

Filociclovir (MBX-400, cyclopropavir) is an antiviral agent with activity against cytomegalovirus (CMV). A phase 1, double-blind, randomized, placebo-controlled (3:1 ratio), single-center, multiple-ascending-dose trial was conducted to assess the safety, tolerability, and pharmacokinetics of filociclovir.
KEYWORDS: filociclovir, MBX-400, pharmacokinetics, safety
ABSTRACT
Filociclovir (MBX-400, cyclopropavir) is an antiviral agent with activity against cytomegalovirus (CMV). A phase 1, double-blind, randomized, placebo-controlled (3:1 ratio), single-center, multiple-ascending-dose trial was conducted to assess the safety, tolerability, and pharmacokinetics of filociclovir. Filociclovir (n = 18) or placebo (n = 6) was administered as a daily oral dose (100 mg, 350 mg, or 750 mg) for 7 days to normal healthy adults (ages, 25 to 65 years) who were monitored for 22 days. Safety assessments included clinical, laboratory, and electrocardiogram monitoring. Plasma and urine samplings were used to determine pharmacokinetic parameters. All study product-related adverse events were mild, most commonly gastrointestinal (17%), nervous system (11%), and skin and subcutaneous tissue (11%) disorders. One subject had reversible grade 3 elevation in serum creatinine and bilirubin, which was associated with an ∼1-log increase in plasma filociclovir exposure compared to levels for other subjects in the same (750-mg) cohort. No other serious adverse events were observed. Plasma exposures (area under the concentration-time curve from 0 to 24 h [AUC0–24]) on days 1 and 7 were similar, suggesting negligible dose accumulation. There was a sublinear increase in plasma exposure with dose, which plateaued at the daily dose of 350 mg. The amount of filociclovir recovered in the urine remained proportional to plasma exposure (AUC). Doses as low as 100 mg achieved plasma concentrations sufficient to inhibit CMV in vitro. (This study has been registered at ClinicalTrials.gov under identifier NCT02454699.)
INTRODUCTION
Cytomegalovirus (CMV) can cause significant morbidity and mortality, with severe infections particularly affecting the fetus and immunocompromised patients (1–3). Prevention and treatment can improve clinical outcomes. While several antiviral drugs targeting CMV have been approved (ganciclovir, valganciclovir, cidofovir, foscarnet, and letermovir), these medications have significant limitations related to toxicity, poor bioavailability, and the development of resistance, particularly in the setting of prolonged antiviral use (4). Better-tolerated and safer drugs are needed for CMV treatment.
Filociclovir (MBX-400, cyclopropavir) is an investigational new oral synthetic methylenecyclopropane nucleoside analog (Z)-9-{[2,2-bis-(hydroxymethyl) cyclopropylidene] methyl} guanine. Filociclovir is relatively hydrophilic. Like other nucleoside derivatives, the oral absorption and cellular accumulation of filociclovir are expected to be mediated by nucleoside and possibly other transporters located on the membranes of various tissues (5). Once internalized, nucleoside derivatives are phosphorylated to their active nucleotide forms. The initial (rate-limiting) phosphorylation step to the monophosphate form is catalyzed by the viral enzyme pUL97. Additional phosphorylation steps are then catalyzed by cellular kinases to the di- and triphosphate forms. The triphosphate form inhibits viral DNA polymerase (expression of both phosphoproteins ppUL44 and pp28, essential for viral assembly and replication), which blocks viral replication (6–9). Since uninfected cells do not contain pUL97, they are not expected to phosphorylate filociclovir.
Filociclovir has potent antiviral activity against human CMV in vitro and in animal models. In vitro median effective concentration (EC50) values determined against wild-type human CMV are ∼1.2 μM (range, 0.36 to 1.91 μM) (10). For ganciclovir-resistant strains, the in vitro EC50 values of filociclovir are generally lower than those for ganciclovir (range, 0.04 to 37.2 μM depending on the strains tested) (10), often by a factor of 5 to 10 or more. These data suggest that filociclovir is more potent than ganciclovir and that it has a role in the treatment of patients that do not respond to ganciclovir (11).
Single oral doses of filociclovir ranging from 35 to 1,350 mg were safe and well tolerated in a cohort of 48 healthy participants enrolled in a prior phase 1 study (https://www.clinicaltrials.gov/ct2/show/NCT01433835). The most commonly observed side effects were gastrointestinal adverse events (AEs). There were no clear trends in frequency or intensity of AEs with increasing doses of filociclovir, and dose-limiting toxicity was not reached. Maximum plasma filociclovir concentrations following single-dose administration peaked 1 to 2 h postdose and returned to low/undetectable levels by 24 h postdose. The maximum concentration of drug in serum (Cmax) and area under the concentration-time curve (AUC) increased as filociclovir doses increased from 35 to 1,000 mg, although not in a dose proportional manner, and exposure peaked at a filociclovir dose of 1,000 mg. Most of the filociclovir excreted in the urine occurred within 4 h postdose, with minor amounts recovered between 8 and 96 h.
The objective of the current phase 1 trial was to evaluate the safety and pharmacokinetics (PK) of filociclovir with dose escalation (100, 350, 750, and 1,000 mg) in healthy adults (ages 18 to 65 inclusive) receiving filociclovir versus placebo (3:1 ratio) once daily for 7 consecutive days with full plasma concentrations time profiles obtained following the first and seventh doses of the drug.
RESULTS
Demographics.
A total of 24 subjects were enrolled into this single-center, multiple-ascending-dose escalation clinical trial (Fig. 1). Eighteen received filociclovir, while six received placebo. The majority receiving filociclovir were female (72%) and African-American (56%). The mean age was 47.4 years (standard deviations [SD] of 13.0 and a range of 25 to 65 years). Their mean height was 169.4 cm (SD of 10.1 and a range of 155 to 189 cm). Their mean weight was 76.5 kg (SD of 14.0 and range of 52 to 103 kg). The average body mass index (BMI) was 26.6 kg/m2 (SD of 3.7 and range of 21 to 32.0 kg/m2). The only notable difference among the treatment groups regarding baseline characteristics (Table 1) was the distribution of genders: placebo recipients were 17% female, while filociclovir recipients were 72% female (and similar across dose levels).
FIG 1.

Consort flow diagram.
TABLE 1.
Demographics and baseline characteristics of subjects in the study
| Parameter | Values for each group |
||||
|---|---|---|---|---|---|
| 100 mg (N = 6) | 350 mg (N = 6) | 750 mg (N = 6) | All PK subjects (N = 18) | Placebo (N = 6) | |
| Age, yr | |||||
| Mean (SD) | 50.8 (16.0) | 40.3 (10.8) | 51.0 (10.5) | 47.4 (13.0) | 39.7 (8.8) |
| Min, max | 25, 64 | 27, 51 | 33, 65 | 25, 65 | 28, 52 |
| Sex, n (%) | |||||
| Female | 4 (66.7) | 4 (66.7) | 5 (83.3) | 13 (72.2) | 1 (16.7) |
| Male | 2 (33.3) | 2 (33.3) | 1 (16.7) | 5 (27.8) | 5 (83.3) |
| Race, n (%) | |||||
| Black or African-American | 3 (50.0) | 5 (83.3) | 2 (33.3) | 10 (55.6) | 2 (33.3) |
| Unknown | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) |
| White | 3 (50.0) | 1 (16.7) | 4 (66.7) | 8 (44.4) | 3 (50.0) |
| Height, cm | |||||
| Mean (SD) | 171.3 (10.2) | 172.6 (8.5) | 164.4 (10.9) | 169.4 (10.1) | 180.4 (5.7) |
| Min, max | 160, 189 | 165, 186 | 155, 183 | 155, 189 | 173, 188 |
| Weight, kg | |||||
| Mean (SD) | 73.7 (7.9) | 84.5 (10.0) | 71.4 (19.7) | 76.5 (14.0) | 82.7 (5.1) |
| Min, max | 65, 84 | 68, 98 | 52, 103 | 52, 103 | 75, 89 |
| Body mass index, kg/m2 | |||||
| Mean (SD) | 25.1 (2.3) | 28.4 (3.5) | 26.1 (4.6) | 26.6 (3.7) | 25.4 (1.8) |
| Min, max | 23, 29 | 24, 32 | 21, 31 | 21, 32 | 23, 28 |
Safety.
No serious adverse events were reported in this study. There were 21 nonserious clinical adverse events reported among 14 out of 18 (78%) subjects receiving filociclovir during the period of enrollment through day 22. None of the clinical adverse events was considered to be severe; one unrelated event (physical assault) was considered moderate, and 20 events were considered mild. The most common nonserious adverse event was skin and subcutaneous tissue disorders (three events, two related [rash and generalized pruritus]) and gastrointestinal disorders (three events all related [nausea, esophageal discomfort, and abdominal distension]). Seven events were considered related to the study product, including headache in 2 subjects. Five unrelated clinical adverse events were observed in 4 out of 6 placebo recipients (67%).
Hematologic adverse events (all grade 2 or lower) were observed in 10 subjects (56%) receiving filociclovir with no difference between dose groups and in 5 subjects (83%) receiving placebo. The most common related hematology adverse events reported among the subjects receiving filociclovir were hemoglobin decrease (in 2 subjects [11%]) and decreased leukocytes (in 2 subjects [11%]).
Biochemical adverse events were observed in 17 subjects (94%) receiving filociclovir with no difference between dose groups and in 6 subjects (100%) receiving placebo. The most commonly reported related biochemistry adverse events (all grade 1) were elevation of aspartate aminotransferase (AST; in 3 subjects [17%]), alanine aminotransferase (ALT; in 2 subjects [11%]), and alkaline phosphatase (in 2 subjects [11%]) and decrease in calcium (in 4 subjects [22%]) or potassium (in 3 subjects [17%]) levels. There were no urinalysis abnormalities reported for this study. Two subjects had abnormal but not clinically significant electrocardiogram (ECG) results that were considered unrelated to the study product.
One subject in the 750-mg group had a grade 3 elevation in creatinine and bilirubin about 24 h after the initial dose, prior to receipt of the second dose of filociclovir. The subject was withdrawn from further drug dosing after the second dose and was monitored for safety. The creatinine peaked at day 4 at 2.57 mg/dl (grade 3 laboratory AE), and the bilirubin level peaked at day 3 at 2.6 mg/dl (grade 3 laboratory AE) with direct bilirubin at 0.3 mg/dl. She had no symptoms and her physical exam was normal, including no jaundice or right upper quadrant tenderness. She was managed with aggressive oral hydration, and her laboratory abnormalities returned to baseline without sequelae. The subject had a drug exposure as measured by the area under the plasma concentration versus time curve (AUC0–24) of 46,716 ng·h/ml, which exceeded the predetermined study safety cutoff (20,000 ng·h/ml). Her AUC and Cmax were 11.3-fold and 6.5-fold higher, respectively, than the mean for the other subjects in the cohort. The sponsor stopped dose escalation after predefined halting rules were met. The safety results are detailed in the supplemental material (Tables S1 and S2).
Pharmacokinetics.
Plasma exposures to filociclovir on dosing days 1 and 7 were similar for each cohort. The mean (±SD) accumulation ratios (Ro) were 0.94 (±0.11), 1.03 (±0.40), and 1.19 (±0.74) for the 100-, 350-, and 750-mg cohorts, respectively, suggesting negligible dose accumulation (Fig. 2). AUC0–24 increased sublinearly with increasing dose (Fig. 3) and plateaued around the 350-mg dose. The mean (±SD) AUC0–24 values on day 7 were 2,170.8 (813.8), 4,211.3 (1,393.9), and 3,962.8 (1,307.0) ng·h/ml for the 100-, 350-, and 750-mg cohorts, respectively, and were similar in the 350-mg and 750-mg dosing cohorts (these calculations did not include the subject that did not receive doses after the second day) (Table 2). The mean oral clearance values (CL/F) were 47.2 (±15.3), 96.7 (±61.3), and 195.7 (±179.5) liters/h for the 100-, 350-, and 750-mg cohorts, respectively, for day 1.
FIG 2.

Plasma concentration versus time profiles of filociclovir recipients by dose level and study day. Solid curves and markers represent day 1 concentrations, each coded by subject identifier, while dashed curves and hollow markers represent day 7 concentrations. There were 24 samples below the limits of quantification; these are plotted at 1 ng/ml on the log10-transformed y axis.
FIG 3.

Boxplot summaries of subject AUC0–24, percentage of oral dose recovered in the urine, and renal clearance (Ae/AUC, where Ae is the amount recovered in urine during the dosing interval). Solid symbols represent averages, while open symbols represent individual data points. Bars extend to the minimum and maximum values for the group, whereas the midline, top, and bottom of the box indicate the median, 75th percentile, and 25th percentile values, respectively.
TABLE 2.
Plasma pharmacokinetic parameters of filociclovir by dose level on days 1 and 7
| Parameter | Value by group and daya
|
|||||
|---|---|---|---|---|---|---|
| Day 1 |
Day 7 |
|||||
| 100 mg (N = 6) | 350 mg (N = 6) | 750 mg (N = 6) | 100 mg (N = 6) | 350 mg (N = 6) | 750 mg (N = 5) | |
| Mean AUC0–24, ng·h/ml (%CV) | 2,281.0 (29.7) | 4,832.2 (55.9) | 11,343.0 (153.5) | 2,170.8 (37.5) | 4,211.3 (33.1) | 3,962.8 (33.0) |
| Mean Cmax, ng/ml (%CV) | 547.8 (26.1) | 1,086.0 (41.9) | 2,084.0 (119.1) | 588.5 (25.3) | 855.5 (49.0) | 882.0 (60.1) |
| Mean Tmax, h (Min, max) | 1.835 (1.00, 3.65) | 2.050 (1.43, 3.97) | 2.500 (1.43, 3.87) | 1.618 (0.95, 1.98) | 2.552 (1.98, 3.80) | 1.882 (1.43, 2.07) |
| Mean t1/2, h (%CV) | 3.2080 (10.3) | 3.3532 (15.5) | 3.5905 (21.4) | 3.6220 (17.2) | 3.2635 (9.4) | 3.8974 (12.3) |
| Mean CL/F, liters/h (%CV) | 47.198 (32.3) | 96.750 (63.4) | 195.658 (91.7) | 52.618 (42.4) | 91.268 (33.1) | 210.970 (40.3) |
| Mean CLr, liters/h (%CV) | 15.359 (29.9) | 15.517 (47.1) | 15.602 (36.1) | 16.090 (31.8) | 17.005 (17.2) | 16.978 (17.0) |
| Mean Pctrec, % (%CV) | 34.25 (30.3) | 22.18 (65.7) | 13.82 (76.3) | 32.46 (30.3) | 19.67 (20.9) | 9.07 (40.7) |
| Mean V/F, liters (%CV) | 220.95 (35.8) | 502.08 (83.2) | 1015.37 (105.8) | 286.53 (53.6) | 430.92 (34.1) | 1,205.16 (49.6) |
N, number of subjects in the PK population; AUC, area under the curve; Cmax, maximum observed concentration; Tmax, time to maximum concentration; t1/2, elimination half-life; CL/F, dose/AUC; CLr, Ae/AUC, where Ae is the amount recovered in urine during the dosing interval; Pctrec, 100 × (Ae/dose); V/F, dose/(AUC × ke), where ke is the elimination rate constant.
The CLr (amount recovered in urine/AUC0–24, means ± SD) were similar on day 1 (15.4 ± 4.6, 15.5 ± 7.3, and 15.6 ± 5.6 liters/h) and day 7 (16.1 ± 5.1, 17.0 ± 2.9, and 17.0 ± 2.9 liters/h) for the 100-, 350-, and 750-mg cohorts, respectively.
DISCUSSION
This phase 1, double-blind, randomized, placebo-controlled, single-center, multiple-ascending-dose trial was the second human study with filociclovir conducted in healthy CMV-uninfected subjects.
While filociclovir was generally well tolerated, one subject receiving 750 mg daily for 2 days developed a grade 3 elevation in creatinine and bilirubin levels that was considered to be related to increased exposure to the drug relative to levels for other subjects in the cohort, which triggered individual halting rules for that subject and prevented dose escalation to the 1,000-mg cohort. The subject fully recovered. In rat toxicology studies (n = 2), there were no clinical or histopathologic changes in the kidney or the liver at any dose administered (up to 300 mg/kg of body weight as a single dose and 100 mg/kg as a multiple dose). In toxicology studies in dogs (n = 4), increased blood urea nitrogen (BUN) and creatinine were observed at higher doses. These changes were associated with histopathologic observations in the kidney. Clinical pathology parameters as well as decreased oral intake showed that the animals were likely dehydrated and that there is likely a prerenal component to the toxicity. There were no changes in liver enzymes observed at any dose following oral administration of filociclovir. These findings identified the kidney as a target organ and suggested that the liver is not a target organ.
Preclinical studies and data from a single-dose human study (https://www.clinicaltrials.gov/ct2/show/NCT01433835) suggest that filociclovir is eliminated primarily via renal excretion. CLr remained similar at the plasma concentrations achieved in the trial, and the subject with the elevated plasma AUC0–24 also had the largest amount of administered drug recovered in the urine (Fig. 3). Since filociclovir is primarily eliminated in the urine and the CL of the subject with the high exposure remained similar to that of the other subjects, the elevated AUC0–24 may result from enhanced absorption rather than reduced drug elimination. Incorrect dosing was excluded and plasma concentration data were verified by reanalysis. The possibility of an unusual nucleoside transporter phenotype in the subject cannot be excluded. Other antiherpetic acyclic nucleosides, including acyclovir and valacyclovir, also demonstrate saturable oral absorption (12). In a study with wild-type and PepT1 knockout mice, Yang et al. reported saturable oral absorption of valacyclovir at clinically relevant doses, which was markedly reduced in knockout mice lacking the PepT1 proton-coupled oligopeptide transporter (13). Although the precise mechanism behind the isolated case of increased exposure remains uncharacterized, physicochemical characterization studies indicated that the solubility of filociclovir increases at low pH. Therefore, it is also possible that the subject’s increased absorption was related to an abnormally low gastric pH. Improving oral bioavailability of filociclovir in the future would minimize the risk of enhanced exposure for certain subjects with this potential interindividual variability.
The in vivo potency of antiviral nucleoside agents is related to the pharmacokinetics of the agent, its cellular accumulation and phosphorylation to the active nucleotide triphosphate (NTP) form, the cellular stability of the NTP, and its binding affinity with the CMV polymerase target (14, 15). The mean plasma concentrations (AUC0–24/dose interval) between doses exceeded the in vitro 90% inhibitory concentration (IC90) for doses of ≥100 mg per day (Fig. 4). Comparison of the median between dose plasma concentrations with in vitro IC90 may be a reasonable predictor of in vivo efficacy, as antiviral nucleoside derivatives do not bind significantly to plasma proteins. However, PK profiles of the unphosphorylated form of the drug in plasma may underestimate in vivo efficacy, as the active triphosphate form of filociclovir in CMV-infected cells (not measureable in uninfected individuals) is expected to decay less rapidly between doses than concentrations of filociclovir in plasma. Compared to that of ganciclovir, filociclovir has a 2-fold lower in vitro IC50 (11, 16, 17), a 2-fold higher peak triphosphate level in infected cells (18), and an ∼10-fold higher affinity of the nucleotide triphosphate form to CMV polymerase (6).
FIG 4.
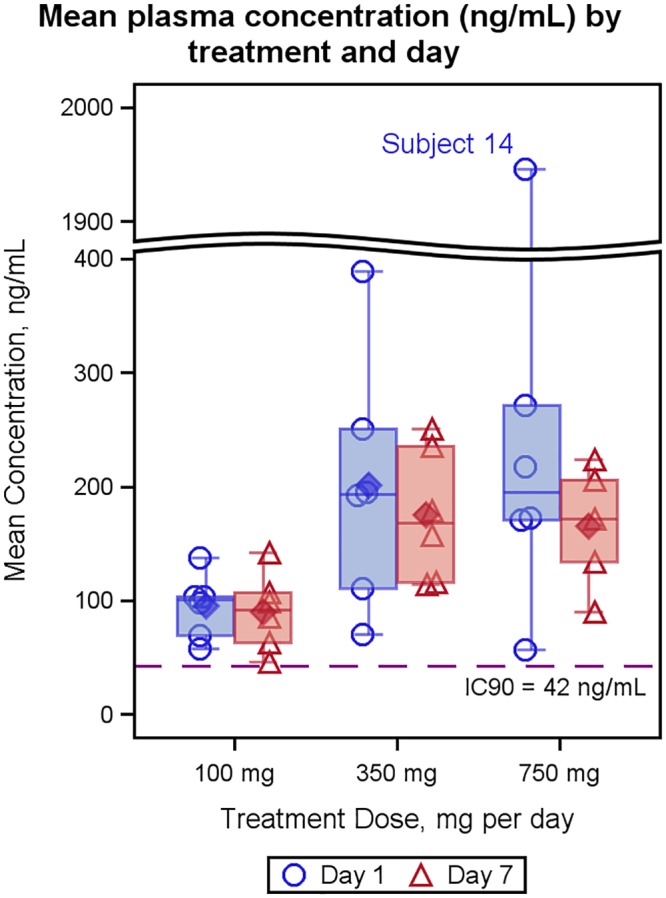
Mean plasma concentration by treatment and day. Solid symbols represent averages, while open symbols represent individual data points. Bars extend to the minimum and maximum values for the group, whereas the midline, top, and bottom of the box indicate the median, 75th percentile, and 25th percentile values, respectively. The IC90 (in nanograms per milliliter) was calculated by multiplying the in vitro IC90 (micromolars) by the molecular weight of filociclovir (MBX-400; 263.257 g/mol).
Seven days of single-dose filociclovir treatment demonstrated that exposures from doses as low as 100 mg may be adequate to achieve efficacy, and doses can be safely administered with close monitoring.
MATERIALS AND METHODS
Participants.
Subjects were required to be between the ages of 18 and 65 years with a BMI between 18.0 and 32.0 kg/m2, with either no ability to reproduce or no sexual behavior that could result in fathering or conceiving a child in the future (while the reproductive risks of filociclovir have not been thoroughly assessed, azoospermia is a possible risk given its occurrence in closely related drugs, such as ganciclovir). Subjects were also required to be in good health, as determined by the absence of clinically significant findings from medical history, electrocardiogram, clinical laboratory assessments, vital sign measurements, and physical exams.
Subjects were excluded if they had active CMV, HIV, or hepatitis B and C infections, known allergy to the components of filociclovir or placebo capsules (microcrystalline cellulose, gelatin, and titanium dioxide), or history of anaphylaxis or other serious adverse reactions to nucleoside analog medications. They were also excluded if they received any drug active against herpesviruses within 30 days, received any chronic medication, had a significant gastrointestinal disorder affecting absorption, or reported ongoing drug abuse/dependence, including alcohol (or a history of this within 5 years of enrollment), smoking (or a history of this within 3 months of enrollment), or a positive urine drug screen. Further reasons for exclusion were significant psychiatric history, blood donation within 30 days before initiation of dosing or planning to donate blood during the study, and receipt of any investigational agent within 28 days before initiation of dosing. Receipt of any drug or alcohol, grapefruit, and any caffeine-containing products was also not allowed within 72 h prior to dosing.
Study design.
The study is a single-center, double-blind, randomized, placebo-controlled, dose escalation phase 1 trial. The study protocol and informed consent forms were reviewed and approved by the Emory University Institutional Review Board and the U.S. Food and Drug Administration. At the screening visit, subjects were educated and provided written consent to participate in the study.
Four cohorts of 8 subjects each (6 receiving filociclovir and 2 receiving placebo) were planned to be enrolled sequentially. Filociclovir or placebo was administered orally once daily between days 1 and 7 by study staff while the subject was in an inpatient research unit. The study doses planned in these sequential cohorts were 100, 350, 750, and 1,000 mg daily. Subjects were admitted to the research unit 1 day prior to the first dosage and remained in the unit until the day after the 7th daily dosage. Subjects returned for outpatient follow-up visits at days 9, 10, 14, and 22. The study duration for each subject was approximately 80 days (screening period, administration of daily dose, and follow-up). Subjects fasted for 1 h prior to each dose until 2 h after each dose.
Study products.
Filociclovir powder is a white to off-white crystalline powder manufactured for Microbiotix (Worcester, MA). Filociclovir was supplied as 100-mg (size 3, white opaque) and 250-mg (size 0, white opaque) hard gelatin capsules, while the placebo capsules were identical in appearance and weight but contained microcrystalline cellulose in place of the active ingredient.
Safety assessments.
Evaluated safety parameters included general safety (adverse events [AEs]), vital sign measurements, physical examination findings, laboratory test results (complete blood count [CBC] with differential, prothrombin time [PT], partial thromboplastin time [PTT], and sodium, potassium, calcium, glucose, creatinine, BUN, albumin, protein, bilirubin, AST, ALT, and alkaline phosphate levels), urinalysis, and ECG findings (PR, QRS, QT, QTcB [Bazett’s formula], and QTcF [Fridericia’s formula]). Laboratory safety tests were performed at screening, predosing on day 1, and on days 2, 4, 7, 14, and 22, with the 12-lead ECG performed at screening, predosing, and 2 h postdosing on day 1 and 2 h postdose on days 4 and 7.
Pharmacokinetic assessments.
Blood samples to measure plasma filociclovir concentrations were collected before day 1 dosing and postdosing at 0.5, 1, 1.5, 2, 4, 6, 8, and 24 h. On day 4, PK samples were obtained at baseline predosing and postdosing at 2 h only. On day 7, PK samples were obtained at baseline predosing and postdosing at 0.5, 1, 1.5, 2, 4, 6, 8, 24 (day 8), 30 (day 8), 48 (day 9), and 72 h (day 10). Subjects were offered the option of venipuncture or placement of an indwelling intravenous catheter for inpatient PK sampling.
Urine PK samples were collected for the day 1 and day 7 doses, at baseline predosing, and across the following intervals: 0 to 4 h, 4 to 8 h, 8 to 12 h, and 12 to 24 h after dosing. In addition, urine for PK samples were also collected 24 to 48 and 48 to 72 h after the last dose.
The derived PK parameters were evaluated for area under the concentration-time curve from 0 to 24 h (AUC0–24), maximum observed concentration (Cmax), time to maximum concentration (Tmax), elimination rate constant (λz), elimination half-life (t1/2), clearance (CL/F), urinary clearance (CLr), volume of distribution (Vz/F), and accumulation ratio (Ro).
The analytical chemistry assays for drug concentrations in plasma and urine were performed by Frontage Laboratories, Inc. (Hackensack, NJ), using a validated method of high-performance liquid chromatography with tandem mass spectrometry (LC-MS/MS). Standard curves were developed using eight nonzero concentrations of filociclovir, ranging from 1 ng/ml to 1,000 ng/ml in plasma and 5 ng/ml to 5,000 ng/ml in urine, with the minimum concentration representing the LLOQ (lower limit of quantification) for each sample type.
Statistical analysis.
All subjects who received study product were included in the safety population. For continuous variables, descriptive statistics included the number of nonmissing values and mean, standard deviation, median, minimum, and maximum values. For categorical variables, descriptive statistics included counts and percentages per category.
The number of subjects experiencing adverse events classified using the Medical Dictionary for Regulatory Activities (MedDRA) System Organ Classes (SOC) and Preferred Terms are reported throughout the study (19). The severity and relationship to study drug of reported adverse events were also assessed. The rate and exact 95% confidence intervals of related AEs in aggregate, and by MedDRA categories, were computed.
Clinical laboratory (chemistry, hematology, coagulation, and urinalysis) and ECG data presented as safety endpoints were summarized for each evaluation time point, as well as for changes from baseline. The clinical laboratory evaluations were graded (mild, moderate, and severe) as laboratory AEs and were considered for the study or individual halting rules for any of the following: sodium, potassium, glucose, blood urea nitrogen, creatinine, calcium, albumin, total protein, aspartate aminotransferase, alanine aminotransferase, total bilirubin, hemoglobin, white blood cell, lymphocytes, neutrophils, platelets, prothrombin time, partial thromboplastin time, and glucose, protein, and blood in urine.
Data for subjects randomized to filociclovir were grouped by dosing cohort. Data for all placebo subjects, regardless of dosing cohort, were treated as one group. All subjects having at least one measurable drug concentration were included in the PK population. Concentration data for plasma were described both graphically and quantitatively. Time-concentration plots were generated for each subject. Additionally, a plot using mean concentrations at protocol time points was generated. Summary statistics for concentrations in plasma and urine were presented in tabular form.
WinNonlin 6.4 was used to perform linear-up log-down noncompartmental analysis of the dose 1 and dose 7 plasma samples collected from predosing to 24 h postdose. Estimation of λz (terminal half-life) was performed according to WinNonlin’s iterative process that selects the set of concentrations collected during the terminal phase (after Tmax) that maximizes the adjusted R2 value. Estimation of λz required at least three concentrations measured above the LLOQ. For estimation of PK parameters using noncompartmental analysis, predose sampling times were set to zero for dose 1. Boxplots of select PK parameters were generated for day 1 and day 7 by dosing cohort where the midline, top, and bottom of the box indicate the median, 75th percentile, and 25th percentile values, respectively, a solid diamond marks the mean, and the whiskers extend to the maximum and minimum values. For calculated summary statistics (mean, median, SD, coefficient of variation [CV], and geometric mean) of plasma and urine concentrations, results below the LLOQ were imputed as LLOQ/2.
Statistical analyses were performed using SAS (version 9.4; SAS Institute, Cary, NC).
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge our participants and the following Hope Clinic staff: Dawn Battle, Mary Bower, Lyngail Cooper, Ellen DeStefano, Michele Paine McCullough, Briyana Domjahn, Melinda Ogilvie, JoAnn Sadowski, Alicarmen Alvarez, Juton Winston, Tigisty Girmay, Jean Winter, Eileen Osinski, Emily Presmanes, Eric Johnson, Bruce Ribner, David Rimland, Talib Sirajud-Deen, Hannah Houston, Dean Kleinhenz, Jianguo Xu, Susan Rogers, April Ross, Pamela Turner, Lilin Lai, the Georgia CTSA nurses and staff, Barry Clark, Nadine Hines, Lucy Kugbila, Monica Lalor, Sandra Leonard, Shawanda Carter, Carmile Jerome, Debora Clem, Jennifer Shamloo, the DMID Medical Monitor, Venus Shahamatdar, pharmacist Michelle Wildman, and Walla Dempsey.
T.L.B. and J.B. are employees of Microbiotix, the manufacturer of the study product. G.D., B.O., and S.Y.C. are employees of the NIH, the study sponsor. All other authors have no conflicts of interest.
All authors contributed to the study design. N.G.R. and S.J.H. wrote the manuscript. N.G.R., S.J.H., M.J.M., C.A., and C.F. analyzed and interpreted data. N.G.R., M.H., A.B., E.J.A., and M.J.M. collected clinical data. N.G.R. and M.J.M. supervised the study. C.F. and C.A. provided statistical analyses.
This work was funded by NIH/NIAID/DMID awards to the Emory Vaccine and Treatment Evaluation Units (VTEU), HHSN272201300018I, HHSN27200003, and HHSN27200018, supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under award number UL1TR002378. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00717-19.
REFERENCES
- 1.Kenneson A, Cannon MJ. 2007. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol 17:253–276. doi: 10.1002/rmv.535. [DOI] [PubMed] [Google Scholar]
- 2.Razonable RR, Humar A, AST Infectious Diseases Community of Practice. 2013. Cytomegalovirus in solid organ transplantation. Am J Transplant 13(Suppl 4):93–106. doi: 10.1111/ajt.12103. [DOI] [PubMed] [Google Scholar]
- 3.Gallant JE, Moore RD, Richman DD, Keruly J, Chaisson RE. 1992. Incidence and natural history of cytomegalovirus disease in patients with advanced human immunodeficiency virus disease treated with zidovudine. The Zidovudine Epidemiology Study Group. J Infect Dis 166:1223–1227. doi: 10.1093/infdis/166.6.1223. [DOI] [PubMed] [Google Scholar]
- 4.Fisher CE, Knudsen JL, Lease ED, Jerome KR, Rakita RM, Boeckh M, Limaye AP. 2017. Risk factors and outcomes of ganciclovir-resistant cytomegalovirus infection in solid organ transplant recipients. Clin Infect Dis 65:57–63. doi: 10.1093/cid/cix259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Young JD, Yao SY, Baldwin JM, Cass CE, Baldwin SA. 2013. The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol Aspects Med 34:529–547. doi: 10.1016/j.mam.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 6.Chen H, Li C, Zemlicka J, Gentry BG, Bowlin TL, Coen DM. 2016. Potency and stereoselectivity of cyclopropavir triphosphate action on human cytomegalovirus DNA polymerase. Antimicrob Agents Chemother 60:4176–4182. doi: 10.1128/AAC.00449-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gentry BG, Kamil JP, Coen DM, Zemlicka J, Drach JC. 2010. Stereoselective phosphorylation of cyclopropavir by pUL97 and competitive inhibition by maribavir. Antimicrob Agents Chemother 54:3093–3098. doi: 10.1128/AAC.00468-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gentry BG, Gentry SN, Jackson TL, Zemlicka J, Drach JC. 2011. Phosphorylation of antiviral and endogenous nucleotides to di- and triphosphates by guanosine monophosphate kinase. Biochem Pharmacol 81:43–49. doi: 10.1016/j.bcp.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 9.Li C, Gentry BG, Drach JC, Zemlicka J. 2009. Synthesis and enantioselectivity of cyclopropavir phosphates for cellular GMP kinase. Nucleosides Nucleotides Nucleic Acids 28:795–808. doi: 10.1080/15257770903172720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chemaly RF, Hill JA, Voigt S, Peggs KS. 2019. In vitro comparison of currently available and investigational antiviral agents against pathogenic human double-stranded DNA viruses: a systematic literature review. Antiviral Res 163:50–58. doi: 10.1016/j.antiviral.2019.01.008. [DOI] [PubMed] [Google Scholar]
- 11.Komazin-Meredith G, Chou S, Prichard MN, Hartline CB, Cardinale SC, Comeau K, Williams JD, Khan AR, Peet NP, Bowlin TL. 2014. Human cytomegalovirus UL97 kinase is involved in the mechanism of action of methylenecyclopropane analogs with 6-ether and -thioether substitutions. Antimicrob Agents Chemother 58:274–278. doi: 10.1128/AAC.01726-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steingrimsdottir H, Gruber A, Palm C, Grimfors G, Kalin M, Eksborg S. 2000. Bioavailability of aciclovir after oral administration of aciclovir and its prodrug valaciclovir to patients with leukopenia after chemotherapy. Antimicrob Agents Chemother 44:207–209. doi: 10.1128/aac.44.1.207-209.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang B, Hu Y, Smith DE. 2013. Impact of peptide transporter 1 on the intestinal absorption and pharmacokinetics of valacyclovir after oral dose escalation in wild-type and PepT1 knockout mice. Drug Metab Dispos 41:1867–1874. doi: 10.1124/dmd.113.052597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hurwitz SJ, Schinazi RF. 2012. Practical considerations for developing nucleoside reverse transcriptase inhibitors. Drug Discov Today Technol 9:e183–e193. doi: 10.1016/j.ddtec.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hurwitz SJ, Schinazi RF. 2013. Prodrug strategies for improved efficacy of nucleoside antiviral inhibitors. Curr Opin HIV AIDS 8:556–564. doi: 10.1097/COH.0000000000000007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kern ER, Kushner NL, Hartline CB, Williams-Aziz SL, Harden EA, Zhou S, Zemlicka J, Prichard MN. 2005. In vitro activity and mechanism of action of methylenecyclopropane analogs of nucleosides against herpesvirus replication. Antimicrob Agents Chemother 49:1039–1045. doi: 10.1128/AAC.49.3.1039-1045.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.James SH, Hartline CB, Harden EA, Driebe EM, Schupp JM, Engelthaler DM, Keim PS, Bowlin TL, Kern ER, Prichard MN. 2011. Cyclopropavir inhibits the normal function of the human cytomegalovirus UL97 kinase. Antimicrob Agents Chemother 55:4682–4691. doi: 10.1128/AAC.00571-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gentry BG, Drach JC. 2014. Metabolism of cyclopropavir and ganciclovir in human cytomegalovirus-infected cells. Antimicrob Agents Chemother 58:2329–2333. doi: 10.1128/AAC.02311-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. 2019. Medical dictionary for regulatory activities. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, Geneva, Switzerland. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.