

Nontuberculous mycobacteria (NTM) are highly drug-resistant, opportunistic pathogens that can cause pulmonary disease. The outcomes of the currently recommended treatment regimens are poor, especially for Mycobacterium abscessus. New or repurposed drugs are direly needed. Auranofin, a gold-based antirheumatic agent, was investigated for Mycobacterium tuberculosis. Here, we test auranofin against NTM in vitro and ex vivo. We tested the susceptibility of 63 NTM isolates to auranofin using broth microdilution.
KEYWORDS: drug discovery, drug susceptibility, mycobacteria, Mycobacterium abscessus, pharmacodynamics
ABSTRACT
Nontuberculous mycobacteria (NTM) are highly drug-resistant, opportunistic pathogens that can cause pulmonary disease. The outcomes of the currently recommended treatment regimens are poor, especially for Mycobacterium abscessus. New or repurposed drugs are direly needed. Auranofin, a gold-based antirheumatic agent, was investigated for Mycobacterium tuberculosis. Here, we test auranofin against NTM in vitro and ex vivo. We tested the susceptibility of 63 NTM isolates to auranofin using broth microdilution. Next, we assessed synergy between auranofin and antimycobacterial drugs using the checkerboard method and calculated the fractional inhibition concentration index (FICI). Using time-kill kinetics assays (TK), we assessed pharmacodynamics of auranofin alone and in combination with drug combinations showing the lowest FICIs for M. abscessus CIP 104536. A response surface analysis was used to assess synergistic interactions over time in TKs. Primary isolated macrophages were infected with M. abscessus and treated with auranofin. Finally, using KEGG Orthology, we looked for orthologues to auranofins drug target in M. tuberculosis. M. abscessus had the lowest auranofin MIC50 (2 μg/ml) among the tested NTM. The lowest average FICIs were observed between auranofin and amikacin (0.45) and linezolid (0.50). Auranofin exhibited concentration-dependent killing of M. abscessus, with >1-log killing at concentrations of >2× MIC. Only amikacin was synergistic with auranofin according to Bliss independence. Auranofin could not lower the intracellular bacterial load in macrophages. Auranofin itself may not be feasible for M. abscessus treatment, but these data point toward a promising, unutilized drug target.
INTRODUCTION
Nontuberculous mycobacteria (NTM) are opportunistic pathogens that mainly cause pulmonary infections in susceptible patients (1). NTM can be divided into rapidly growing mycobacteria (RGM) and slowly growing mycobacteria (SGM), of which Mycobacterium abscessus and Mycobacterium avium complex (MAC) most frequently cause human disease worldwide (2, 3). Diseases caused by NTM are challenging to treat, as NTM exhibit high levels of inherent resistance to most antibiotics (1, 4, 5).
The recommended regimens for treatment of M. abscessus, as well as MAC, are based on experience or single-center observational studies and trials rather than pharmacokinetic/pharmacodynamic studies (6–9). Because of low cure rates (MAC, 50 to 70%; M. abscessus, <50%) and high recurrence rates (50% for both MAC and M. abscessus), regimens need to be improved (8, 10). Evidence found in in vitro studies shows that especially in treating M. abscessus infections, antimicrobial chemotherapy fails both because of low bactericidal activity (11) and because of the swift emergence of antibiotic resistance (12). Thus, new antibiotics exploiting new drug targets are direly needed (13).
In a recent publication, Maier et al. (14) screened commonly used pharmaceuticals that do not have clear antibiotic indication for activity against 38 strains of human gut-associated bacteria. In this screening, these authors identified auranofin (AUR; a gold complex used as an antirheumatic agent) as an antimicrobial agent inhibiting 36 of the 38 tested strains. AUR has been in investigated for other indications earlier and shows a broad activity against a variety of diseases, including cancer (15). The potential of AUR to act as an antimycobacterial agent was suggested in another study that demonstrated its activity against Mycobacterium tuberculosis (H37Ra reference strain), for which the authors report an MIC of 0.5 μg/ml (16).
AUR inhibits the thioredoxin reductase enzyme (TrxR) in vitro (17). The active site of TrxR has a redox-active dithiol group that is essential for the survival of M. tuberculosis under oxidative stress. Inhibition of TrxR by AUR leads to a disruption of the thiol-redox homeostasis (17, 18). This balance is responsible for the maintenance of the intracellular environment and the regulation of redox enzymes and proteins by oxidoreduction, as well as the detoxification of reactive oxygen species (19). Therefore, AUR may be a sensitizing agent for oxidative stress (17, 18, 20). This unique mechanism of action, as well as the proven susceptibility of M. tuberculosis to AUR, encourages further investigation of AUR’s potential activity against other mycobacteria. In the present study, we investigated the effect of AUR on clinically relevant NTM species.
RESULTS
MIC/MBC determination.
The MIC range and the MICs of reference strains are given in Table 1 . A detailed per-isolate overview can be found in Table S1 in the supplemental material. Generally, RGM were more susceptible to auranofin than SGM. The AUR MIC50 for M. abscessus is 2 μg/ml, and the AUR MIC90 is 4 μg/ml. The MIC distribution for M. abscessus is shown in Fig. 1. The MIC50 and MIC90 for MAC are 64 μg/ml. For both MAC and M. abscessus, bacterial growth was visible on all MBC plates in MBC determination assays. The MBC/MIC ratios are >64 for M. abscessus and >8 for MAC, indicating a bacteriostatic effect. Stability testing revealed AUR to be stable for 7 days but showed a 4-fold-higher MIC after preincubation for 14 days (Table S2).
TABLE 1.
MIC ranges of auranofin for different strains of nontuberculous mycobacteria and M. tuberculosis
| Species | No. of isolates | MIC (μg/ml) in CAMHa
|
|
|---|---|---|---|
| Reference strain MIC | Clinical isolate MIC range | ||
| M. abscessus | 39 | 4 | <0.25–32 |
| M. fortuitum | 4 | 4 | 1 |
| M. chelonae | 2 | 0.5–4 | |
| M. peregrinum | 1 | 4 | |
| M. avium | 5 | 32 | 32–64 |
| M. intracellulare | 3 | 64 | 8−128 |
| M. chimaera | 4 | 64 | 64 |
| M. simiae | 1 | 256 | |
| M. kansasii | 4 | 32–64 | |
| M. tuberculosisb | 4 | 4 | 4–8 |
CAMH, cation-adjusted Muller-Hinton broth.
M. tuberculosis isolates were tested in Middlebrook 7H9 broth.
FIG 1.
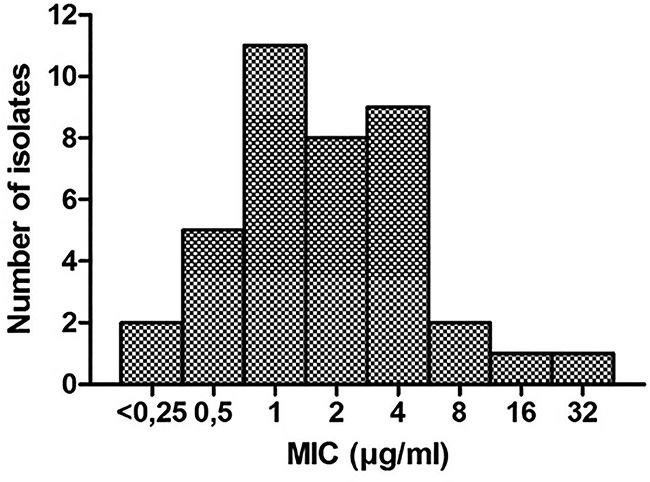
MIC distribution of auranofin for 39 M. abscessus isolates.
Synergy determination.
Calculated fractional inhibition concentration index (FICI) values for auranofin and selected antimycobacterial drugs are shown in Table 2 for RGM and in Table 3 for SGM. We found unadulterated synergy only in combination with AMK for M. abscessus (FICI = 0.45), but other individual isolates showed synergistic interactions, especially for RGM. Synergy between AUR and CLR was rapidly abolished by induction of CLR resistance mediated by the erm(41) gene in M. abscessus.
TABLE 2.
FICI values for auranofin combinations against four M. abscessus isolates (including reference strain CIP 104536), as well as M. fortuitum and M. peregrinum reference strains
| Combination | FICI |
|||
|---|---|---|---|---|
|
M. abscessus |
M. fortuitum ATCC 6841 | M. peregrinum ATCC 700686 | ||
| Avg FICI | Range | |||
| Auranofin-clarithromycin | 0.61 | 0.38−1.00 | 1 | 0.25 |
| Auranofin-amikacin | 0.45 | 0.31–0.5 | 0.53 | 0.50 |
| Auranofin-clofazimine | 0.56 | 0.38−1.50 | 1.5 | 0.75 |
| Auranofin-cefoxitin | 0.63 | 0.38–0.75 | 0.28 | 0.75 |
| Auranofin-linezolid | 0.50 | 0.38–0.75 | 1 | 1 |
TABLE 3.
FICI values for auranofin combinations against MAC reference strains
| Combination | FICI |
||
|---|---|---|---|
| M. avium ATCC 700898 | M. intracellulare DSM 43223 | M. chimaera DSM 44623 | |
| Auranofin-clarithromycin | 1.5 | 0.75 | 1.25 |
| Auranofin-clofazimine | 1.5 | 1 | 0.53 |
| Auranofin-amikacin | 1.5 | 1.25 | 1.25 |
| Auranofin-ethambutol | 1.25 | 1.25 | 1.125 |
| Auranofin-rifampin | 1 | 0.53 | 0.375 |
Time-kill kinetics assays.
Kill curves of the dose-response time-kill kinetics of AUR against M. abscessus are shown in Fig. 2. For concentrations of 1× MIC or higher, we observe an initial drop of at least 1 log in the bacterial load (>90% killing). A subsequent increase in CFU counts occurred under all conditions.
FIG 2.

Dose-response time-kill kinetics of AUR against M. abscessus CIP 104536 at an MIC of 4 μg/ml.
The time-kill curves for the AUR and linezolid (LNZ) combination TK assay are shown in Fig. 3a. Although the 1× MIC and 2× MIC combination conditions consistently performed better than the single-drug conditions, only the 2× MIC combination could lower the bacterial burden below stasis. Sustained growth was observed under all conditions by day 10. The proportion of the 5× MIC LNZ-resistant population is shown in Fig. 3b. M. abscessus has a 1 to 10% baseline resistance against 5× MIC LNZ. Here again, the combination conditions have the highest percentage of resistant CFU, and the AUR conditions show the smallest LNZ-resistant population, indicating a protective effect of AUR against LNZ resistance. The proportion of the 3× MIC AUR-resistant population is shown in Fig. 3c. M. abscessus shows a baseline resistance against 3× MIC AUR of around 40%. It is also apparent that the combination therapy has most resistant CFU against AUR, but no distinct clusters are apparent.
FIG 3.

Auranofin-linezolid combination time-kill kinetics against M. abscessus CIP 104536. (a) CFU counts of the monotherapies and combination therapy of AUR (MIC = 4 μg/ml) and LNZ (MIC = 32 μg/ml). (b) Percentages of CFU that are resistant to 5× MIC LNZ. (c) Percentages of CFU that are resistant to 3× MIC AUR.
The time-kill curves of the AUR and AMK combination TK assay are shown in Fig. 4a. Until day 10, the 1× MIC and 2× MIC drug combinations remained below stasis, with a maximum 2-log10 CFU drop (>99% kill). After day 10, the CFU counts steadily increased under all conditions. The proportion of the 5× MIC AMK-resistant population is shown in Fig. 4b. M. abscessus has low baseline resistance against AMK; auranofin does not seem to induce AMK resistance but also does not protect against it in AUR-AMK combination conditions. The proportion of the 3× MIC AUR-resistant population is shown in Fig. 4c. As for LNZ, no clustering of resistance patterns between different conditions is observable; all conditions have highly AUR-resistant populations of around 100%. Also, AMK seems to induce AUR resistance slightly.
FIG 4.

Auranofin-amikacin combination time-kill kinetics against M. abscessus CIP 104536. (a) CFU counts of the monotherapies and combination therapy of AUR (MIC = 4 μg/ml) and AMK (MIC = 32 μg/ml). (b) Percentages of CFU that are resistant to 5× MIC AMK. (c) Percentages of CFU that are resistant to 3× MIC AUR.
Response surface analysis.
The sigmoidal Emax curve for auranofin on M. abscessus is shown in Fig. S1 in the supplemental material. The fitted Emax was 212.01 log10 CFU/ml ⋅ day, the 50% effective concentration was 9.11× MIC, and the estimated Hill slope was 0.63. The calculated ΔE percentages, as well as Ecomb,obs, Ecomb,BI, and the effect sizes of the companion drugs, in the combination TK assays are shown in Table 4. The combination of AUR and LNZ had no interaction, showing an ΔE of ±10%. The 0.5× MIC combination of AUR and AMK was antagonistic, with an ΔE of −20%, but both the 1× MIC and the 2× MIC combinations were synergistic, with ΔE values of 77.24 and 31.61%, respectively.
TABLE 4.
Response surface analysis results, as well as the interaction effect size, as determined by deviation from the expected combination effect under Bliss independence in percent ΔE
| Combination (MIC) | Effect (log10 CFU/ml ⋅ day) |
||||
|---|---|---|---|---|---|
| AUR | Companion | Observed, combination | Expected, combinationa | ΔE (%) | |
| AUR-LNZ (0.5×) | 14.19 | 14.64 | 24.80 | 27.85 | −10.95 |
| AUR-LNZ (1×) | 23.74 | 21.78 | 42.29 | 43.09 | −1.85 |
| AUR-LNZ (2×) | 37.50 | 35.62 | 74.79 | 66.82 | 11.93 |
| AUR-AMK (0.5×) | 18.41 | 12.04 | 23.44 | 29.41 | −20.30 |
| AUR-AMK (1×) | 21.00 | 19.67 | 68.62 | 38.71 | 77.24 |
| AUR-AMK (2×) | 40.69 | 29.45 | 84.87 | 64.48 | 31.61 |
Under Bliss independence.
Ex vivo intracellular assays.
Intracellular CFU counts of M. abscessus are shown in Fig. 5. On day 1, the CFU counts of M. abscessus inside macrophages treated with AUR decreased slightly, but by days 2 and 3, that difference was no longer visible. Also, the proportion of extracellular M. abscessus is higher under conditions treated with AUR than under control conditions.
FIG 5.
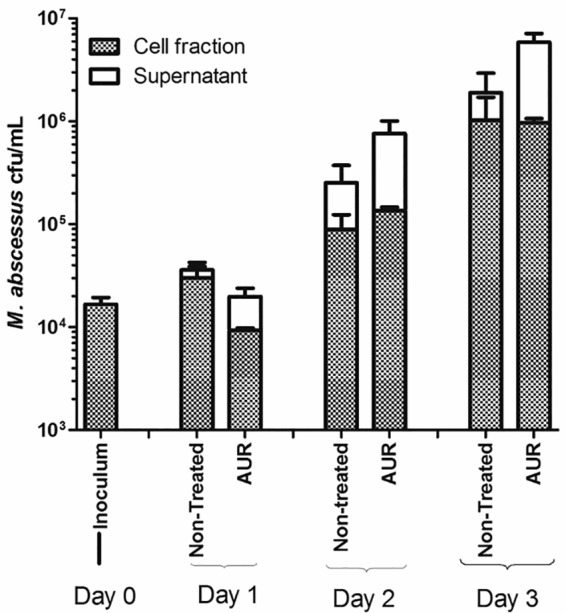
Intracellular (cell fraction) and extracellular CFU counts (in the supernatant) of macrophages infected with M. abscessus CIP 104536 (MOI of 1) and treated with 2× MIC of AUR (8 μg/ml).
Orthologue search.
Orthologues in the KEGG database were found under entries MAB_4940 for M. abscessus, MSMEG_1516 for M. smegmatis, b0888 for E. coli, OCU_21510 for M. intracellulare, ML2703 for M. leprae, MKAN_14755 for M. kansasii, and MAV_5301 for M. avium. The calculated phylogeny tree is given in Fig. S2 in the supplemental material. Generally, all mycobacterial TRXB2 orthologues are very similar, with E. coli being very distinct.
DISCUSSION
We determined the potential of auranofin as a new antibiotic modality in NTM treatment. In vitro, AUR shows promising activity against M. abscessus but not against M. avium complex. Against M. tuberculosis, we determined MICs higher than the 0.5 μg/ml reported by Harbut et al. (17). This discrepancy might be due to batch variations of auranofin or slight deviations in MIC determination, which are inherent to these assays (21). Surprisingly, auranofin is especially effective against M. abscessus, but not M. avium isolates, as evidenced by our MIC data. Both organisms have a Trxb2 orthologue, which proves well conserved within mycobacteria and serves a similar function in a similar pathway (see Fig. S2 in the supplemental material). The only NTM where evidence for the TRXB protein is available on protein level is M. smegmatis, for which high similarity both in gene clustering and in protein sequence is found compared to M. leprae and M. tuberculosis (22). Since the target is conserved throughout different species of mycobacteria, differences in cell wall structure, content, and therefore permeability may be the prime reason we observed such large differences in AUR susceptibility between NTM species (23).
The MIC50 of auranofin against M. abscessus is 2 μg/ml. To exert its bacteriostatic effect in vivo, these or higher concentrations need to be reached at the site of infection, for example in bronchial epithelial lining fluid (ELF) in the case of M. abscessus pulmonary disease. For AUR, pharmacokinetic studies are largely absent, and the plasma/ELF penetration ratio is unknown. Capparelli et al. performed a small phase 1 clinical trial on 15 healthy individuals, administering 6 mg of auranofin orally daily for 7 days and measuring plasma gold concentrations. These researchers found a mean auranofin Cmax of 0.312 μg/ml in plasma (24), which is far below the MIC50 of 2 μg/ml, but this plasma concentration might still be effective for individual M. abscessus isolates. Using Monte-Carlo simulations, Capparelli et al. were able to project a Cmax gold plasma concentration for a 21-mg daily oral auranofin dose and determined the Cmax to be between 0.4 and 1.6 μg/ml (24), which might be enough to inhibit 40 to 50% of M. abscessus isolates, according to our MIC distribution and provided that similar concentrations can be reached at the site of infection. This dose has never been tested clinically.
Antimycobacterial drugs should accumulate in, or at least penetrate, macrophages and, more specifically, their lysosomes, where mycobacteria also reside (25, 26). Based on our ex vivo monocyte infection experiments, auranofin does not do so; it failed to reduce the intracellular M. abscessus burden below that of the growth control. A study performed by Molina-Torres et al. assessed the intracellular killing capacity of antimycobacterial drugs against M. abscessus in THP-1 cells, showing that drugs can lower the intracellular burden below growth control (40). Although AUR may still inhibit extracellular bacteria, these results, combined with low achievable plasma concentrations, render auranofin a poor candidate for novel treatment regimens. Exploiting new targets with metal ions such as gold might open potent new treatment modalities previously unexplored. Indeed, targeting the thioredoxin reductase-mediated stress response in M. tuberculosis might hamper its survival in macrophages, as a group of thioredoxin reductases is upregulated during macrophage infection, possibly enhancing antibiotic effectiveness under acidic stress (27) but again emphasizing the need to penetrate into infected cells.
Any new treatment modality for NTM, especially M. abscessus, needs to be integrated into the current multidrug regimen, preferably exploiting synergistic interactions. We found that auranofin can act as a sensitizing agent for drugs already used in M. abscessus treatment, especially for amikacin, but for the combinations tested in time-kill kinetics assays only high-dose combinations with amikacin are synergistic; according to Bliss independence, lower combination doses (0.5× MIC) are antagonistic. This is not reflected in the resistance formation in our time-kill assays, where the 0.5× MIC auranofin-amikacin combination has a smaller resistant population to both auranofin and amikacin than higher concentration combinations, indicating that the observed antagonism is not based on increased resistance to either of the two drugs. Although the AUR combination with LNZ borders antagonism/synergism relative to Bliss independence, we interpret the combination between auranofin and linezolid as having no interaction. The combination of auranofin, colistin, and rifabutin was found to inhibit other MDR bacteria in a large compound combination screening (28). The target of auranofin is thus interesting and exploitable, but auranofin itself may not be effective clinically, based on our data. This also emphasizes the need for assays escalating in complexity to screen compounds effectively when trying to repurpose them for different indications.
Our study has some limitations to consider. Most importantly, all our assays are static assays wherein the drug is added only at the beginning and not continuously, making human pharmacokinetics impossible to model. For this, a hollow-fiber system might need to be employed (11) to assess auranofins effect under more physiological conditions. This would also counteract the stability issues of auranofin that might have influenced our time-kill assays. Without the addition of fresh antibiotics, it is unclear whether the microbial response might even be under estimated because of drug degradation after 7 days, which has also been shown for linezolid (29). This might also explain some of the regrowth observed after day 7. Also, though an auranofin-amikacin combination shows some activity, the drug-drug and toxicity interactions need to be carefully assessed before one could implement this in clinical practice. We found that at lower concentrations, this combination becomes antagonistic as determined by Bliss independence, further complicating a translation into clinical practice. It is unknown how these interactions affect treatment efficacy. We might have overestimated resistance against auranofin using only 3× MIC in the plates. Culture on plates with higher concentrations of auranofin might provide a more clear-cut view on the emergence of resistance. Lastly, the estimated Emax in our sigmoid curve fitting was not near the highest observed data points, making this estimate somewhat uncertain.
In summary, auranofin points to an unexploited, potent drug target in mycobacteria, particularly in M. abscessus: the thioredoxin reductase system. Auranofin itself is not likely to be effective clinically due to low plasma levels and failure to penetrate macrophages. Repurposing old drugs for new indications or using them as scaffolds for subsequent development might lead to important new NTM treatment modalities.
MATERIALS AND METHODS
Strains.
Reference strains of M. abscessus (CIP 104536; Collection of Institute Pasteur), M. fortuitum (ATCC 6841; American Type Culture Collection), M. peregrinum (ATCC 700686), M. avium (ATCC 700898), M. intracellulare (DSM 43223), M. chimera (DSM 44623), and M. simiae (ATCC 25275) were used. Clinical isolates were acquired from the collection of the Department of Medical Microbiology at Radboud University Medical Center, Nijmegen, The Netherlands (see Table S1 in the supplemental material). For the M. tuberculosis reference strain, H37Rv was used along with three clinical isolates proven to be multidrug resistant.
Antibiotics.
AUR (batch 0000019301), clarithromycin (CLR), amikacin (AMK), clofazimine (CLZ), cefoxitin (FOX), rifampin (RIF), and ethambutol (EMB) were obtained from Sigma-Aldrich (Zwijndrecht, The Netherlands). Linezolid (LNZ) was obtained from Pfizer as Zyvoxid (2 mg/ml; Pfizer, Capelle aan den Ijssel, The Netherlands).
Susceptibility testing.
The MIC of AUR against mycobacteria was determined by broth microdilution in cation-adjusted Mueller-Hinton broth (CAMH) as recommended by Clinical and Laboratory Standards Institute (CLSI) guidelines (30), and in Middlebrook 7H9 broth (M7H9). Twofold serial dilutions were made in the range of 0.25 to 256 μg/ml and inoculated using a 1:100 dilution of a 0.5 McFarland bacterial suspension. RGM were incubated at 30°C for 3 days, and SGM were incubated at 37°C for 7 days as described previously (31, 32). M. tuberculosis was incubated for 10 days. The minimum bactericidal concentration (MBC) was determined for reference strains of M. abscessus and M. avium, by quantification of CFU (CFU for auranofin concentrations that showed no visible growth in microdilution, on Middlebrook 7H10 agar plates (Becton Dickinson, Drachten, The Netherlands). The first concentration without visible CFU was considered to be the MBC. To determine whether auranofin should be considered bacteriostatic or bactericidal, we calculated the MBC/MIC ratio and considered a ratio of >8 to indicate a bacteriostatic effect, as previously defined (32).
We tested auranofin stability by preincubating uninoculated MIC plates for 7 and 14 days at 30°C (M. abscessus) and 37°C (MAC). After this incubation period, the plates were inoculated and incubated as for the susceptibility tests and then checked for higher MICs relative to the previously found MIC (29).
Synergy testing.
We assessed the synergy between auranofin and other antimycobacterial drugs by the broth microdilution checkerboard method (33). We performed all synergy tests in CAMH; only clofazimine/auranofin synergy tests were performed in M7H9 since clofazimine is insoluble in CAMH (12). Drug interactions were interpreted according to the fractional inhibitory concentration index (FICI) and classified as either synergistic (FICI ≤ 0.5), having no interaction (FICI > 0.5 to 4.0), or antagonistic (FICI > 4.0) (33). For synergy testing on RGM with CLR, the checkerboards were read after 3 and 14 days to check for inducible macrolide resistance. Combinations with the lowest average FICI values were selected for a combination time-kill assay.
Dose-response time-kill kinetics assay.
A range of 0.25× MIC to 32× MIC AUR was 2-fold serially diluted in CAMH with 0.05% Tween 80 (Sigma-Aldrich, Zwijndrecht, The Netherlands) and inoculated with 0.5 McFarland of M. abscessus, alongside an antibiotic-free growth control, all in duplicate. Conditions were kept at 30°C under constant shaking and ventilation through a filtered needle. On days 0 to 5, 7, 10, 14, and 21, samples were taken and 10-fold serially diluted. Then, 10-μl triplicate samples of each dilution were brought on Middlebrook 7H10 (Becton Dickinson) plates, followed by incubation for 3 days at 30°C (32, 34).
For the combination time-kill assays, 2-fold dilutions ranging from 0.5× MIC to 2× MIC were diluted in CAMH for each antibiotic and their combinations, inoculated with a 0.5 McFarland bacterial suspension of M. abscessus, and incubated and sampled as described above (32, 34). Samples were also brought on Middlebrook 7H11 plates containing a 3× MIC AUR or a 5× LNZ or AMK MIC, based on economic considerations. The plates were freshly prepared according to the manufacturers’ recommendation to counteract possible stability issues. These plates were incubated until colonies were clearly visible with a maximal incubation period of 10 days. The MICs of resistant colonies were not assessed. From these plates the percentages of resistant CFU were calculated as previously described by dividing CFU counts from M7H11 plates by CFU counts from drug-free plates (32).
Response surface analysis.
Response surface analysis to assess interaction according to Bliss independence was performed as previously described (32, 35). The area under the curve (AUC) was calculated from log CFU-versus-time plots using the trapezoidal rule after averaging the result from the two replicates and normalizing that value to the baseline colony count. The drug effect was calculated according to the following equations: effectx = AUCgrowth control – AUCx, where x is any given curve other than the growth control.
To assess potential interactions in the TK experiments, we calculated the expected effect for a combination under Bliss independence (Ecomb,BI) to be compared to the observed effect (Ecomb,obs). Since Bliss independence builds on probability theory, the maximum effect to be evaluated is limited to 1, and therefore all effects were normalized to the Emax of the most potent drug (auranofin). The following formula defines the expected effect: Ecomb,BI = EA + EB – (EA| × EB)/Emaxhigh, where EA and EB are the observed effects of drug A and B separately and Emaxhigh is the highest maximum effect. The Emax of auranofin was determined by fitting a sigmoidal Emax model to the concentration effect data using ordinary least-squares.
The difference between the observed and expected effect (ΔE) was quantified as a percentage difference relative to the expected effect: ΔE = (Ecomb,obs – Ecomb,BI)/Ecomb,BI. We defined an ΔE of 0% ± 10% as no interaction, anything less than −10% was defined as antagonistic, and anything more than 10% was defined as synergistic.
Ex vivo intracellular infection assays.
To assess auranofin’s intracellular efficacy, peripheral blood mononuclear cells were isolated from buffy coats obtained from three healthy volunteers (Sanquin Bloodbank, Nijmegen, The Netherlands) as described before (36). Informed consent from healthy volunteers was obtained for use of their blood for scientific purposes, as approved by the Ethics Committee of Radboud University Medical Centre (Nijmegen, The Netherlands). Isolation was performed using Ficoll-Paque, involving separation by a density gradient, followed by three wash steps in cold PBS and resuspension in RPMI 1640 (Life Technologies, Paisley, UK) supplemented with 10 μg/ml gentamicin, 2 mM GlutaMAX, and 1 mM pyruvate. The mononuclear cells were seeded in petri dishes and allowed to adhere for 2 h before the nonadherent lymphocytes were washed away. The adherent monocytes could differentiate into monocyte-derived macrophages at 37°C for 6 days in RPMI 1640 supplemented with 5 ng/ml granulocyte-macrophage colony-stimulating factor and 10% human pooled serum. One day prior to the experiment, cells were dissociated with EDTA and reseeded in antibiotic-free medium in a 96-well plate (105 cells/well).
On the day of infection, the cell culture medium was removed, and M. abscessus was added to the macrophages at a multiplicity of infection of 1. The macrophages were incubated with mycobacteria for 1 h at 37°C to allow phagocytosis before the medium was changed to RPMI 1640 supplemented with 10% human pooled serum. Auranofin was added at 2× MIC after 1 h of incubation and was present for the remainder of the experiment. After 1, 2, and 3 days, the numbers of extracellular bacteria were quantified by plating on Middlebrook 7H10 agar for CFU counts. In addition, macrophages were washed with warm PBS before they were subjected to hypotonic lysis in sterile water for 10 min. The cell-associated fraction was then also quantified through serial dilutions and plating of the different dilutions on Middlebrook 7H10 plates.
TRX2B ortholog search.
To determine whether the target of AUR is present and conserved between different species of NTM, we performed an ortholog search. The Kyoto Encyclopedia of Genes and Genomes (KEGG; www.kegg.jp) orthology database was used to find orthologues of the M. tuberculosis gene TRXB2 using entry number K00384. The KEGG genes database was then used to look for orthologs in M. abscessus, M. avium, M. smegmatis, M. intracellulare, M. kansasii, M. leprae, and E. coli (37, 38). From these protein entries, we built a phylogeny tree using phylogeny.fr (39), using standard MUSCLE sequence alignment, allowing for less strict flanking positions in the Gblocks curation step, and using 100 bootstraps based on the Whelan and Goldman substitution matrix.
Statistics.
Calculations were performed using Prism version 5.03 (GraphPad Software, Inc., La Jolla, CA) or R version 3.1.2 (R Foundation for Statistical Computing, Vienna, Austria; https://www.R-project.org/). Error bars show the standard errors of the mean.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jodie Schildkraut for assistance with the TRX2B orthologue search.
J.V.I. is supported by a personal grant from the Netherlands Organization for Scientific Research (NWO/ZonMW grant Veni 016.176.024).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00449-19.
REFERENCES
- 1.Falkinham JO. 2018. Challenges of NTM drug development. Front Microbiol 9:1613. doi: 10.3389/fmicb.2018.01613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kendall BA, Winthrop KL. 2013. Update on the epidemiology of pulmonary nontuberculous mycobacterial infections. Semin Respir Crit Care Med 34:087–094. doi: 10.1055/s-0033-1333567. [DOI] [PubMed] [Google Scholar]
- 3.Hoefsloot W, van Ingen J, Andrejak C, Ängeby K, Bauriaud R, Bemer P, Beylis N, Boeree MJ, Cacho J, Chihota V, Chimara E, Churchyard G, Cias R, Daza R, Daley CL, Dekhuijzen PNR, Domingo D, Drobniewski F, Esteban J, Fauville-Dufaux M, Folkvardsen DB, Gibbons N, et al. 2013. The geographic diversity of nontuberculous mycobacteria isolated from pulmonary samples: an NTM-NET collaborative study. Eur Respir J 42:1604. doi: 10.1183/09031936.00149212. [DOI] [PubMed] [Google Scholar]
- 4.van Ingen J, Ferro BE, Hoefsloot W, Boeree MJ, van Soolingen D. 2013. Drug treatment of pulmonary nontuberculous mycobacterial disease in HIV-negative patients: the evidence. Expert Rev anti Infect Ther 11:1065–1077. doi: 10.1586/14787210.2013.830413. [DOI] [PubMed] [Google Scholar]
- 5.Hurst-Hess K, Rudra P, Ghosh P. 2017. Mycobacterium abscessus WhiB7 regulates a species-specific repertoire of genes to confer extreme antibiotic resistance. Antimicrob Agents Chemother 61:e01347-17. doi: 10.1128/AAC.01347-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, Holland SM, Horsburgh R, Huitt G, Iademarco MF, Iseman M, Olivier K, Ruoss S, von Reyn CF, Wallace RJ, Winthrop K. 2007. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med 175:367–416. doi: 10.1164/rccm.200604-571ST. [DOI] [PubMed] [Google Scholar]
- 7.Ruth MM, Ingen J. v. 2017. New insights in the treatment of nontuberculous mycobacterial pulmonary disease. Future Microbiol 12:1109–1112. doi: 10.2217/fmb-2017-0144. [DOI] [PubMed] [Google Scholar]
- 8.Pasipanodya JG, Ogbonna D, Ferro BE, Magombedze G, Srivastava S, Deshpande D, Gumbo T. 2017. Systematic review and meta-analyses of the effect of chemotherapy on pulmonary Mycobacterium abscessus outcomes and disease recurrence. Antimicrob Agents Chemother 61:e01206-17. doi: 10.1128/AAC.01206-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Novosad S, Henkle E, Winthrop KL. 2015. The challenge of pulmonary nontuberculous mycobacterial infection. Curr Pulmonol Rep 4:152–161. doi: 10.1007/s13665-015-0119-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pasipanodya JG, Ogbonna D, Deshpande D, Srivastava S, Gumbo T. 2017. Meta-analyses and the evidence base for microbial outcomes in the treatment of pulmonary Mycobacterium avium-intracellulare complex disease. J Antimicrob Chemother 72:i3–i19. doi: 10.1093/jac/dkx311. [DOI] [PubMed] [Google Scholar]
- 11.Ferro BE, Srivastava S, Deshpande D, Pasipanodya JG, van Soolingen D, Mouton JW, van Ingen J, Gumbo T. 2016. Failure of the amikacin, cefoxitin, and clarithromycin combination regimen for treating pulmonary Mycobacterium abscessus infection. Antimicrob Agents Chemother 60:6374–6376. doi: 10.1128/AAC.00990-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferro BE, Meletiadis J, Wattenberg M, de Jong A, van Soolingen D, Mouton JW, van Ingen J. 2016. Clofazimine prevents the regrowth of Mycobacterium abscessus and Mycobacterium avium type strains exposed to amikacin and clarithromycin. Antimicrob Agents Chemother 60:1097–1105. doi: 10.1128/AAC.02615-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Ingen J. 2015. Treatment of pulmonary disease caused by non-tuberculous mycobacteria. Lancet Respir Med 3:179–180. doi: 10.1016/S2213-2600(15)00033-8. [DOI] [PubMed] [Google Scholar]
- 14.Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson EE, Brochado AR, Fernandez KC, Dose H, Mori H, Patil KR, Bork P, Typas A. 2018. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 555:623–628. doi: 10.1038/nature25979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roder C, Thomson MJ. 2015. Auranofin: repurposing an old drug for a golden new age. Drugs R D 15:13–20. doi: 10.1007/s40268-015-0083-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thangamani S, Mohammad H, Abushahba MFN, Sobreira TJP, Hedrick VE, Paul LN, Seleem MN. 2016. Antibacterial activity and mechanism of action of auranofin against multidrug resistant bacterial pathogens. Sci Rep 6:22571. doi: 10.1038/srep22571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harbut MB, Vilchèze C, Luo X, Hensler ME, Guo H, Yang B, Chatterjee AK, Nizet V, Jacobs WR, Schultz PG, Wang F. 2015. Auranofin exerts broad-spectrum bactericidal activities by targeting thiol-redox homeostasis. Proc Natl Acad Sci U S A 112:4453–4458. doi: 10.1073/pnas.1504022112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.May HC, Yu J-J, Guentzel MN, Chambers JP, Cap AP, Arulanandam BP. 2018. Repurposing auranofin, ebselen, and PX-12 as antimicrobial agents targeting the thioredoxin system. Front Microbiol 9:336. doi: 10.3389/fmicb.2018.00336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar A, Farhana A, Guidry L, Saini V, Hondalus M, Steyn AJ. 2011. Redox homeostasis in mycobacteria: the key to tuberculosis control? Expert Rev Mol Med 13:e39. doi: 10.1017/S1462399411002079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Casini A, Hartinger C, Gabbiani C, Mini E, Dyson PJ, Keppler BK, Messori L. 2008. Gold(III) compounds as anticancer agents: relevance of gold-protein interactions for their mechanism of action. J Inorg Biochem 102:564–575. doi: 10.1016/j.jinorgbio.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 21.Woods GL. 2000. Susceptibility testing for mycobacteria. Clin Infect Dis 31:1209–1215. doi: 10.1086/317441. [DOI] [PubMed] [Google Scholar]
- 22.Asano RL, Davies J. 1998. Molecular characterization of the thioredoxin system of Mycobacterium smegmatis. Res Microbiol 149:567–576. doi: 10.1016/S0923-2508(99)80004-7. [DOI] [PubMed] [Google Scholar]
- 23.van Ingen J, Boeree MJ, van Soolingen D, Mouton JW. 2012. Resistance mechanisms and drug susceptibility testing of nontuberculous mycobacteria. Drug Resist Updat 15:149–161. doi: 10.1016/j.drup.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 24.Capparelli EV, Bricker-Ford R, Rogers MJ, McKerrow JH, Reed SL. 2016. Phase I clinical trial results of auranofin, a novel antiparasitic agent. Antimicrob Agents Chemother 61:e01947-16. doi: 10.1128/AAC.01947-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bryant JM, Grogono DM, Rodriguez-Rincon D, Everall I, Brown KP, Moreno P, Verma D, Hill E, Drijkoningen J, Gilligan P, Esther CR, Noone PG, Giddings O, Bell SC, Thomson R, Wainwright CE, Coulter C, Pandey S, Wood ME, Stockwell RE, Ramsay KA, Sherrard LJ, Kidd TJ, Jabbour N, Johnson GR, Knibbs LD, Morawska L, Sly PD, Jones A, Bilton D, Laurenson I, et al. 2016. Emergence and spread of a human-transmissible multidrug-resistant nontuberculous mycobacterium. Science 354:751–757. doi: 10.1126/science.aaf8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Houben D, Demangel C, van Ingen J, Perez J, Baldeón L, Abdallah AM, Caleechurn L, Bottai D, van Zon M, de Punder K, van der Laan T, Kant A, Bossers-de Vries R, Willemsen P, Bitter W, van Soolingen D, Brosch R, van der Wel N, Peters PJ. 2012. ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol 14:1287–1298. doi: 10.1111/j.1462-5822.2012.01799.x. [DOI] [PubMed] [Google Scholar]
- 27.Coulson GB, Johnson BK, Zheng H, Colvin CJ, Fillinger RJ, Haiderer ER, Hammer ND, Abramovitch RB. 2017. Targeting Mycobacterium tuberculosis sensitivity to thiol stress at acidic pH kills the bacterium and potentiates antibiotics. Cell Chem Biol 24:993–1004. doi: 10.1016/j.chembiol.2017.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun W, Weingarten RA, Xu M, Southall N, Dai S, Shinn P, Sanderson PE, Williamson PR, Frank KM, Zheng W. 2016. Rapid antimicrobial susceptibility test for identification of new therapeutics and drug combinations against multidrug-resistant bacteria. Emerg Microbes Infect 5:e116. doi: 10.1038/emi.2016.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schoutrop ELM, Brouwer MAE, Jenniskens JCA, Ferro BE, Mouton JW, Aarnoutse RE, van Ingen J. 2018. The stability of antimycobacterial drugs in media used for drug susceptibility testing. Diagn Microbiol Infect Dis 92:305–308. doi: 10.1016/j.diagmicrobio.2018.06.015. [DOI] [PubMed] [Google Scholar]
- 30.Clinical and Laboratory Standards Institute. 2011. Susceptibility testing of mycobacteria, nocardia, and other aerobic actinomycetes: approved standard, 2nd ed . CLSI document M24-A2 Clinical and Laboratory Standards Institute, Wayne, PA. [PubMed] [Google Scholar]
- 31.Ruth MM, Sangen JJN, Pennings LJ, Schildkraut JA, Hoefsloot W, Magis-Escurra C, Wertheim HFL, van Ingen J. 2018. Minocycline Has No Clear Role in the Treatment of Mycobacterium abscessus Disease. Antimicrob Agents Chemother 62:e01208-18. doi: 10.1128/AAC.01208-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruth MM, Sangen JJN, Remmers K, Pennings LJ, Svensson E, Aarnoutse RE, Zweijpfenning SMH, Hoefsloot W, Kuipers S, Magis-Escurra C, Wertheim HFL, van Ingen J. 2019. A bedaquiline/clofazimine combination regimen might add activity to the treatment of clinically relevant non-tuberculous mycobacteria. J Antimicrob Chemother 74:935–943. doi: 10.1093/jac/dky526. [DOI] [PubMed] [Google Scholar]
- 33.Odds FC. 2003. Synergy, antagonism, and what the chequerboard puts between them. J Antimicrob Chemother 52:1–1. doi: 10.1093/jac/dkg301. [DOI] [PubMed] [Google Scholar]
- 34.Ferro BE, van Ingen J, Wattenberg M, van Soolingen D, Mouton JW. 2015. Time-kill kinetics of antibiotics active against rapidly growing mycobacteria. J Antimicrob Chemother 70:811–817. doi: 10.1093/jac/dku431. [DOI] [PubMed] [Google Scholar]
- 35.Wicha SG, Kees MG, Kuss J, Kloft C. 2015. Pharmacodynamic and response surface analysis of linezolid or vancomycin combined with meropenem against Staphylococcus aureus. Pharm Res 32:2410–2418. doi: 10.1007/s11095-015-1632-3. [DOI] [PubMed] [Google Scholar]
- 36.Ruth MM, Magombedze G, Gumbo T, Bendet P, Sangen JJN, Zweijpfenning S, Hoefsloot W, Pennings L, Koeken V, Wertheim HFL, Lee PS, van Ingen J, Deshpande D. 2019. Minocycline treatment for pulmonary Mycobacterium avium complex disease based on pharmacokinetics/pharmacodynamics and Bayesian framework mathematical models. J Antimicrob Chemother 74:1952–1961. doi: 10.1093/jac/dkz143. [DOI] [PubMed] [Google Scholar]
- 37.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. 2016. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 44:D457–D462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kanehisa M, Sato Y, Morishima K. 2016. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol 428:726–731. doi: 10.1016/j.jmb.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 39.Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard J-F, Guindon S, Lefort V, Lescot M, Claverie J-M, Gascuel O. 2008. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36:W465–W469. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Molina-Torres CA, Tamez-Peña L, Castro-Garza J, Ocampo-Candiani J, Vera-Cabrera L. 2018. Evaluation of the intracellular activity of drugs against Mycobacterium abscessus using a THP-1 macrophage model. J Microbiol Methods 148:29–32. doi: 10.1016/j.mimet.2018.03.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.