S2 in the supplemental material). Similarly, CSP activated the carbohydrate metabolism genes and inhibited DNA synthesis and replication genes (see Tables S3 and S4 in the supplemental material). On the other hand, VOR increased the levels of meiosis genes rather than energy metabolism and also inhibited lipid metabolism (see Tables S5 and S6 in the supplemental material).
Membrane-related physiological and morphological analyses.
The plasma membrane forms the first line of defense against external stressors, and its lipid composition plays essential roles in its integrity and functionality. Both the Pmp3 and Pdr16 proteins are associated with the cell membrane and have roles in lipid dynamics. Additionally, antifungal drugs mainly target the plasma membrane in pathogenic fungi. Therefore, we wanted to determine membrane-related functions, such as membrane potential, cytosolic pH, and cytotoxic-cation tolerance, in the mutants and the wild-type cells overexpressing these genes.
First, we monitored the membrane potentials of the cells using a fluorescence assay based on the membrane potential-dependent distribution of the dye diS-C3(3) (3,3′-dipropylthiacarbocyanine iodide). As shown in Fig. 3A, most of the mutants had membrane potentials similar to that of the wild-type cells, except the Δrmd9 mutants, which were hyperpolarized. When the RMD9 gene was overexpressed, the membrane potential was returned to the normal value (Fig. 3C). Overexpression of the PDR16, PMP3, MRS3, and TRI1 genes led to normal membrane potentials (Fig. 3B and D). However, SWH1 overexpression hyperpolarized the cells (Fig. 3C).
FIG 3.

Plasma membrane potential measurements. (A) Relative membrane potentials of the deletion mutants. BY4741 wild-type cells were used as the control. (B) Membrane potentials of wild-type cells overexpressing PDR16 and PMP3 genes. Cells carrying empty plasmids (p426) were used as the control. (C) Membrane potentials of wild-type cells overexpressing RMD9 and SWH1 genes. Cells carrying empty plasmids (pAG425) were used as the control. (D) Membrane potentials of wild-type cells overexpressing MRS3 and TRI1 genes. Cells carrying empty plasmids (pAG425) were used as the control.
Next, we exposed cells to NaCl, LiCl, and the cationic drugs hygromycin B (HygB), tetramethyl ammonium (TMA), and spermine. As shown in Fig. S1A in the supplemental material, Δpmp3 cells were salt, HygB, and TMA susceptible, but Δpdr16 cells showed no susceptibility to any of the conditions tested. The hypersensitivity of Δpmp3 mutants to salt, HygB, and TMA was in close agreement with previously published data (19). The other deletion mutants were not susceptible to the cationic agents. Similarly, overexpression of these genes in wild-type cells caused no observable phenotypes on plates containing cations and drugs, except that SWH1 overexpression caused TMA resistance (see Fig. S1B and C).
As the next step, we tested whether the drug tolerance of the mutants was correlated with changes in intracellular pH. When we measured the intracellular pH with pHluorin expression (20), we did observe that the Δpmp3, Δpdr16, and Δrmd9 mutants were more acidic than wild-type cells (Fig. 4).
FIG 4.
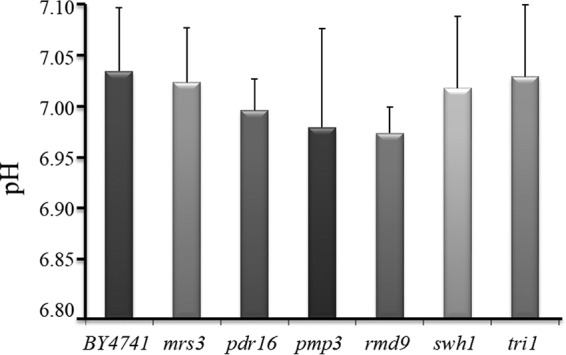
Intracellular pH values of the mutants. The error bars indicate SD.
Expression of Candida orthologous genes in budding yeast reduces susceptibility to the drugs.
To test if the susceptibility genes had similar roles in pathogenic C. albicans, we amplified the open reading frames of C. albicans orthologs by PCR and expressed them in wild-type S. cerevisiae cells. There were no C. albicans orthologs for the CSP resistance genes RMD9 and SWH1, and thus, we did not include these genes in analyses. Expression of both C. albicans PMP3 (CaPMP3) and CaPDR16 reduced susceptibility to AmB treatment (Fig. 5A). The CaPMP3 gene was previously shown to reduce AmB susceptibility in S. cerevisiae (13, 14). However, the CaPDR16 gene was not associated with reduced AmB susceptibility previously. This study showed that the CaPDR16 gene was also crucial to reduce AmB susceptibility in the heterologous host system. Similarly, the orthologs of the TRI1 and MRS3 (CaTRI1 and CaMRS3) genes also provided reduced VOR susceptibility when expressed in yeast cells (Fig. 5B).
FIG 5.
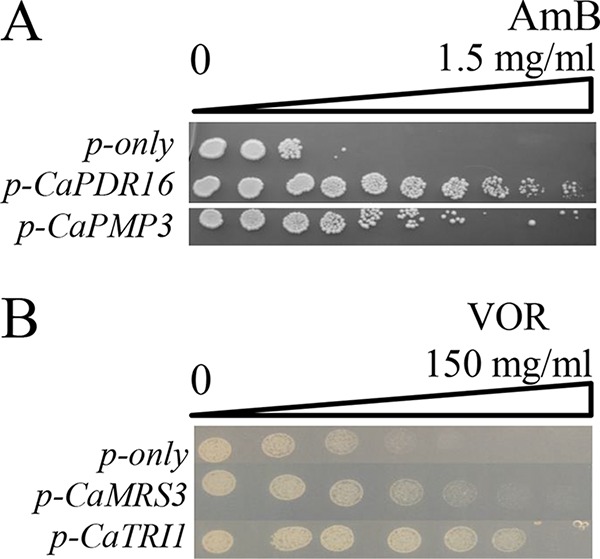
Test of C. albicans orthologous genes. Candida genes were cloned into yeast expression vectors and expressed in S. cerevisiae. (A) Cells were grown on an AmB gradient between 0 μg/ml and 1.5 μg/ml. (B) Cells were grown on a VOR gradient between 0 μg/ml and 150 μg/ml.
DISCUSSION
In this study, we screened a high-copy-number gDNA library to find the genes that support cell growth in the presence of lethal doses of systemically used antifungals: AmB, CSP, and VOR.
We identified the PDR16 and PMP3 genes for their ability to reduce AmB susceptibility. Their overexpression from a plasmid reduced susceptibility, and their deletion rendered cells susceptible to AmB treatment. Moreover, the genes were transcriptionally responsive to AmB treatment. Thus, our findings confirmed the role of PMP3 and suggested a new gene, PDR16, for AmB tolerance. Both of these genes have known roles in plasma membrane lipid dynamics.
A recent study suggested that AmB binds to ergosterol and forms extramembranous and fungicidal sponge structures (21). Although AmB resistance is not common, it is observed in Candida spp. and other fungal species (22, 23). AmB resistance mechanisms commonly involve alterations in the cell membrane and reduction in ergosterol levels (22–25). Sphingolipids are also involved in AmB resistance. Deletion of the sphingolipid-biosynthetic genes FEN1 and SUR4 renders cells susceptible to AmB (26). The role of PMP3 in AmB tolerance might be specific to AmB, since it does not provide vulnerability to AmB-related drugs, such as natamycin and filipin (13). Overexpression of PMP3 directly or indirectly antagonizes the membrane permeation effect of AmB. A recent study showed that AmB binds to ergosterol and forms extramembranous aggregates (21). On the other hand, it is known that ergosterol biosynthesis is required for AmB susceptibility (13). A possible mechanism for Pmp3 action might be that it interferes with AmB-ergosterol interactions, which needs to be further investigated using biophysical assays.
A previous study suggested that the absence of PMP3 hyperpolarizes the plasma membrane and sensitizes cells to salt (19). We also noticed that Δpmp3 cells were salt susceptible (see Fig. S1), but their membranes were not hyperpolarized. The observation of different results for the membrane potential might be due to the methods used for membrane potential prediction or to the cell background. Navarre and Goffeau used a [14C]methylammonium uptake assay as the indicator of the plasma membrane potential, whereas we utilized a recently developed diS-C3(3) method (27).
PDR16 affects ergosterol biosynthesis (28) and facilitates the movement of phosphatidylinositol between membranes (29). The drug tolerance of yeast cells overexpressing PDR16 may be related to the alterations in membrane composition so that AmB is not able to interfere with membrane permeability. Further studies comparing the permeabilities and lipid compositions in the cellular membranes of PDR16-overexpressing and PDR16-deficient cells may provide a better understanding of its role in drug tolerance.
Echinocandin resistance is mainly associated with the FKS1 gene in different fungal species. Spontaneous mutations can arise in hot-spot regions of Fks1 and can reduce the enzyme’s sensitivity to the drug and thus lead to drug resistance. Moreover, fungal strains have been isolated in which the sequence of FKS1 is unaltered yet the fungus has decreased susceptibility to echinocandins (30, 31), suggesting that there are additional mechanisms governing CSP resistance. CSP was the first member of the new class of echinocandins, and we identified two novel genes, RMD9 and SWH1, whose overexpression reduces susceptibility to CSP. Rmd9 has a role in the processing and stability of mRNAs encoding the subunits of the electron transport chain (ETC) (15). The RMD9 gene was not transcriptionally activated by CSP treatment and reduced susceptibility only when expressed ectopically. The Δrmd9 deletion mutants are respiration deficient (15), and production of organic acids via fermentation (32) could be a reason why these cells had lower pH values. The other CSP tolerance gene that we identified was SWH1. The protein encoded by this gene has homology with a mammalian oxysterol binding protein (33). The primary function of Swh1 is still unknown, although it may have a role in nucleus-vacuole (NV) junction and endoplasmic reticulum (ER) trafficking as a result of its structural similarity to oxysterol binding protein (34). Mutations in the SWH1 gene result in a reduction in membrane ergosterol levels (35). The reduced susceptibility that SWH1 provides may be related to membrane ergosterol dynamics, membrane fusion pathways, and ER stress response (36).
Voriconazole prevents ergosterol biosynthesis by inhibiting the cytochrome P450-dependent 14-α-demethylase (CYP51) enzyme (37). Triazoles like voriconazole directly target the ferric ions of the heme group that exists in CYP51 (11). Fungi can overcome these effects by overproducing Erg11, by mutational disruption of the azole-Erg11 interaction, or by effluxing the azole from the cytoplasm (38). Voriconazole is active against many Candida species that are resistant to fluconazole (37). However, recent reports show that gain-of-function mutations in the MRR1 transcription factor provide voriconazole resistance (39). In this study, the genes we found for reduced VOR susceptibility are novel genes and have not been associated with known drug resistance mechanisms previously. MRS3 was first identified as a mitochondrial-RNA splicer (40) but was then defined as a member of the mitochondrial transport family and as responsible for iron transportation (41). In cells, ferric ions are held in the intermembrane space of the mitochondria, and the cytoplasm is free of these ions. When the cells need iron, the FeS complexes of mitochondria process iron ions and transport them to the cytoplasm (42). We found that cells significantly (17-fold) upregulate the MRS3 gene (Fig. 2B) in response to voriconazole treatment, probably to compensate for the iron requirement. Thus, if azole drugs remove the iron ions from the CYP51 enzyme, high expression of MRS3 may replenish the iron requirement of CYP51 and therefore could reduce drug susceptibility. TRI1 has no known function and encodes a SUMOylated protein (18). Expression of its Candida homolog significantly reduced susceptibility to VOR; however, its mechanism is unknown.
The genes for CSP and VOR tolerance we have identified may be less active than the genes we found for AmB tolerance. This is especially evident when we consider the susceptibilities of the deletion mutants to the corresponding drugs. Therefore, the roles of CSP and VOR genes in drug resistance may not be physiological and may have resulted from an indirect effect of ectopic overexpression. The clinical significance of these genes should be investigated with additional experiments.
We used toxic concentrations of drugs in our gDNA screening in the hope of finding only robust genes that can reduce drug susceptibility. The doses tested here and the MIC values were in agreement with those in previous studies (43–46). We used 150 μg/ml VOR as the cutoff value for the initial screening analyses because of residual growth at high doses. This range for VOR was also observed by others for BY4741 cells (46).
In summary, our genomewide genetic screening for the identification of genes involved in reduced drug susceptibility resulted in new potential target genes for AmB, CSP, and VOR. We limited our study to budding yeast cells as a model because of technical advantages and genetic stability. Further characterization of these genes in pathogenic fungal strains or clinical isolates may shed light on the drug resistance mechanisms and may lead to novel diagnostic tools that would allow identification of specific resistance profiles from genetic hallmarks.
MATERIALS AND METHODS
Drugs, yeast strains, cell growth and gDNA library screening.
Amphotericin B (Fungizone; Bristol-Myers, Squibb), caspofungin (Sigma), and voriconazole (Vfend IV; Pfizer) were used in the library-screening, resistance, and susceptibility tests. The S. cerevisiae strain BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0; EUROSCARF) and its isogenic deletion mutants were used in the study. Genomic DNA of C. albicans strain SC5314 (47) was used to amplify and clone the orthologous genes. A high-copy-number yeast genomic DNA library (ATCC no. 37323) was used for the screening. Yeast transformations were performed by the standard LiAc method (48). Unless otherwise indicated, all experiments were performed on YNB medium supplemented with appropriate amino acids and bases (49). YPD rich medium (1% yeast extract, 2% peptone, 2% dextrose) was also used to grow the wild type and deletion mutants before the transformation process. The Candida cells were grown in Sabouraud dextrose agar (SDA) medium (2% dextrose, 1% peptone, 1.5% agar, pH 5.6). Plates were incubated at 30°C for 3 days and photographed.
MIC analyses.
For MIC analyses, EUCAST broth microdilution testing was performed as described in document EDEF 7.3.1 (50) with some modifications to adapt to yeast growth and screening conditions. In brief, we first prepared the yeast inoculum (5 × 106 CFU/ml) in sterile water. Instead of RPMI medium, we used YNB medium (0.67 g YNB, 2 g glucose 100 ml−1) including the necessary amino acids and nucleobases (5 mg l-methionine, 10 mg l-leucine, 5 mg l-histidine, 1 mg uracil 100 ml−1) for the auxotrophic requirements of the BY4741 strain. The antifungal stock solutions were prepared in 100% dimethyl sulfoxide (DMSO). Serial dilutions of antifungal drugs were prepared by using YNB medium in flat-bottom 96-well plates. After the inoculum was added, the final concentrations of antifungal drugs ranged from 0.1 to 1 mg/liter for amphotericin B, 0.0125 to 0.5 mg/liter for caspofungin, and 0.025 to 100 mg/liter for voriconazole. The plates were incubated at 30°C, and the MICs were determined spectrophotometrically at 600 nm after 24 h of incubation. The MIC100 for AmB and the MIC50 values for CSP and VOR were calculated.
Gradient plate preparation and spotting assay.
Gradient plates were prepared as described by Metzenberg and Grotelueschen (51). In brief, 50 ml of agar medium without drug was poured into 120-mm by 120-mm square petri dishes and slanted at an angle of 45°. When the agar had solidified, 50 ml of the same medium including the lethal dose of the drug was poured, and the plate was left horizontal until all the agar had solidified.
For spotting assays, suspensions of fresh cells (optical density at 600 nm [OD600] = 0.2) were serially diluted and dropped onto the related agar media. For gradient plates, 5 μl of cells at an OD600 of 0.02 were dropped one by one on the same line. The plates were incubated at 30°C for 3 days.
Plasmid isolation, sequencing, and gene cloning.
Yeast cells were digested with lyticase (5 U/ml) for 30 min in Tris-EDTA (TE) buffer before the isolation. Plasmids were isolated from yeast cells using a GeneJet plasmid miniprep kit (ThermoScientific) as described by the manufacturer. The isolated plasmids were amplified using Escherichia coli JM109 competent cells and sequenced from both sides with a pair of vector-specific primers at the Biotechnology Center of the Izmir Institute of Technology.
The S. cerevisiae genes were subcloned into p425GPD (ATCC 87359) overexpression vector from the ThermoScientific OpenBiosystems Yeast ORF Collections (52). The PCR-amplified C. albicans genes CaPDR16, CaPMP3, CaMRS3, and CaTRI1 were cloned into the pAG426GPD-ccdB destination vector by Gateway Technology (52). The primers used to amplify Candida (SC5314) genes are shown in Table 2. PCR conditions for the CaPDR16, CaMRS3, and CaTRI1 genes were 94°C for 5 min, 94°C for 30 s, 46°C for 30 s, 72°C for 1.5 min, and 72°C for 7 min for 30 cycles; the conditions for CaPMP3 were almost the same, but the annealing temperature was 45°C and the elongation time was 45 s.
TABLE 2.
Primers used to amplify C. albicans (SC5314) genes

Transcriptional analyses.
Total RNA samples from logarithmically growing cells were isolated using a ThermoScientific GeneJet RNA purification kit. Genomic-DNA contaminations were removed by DNase treatment (DNaseRQ1; Promega).
cDNA synthesis was performed using a First Strand cDNA synthesis kit (ThermoScientific) according to the manufacturer’s instructions. The cDNAs were used as templates to amplify internal parts of the selected genes. The ACT1 gene was used as an internal control. RNA samples were obtained from two independent yeast samples, and triple reactions were set for each one. Statistical significance was evaluated with Student's t test. Real-time PCR assays were performed with the IQ5 real-time PCR system (Bio-Rad).
Transcriptional profiling of the whole genome was performed by microarray analysis with Agilent’s single-channel array. After background correction, the data were normalized via a quantile procedure. The consistency of each sample was measured by principal-component analysis (PCA). Then, the similarities of the samples were monitored by cluster analysis. Discrimination of the varied genes was detected by fold change (log2) analysis instead of classical statistical methods. Each gene detected was classified by MIPS functional analysis using the FunSpec tools (http://funspec.med.utoronto.ca).
Relative membrane potential measurement.
Relative membrane potential measurement was performed as described previously (27, 53). The wild type and deletion mutants were grown in YPD medium; strains with overexpression of the genes were grown in YNB medium, as stated above. Exponential-phase cells were harvested by centrifugation, washed twice with sterile distilled water, and resuspended in 10 mM MES (morpholineethanesulfonic acid) buffer (pH 6.0, adjusted with triethanolamine) to an OD600 of 0.2 for each sample. Measurements were performed in UV grade cuvettes (Kartell). The probe diS-C3(3) was added to a final concentration of 4 × 10−8 M (10−5 M stock solution in ethanol). The fluorescence emission spectra (λex [excitation wavelength] = 531 nm; λem [emission wavelength] = 560 to 590 nm) of the cell suspensions were measured with an ISS PC1 (Photon Counting) spectrofluorimeter. The staining curves recorded the dependence of the fluorescence emission maximum wavelength (λmax) at the time of staining. Measurements were done with three independent biological samples.
Fluorescent imaging and cytosolic-pH measurement.
Cells expressing pHluorin (transformed with the plasmid pHl-U) (54) were grown in a specific selective medium [0.175% YNB (minimal medium without ammonium sulfate, folic acid, riboflavin, and KCl; MP Biomedicals), 0.4% (NH4)2SO4, 2% glucose, and auxotrophic supplements, including leucine, methionine, and histidine] to an OD600 of ∼0.5.
Measurement of the cytosolic pH was based on a polynomic calibration curve generated according to the method of Orij et al. (55) and Duškova et al. (56). The pH standards were 5.67, 5.97, 6.44, 6.59, 6.80, 7.01, 7.3, and 7.76. The intensity of the fluorescence was measured in a fluorescence reader (Synergy HT; BioTek) at 400 nm and 485 nm; emission was at 516 nm. To eliminate the background fluorescence, a culture of a nontransformed strain was grown in parallel, and the corresponding values were subtracted from the fluorescence at each excitation wavelength (Gen5 software; BioTek Instruments). The I400/I485 ratio was used to calculate the intracellular pH from the calibration curve. Each strain was measured in 8 wells (100 μl of cells per well) within one experiment (technical replicates), and the data presented are means and standard deviations (SD) of the results of two independent experiments (biological replicates).
Supplementary Material
ACKNOWLEDGMENTS
This study was financially supported by the Scientific and Technological Research Council of Turkey (TUBITAK 212T186 to A.K.) and the Academy of Sciences of the Czech Republic (ASCR).
We declare no conflict of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02268-18.
REFERENCES
- 1.Bow EJ, Loewen R, Cheang MS, Schacter B. 1995. Invasive fungal disease in adults undergoing remission-induction therapy for acute myeloid leukemia: the pathogenetic role of the antileukemic regimen. Clin Infect Dis 21:361–369. doi: 10.1093/clinids/21.2.361. [DOI] [PubMed] [Google Scholar]
- 2.George MJ, Snydman DR, Werner BG, Griffith J, Falagas ME, Dougherty NN, Rubin RH, Boston Center for Liver Transplantation CMVIG-Study Group, Cytogam, MedImmune, Inc. Gaithersburg, Maryland. 1997. The independent role of cytomegalovirus as a risk factor for invasive fungal disease in orthotopic liver transplant recipients. Am J Med 103:106–113. doi: 10.1016/S0002-9343(97)80021-6. [DOI] [PubMed] [Google Scholar]
- 3.De Pauw B, Walsh TJ, Donnelly JP, Stevens DA, Edwards JE, Calandra T, Pappas PG, Maertens J, Lortholary O, Kauffman CA, Denning DW, Patterson TF, Maschmeyer G, Bille J, Dismukes WE, Herbrecht R, Hope WW, Kibbler CC, Kullberg BJ, Marr KA, Munoz P, Odds FC, Perfect JR, Restrepo A, Ruhnke M, Segal BH, Sobel JD, Sorrell TC, Viscoli C, Wingard JR, Zaoutis T, Bennett JE, European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group, National Institute of Allergy, Infectious Diseases Mycoses Study Group Consensus Group. 2008. Revised definitions of invasive fungal disease from the European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group and the National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. Clin Infect Dis 46:1813–1821. doi: 10.1086/588660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cutler JE, Deepe GS Jr, Klein BS. 2007. Advances in combating fungal diseases: vaccines on the threshold. Nat Rev Microbiol 5:13–28. doi: 10.1038/nrmicro1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis RE, Cahyame-Zuniga L, Leventakos K, Chamilos G, Ben-Ami R, Tamboli P, Tarrand J, Bodey GP, Luna M, Kontoyiannis DP. 2013. Epidemiology and sites of involvement of invasive fungal infections in patients with haematological malignancies: a 20-year autopsy study. Mycoses 56:638–645. doi: 10.1111/myc.12081. [DOI] [PubMed] [Google Scholar]
- 6.Cohen BE. 2010. Amphotericin B membrane action: role for two types of ion channels in eliciting cell survival and lethal effects. J Membrane Biol 238:1–20. doi: 10.1007/s00232-010-9313-y. [DOI] [PubMed] [Google Scholar]
- 7.Gray KC, Palacios DS, Dailey I, Endo MM, Uno BE, Wilcock BC, Burke MD. 2012. Amphotericin primarily kills yeast by simply binding ergosterol. Proc Natl Acad Sci U S A 109:2234–2239. doi: 10.1073/pnas.1117280109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walsh TJ, Teppler H, Donowitz GR, Maertens JA, Baden LR, Dmoszynska A, Cornely OA, Bourque MR, Lupinacci RJ, Sable CA, dePauw BE. 2004. Caspofungin versus liposomal amphotericin B for empirical antifungal therapy in patients with persistent fever and neutropenia. N Engl J Med 351:1391–1402. doi: 10.1056/NEJMoa040446. [DOI] [PubMed] [Google Scholar]
- 9.Letscher-Bru V, Herbrecht R. 2003. Caspofungin: the first representative of a new antifungal class. J Antimicrob Chemother 51:513–521. doi: 10.1093/jac/dkg117. [DOI] [PubMed] [Google Scholar]
- 10.McGinnis MR, Pasarell L, Sutton DA, Fothergill AW, Cooper CR Jr, Rinaldi MG. 1997. In vitro evaluation of voriconazole against some clinically important fungi. Antimicrob Agents Chemother 41:1832–1834. doi: 10.1128/AAC.41.8.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu S, Chai X, Hu H, Yan Y, Guan Z, Zou Y, Sun Q, Wu Q. 2010. Synthesis and antifungal evaluation of novel triazole derivatives as inhibitors of cytochrome P450 14alpha-demethylase. Eur J Med Chem 45:4435–4445. doi: 10.1016/j.ejmech.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 12.Zhang C, Milunsky JM, Newton S, Ko J, Zhao G, Maher TA, Tager-Flusberg H, Bolliger MF, Carter AS, Boucard AA, Powell CM, Sudhof TC. 2009. A neuroligin-4 missense mutation associated with autism impairs neuroligin-4 folding and endoplasmic reticulum export. J Neurosci 29:10843–10854. doi: 10.1523/JNEUROSCI.1248-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang Z, Chen K, Zhang JH, Li YX, Wang H, Cui DD, Tang JW, Liu Y, Shi XM, Li W, Liu D, Chen R, Sucgang RS, Pan XW. 2013. A functional variomics tool for discovering drug-resistance genes and drug targets. Cell Rep 3:577–585. doi: 10.1016/j.celrep.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bari VK, Sharma S, Alfatah M, Mondal AK, Ganesan K. 2015. Plasma membrane proteolipid 3 protein modulates amphotericin B resistance through sphingolipid biosynthetic pathway. Sci Rep 5:9685. doi: 10.1038/srep09685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nouet C, Bourens M, Hlavacek O, Marsy S, Lemaire C, Dujardin G. 2007. Rmd9p controls the processing/stability of mitochondrial mRNAs and its overexpression compensates for a partial deficiency of oxa1p in Saccharomyces cerevisiae. Genetics 175:1105. doi: 10.1534/genetics.106.063883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Kruglyak L. 2014. Genetic basis of haloperidol resistance in Saccharomyces cerevisiae is complex and dose dependent. PLoS Genet 10:e1004894. doi: 10.1371/journal.pgen.1004894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Froschauer EM, Schweyen RJ, Wiesenberger G. 2009. The yeast mitochondrial carrier proteins Mrs3p/Mrs4p mediate iron transport across the inner mitochondrial membrane. Biochim Biophys Acta 1788:1044–1050. doi: 10.1016/j.bbamem.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 18.Chen XL, Silver HR, Xiong L, Belichenko I, Adegite C, Johnson ES. 2007. Topoisomerase I-dependent viability loss in Saccharomyces cerevisiae mutants defective in both SUMO conjugation and DNA repair. Genetics 177:17–30. doi: 10.1534/genetics.107.074708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Navarre C, Goffeau A. 2000. Membrane hyperpolarization and salt sensitivity induced by deletion of PMP3, a highly conserved small protein of yeast plasma membrane. EMBO J 19:2515–2524. doi: 10.1093/emboj/19.11.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miesenbock G, De Angelis DA, Rothman JE. 1998. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- 21.Anderson TM, Clay MC, Cioffi AG, Diaz KA, Hisao GS, Tuttle MD, Nieuwkoop AJ, Comellas G, Maryum N, Wang S, Uno BE, Wildeman EL, Gonen T, Rienstra CM, Burke MD. 2014. Amphotericin forms an extramembranous and fungicidal sterol sponge. Nat Chem Biol 10:400–406. doi: 10.1038/nchembio.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Young LY, Hull CM, Heitman J. 2003. Disruption of ergosterol biosynthesis confers resistance to amphotericin B in Candida lusitaniae. Antimicrob Agents Chemother 47:2717–2724. doi: 10.1128/AAC.47.9.2717-2724.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hull CM, Parker JE, Bader O, Weig M, Gross U, Warrilow AG, Kelly DE, Kelly SL. 2012. Facultative sterol uptake in an ergosterol-deficient clinical isolate of Candida glabrata harboring a missense mutation in ERG11 and exhibiting cross-resistance to azoles and amphotericin B. Antimicrob Agents Chemother 56:4223–4232. doi: 10.1128/AAC.06253-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vandeputte P, Tronchin G, Larcher G, Ernoult E, Berges T, Chabasse D, Bouchara JP. 2008. A nonsense mutation in the ERG6 gene leads to reduced susceptibility to polyenes in a clinical isolate of Candida glabrata. Antimicrob Agents Chemother 52:3701–3709. doi: 10.1128/Aac.00423-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martel CM, Parker JE, Bader O, Weig M, Gross U, Warrilow AG, Kelly DE, Kelly SL. 2010. A clinical isolate of Candida albicans with mutations in ERG11 (encoding sterol 14alpha-demethylase) and ERG5 (encoding C22 desaturase) is cross resistant to azoles and amphotericin B. Antimicrob Agents Chemother 54:3578–3583. doi: 10.1128/AAC.00303-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma S, Alfatah M, Bari VK, Rawal Y, Paul S, Ganesan K. 2014. Sphingolipid biosynthetic pathway genes FEN1 and SUR4 modulate amphotericin B resistance. Antimicrob Agents Chemother 58:2409–2414. doi: 10.1128/AAC.02130-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaskova D, Brodska B, Herman P, Vecer J, Malinsky J, Sigler K, Benada O, Plasek J. 1998. Fluorescent probing of membrane potential in walled cells: diS-C3(3) assay in Saccharomyces cerevisiae. Yeast 14:1189–1197. doi:. [DOI] [PubMed] [Google Scholar]
- 28.van den Hazel HB, Pichler H, do Valle Matta MA, Leitner E, Goffeau A, Daum G. 1999. PDR16 and PDR17, two homologous genes of Saccharomyces cerevisiae, affect lipid biosynthesis and resistance to multiple drugs. J Biol Chem 274:1934–1941. doi: 10.1074/jbc.274.4.1934. [DOI] [PubMed] [Google Scholar]
- 29.Li X, Routt SM, Xie Z, Cui X, Fang M, Kearns MA, Bard M, Kirsch DR, Bankaitis VA. 2000. Identification of a novel family of nonclassic yeast phosphatidylinositol transfer proteins whose function modulates phospholipase D activity and Sec14p-independent cell growth. Mol Biol Cell 11:1989–2005. doi: 10.1091/mbc.11.6.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Desnos-Ollivier M, Dromer F, Dannaoui E. 2008. Detection of caspofungin resistance in Candida spp. by Etest. J Clin Microbiol 46:2389–2392. doi: 10.1128/JCM.00053-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walker LA, Gow NA, Munro CA. 2010. Fungal echinocandin resistance. Fungal Genet Biol 47:117–126. doi: 10.1016/j.fgb.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawahata M, Masaki K, Fujii T, Iefuji H. 2006. Yeast genes involved in response to lactic acid and acetic acid: acidic conditions caused by the organic acids in Saccharomyces cerevisiae cultures induce expression of intracellular metal metabolism genes regulated by Aft1p. FEMS Yeast Res 6:924–936. doi: 10.1111/j.1567-1364.2006.00089.x. [DOI] [PubMed] [Google Scholar]
- 33.Schmalix WA, Bandlow W. 1994. SWH1 from yeast encodes a candidate nuclear factor containing ankyrin repeats and showing homology to mammalian oxysterol-binding protein. Biochim Biophys Acta 1219:205–210. doi: 10.1016/0167-4781(94)90273-9. [DOI] [PubMed] [Google Scholar]
- 34.Levine TP, Munro S. 2001. Dual targeting of Osh1p, a yeast homologue of oxysterol-binding protein, to both the Golgi and the nucleus-vacuole junction. Mol Biol Cell 12:1633–1644. doi: 10.1091/mbc.12.6.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang B, Brown JL, Sheraton J, Fortin N, Bussey H. 1994. A new family of yeast genes implicated in ergosterol synthesis is related to the human oxysterol binding protein. Yeast 10:341–353. doi: 10.1002/yea.320100307. [DOI] [PubMed] [Google Scholar]
- 36.Boyce M, Yuan J. 2006. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death Differ 13:363–373. doi: 10.1038/sj.cdd.4401817. [DOI] [PubMed] [Google Scholar]
- 37.Johnson LB, Kauffman CA. 2003. Voriconazole: a new triazole antifungal agent. Clın Infect Dıs 36:630–637. doi: 10.1086/367933. [DOI] [PubMed] [Google Scholar]
- 38.Cuenca-Estrella M. 2014. Antifungal drug resistance mechanisms in pathogenic fungi: from bench to bedside. Clin Microbiol Infect 20(Suppl 6):54–59. doi: 10.1111/1469-0691.12495. [DOI] [PubMed] [Google Scholar]
- 39.Branco J, Silva AP, Silva RM, Silva-Dias A, Pina-Vaz C, Butler G, Rodrigues AG, Miranda IM. 2015. Fluconazole and voriconazole resistance in Candida parapsilosis ıs conferred by gain-of-function mutations in MRR1 transcription factor gene. Antimicrob Agents Chemother 59:6629–6633. doi: 10.1128/AAC.00842-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sollner T, Schmidt C, Schmelzer C. 1987. Amplification of the yeast nuclear gene MRS3 confers suppression of a mitochondrial RNA splice defect. Curr Genet 12:497–501. doi: 10.1007/BF00419558. [DOI] [PubMed] [Google Scholar]
- 41.Muhlenhoff U, Stadler JA, Richhardt N, Seubert A, Eickhorst T, Schweyen RJ, Lill R, Wiesenberger G. 2003. A specific role of the yeast mitochondrial carriers MRS3/4p in mitochondrial iron acquisition under iron-limiting conditions. J Biol Chem 278:40612–40620. doi: 10.1074/jbc.M307847200. [DOI] [PubMed] [Google Scholar]
- 42.Nyhus KJ, Ozaki LS, Jacobson ES. 2002. Role of mitochondrial carrier protein Mrs3/4 in iron acquisition and oxidative stress resistance of Cryptococcus neoformans. Med Mycol 40:581–591. doi: 10.1080/714031152. [DOI] [PubMed] [Google Scholar]
- 43.Lovero G, De Giglio O, Rutigliano S, Diella G, Caggiano G, Montagna MT. 2017. In vitro antifungal susceptibilities of Candida species to liposomal amphotericin B, determined using CLSI broth microdilution, and amphotericin B deoxycholate, measured using the Etest. J Med Microbiol 66:213–216. doi: 10.1099/jmm.0.000402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Powderly WG, Kobayashi GS, Herzig GP, Medoff G. 1988. Amphotericin B-resistant yeast infection in severely immunocompromised patients. Am J Med 84:826–832. doi: 10.1016/0002-9343(88)90059-9. [DOI] [PubMed] [Google Scholar]
- 45.Silver PM, Silver PM, Oliver BG, White TC. 2008. Characterization of caspofungin susceptibilities by broth and agar in Candida albicans clinical isolates with characterized mechanisms of azole resistance. Med Mycol 46:231–239. doi: 10.1080/13693780701816557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parker JE, Merkamm M, Manning NJ, Pompon D, Kelly SL, Kelly DE. 2008. Differential azole antifungal efficacies contrasted using a Saccharomyces cerevisiae strain humanized for sterol 14 alpha-demethylase at the homologous locus. Antimicrob Agents Chemother 52:3597–3603. doi: 10.1128/AAC.00517-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones T, Federspiel NA, Chibana H, Dungan J, Kalman S, Magee BB, Newport G, Thorstenson YR, Agabian N, Magee PT, Davis RW, Scherer S. 2004. The diploid genome sequence of Candida albicans. Proc Natl Acad Sci U S A 101:7329–7334. doi: 10.1073/pnas.0401648101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gietz RD, Schiestl RH, Willems AR, Woods RA. 1995. Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11:355–360. doi: 10.1002/yea.320110408. [DOI] [PubMed] [Google Scholar]
- 49.Amberg DC, Burke D, Strathern J. 2005. Methods in yeast genetics: a Cold Spring Harbor Laboratory course manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 50.Arendrup MC, Meletiadis J, Mouton JW, Lagrou K, Hamal P, Guinea J. 2017. Method for the determination of broth dilution minimum inhibitory concentrations of antifungal agents for fermentative yeasts. EUCAST defınıtıve document EDEF 7.3.1 https://www.ncbi.nlm.nih.gov/pubmed/18190574.
- 51.Metzenberg RL, Grotelueschen JS. 1992. Disruption of essential genes in Neurospora by RIP. Fungal Genet Newsl 39:37–49. [Google Scholar]
- 52.Alberti S, Gitler AD, Lindquist S. 2007. A suite of Gateway cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast 24:913–919. doi: 10.1002/yea.1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kodedova M, Sychrova H. 2015. Changes in the sterol composition of the plasma membrane affect membrane potential, salt tolerance and the activity of multidrug resistance pumps in Saccharomyces cerevisiae. PLoS One 10:e0139306. doi: 10.1371/journal.pone.0139306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maresova L, Hoskova B, Urbankova E, Chaloupka R, Sychrova H. 2010. New applications of pHluorin-measuring intracellular pH of prototrophic yeasts and determining changes in the buffering capacity of strains with affected potassium homeostasis. Yeast 27:317–25. doi: 10.1002/yea.1755. [DOI] [PubMed] [Google Scholar]
- 55.Orij R, Postmus J, Ter Beek A, Brul S, Smits GJ. 2009. In vivo measurement of cytosolic and mitochondrial pH using a pH-sensitive GFP derivative in Saccharomyces cerevisiae reveals a relation between intracellular pH and growth. Microbiology 155:268–278. doi: 10.1099/mic.0.022038-0. [DOI] [PubMed] [Google Scholar]
- 56.Duskova M, Borovikova D, Herynkova P, Rapoport A, Sychrova H. 2015. The role of glycerol transporters in yeast cells in various physiological and stress conditions. FEMS Microbiol Lett 362:1–8. doi: 10.1093/femsle/fnu041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.