

Yellow fever virus (YFV) is a human Flavivirus reemerging in parts of the world. While a vaccine is available, large outbreaks have recently occurred in Brazil and certain African countries. Development of an effective antiviral against YFV is crucial, as there is no available effective drug against YFV.
KEYWORDS: Flaviviridae, yellow fever virus, antiviral agents, nucleoside analogs
ABSTRACT
Yellow fever virus (YFV) is a human Flavivirus reemerging in parts of the world. While a vaccine is available, large outbreaks have recently occurred in Brazil and certain African countries. Development of an effective antiviral against YFV is crucial, as there is no available effective drug against YFV. We have identified several novel nucleoside analogs with potent antiviral activity against YFV with 50% effective concentration (EC50) values between 0.25 and 1 μM with selectivity indices over 100 in culture.
TEXT
Yellow fever virus (YFV) is the prototype virus of the genus Flavivirus from the Flaviviridae family with a single-stranded, positive-sense RNA genome consisting of approximately 11 kb that encode three structural and seven nonstructural proteins (1). Yellow fever is endemic to tropical areas of Africa and South America and is primarily transmitted between nonhuman primates by mosquitoes (mainly from two genera, Haemagogus and Aedes). While YFV infection in most nonhuman primates is asymptomatic (with the exception of some New World monkeys), severe clinical illness can result in humans, with reported mortality rates of ∼20% in Africa and up to 60% in South America (2).
Currently, there are no available antiviral drugs to combat YFV. An effective vaccine against YFV was developed in 1937 (3), but it is not recommended for persons over the age of 60. Despite the available vaccine, outbreaks continue to occur in areas where large-scale vaccination efforts have not been successfully implemented. Furthermore, according to the World Health Organization (http://www.who.int/csr/don/22-january-2018-yellow-fever-brazil/en/), the possibility exists for a viremic traveler to spread YFV from its current endemic range to new areas harboring significant Aedes aegypti populations, such as India, Southeast Asia, and even some southern regions of the United States. Recent outbreaks of YFV in both Brazil and Nigeria (between 2017 and 2018) resulting in a substantial number of deaths further illustrate the persistent threat to humans and potential spreading of the virus if not carefully managed and contained (http://www.who.int/csr/don/22-december-2017-yellow-fever-nigeria/en/). Because YFV continues to be a significant public health threat, the development of antiviral therapies to treat infected individuals remains highly pertinent.
In this study, we evaluated an in-house library of 21 synthesized nucleoside analogs for their potential in vitro antiviral activity against YFV. Compounds 1 to 13 and 16 to 21 were prepared following published protocols (4–14) while compounds 14 and 15 were synthesized according to the chemistry protocols that are described in the supplemental material. The focused library (Fig. 1) was evaluated for toxicity using an MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) method (15) (Table 1). Compound 16 (sofosbuvir, an FDA approved hepatitis C virus [HCV] polymerase inhibitor) was used as our positive control with reported activity against YFV (16). Briefly, the cytotoxic concentration of drug required to reduce cell viability by 50% (CC50) values were determined for each test compound in the following two cell types: human rhabdomyosarcoma (RD) cell line and Huh-7 cells. Overall, 13 of the 21 compounds were found to have CC50 values of >50 μM in both cell types. In addition, the maximum nontoxic concentrations (MNTCs) were also determined for each compound.
FIG 1.

Chemical structures of compounds 1 to 21.
TABLE 1.
Cytotoxicity and anti-YFV activity of nucleoside analogs
| Compound | Huh-7 cells |
RD cells |
Virus CPE inhibition in RD cells (%) | ||
|---|---|---|---|---|---|
| CC50a (μM) | MNTCa (μM) | CC50a (μM) | MNTCa (μM) | ||
| 1 | >100 | >20 | 56 | >20 | 44.7 ± 0.4 |
| 2 | 81 | >20 | 94 | >20 | 54.3 ± 0.4 |
| 3 | 60 | >20 | >100 | >20 | 26 ± 1.1 |
| 4 | >100 | >20 | 30.9 | >20 | 99.5 ± 0.3 |
| 5 | >100 | >20 | >100 | >20 | <2 |
| 6 | >100 | >20 | >100 | >20 | <5 |
| 7 | >100 | >20 | >100 | >20 | <2 |
| 8 | 37 | >20 | 80 | >20 | <2 |
| 9 | >100 | >20 | >100 | >20 | 95 ± 1 |
| 10 | 70 | >20 | >100 | >20 | 99.5 ± 0.3 |
| 11 | 22 | 15 | >100 | >20 | 99.9 ± 0.1 |
| 12 | >100 | >20 | >100 | >20 | <5 |
| 13 | 17 | >10 | 77 | >20 | 99 ± 0.4 |
| 14 | 10 | 5 | 11 | 7 | 99.8 ± 0.1d |
| 15 | 7 | 5 | 12 | 7.5 | 99.9 ± 0.1d |
| 16b | >100 | >20 | >100 | >20 | 98 ± 0.7 |
| 17 | 2.9 | 2 | 10.5 | 6.5 | 99.5 ± 0.8c |
| 18 | 11.5 | 5.5 | 32.5 | 16 | 90.5 ± 1d |
| 19 | >100 | >20 | >100 | >20 | <5 |
| 20 | 62 | >20 | >100 | >20 | <2 |
| 21 | >100 | >20 | >100 | >20 | 99 ± 0.2 |
Cytotoxicity of all nucleoside analogs was measured using MTS assay to calculate the CC50 and maximum nontoxic concentration (MNTC) of each compound.
Sofosbuvir (positive control).
Compound 17 was tested at 1.5 μM for its anti-YFV activity because of its MNTC.
Compounds 14, 15, and 18 were tested in 5 μM for their anti-YFV activity because of their MNTCs. All other compounds were tested in 20 μM against YFV in a CPE inhibition assay using MTS assay.
Next, 21 compounds were selected for further evaluation to determine their anti-YFV activity. To avoid the potential of drug toxicity that would interfere with viral cytopathic effect (CPE), compounds were evaluated at nontoxic concentrations. The resultant inhibitory effect of each test compound was calculated as a percentage of decrease in YFV CPE. In this case, the RD cell line was chosen for our antiviral evaluation, as YFV exhibits significant and visible CPE in this cell line compared to that in Huh-7 cells. Briefly, a monolayer of RD cells was prepared in 96-well plates. The cells were then infected with 1 multiplicity of infection (MOI) of YFV (17D strain) followed by treatment with a single nontoxic dose of each compound in triplicate. The vehicle control wells were treated with 0.1% dimethyl sulfoxide (DMSO) diluted in the working medium. The plate was then incubated for 96 h at which time the virus-control wells produced detectable CPE. To determine the probable inhibitory CPE of test compounds, an MTS assay was performed (15). We identified 10 nucleoside analogs with significant inhibitory effect (>80% CPE inhibition) against YFV replication (summarized in Table 1). Therefore, these compounds were chosen for further studies to identify the potency of the compounds and their concentration-dependent manner.
The dose-response antiviral activity of each effective drug was determined by initially measuring the reduction in the number of viral foci as a semiquantitative method using two different cell lines (RD and Huh-7). Briefly, confluent monolayers of RD cells in a 96-well microplate were infected with 0.1 MOI of YFV followed by treatment with increasing concentrations of each compound up to its respective MNTC dose. Then, cells were overlaid with 1.5% carboxymethyl cellulose (CMC) containing minimal essential medium (MEM) with 2% fetal bovine serum (FBS). Viral foci were stained and visualized using antiflavivirus group antigen (4G2; Millipore), followed by horseradish peroxidase (HRP) anti-mouse IgG and TrueBlue substrate, and imaged using CTL ImmunoSpot S6 micro analyzer 2 on day 2 after treatment (Fig. 2).
FIG 2.

Foci-forming unit reduction assay. Compounds 9, 10, and 21 as the most promising showed significant inhibitory effect against YFV compared to that of sofosbuvir (compound 16) as a drug control.
The effective compounds were further quantified and confirmed with a secondary, virus yield reduction assay using an optimized in-house reverse transcription-quantitative PCR (qRT-PCR) to determine the YFV RNA copy number after 3 days posttreatment from collected supernatants compared to the results from infected nontreated cells and noninfected and nontreated cells as controls (Table 2). qRT-PCR was performed using the probe/primer mix (forward primer, 5′-GCA CGG ATG TAA CAG ACT GAA GA-3′; reverse primer, 5′-CCA GGC CGA ACC TGT CAT-3′; and probe, 5′-56-FAM/CG ACT GTG T/ZEN/G GTC CGG CCC TC/3IABkFQ-3′) and qScript Tough master mix (Quantabio, USA). Quantitative PCR measurement was performed using the StepOnePlus real-time PCR system (Roche, Germany) according to the manufacturer’s protocol.
TABLE 2.
Anti-YFV activity in RD and Huh-7 cells
| Compound | RD cellsa
|
Huh-7 cellsa
|
||||
|---|---|---|---|---|---|---|
| EC50 (μM) | EC90 (μM) | SI | EC50 (μM) | EC90 (μM) | SI | |
| 10 | 0.25 ± 0.01 | 0.49 ± 0.03 | >400 | 0.55 ± 0.03 | 3.8 ± 0.1 | >100 |
| 16 | 0.34 ± 0.01 | 3.3 ± 0.1 | 30 | 1.2 ± 0.07 | 5.3 ± 0.2 | 83 |
| 11 | 0.87 ± 0.03 | 2.3 ± 0.2 | >100 | 1.1 ± 0.01 | 2.1 ± 0.1 | 20 |
| 17 | 0.17 ± 0.01 | 0.5 ± 0.08 | 61 | 0.16 ± 0.01 | 0.42 ± 0.07 | 18 |
| 18 | 1.36 ± 0.03 | 5.3 ± 0.2 | 23 | 1.43 ± 0.08 | 6.2 ± 0.15 | 8 |
| 15 | 0.25 ± 0.001 | 0.5 ± 0.1 | 48 | 0.18 ± 0.01 | 0.47 ± 0.02 | 38 |
| 14 | 0.66 ± 0.003 | 1.1 ± 0.02 | 16 | 0.37 ± 0.03 | 1.3 ± 0.17 | 27 |
| 13 | 1.1 ± 0.01 | 2.6 ± 0.15 | 70 | 1.2 ± 0.05 | 10 ± 0.7 | 14 |
| 9 | 0.51 ± 0.01 | 3.9 ± 0.2 | >200 | 1 ± 0.03 | 6.8 ± 0.43 | >100 |
| 21 | 2.1 ± 0.1 | 10.1 ± 0.2 | >50 | 0.97 ± 0.01 | 1.3 ± 0.12 | >100 |
All EC50 and EC90 values are presented as mean ± SD.
The median effective concentration (EC50) and the concentration with 90% of inhibitory effect (EC90) were calculated using GraphPad Prism for Windows version 5 (GraphPad Software Inc., San Diego, CA) as the means ± standard deviation (SD) of the mean from triplicate assay from three independent experiments. The selectivity index (SI) values for effective compounds also have been calculated by the ratio of CC50/EC50 for each compound (Table 2). As it is shown in Table 2, compounds 10, 9, and 21 showed significant activity with great selectivity index values higher than 100 in Huh-7 cells.
Based on the primary antiviral assays, the 2′-F derivatives (e.g., compounds 5, 6, and 7) did not exhibit any in vitro anti-YFV activity up to 20 μM (Table 1). Compound 8, another 2′-F nucleoside, known to inhibit respiratory syncytial virus (RSV) replication, was also inactive at that concentration. More interestingly, compound 4 (β-d-N4-hydroxycytidine), a known inhibitor of chikungunya virus (CHIKV) and HCV replication, displayed an EC50 of 0.5 μM and 0.63 μM in RD and Huh-7 cells, respectively. In a similar manner, FDA-approved HCV polymerase inhibitor compound 16 (sofosbuvir) was active in both cell lines with EC50 values of 0.25 and 1.2 μM in RD and Huh-7 cells, respectively. However, De Freitas and colleagues reported the activity of sofosbuvir against YFV 17D strain in Huh-7 with an EC50 of 4.8 μM (16). The difference between their EC50 and ours could be due to the methods used for virus yield reduction, as they used a plaque reduction assay and we used qRT-PCR as a more sensitive and quantitative method. Novel thio prodrugs of sofosbuvir that were previously reported to be more potent than sofosbuvir itself against HCV were also evaluated in the YF assays, but neither the RP nor the SP isomer (where R stands for rectus [right], S stands for sinister [left], and p stands for phosphorus center) appeared markedly more potent than sofosbuvir (Table 2). In addition, both compounds 17 and 18 displayed cytotoxicity in the low micromolar range in both RD and Huh-7 cells, which seems to be a severe limitation for this type of monophosphate prodrug. The 2′-C-methyl-uridine prodrugs were then tested as an RP/SP mixture (compound 13) and as pure RP and SP isomers (compounds 11 and 12). It is worth noting that both compounds 11 and 13 displayed anti-YF activity in the submicromolar range while compound 12 was inactive up to 20 μM. This difference of potency between RP and SP isomers is a phenomenon that may be explained by the fact that the enzymes responsible for the cleavage of the prodrug moiety are preferentially selective for one phosphorous isomer over the other. As for sofosbuvir (compound 16), we also tested the corresponding thio prodrugs of 2′-C-methyl-uridine but again, despite both isomers displaying EC50 values in the submicromolar range, they also proved to be toxic in both RD and Huh-7 cells. Other 2′-C-methyl-ribonucleosides known to inhibit HCV replication (e.g., compounds 1, 2, 3, 9, and 10) were also examined along with the 7-deaza purine analogs (compounds 9 and 10), which showed anti-YF activity with EC50 values ranging between 0.25 and 1 μM. In contrast, the 2′-C-methyl-cytidine analogs (including compounds 1, 2, and 3) did not exhibit significant anti-YFV activity up to 20 μM (Table 2), whereas Julander and colleagues reported the activity of 2′-C-methyl-cytidine against YFV 17D with an EC50 of 10 μM in Vero cells (17). This difference could be due to the use of different MOIs of virus. We have used 1 MOI of YFV 17D for our primary antiviral assays, and they used a markedly lower 0.0001 MOI of the same virus that could lead to the different EC50 values. The other affecting factor could be the difference between the types of cells used in their studies and ours. However, according to our data, the EC50 value for that compound (compound 1) is ≈20 μM, which is close to their finding.
However, in several studies, the effect of different nucleoside analogs has been discussed against flaviviruses. The activity of 3′,5′di-O-trityluridine was shown against dengue virus (DENV) and YFV in cell culture systems with EC50 values of 1.5 and 0.83 μM, respectively (18). In the case of adenosine analogs, several compounds have been developed and identified, such as NITD008 and NITD203, through in vitro and in vivo studies, but none were considered for further development (19–21). In one recent study, 2′-modified nucleoside analogs were developed and evaluated against DENV and Zika virus (ZIKV) in cell culture systems with significant antiviral activity (22). Several nucleoside analogs and their novel derivatives also have shown antiviral activity against tick-borne encephalitis virus and West Nile virus as other flaviviruses but have yet to be developed as antiviral drugs (23, 24).
The intracellular metabolisms of the following four compounds in RD cells were evaluated: compound 10 as our main lead compound, sofosbuvir (compound 16) as a drug control, together with compounds 14 and 15 as potent novel compounds but with marked toxicity. Briefly, RD cells were seeded at 1 × 106 cells per well in 12-well plates and incubated with 10 or 50 μM of the four compounds, including compounds 10, 16, 14, and 15. At 4 h, cells were washed twice by ice-cold phosphate-buffered saline (PBS) and resuspended in 70% ice-cold methanol overnight at −20°C. The supernatants were then dried and reconstituted with the high-pressure liquid chromatography (HPLC) mobile phase before being subjected to liquid chromatography-mass spectrometry (LC-MS) analysis. All of the four compounds were metabolized and phosphorylated to their corresponding nucleoside triphosphates (Fig. 3). For compounds 14 and 15, a small amount of 2′-C-methyl-CTP was also detected in addition to the main metabolite, the nucleoside triphosphate 2′-C-methyl-UTP. Also, for those compounds (including 14 and 15) that generated the same nucleoside triphosphates, the levels of nucleoside 5′-triphosphates were positively correlated with their anti-YFV potency in RD cells. Nevertheless, the respective concentrations for the 4 natural ribonucleotide triphosphates (rNTPs) in RD and Huh-7 cells were comparable without any significant difference (data not shown).
FIG 3.
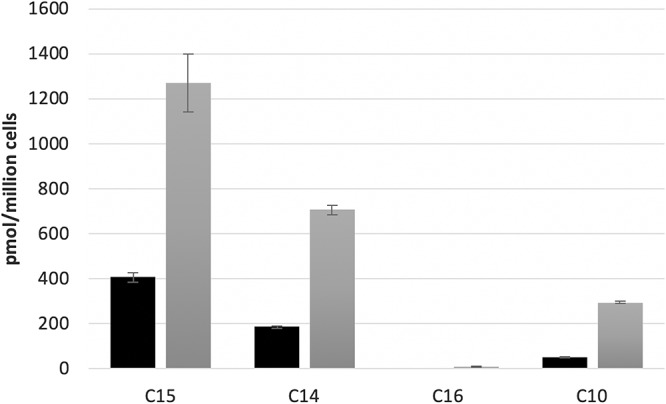
Total level of active nucleoside triphosphate produced for compounds 10, 14, 15, and 16 in RD cells. Ten (black bar) or 50 μM (gray bar) of each compound was incubated with RD cells for 4 h at 37°C. Values represent the means of three replicates.
In conclusion, we have identified compounds 9, 10, and 21 as our first line of potential candidate drugs with antiviral activity to YFV that warrant further investigation. It is worth noting that compounds 9 and 10 as 2′-C-Me ribonucleosides and compound 21 as a prodrug for 2′-di-Cl-uridine showed the most potent (submicromolar) activity with the greatest selectivity index (SI > 100) among the effective compounds, even compared to sofosbuvir as our drug control. As a future plan, we will evaluate the in vivo anti-YFV efficacy of our lead compounds as well as perform pharmacokinetic studies. Therefore, the approaches and findings presented here and our future plan offer promise to reduce the global burden of YFV as an emerging/reemerging Flavivirus.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by NIH grant R21-AI-129607 and by the Emory University CFAR NIH grant 2P30-AI-050409.
We thank James Kohler for his critical review of the paper.
We declare no conflicts of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00889-19.
REFERENCES
- 1.Patkar CG, Larsen M, Owston M, Smith JL, Kuhn RJ. 2009. Identification of inhibitors of yellow fever virus replication using a replicon-based high-throughput assay. Antimicrob Agents Chemother 53:4103–4114. doi: 10.1128/AAC.00074-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Monath TP, Vasconcelos PF. 2015. Yellow fever. J Clin Virol 64:160–173. doi: 10.1016/j.jcv.2014.08.030. [DOI] [PubMed] [Google Scholar]
- 3.Barrett AD, Teuwen DE. 2009. Yellow fever vaccine—ow does it work and why do rare cases of serious adverse events take place? Curr Opin Immunol 21:308–313. doi: 10.1016/j.coi.2009.05.018. [DOI] [PubMed] [Google Scholar]
- 4.Lee J-C, Tseng C-K, Wu Y-H, Kaushik-Basu N, Lin C-K, Chen W-C, Wu H-N. 2015. Characterization of the activity of 2′-C-methylcytidine against dengue virus replication. Antiviral Res 116:1–9. doi: 10.1016/j.antiviral.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 5.Eldrup AB, Prhavc M, Brooks J, Bhat B, Prakash TP, Song Q, Bera S, Bhat N, Dande P, Cook PD, Bennett CF, Carroll SS, Ball RG, Bosserman M, Burlein C, Colwell LF, Fay JF, Flores OA, Getty K, LaFemina RL, Leone J, MacCoss M, McMasters DR, Tomassini JE, Von Langen D, Wolanski B, Olsen DB. 2004. Structure-activity relationship of heterobase-modified 2′-C-methyl ribonucleosides as inhibitors of hepatitis C virus RNA replication. J Med Chem 47:5284–5297. doi: 10.1021/jm040068f. [DOI] [PubMed] [Google Scholar]
- 6.Clark JL, Hollecker L, Mason JC, Stuyver LJ, Tharnish PM, Lostia S, McBrayer TR, Schinazi RF, Watanabe KA, Otto MJ, Furman PA, Stec WJ, Patterson SE, Pankiewicz KW. 2005. Design, synthesis, and antiviral activity of 2′-deoxy-2′-fluoro-2′-C-methylcytidine, a potent inhibitor of hepatitis C virus replication. J Med Chem 48:5504–5508. doi: 10.1021/jm0502788. [DOI] [PubMed] [Google Scholar]
- 7.Zhou S, Mahmoud S, Liu P, Zhou L, Ehteshami M, Bassit L, Tao S, Domaoal RA, Sari O, Schutter C, Amiralaei S, Khalil A, Ollinger Russell O, McBrayer T, Whitaker T, Abou-Taleb N, Amblard F, Coats SJ, Schinazi RF. 2017. 2′-Chloro,2′-fluoro ribonucleotide prodrugs with potent pan-genotypic activity against hepatitis C virus replication in culture. J Med Chem 60:5424–5437. doi: 10.1021/acs.jmedchem.7b00067. [DOI] [PubMed] [Google Scholar]
- 8.Wang G, Deval J, Hong J, Dyatkina N, Prhavc M, Taylor J, Fung A, Jin Z, Stevens SK, Serebryany V, Liu J, Zhang Q, Tam Y, Chanda SM, Smith DB, Symons JA, Blatt LM, Beigelman L. 2015. Discovery of 4′-chloromethyl-2′-deoxy-3′,5′-di-O-isobutyryl-2′-fluorocytidine (ALS-8176), a first-in-class RSV polymerase inhibitor for treatment of human respiratory syncytial virus infection. J Med Chem 58:1862–1878. doi: 10.1021/jm5017279. [DOI] [PubMed] [Google Scholar]
- 9.Gardelli C, Attenni B, Donghi M, Meppen M, Pacini B, Harper S, Di Marco A, Fiore F, Giuliano C, Pucci V, Laufer R, Gennari N, Marcucci I, Leone JF, Olsen DB, MacCoss M, Rowley M, Narjes F. 2009. Phosphoramidate prodrugs of 2′-C-methylcytidine for therapy of hepatitis C virus infection. J Med Chem 52:5394–5407. doi: 10.1021/jm900447q. [DOI] [PubMed] [Google Scholar]
- 10.Pierra C, Amador A, Benzaria S, Cretton-Scott E, D'Amours M, Mao J, Mathieu S, Moussa A, Bridges EG, Standring DN, Sommadossi JP, Storer R, Gosselin G. 2006. Synthesis and pharmacokinetics of valopicitabine (NM283), an efficient prodrug of the potent anti-HCV agent 2′-C-methylcytidine. J Med Chem 49:6614–6620. doi: 10.1021/jm0603623. [DOI] [PubMed] [Google Scholar]
- 11.Pinho P, Kalayanov G, Westerlind H, Rosenquist Å, Wähling H, Sund C, Almeida M, Ayesa S, Tejbrant J, Targett-Adams P, Eneroth A, Lindqvist A. 2017. Discovery of β-d-2′-deoxy-2′-dichlorouridine nucleotide prodrugs as potent inhibitors of hepatitis C virus replication. Bioorg Med Chem Lett 27:3468–3471. doi: 10.1016/j.bmcl.2017.05.075. [DOI] [PubMed] [Google Scholar]
- 12.Negishi K, Harada C, Ohara Y, Oohara K, Nitta N, Hayatsu H. 1983. N4-aminocytidine, a nucleoside analog that has an exceptionally high mutagenic activity. Nucleic Acids Res 11:5223–5233. doi: 10.1093/nar/11.15.5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cho JH, Coats SJ, Schinazi RF. 2015. Synthesis of carbocyclic nucleoside analogs with five-membered heterocyclic nucleobases. Tetrahedron Lett 56:3587–3590. doi: 10.1016/j.tetlet.2015.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Z, Cox BD, Garnier-Amblard EC, McBrayer TR, Coats SJ, Schinazi RF, Amblard F. 2017. Synthesis and anti-HCV activity of a series of β-d-2′-deoxy-2′-dibromo nucleosides and their corresponding phosphoramidate prodrugs. Bioorg Med Chem Lett 27:5296–5299. doi: 10.1016/j.bmcl.2017.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cory AH, Owen TC, Barltrop JA, Cory JG. 1991. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun 3:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- 16.De Freitas CS, Higa LM, Sacramento CQ, Ferreira AC, Reis PA, Delvecchio R, Monteiro FL, Barbosa-Lima G, James Westgarth H, Vieira YR, Mattos M, Rocha N, Hoelz LVB, Leme RPP, Bastos MM, Rodrigues GOL, Lopes CEM, Queiroz-Junior CM, Lima CX, Costa VV, Teixeira MM, Bozza FA, Bozza PT, Boechat N, Tanuri A, Souza T. 2019. Yellow fever virus is susceptible to sofosbuvir both in vitro and in vivo. PLoS Negl Trop Dis 30:13. doi: 10.1371/journal.pntd.0007072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Julander JG, Jha AK, Choi JA, Jung KH, Smee DF, Morrey JD, Chu CK. 2010. Efficacy of 2′-C-methylcytidine against yellow fever virus in cell culture and in a hamster model. Antiviral Res 86:261–267. doi: 10.1016/j.antiviral.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Burghgraeve T, Selisko B, Kaptein S, Chatelain G, Leyssen P, Debing Y, Jacobs M, Van Aerschot A, Canard B, Neyts J. 2013. 3′,5′Di-O-trityluridine inhibits in vitro flavivirus replication. Antiviral Res 98:242–247. doi: 10.1016/j.antiviral.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 19.Chen YL, Yokokawa F, Shi PY. 2015. The search for nucleoside/nucleotide analog inhibitors of dengue virus. Antiviral Res 122:12–19. doi: 10.1016/j.antiviral.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 20.Lo MK, Shi PY, Chen YL, Flint M, Spiropoulou CF. 2016. In vitro antiviral activity of adenosine analog NITD008 against tick-borne flaviviruses. Antiviral Res 130:46–49. doi: 10.1016/j.antiviral.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yin Z, Chen YL, Schul W, Wang QY, Gu F, Duraiswamy J, Kondreddi RR, Niyomrattanakit P, Lakshminarayana SB, Goh A, Xu HY, Liu W, Liu B, Lim JY, Ng CY, Qing M, Lim CC, Yip A, Wang G, Chan WL, Tan HP, Lin K, Zhang B, Zou G, Bernard KA, Garrett C, Beltz K, Dong M, Weaver M, He H, Pichota A, Dartois V, Keller TH, Shi PY. 2009. An adenosine nucleoside inhibitor of dengue virus. Proc Natl Acad Sci U S A 106:20435–20439. doi: 10.1073/pnas.0907010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Potisopon S, Ferron F, Fattorini V, Selisko B, Canard B. 2017. Substrate selectivity of dengue and Zika virus NS5 polymerase towards 2′-modified nucleotide analogues. Antiviral Res 140:25–36. doi: 10.1016/j.antiviral.2016.12.021. [DOI] [PubMed] [Google Scholar]
- 23.Eyer L, Zouharová D, Širmarová J, Fojtíková M, Štefánik M, Haviernik J, Nencka R, de Clercq E, Růžek D. 2017. Antiviral activity of the adenosine analogue BCX4430 against West Nile virus and tick-borne flaviviruses. Antiviral Res 142:63–67. doi: 10.1016/j.antiviral.2017.03.012. [DOI] [PubMed] [Google Scholar]
- 24.Eyer L, Šmídková M, Nencka R, Neča J, Kastl T, Palus M, De Clercq E, Růžek D. 2016. Structure-activity relationships of nucleoside analogues for inhibition of tick-borne encephalitis virus. Antiviral Res 133:119–129. doi: 10.1016/j.antiviral.2016.07.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.