

Acinetobacter baumannii is a major cause of nosocomial infections especially hospital-acquired pneumonia. This bacterium readily acquires antibiotic resistance traits and therefore, new treatment alternatives are urgently needed. The virulence of A. baumannii linked to iron acquisition suggests a potential for new anti-infectives that target its iron acquisition. DIBI, a 3-hydroxypyridin-4-one chelator, is a purpose-designed, iron-sequestering antimicrobial that has shown promise for treating microbial infection.
KEYWORDS: anti-infective, antibiotic resistance, iron acquisition, iron sequestration
ABSTRACT
Acinetobacter baumannii is a major cause of nosocomial infections especially hospital-acquired pneumonia. This bacterium readily acquires antibiotic resistance traits and therefore, new treatment alternatives are urgently needed. The virulence of A. baumannii linked to iron acquisition suggests a potential for new anti-infectives that target its iron acquisition. DIBI, a 3-hydroxypyridin-4-one chelator, is a purpose-designed, iron-sequestering antimicrobial that has shown promise for treating microbial infection. DIBI was investigated for its in vitro and in vivo activities against clinical A. baumannii isolates. DIBI was inhibitory for all isolates tested with very low MICs (2 μg/ml, equivalent to 0.2 μM), i.e., at or below the typical antibiotic MICs reported for antibiotic-sensitive strains. DIBI inhibition is Fe specific, and it caused an iron-restricted bacterial physiology that led to enhanced antibiotic killing by several discrete antibiotics. DIBI also strongly suppressed recovery growth of the surviving population following antibiotic exposure. A low intranasal dose (11 μmol/kg) of DIBI after intranasal challenge with hypervirulent ciprofloxacin (CIP)-resistant A. baumannii LAC-4 significantly reduced bacterial burdens in mice, and DIBI also suppressed the spread of the infection to the spleen. Treatment of infected mice with CIP alone (20 mg/kg, equivalent to 60 μmol/kg) was ineffective given LAC-4’s CIP resistance, but if combined with DIBI, the treatment efficacy improved significantly. Our evidence suggests that DIBI restricts host iron availability to A. baumannii growing in the respiratory tract, bolstering the host innate iron restriction mechanisms. DIBI has potential as a sole anti-infective or in combination with conventional antibiotics for the treatment of A. baumannii pneumonia.
INTRODUCTION
Acinetobacter baumannii is a Gram-negative opportunistic bacterial pathogen causing various community- and hospital-acquired infections, including pneumonia and urinary tract and surgical site infections (1). Hospital-acquired pneumonia (HAP), especially with patients on ventilators, is the most prevalent A. baumannii nosocomial infection, and its mortality rate can approach 70% (2). This bacterium has a remarkable ability to take on multiple-drug resistance (MDR) attributes and consequently has become a major treatment challenge (3, 4). With carbapenem resistance now common, last-resort antibiotic choices have been reduced to tigecycline (TGC) and the polymyxins, especially polymyxin E, also known as colistin (5). Resistance to the polymyxins has been infrequent, but these polypeptide membrane-active agents pose problems due to their toxicity in humans, especially with systemic administration (5–7). Consequently, new therapeutic approaches are urgently needed to meet the MDR threat from A. baumannii.
Iron acquisition is a virulence determinant for A. baumannii with its acinetobactin iron siderophore acquisition system being more prevalent in MDR isolates (1, 8, 9). Gallium, a nonfunctional analogue of Fe that inhibits A. baumannii growth and biofilm formation in vitro by displacing functional iron (10, 11), has also been shown to suppress A. baumannii experimental pneumonia in mice (12). However, gallium poses potential toxicity issues that have not yet been adequately studied (13).
Rather than attempting to displace microbial iron supply with an excess of its nonfunctional analogue gallium, our approach has been to specifically address the relevant host iron pools as accessed by microbial pathogens during infection and selectively take these host iron pools out of play through sequestration with novel antimicrobial iron sequestrants. The rationale for this approach is that microbial iron needs are heightened during infection and vertebrate hosts already mount an innate iron sequestration defense, i.e., attempting to restrict microbial iron access. New iron-selective sequestrants that are not accessible by microbes might bolster the natural host sequestration of key iron pools, i.e., those pivotal for infection. This approach has led to the synthesis of a novel platform of chelating anti-infectives (14–17). The current lead compound DIBI, a 3-hydroxypyridin-4-one chelator, is a 9 kDa, relatively low-molecular-weight copolymer containing nine broadly spaced hydroxypyridinone metal-binding groups that can fully coordinate three iron molecules, and its synthesis and molecular structure have been reported (15). The mechanism of action and characteristics of DIBI are in sharp contrast to conventional chelation therapy agents that were not developed as anti-infectives but for treating hematological disorders. Conventional chelation therapy agents require high concentrations to exert antimicrobial activity, but DIBI has been shown to be anti-infective in the very low micromolar range and is, for example, >250-fold more inhibitory to A. baumannii than deferiprone, one such clinically used hematological iron chelator (15). Conventional chelation therapy agents also have potential issues of toxicity. In contrast, DIBI has a low toxicity profile. The relatively low cytotoxicity for DIBI has been shown with both normal human and mouse fibroblasts and with normal human epithelial cells, with 50% effective concentrations of 8,928, 793, and 691 μg/ml, respectively (18). Importantly, Organization for Economic Cooperation and Development (OECD)-prescribed oral and systemic toxicity testing in rats has shown no observable adverse effects (NOAEL) with 14-day repeated dosing at the highest DIBI doses (1,000 mg/kg/day [oral] and 500 mg/kg/day [intravenous]) tested (unpublished results). DIBI also displays broad-spectrum antimicrobial activities (14–17) and has been shown to enhance the activities of conventional antibacterial and antifungal antibiotics when applied with these at relatively low concentrations (16, 17).
DIBI is being developed initially as a topical anti-infective primarily for use on mucosal surfaces. Here, we report its in vitro and in vivo activities against A. baumannii clinical isolates, including high-virulence MDR LAC-4 (19) and low-virulence ATCC 17978 isolates.
RESULTS
Sensitivity of A. baumannii isolates to DIBI.
All seven of a relatively diverse group of A. baumannii isolates tested, including two ATCC reference strains and various MDR clinical strains were found to display high sensitivity to DIBI, with MICs of 2 μg/ml, as shown in Table 1 . This MIC expressed on a molecular concentration basis corresponds to 0.2 μM DIBI. This relatively low concentration of DIBI was compared to the various antibiotics, whose MICs were also tested (both in μg/ml and μM), as shown in Table 1 . MIC expression in molar concentration is important because with antibiotics, reference is usually made to MIC cutoffs for resistance on a mass concentration basis (i.e., μg/ml) and not on a molecular concentration basis (μmol/ml or μM). For most antibiotics, this has little impact because with conventional antibiotics, the molecular weights fall within a relatively low and narrow range (300 to 1,000 g/mol). However, DIBI is approximately 10 times larger (9 kDa) than a typical antibiotic, and it is thus also important to compare its activity to antibiotics on a molar concentration basis. as shown in Table 1.
TABLE 1.
A. baumannii isolates tested and their antibiotic and DIBI sensitivities
| Isolate | MIC |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| GEN |
CIP |
CST |
TGC |
DIBI |
||||||
| μg/ml | μM | μg/ml | μM | μg/ml | μM | μg/ml | μM | μg/ml | μM | |
| LAC-1 | 64 | 134 | 128 | 386 | 2 | 1.7 | 4 | 6.8 | 2 | 0.2 |
| LAC-4 | 64 | 134 | 64 | 193 | 2 | 1.7 | 1 | 1.7 | 2 | 0.2 |
| LAC-10 | 4 | 8.4 | >256 | >772 | 16 | 13 | 4 | 6.8 | 2 | 0.2 |
| LAC-15 | 2 | 4.2 | >256 | >772 | >512 | >443 | 4 | 6.8 | 2 | 0.2 |
| AYE | >256 | >536 | 256 | 772 | 1 | 0.9 | 2 | 3.4 | 2 | 0.2 |
| ATCC 17978 | 1 | 2.1 | 1 | 3 | 2 | 1.7 | 1 | 1.7 | 2 | 0.2 |
| ATCC 19606 | 8 | 16.7 | 1 | 3 | 8 | 6.9 | 4 | 6.8 | 2 | 0.2 |
The results of Table 1 show that sensitivity of the isolates to DIBI was high, with MICs for all isolates at only 0.2 μM, i.e., below the MICs for each of the four antibiotics tested even in the cases of antibiotic-sensitive isolates that had antibiotic MICs of 1 to 2 μM. The MICs for DIBI and the antibiotics were determined in RPMI given its chemically defined makeup and more predictable iron content (see further discussion below). The MICs for the antibiotics as determined in RPMI were found to be similar or slightly higher to those determined using Mueller-Hinton broth (MHB), as were reported previously for our key isolates, LAC-4 and ATCC 17978 (19).
MDR isolates with high antibiotic resistance to gentamicin (GEN), ciprofloxacin (CIP), or colistin (CST), e.g., LAC-4 for CIP with an MIC of 64 μg/ml or 193 μM, were highly sensitive to DIBI. Importantly, MDR and antibiotic-sensitive isolates had similarly high sensitivities to DIBI.
DIBI inhibition is Fe specific.
DIBI inhibition of microbial growth has been shown previously to be Fe specific and Fe reversible (14–17). The nature of growth inhibition by DIBI was investigated using the LAC-4 clinical isolate, as well as the two reference strains, ATCC 17978 and ATCC 19606. For these experiments, cells from an overnight early-stationary-phase culture were inoculated at a low cell density (i.e., approximately 105 CFU/ml) into fresh RPMI that had first been specifically stripped of its Fe content and then reconstituted with a known amount of iron. Stripping RPMI to a residual Fe content of ≤0.08 μM was done as described previously (16), and the readdition of iron was made with iron-citrate to ensure its availability to the bacterium. The low inoculum density ensured minimal Fe carryover into the fresh culture. The deferrated RPMI provided a control for Fe-dependent growth, and it did not support growth for any of the isolates as shown in Fig. 1. However, the addition of 1 μM Fe to the stripped RPMI to obtain referrated RPMI with an iron content in excess of the minimum required for full bacterial growth allowed robust growth for all three isolates, with Ymax optical density values at 600 nm (OD600) of >1 observed by 24 h of incubation.
FIG 1.

A. baumannii strains LAC-4 (A), ATCC 17978 (B), and ATCC 19606 (C) were cultured in deferrated RPMI (open circles), deferrated RPMI + 1 μM iron citrate (solid triangles), deferrated RPMI + 1 μM iron citrate + 0.1μM DIBI (inverted solid triangles), and deferrated RPMI + 1 μM iron citrate + 1 μM DIBI (solid diamonds). Cultures were shaken continuously at 35°C, and growth was measured periodically by measuring the OD600. Data points are reported as means ± SEM from at least two independent experiments. *, P < 0.05 for deferrated RPMI + 1 μM iron citrate + 1 μM DIBI versus deferrated RPMI + 1 μM iron citrate at 12, 24, and 36 hpi.
Addition of DIBI to the referrated medium suppressed growth in a dose-dependent manner for the three isolates (Fig. 1). LAC-4 appeared somewhat less Fe fastidious compared to ATCC 17978 or ATCC 19606. The low dose of DIBI (0.1 μM) allowed growth of LAC-4 to a Ymax comparable to the iron-replete control by 24 h. DIBI added at 1 μM, however, completely inhibited LAC-4 growth to 24 h and resulted in minimal growth by 36 h. Based on DIBI’s known Fe-binding capacity (15), the 0.1 μM addition would have sequestered approximately 30% of the added iron, while the 1 μM DIBI addition was in 3-fold excess of the amount of DIBI theoretically required to fully sequester the 1 μM Fe added to the deferrated RPMI.
DIBI enhances activity of antibiotics in vitro.
The influence of DIBI iron restriction on antibiotic killing of LAC-4 and ATCC 17978 was investigated. Prototypical representatives of four discrete antibiotic classes of clinical relevance for A. baumannii infection treatment—i.e., GEN, an aminoglycoside; CIP, a fluoroquinolone; CST, a polymyxin; and TGC, a glycylcycline—were investigated. Log-phase cells grown in MHB were transferred to regular iron-replete RPMI and exposed to the different antibiotics both alone and with DIBI present. The cultures were examined using conventional time-kill kinetic assays. It is important to note that RPMI was used for these studies since it is fully chemically defined, it displays less batch-to-batch variation as to iron content, and it has sufficient iron for unrestricted growth (16), but RPMI is still low enough in iron to have some relevance to the low iron availability that predominates in vivo. Although MHB was used to grow the inocula, it was not used in the kill assays since it is not well defined, it varies in iron content from batch to batch, and it typically contains 6 to 10 μM Fe, i.e., around 10× the minimum iron concentration needed for unrestricted bacterial growth (16). Thus, the iron content of MHB has less relevance than RPMI as to in vivo conditions of iron availability for bacterial growth. We wanted to ensure low iron availability to the isolates for testing their sensitivity to DIBI to better ensure their potentially competing iron acquisition mechanisms, i.e., acinetobactin production would not be repressed by excess medium iron.
A concentration of antibiotic that did not provide complete killing on its own (1/8 to 1× MIC) was first determined and utilized to achieve an initial incomplete killing phase, followed by recovery growth of survivors. This approach allowed the assessment of how DIBI might affect the initial killing kinetics of the various antibiotics, as well as its possible effects on the subsequent recovery growth.
For these studies, DIBI on its own was not expected to provide significant killing or to markedly affect overall bacterial replication. DIBI at 9 kDa is too large to be taken up into bacterial cells, where it could directly affect intracellular iron contents. The relatively high bacterial inoculum of log-phase Fe-replete cells (approximately 5 × 106 CFU/ml) would not require immediate supply of exogenous medium Fe for initial bacterial replication. The DIBI concentration utilized (2.2 μM) would ensure maximal sequestration of extracellular medium Fe sources to ensure iron restriction, i.e., once endogenous bacterial iron reserves were exhausted. DIBI would also serve as a sink for any bacterial cell iron reserves lost or freed to the medium from bacterial lysis caused by antibiotic killing. However, the relatively high concentration of DIBI used for these experiments was still well within the concentration range of the various antibiotics tested. For example, GEN was used at as low as 1 μg/ml (2 μM) for ATCC 17978 and as high as 32 μg/ml (67 μM) for LAC-4.
Comparative testing of LAC-4 and ATCC 17978 was performed with GEN, CIP, CST, and TGC both alone and together with DIBI. LAC-4 is resistant to GEN (Table 1) and 1/2× GEN MIC (32 μg/ml=67 μM) provided only modest short-term killing with rapid recovery growth observed by 4 h, reaching a near control 24-h Ymax CFU/ml level (Fig. 2A). DIBI with GEN did not suppress but prolonged the initial killing phase, and it strongly suppressed recovery growth, which remained below the initial inoculum CFU/ml at 24 h. In a somewhat similar manner, GEN-susceptible ATCC 17978 at 1× the GEN MIC on its own displayed only moderate killing to 6 h, and this was also followed by strong recovery growth to a near control CFU/ml level by 24 h (Fig. 2B). However, DIBI together with GEN promoted much stronger killing of ATCC 17978, with a complete >6-log killing by 24 h. No viable bacteria could be recovered at 24 h for ATCC 17978 exposed to 2.1 μM GEN plus 2.2 μM DIBI (Fig. 2B).
FIG 2.

Influence of DIBI on GEN and CIP killing and recovery growth for A. baumannii LAC-4 and ATCC 17978. Cultures of LAC-4 (A and C) or ATCC 17978 (B and D) were inoculated at a final concentration of approximately 5 × 106 CFU/ml in RPMI or RPMI containing GEN (A and B) or CIP (C and D) alone or with DIBI either alone or combined with the antibiotics. Cultures were grown at 35°C, with bacterial counts (CFU/ml) being determined at intervals over 24 h. Symbols: control (open circles), DIBI added (open triangles) at 20 μg/ml, antibiotic added (solid circles, GEN at 32 μg/ml [A] or 1 μg/ml [B] or CIP at 32 μg/ml [C] or 0.5 μg/ml [D]), or DIBI + antibiotic (solid triangles). Data points are reported as means ± SEM. *, P < 0.05 for DIBI plus antibiotic combinations versus DIBI and antibiotic treatments alone.
Both LAC-4 and ATCC 17978 displayed similar killing with 1/2× CIP MIC despite the higher concentration needed for CIP-resistant LAC-4 (i.e., 32 μg/ml [96.6 μM] versus 0.5 μg/ml [1.5 μM] for ATCC 17978), as shown in Fig. 2C and D. DIBI with CIP appeared to slightly slow the initial killing for both isolates, but nevertheless there was substantial overall killing with CIP+DIBI and no net recovery growth by 24 h. The final CFU/ml at 24 h remained at or below the initial inoculum CFU/ml. A higher DIBI concentration (4.4 or 6.6 μM) with CIP exhibited somewhat similar responses for LAC-4 to those shown for 2.2 μM DIBI (results not shown), which is consistent with DIBI’s sequestration of iron supply and the resulting physiological effects, as discussed below.
In the case of CST, 1/4× MIC (0.43 μM) provided substantial initial killing of LAC-4, followed by strong recovery growth approaching control Ymax CFU/ml values by 24 h. DIBI (2.2 μM) with 0.43 μM CST substantially increased killing (>3.5-log killing) for LAC-4, and there was no subsequent recovery growth observed after 6 h (Fig. 3A). For ATCC 17978, 1/8× CST MIC (0.22 μM) did not provide significant initial killing, but there was extensive and continued killing observed with 0.22 μM CST plus 2.2 μM DIBI with a >4-log killing obtained by 24 h (Fig. 3B).
FIG 3.

Influence of DIBI on CST and TGC killing and recovery growth for A. baumannii LAC-4 and ATCC 17978. Cultures of LAC-4 (A and C) or ATCC 17978 (B and D) were inoculated at a final concentration of approximately 5 × 106 CFU/ml in RPMI or in RPMI containing CST (A and B) or TGC (C and D) alone or with DIBI either alone or combined with the antibiotics. Cultures were grown at 35°C with bacterial counts (CFU/ml) being determined at intervals over 24 h. Symbols: control (open circles), DIBI added (open triangles) at 20 μg/ml, antibiotic added (solid circles, CST at 0.5 μg/ml [A] or 0.25 μg/ml [B] or TGC at 0.5 μg/ml [C] or 0.5 μg/ml [D]), or DIBI + antibiotic (solid triangles). Data points are reported as means ± SEM. *, P < 0.05 for DIBI plus antibiotic combinations versus DIBI and antibiotic treatments alone.
Responses to 1/2× TGC MIC (0.5 μg/ml [0.86 μM]) were somewhat similar for both LAC-4 and ATCC 17978 (Fig. 3C and D). With TGC alone, both isolates displayed only modest early killing, followed by strong recovery growth to near control Ymax levels by 24 h. DIBI (2.2 μM) with TGC (0.86 μM) improved killing for both isolates and killing continued over the 24 h of the incubation period, as shown in Fig. 3C and D.
A similar pattern of results was also observed with ATCC 19606. This strain has low-level resistance to both GEN and CST. GEN on its own at 2× MIC (16 μg/ml = 34 μM) provided <2-log CFU/ml killing by 24 h, as shown in Table 2. DIBI (2.2 μM) with GEN (34 μM) improved killing with a 2.85-log additional kill reduction observed with the combination over that obtained for GEN alone. CIP alone at 1× MIC (1 μg/ml = 3 μM) provided only a slight net 24-h kill (−0.44 log) on its own, but when combined with DIBI (2.2 μM), a >3-log increased kill reduction was observed. In the case of CST alone (1/8 MIC, 1 μg/ml = 0.9 μM), net growth of 1.22 log CFU/ml occurred by 24 h. DIBI (2.2 μM), along with CST, resulted in >6-log killing. TGC results for ATCC 19606 were quite similar to those observed with both LAC-4 and ATCC 17978, with an additional 2.5-log kill reduction observed when DIBI (2.2 μM) was present with TGC (0.5 μg/ml = 0.86 μM).
TABLE 2.
Synergy of DIBI with various antibiotics for ATCC 19606
| Treatment | Avg 24-h CFU/ml as –log10 reduction or +log10 increase for various antibioticsa
: |
|||
|---|---|---|---|---|
| GEN, 16.0 μg/ml | CIP, 1.0 μg/ml | CST, 1.0 μg/ml | TGC, 0.5 μg/ml | |
| Antibiotic + DIBI | −2.85 | −3.30 | −6.40 | −2.56 |
| Antibiotic alone | −1.72 | –0.44 | +1.22 | −1.83 |
| DIBI alone | −0.12 | −0.08 | −0.06 | +0.01 |
Average (n = 2) 24-h CFU/ml log10 reductions (–) or increases (+) for antibiotic plus DIBI versus antibiotic alone and for DIBI alone and antibiotic alone versus 24-h-untreated control growth.
Overall, our results indicated that DIBI did not impede antibiotic killing, that it provided substantial synergy of killing with each of the antibiotics tested, and that it also suppressed recovery growth of survivors following the initial antibiotic kill phase, in all cases. Synergy of DIBI with the antibiotics was evident with the three bacterial isolates tested. Importantly, DIBI synergy with the antibiotics was observed for both antibiotic-susceptible and antibiotic-resistant A. baumannii isolates.
DIBI reduces virulent Lac-4 infection burden.
Our in vitro experiments demonstrated that DIBI sensitivity of the A. baumannii isolates was not related to their antibiotic resistance profiles (Table 1). We next investigated the in vivo effect of DIBI administration on A. baumannii infection in a mouse pneumonia model that had previously established marked differences in the pathogenesis between virulent isolates such as LAC-4 compared to low-virulence isolates such as ATCC 17978 (12, 19). LAC-4 and ATCC 17978 were both utilized for our in vivo testing, LAC-4 also being an MDR isolate. Given the initial site of infection was the airway aspect of the respiratory tract, we also examined for DIBI activity by intranasal (i.n.) delivery.
Control lung and spleen infection burdens observed at 24 h postinfection (hpi) were similar to those previously established for this isolate (19). As shown in Fig. 4, the administration of DIBI (100 mg/kg, equivalent to 11 μmol/kg) 3 h after i.n. challenge with A. baumannii LAC-4 reduced the 24-hpi lung bacterial burden significantly (P < 0.05). DIBI also suppressed (∼3-log reduction) the extrapulmonary spread of infection to the spleen (Fig. 4).
FIG 4.
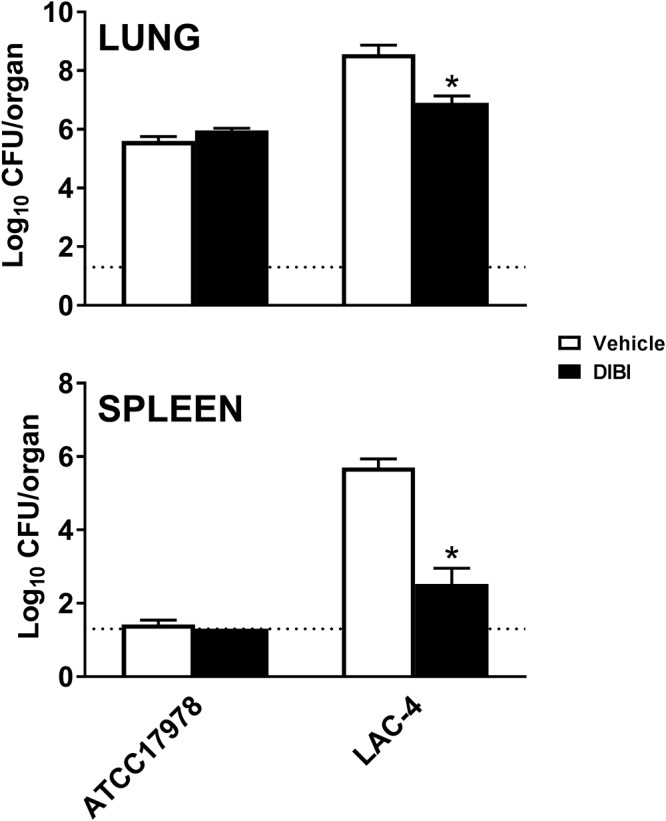
Bacterial burdens in the lungs and spleen of DIBI or vehicle-treated BALB/c mice after i.n. challenge with either A. baumannii ATCC 17978 or LAC-4. DIBI (closed bars)- and diluent vehicle (open bars)-treated mice were infected i.n. with approximately 3.6 × 107 CFU of A. baumannii ATCC 17978 or 3.0 × 106 CFU of LAC-4 at 0 h. The mice were treated 3 and 10 h postchallenge i.n. with DIBI (one half at each time of a total treatment of 100 mg/kg = 11 μmol/kg). Bacterial burdens in the lungs and spleen were determined by quantitative bacteriology 24 h after challenge. The data are presented as means ± SEM (n = 5 to 10) and represent one of at least two independent experiments with similar results. The detection limit (dotted lines) for bacterial burdens was 1.3 log10 CFU/organ. *, P < 0.05 versus vehicle-treated mice.
In contrast, challenge with a much higher (10×) inoculum of the low-virulence ATCC 17978 strain produced only a modest lung bacterial burden in untreated mice. Reductions in lung bacterial burden with DIBI were comparable to the reductions observed for LAC-4 with effective antibiotics, including TGC, as previously reported when this infection model was first developed (19).
The lung burden with ATCC 17978 was >2 log lower than observed for LAC-4, and there was no evidence of dissemination of ATCC 17978 infection to the spleen. DIBI administration did not appear to affect the more modest lung infection burden observed with ATCC 17978 (Fig. 4).
DIBI enhances in vivo CIP efficacy for CIP-resistant LAC-4.
Because DIBI had enhanced antibiotic activity for LAC-4 in vitro, we also examined whether DIBI might enhance CIP efficacy against LAC-4 infection in mice, noting that this isolate is CIP resistant (MIC = 64 μg/ml [193 μM]). For these experiments, we used a substantially increased challenge inoculum (approximately 1.7 × 107 CFU) and compared this to a lower challenge dose (approximately 3 × 106 CFU), i.e., the latter being somewhat reduced over that used for the challenge experiments shown in Fig. 4.
As expected, bacterial burdens in control infected mice were correspondingly higher with the higher challenge inoculum than those observed with the lower challenge inoculum (Fig. 5). CIP alone at 20 mg/kg (60 μmol/kg) administered intraperitoneally (i.p.) did not affect bacterial burdens in the lungs, spleen, or blood, as shown in Fig. 5, an observation consistent with LAC-4’s CIP resistance. However, 11 μmol/kg DIBI administered i.n. improved the treatment efficacy of CIP in mice that had received a higher bacterial challenge inoculum. The tissue (lung and spleen) and blood bacterial burdens were significantly reduced with the CIP/DIBI combination (P < 0.05 for lung, spleen, and blood).
FIG 5.

Effect of DIBI and DIBI/CIP treatment on bacterial burdens in the lungs, spleens, and blood of BALB/c mice after i.n. challenge with A. baumannii LAC-4. Groups of five mice were infected i.n. with 3.0 × 106 or 1.7 × 107 CFU of A. baumannii LAC-4 at 0 h. Treatments were administered to mice i.n. 3 and 10 h later, with 100 mg/kg (11 μmol/kg) DIBI, 100 mg/kg DIBI + 20 mg/kg (60 μmol/kg) CIP, 20 mg/kg CIP, or diluent. Bacterial burdens in the lungs, spleens, and blood were determined by quantitative bacteriology 24 h after inoculation. The data are presented as means ± SEM (n = 5 to 10) and represent one of at least two independent experiments with similar results. The detection limits for the tissue and blood are indicated by dotted lines. *, P < 0.05 versus diluent vehicle-treated mice.
The tissue and blood bacterial burdens in DIBI/CIP-treated mice infected with the lower challenge dose of LAC-4 were reduced over controls, but these reductions were more modest and not statistically significant (Fig. 5). CIP treatment alone at this lower infecting dose also showed no effect on the tissue or blood bacterial burdens, whereas DIBI administration alone substantially reduced the tissue and bacterial burdens of the infected mice. The bacterial burden reduction with DIBI was similar to that shown in Fig. 4, i.e., as had been observed with a slightly higher (1.7×) challenge inoculum.
Our results indicate that DIBI improved CIP efficacy against CIP-resistant LAC-4 in vivo, which is consistent with our results showing DIBI/CIP synergy in vitro (Fig. 2C). DIBI was effective for both the low and high challenge doses, and DIBI enhanced CIP efficacy, suppressing the systemic spread of the infection.
Effect of DIBI and CIP on cytokine and chemokine responses.
Our experiments had shown that i.n. administration of DIBI alone and in combination with CIP reduced lung bacterial burdens and suppressed the systemic dissemination of LAC-4 infection. To further characterize the in vivo effect of DIBI on LAC-4 infection and in conjunction with CIP, we compared the levels of a panel of proinflammatory cytokines and chemokines that had been established in relation to the host response to this infection. The lung and serum titers of the panel in infected control (vehicle-treated) mice were similar to those previously reported (19, 20). Treatment of LAC-4-infected mice with CIP+DIBI significantly reduced granulocyte-macrophage colony-stimulating factor, keratinocyte-derived chemokine, and monocyte chemoattractant protein 1 (MCP-1) levels over control levels in both lung tissue and blood serum as shown in Fig. 6. The CIP/DIBI treatment also significantly reduced the levels of interleukin-1α (IL-1α), IL-1β, and tumor necrosis factor alpha levels in the lung and IL-6, macrophage inflammatory protein 1β (MIP-1β), MIP-2, and RANTES (regulated on activation, normal T cell expressed and secreted) in the serum (P < 0.05). Similar reductions in these cytokines and chemokines were also observed, although to lesser magnitudes, in the lungs and sera of LAC-4-infected mice treated with DIBI alone. Overall, these results indicated that the synergistic effect of DIBI/CIP is associated with the local (lung) and systemic (serum) reductions of a panel of key proinflammatory cytokines and chemokines, and these reductions were correlated with reduced tissue and blood bacterial burdens.
FIG 6.

Effect of DIBI and DIBI/CIP treatment on the cytokine and chemokine levels in the sera (A) and lungs (B) of BALB/c mice after i.n. challenge with A. baumannii. Groups of BALB/c mice were i.n. inoculated with 1.7 × 107 CFU of A. baumannii LAC-4. Serum and lung homogenate supernatant samples were collected and processed at 24 h, and cytokine and chemokine levels were determined using the mouse panel of 12-plex multiplex kits (Millipore) on a Luminex Magpix instrument. The data are expressed as means ± SEM of five mice. The detection limits of the assays were 2.5 to 15 pg/ml. *, P < 0.05 versus diluent vehicle-treated mice.
DISCUSSION
DIBI was strongly inhibitory to growth for all seven A. baumannii isolates with a similarly low MIC of 2 μg/ml, i.e., equivalent to only 0.2 μM DIBI for each isolate. MIC expression in molar concertation units is important with larger molecules such as DIBI (at 9 kDa) to allow a proper comparison to the MICs of conventional antibiotics as antibiotics are typically low-molecular-weight molecules (range, 300 to 1,100 Da). On this basis, DIBI with an MIC of 0.2 μM is more inhibitory to A. baumannii than the antibiotics we tested. For example, our antibiotic-susceptible isolate ATCC 17978 displayed MICs for GEN, CIP, CST, and TGC of 2.1, 3, 1.7, and 1.7 μM, respectively (see Table 1). Thus, these antibiotic MICs were approximately 10× higher than the 0.2 μM MIC found for DIBI.
DIBI’s antimicrobial activities (bacterial and fungal), as well as its anticancer activities, have been shown to be iron specific (14–18). The addition of DIBI to RPMI medium sufficient to fully chelate its known Fe content caused complete growth inhibition, whereas a smaller DIBI addition yielded correspondingly reduced inhibition. These results indicated that DIBI inhibition was Fe specific and that the Fe-DIBI chelate was not readily accessible as an iron source for this bacterium. Conventional iron chelators, including deferiprone (Ferriprox) and desferrioxamine (Desferal), that are used clinically for chelation therapy of human transfusional iron overload are not or only poorly inhibitory to A. baumannii and can actually serve as iron sources for this and other bacteria (15).
The production of the siderophore acinetobactin and its role in Fe acquisition and the virulence of LAC-4 and ATCC 19606 has been described (8, 21), and acinetobactin synthesis and associated transport genes have been mapped in various isolates, including LAC-4, ATCC 17978, and ATCC 19606 (22, 23). It is interesting to note in this regard that MDR isolates have been reported to typically possess the acinetobactin Fe acquisition virulence system (9).
Reduced growth caused by DIBI addition would be expected to induce siderophore production for the acinetobactin-competent isolates. Other studies involving addition of different chelators to sequester iron for A. baumannii ATCC 17978 have shown its upregulation of siderophore synthesis and related iron acquisition gene product mRNAs (24). On this basis, DIBI appears to circumvent Fe acquisition by the A. baumannii acinetobactin iron acquisition system through DIBI’s more avid sequestration of iron and also the bacterium’s inability to readily access iron when bound to DIBI.
It is also important to note that DIBI sensitivity of the isolates was independent of their antibiotic sensitivity/resistance profiles. MDR isolates with high-level resistance to CIP, GEN, and CST (e.g., LAC-4, LAC-10, and AYE) all had similar high sensitivities to DIBI, as did the antibiotic-susceptible isolates (e.g., ATCC 17978 and ATCC 19606). Given this, DIBI targeting of iron acquisition appears unrelated to the antibiotic sensitivity or resistance attributes of A. baumannii.
DIBI, when added to a relatively large population (5 × 106 CFU/ml) of actively dividing, log-phase, iron-replete bacterial cells, did not overtly affect growth, as measured by net CFU increases over the 24-h observation period. However, DIBI evidently induced an Fe-restricted physiology for such cultures. This Fe-restricted physiology became evident when DIBI was present along with various chemically distinct bactericidal antibiotics. When DIBI was tested with any of the four antibiotics we tested, an increased and/or extended antibiotic killing phase was observed, and there was also little or no subsequent recovery growth of survivors following the initial antibiotic killing phase. In contrast, in all cases with antibiotic alone, there was robust recovery growth following the initial killing phase.
DIBI is not like a typical antibiotic in terms of its mechanism of action because an antibiotic typically has a singular or relatively few cellular target(s). Rather, DIBI has multiple potential targets and would be expected to affect a number of Fe-dependent targets, these being essential for diverse essential physiological functions, such as DNA synthesis (ribonucleotide reductase), respiration (cytochromes), and defense (catalase). These examples of critical enzymes require iron for their activity, and this iron requirement is not replaceable. Thus, iron restriction by DIBI would exert a broad-based iron deprivation physiological stress conceivably resulting initially in unbalanced bacterial growth from reduced supply of iron, progressing to impaired repair and defense and then eventually progressing to cessation of replication and ultimately to bacterial apoptosis.
We have shown previously that Staphylococcus aureus is highly susceptible to DIBI and that iron restriction from DIBI inhibits growth and results in enhanced sensitivity to various antibiotics (16). In the present study, we have found a somewhat similar pattern of enhancement of antibiotic activity with DIBI for both MDR (LAC-4) and antibiotic-susceptible (ATCC 17978) A. baumannii isolates. DIBI increased the initial killing phase for the antibiotics, except in the case of CIP. Enhanced antibiotic killing with DIBI was pronounced for GEN for ATCC 17978, where the combination of GEN+DIBI caused complete killing, in contrast to GEN alone, which provided only an early 2-log kill, followed by robust recovery growth to untreated control bacterial numbers by 24 h. Strongly enhanced and continued killing was also evident for CST and TGC when combined with DIBI. For CIP, there was a modest slowing of the initial rate of killing with DIBI, although the overall extent of killing was maintained, and in general, bacterial numbers remained low, i.e., at least 2 log CFU below initial numbers at 24 h. For all the antibiotics when combined with DIBI, the 24-h total CFU/ml reductions compared to treatment with either antibiotic alone or DIBI alone were ≥2 log. On this basis, DIBI was synergistic in action with these antibiotics.
Virulent A. baumannii LAC-4 produced a rapid initial pneumonia in mice with lung bacterial burdens of approximately 109 CFU and with dissemination of the infection to the spleen by 24 hpi. In contrast, the low-virulence ATCC 17978 isolate produced a much lower (3 log less) lung bacterial burden without any evidence of infection spread to spleen, even with a 10× higher initial bacterial challenge inoculum. These findings agreed with earlier results (19) and provided the basis for a model infection useful to investigate experimental DIBI therapy of A. baumannii infection. Our testing with the low-virulence ATCC 17978 that had been previously demonstrated not to cause appreciable infection in this model, i.e., essentially avirulent for normal mice, provided additional evidence that DIBI administration did not inappropriately support infection with a nonvirulent strain. DIBI suppressed infection with the virulent LAC-4 strain, which had been shown previously to be hypervirulent. The iron acquisition and other virulence determinants for these virulent and nonvirulent strains of A. baumannii are part of our ongoing studies.
We examined 24-hpi bacterial burdens prior to full dissemination of the infection and well before any mortality to allow assessment of anti-infective treatments, i.e., as might be effective early during the course of the establishment and early spread of infection. DIBI administered i.n. at 11 μmol/kg and 3 hpi significantly decreased 24-hpi bacterial lung burdens and suppressed the spread of the LAC-4 infection to spleen. Lung burdens were approximately 100× lower as a result of DIBI treatment, and there was an even greater reduced (3 log) spleen burden observed. The reductions in lung bacterial burden, as achieved with DIBI, were comparable to those previously obtained with TGC for LAC-4 using this infection model (19). The high sensitivity of LAC-4 to TGC (see Table 1) is important in this regard, i.e., for comparison to the observed DIBI efficacy. Lung and serum levels of key cytokines and chemokines also correlated well with the infection burdens.
Our results are consistent with DIBI acting as an anti-infective in vivo by restricting availability of iron supply as needed by A. baumannii for growth in the respiratory tract. We had previously shown that DIBI suppressed the growth of S. aureus in the nares of mice (16). The results of our studies provide strong additional direct evidence that host iron pools associated with the mucosal surfaces of the respiratory tract, are in play during infection and that these host iron pools appear pivotal for supply of infection-essential iron to both Gram-positive (S. aureus) and Gram-negative (A. baumannii) bacteria.
Importantly, our findings suggest that these iron pools can be sequestered by DIBI to suppress respiratory infection and therefore indicate a potential for DIBI for the treatment of pneumonia noting that HAP is often caused by A. baumannii or S. aureus. This potential for DIBI bolstering host innate iron restriction defenses warrants further investigation.
DIBI also enhanced the efficacy of CIP for the MDR CIP-resistant LAC-4 isolate both in vitro and in vivo. Infection was not reduced by CIP alone consistent with LAC-4’s CIP resistance; however, CIP with DIBI reduced infection burdens substantially. The reduced infection burdens with CIP+DIBI treatment were also reflected in reduced levels of various key lung and blood serum cytokines and chemokines. The cytokine/chemokine responses observed in control LAC-4-infected mice were consistent with those reported previously with this model (20). These previous studies had suggested that the magnitude of the tissue and blood cytokine responses to A. baumannii infection are largely driven by the intensity of bacterial antigen stimulation (thus bacterial burden) and tissue damage caused by the infection (20).
The reductions in several key cytokines and chemokines, such as IL-6, KC, MCP-1, RANTES, etc., in DIBI- and DIBI+CIP-treated mice are consistent with the effects of DIBI suppressing pneumonia and the systemic spread of infection, as well as with the synergy observed for the CIP+DIBI combination. Overall, our results indicate that DIBI does not interfere with the activity of conventional antibiotics but rather has potential for working in conjunction with antibiotics to improve their efficacies, including against antibiotic-resistant A. baumannii.
MATERIALS AND METHODS
Bacterial strains and cultivation.
Bacterial isolates used in this study are listed in Table 1, and these represented a relatively diverse group for study. A. baumannii ATCC 17978 and type strain ATCC 19606 were originally isolated from a fatal case of meningitis and a case of urinary tract infection (UTI), respectively, and were obtained either from the ATCC (Manassas, VA) or the Capital Health District, Halifax, Nova Scotia, Canada. The LAC clinical isolates (1, 4, 10, and 15) were originally isolated from hospital outbreaks over an 8-year period in Los Angeles County, CA, while AYE is a UTI isolate from France. These clinical isolates were obtained from Howard Xu, Department of Biological Sciences, California State University, Los Angeles, CA. All isolates were routinely cultured from frozen stocks (−80°C) and maintained on Trypticase soy agar (TSA; Sigma-Aldrich) or TSA with 5% sheep blood (BA; Becton Dickinson). Liquid cultures were grown in MHB (Oxoid). Partially deferrated RPMI, extracted with FEC1 to remove excess Fe (RPMI-FEC1), was prepared using FEC1 as previously described (16). Cultures were grown at 35°C with shaking. All experiments were performed following biosafety and biosecurity standards (Public Health Agency of Canada) in a biosafety level 2 containment laboratory.
Iron-chelators, antimicrobials, and iron supplementation.
DIBI and FEC1 were supplied by Chelation Partners, Inc. GEN, CIP, and CST (Sigma-Aldrich) were prepared as 10-mg/ml stocks, but TGC (Sigma-Aldrich) was prepared as a 5-mg/ml stock. DIBI stocks were prepared in RPMI at a concentration of 200 mg/ml. Iron was added to growth media as ferric citrate (Sigma-Aldrich) in RPMI. All stock solutions were filter sterilized (0.2-μm-pore-size filter) before use.
Antibiotic susceptibility testing.
MICs of DIBI, GEN, CIP, CST, and TGC were determined in RPMI using the broth microdilution method in 96-well round-bottom plates. Bacterial cultures grown overnight in MHB were diluted to an OD600 of 0.1, and the diluted bacterial suspension was used to inoculate MIC plates at a final dilution of 1/200 (approximately 1 × 105 to 5 × 105 CFU/ml). Chelator and antibiotic stocks were diluted in RPMI with serial 1/2 dilutions made. Negative and positive controls were tested in parallel. MIC plates were incubated at 35°C for 48 h, and the MIC value was defined as the lowest concentration of chelator or antibiotic required to inhibit visible growth at 24 h of incubation. At least two independent experiments with two replicates in each were performed.
Effect of Fe and DIBI on bacterial growth.
A. baumannii grows well in RPMI but does not grow in RPMI that was extracted of its iron content using FEC1. Ferric citrate at 1 μM was used to supplement this deferrated medium. DIBI was added at concentrations 0.1 μM (0.9 μg/ml) and 1 μM (9 μg/ml) into referrated RPMI to assess the effect of iron sequestration on A. baumannii growth. Cultures were incubated at 35°C with shaking, and OD600 readings were taken over a 36-h incubation period for two independent experiments. For inoculum preparation, MHB-grown overnight cultures of A. baumannii strains LAC-4, ATCC 17978, and ATCC 19606 were diluted to an OD600 of 0.1, and growth cultures were inoculated at a final dilution of 1/200.
DIBI synergy with antibiotics.
Time-kill assays were performed to determine the effect of DIBI on antibiotic activity. A. baumannii strains LAC-4, ATCC 17978, and ATCC 19606 were grown overnight in MHB and log-phase bacterial were inoculated to approximately 5 × 106 CFU/ml into RPMI medium (control) and RPMI containing either antibiotic alone, DIBI alone, or a combination of DIBI and antibiotic. A concentration of DIBI that did not affect bacterial growth on its own was first determined and used for further testing. GEN, CIP, CST, and TGC were each tested at concentrations equivalent to 1/8× to 1× MIC to obtain an initial killing phase of activity, followed by recovery growth. Cultures were incubated 24 h at 35°C, during which viable bacteria were enumerated at various time points with 10-fold serial dilutions in phosphate-buffered saline and plating onto TSA or BA. Colony counts were taken after overnight incubation at 35°C. At least two independent experiments were performed for each antibiotic and DIBI pair.
Infection testing in mice.
Eight- to ten-week-old, specific-pathogen-free, female BALB/c mice were purchased from Charles River Laboratories (St. Constant, Quebec, Canada). The animals were maintained and used in accordance with the recommendations of the Canadian Council on Animal Care Guide to the Care and Use of Experimental Animals. All the experimental procedures were approved by the institutional animal care committee (NRC, Human Health Therapeutics, Ottawa, Canada).
For i.n. inoculation of mice, fresh A. baumannii (LAC-4 and ATCC 17978) inocula were prepared for each experiment from frozen stocks as previously described (19). Mice were anesthetized by isoflurane inhalation and then inoculated i.n. with appropriate numbers of various A. baumannii isolates in 50 μl of saline. Actual inocula in each experiment were determined by plating 10-fold serial dilutions on brain heart infusion agar plates.
In vivo DIBI and antibiotic treatment efficacy studies.
Groups of 5 to 10 mice were i.n. inoculated with LAC-4 and treated twice with DIBI (100 mg [11 μmol]/kg/day, twice daily [BID], i.n.), CIP (20 mg [60 μmol]/kg/day, BID, i.p.), CIP+DIBI (20 + 100 mg/kg/day, BID, i.n./i.p.), or diluent (placebo, i.p.) starting 3 h after the bacterial challenge inoculation. The blood and tissue (lungs and spleen) bacterial burdens at 24 hpi were determined.
Quantitative bacteriology.
Groups of five infected mice were sacrificed at 24 h postinoculation. Lungs and spleens were aseptically harvested and homogenized in sterile saline using aerosol-proof homogenizers. Aliquots (100 μl) of 10-fold serial dilutions of the homogenates were cultured on brain heart infusion agar plates to quantify the number of viable A. baumannii in the respective organs. In some experiments, blood samples were collected, and 0.1-ml portions were similarly cultured with the rest used for serum separation.
Cytokine and chemokine determination.
The levels of cytokines and chemokines in the sera and lung homogenate supernatants were measured using the 12-plex Milliplex MAP mouse cytokine/chemokine kits (MCYTOMAG-70K; Millipore, Ltd., Billerica, MA) on a Luminex Magpix system (Luminex, Austin, TX) as specified by the manufacturer. Samples were assayed in duplicate, and cytokine/chemokine concentrations were calculated against the standards using Beadview software (v1.03; Upstate) (19).
Data analysis.
Growth curves and time-kill assay curves are reported as means ± standard errors of the mean (SEM) for at least two independent replicate experiments. For infection studies, data are reported as means ± SEM for each group unless otherwise specified. For multiple group comparisons of results, the one-way analysis of variance, followed by Bonferroni’s post hoc multiple-comparison tests, when appropriate, was used. Significant differences were defined as P < 0.05.
ACKNOWLEDGMENTS
We thank Rhonda KuoLee for project management and discussion, and we thank Annie Aubry and Greg Harris for technical assistance with some of the in vitro and the in vivo experiments.
Funding from NRC-IRAP is gratefully acknowledged.
B.E.H. has a beneficial interest in Chelation Partners, Inc., which has contributed financially to this study. M.D.C.P., K.A.S., D.S.A., and M.T.C.A. are employees of Chelation Partners, Inc. W.C. and S.M.L. have no conflicts or competing interests in relation to this study.
REFERENCES
- 1.Lee CR, Lee JH, Park M, Park KS, Bae IK, Kim YB, Cha CJ, Jeong BC, Lee SH. 2017. Biology of Acinetobacter baumannii: pathogenesis, antibiotic resistance mechanisms, and prospective treatment options. Front Cell Infect Microbiol 7:55. doi: 10.3389/fcimb.2017.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McConnell MJ, Actis L, Pachón J. 2013. Acinetobacter baumannii: human infections, factors contributing to pathogenesis and animal models. FEMS Microbiol Rev 37:130–155. doi: 10.1111/j.1574-6976.2012.00344.x. [DOI] [PubMed] [Google Scholar]
- 3.Gordon NC, Wareham DW. 2010. Multidrug-resistant Acinetobacter baumannii: mechanisms of virulence and resistance. Int J Antimicrob Agents 35:219–226. doi: 10.1016/j.ijantimicag.2009.10.024. [DOI] [PubMed] [Google Scholar]
- 4.Clark NM, Zhanel GG, Lynch JP III, 2016. Emergence of antimicrobial resistance among Acinetobacter species: a global threat. Curr Opin Crit Care 22:491–499. doi: 10.1097/MCC.0000000000000337. [DOI] [PubMed] [Google Scholar]
- 5.Cikman A, Gulhan B, Aydin M, Ceylan MR, Parlak M, Karakecili F, Karagoz A. 2015. In vitro activity of colistin in combination with tigecycline against carbapenem-resistant Acinetobacter baumannii strains isolated from patients with ventilator-associated pneumonia. Int J Med Sci 12:695–700. doi: 10.7150/ijms.11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Landman D, Georgescu C, Martin DA, Quale J. 2008. Polymyxins revisited. Clin Microbiol Rev 21:449–465. doi: 10.1128/CMR.00006-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dhariwal AK, Tullu MS. 2013. Colistin: re-emergence of the “forgotten” antimicrobial agent. J Postgrad Med 59:208–215. doi: 10.4103/0022-3859.118040. [DOI] [PubMed] [Google Scholar]
- 8.Gaddy JA, Arivett BA, McConnell MJ, López-Rojas R, Pachón J, Actis LA. 2012. Role of acinetobactin-mediated iron acquisition functions in the interaction of Acinetobacter baumannii strain ATCC 19606T with human lung epithelial cells, Galleria mellonella caterpillars and mice. Infect Immun 80:1015–1024. doi: 10.1128/IAI.06279-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ali HM, Salem MZM, El-Shikh MS, Megeed AA, Alogaibi YA, Talea IA. 2017. Investigation of the virulence factors and molecular characterization of the clonal relations of multidrug-resistant Acinetobacter baumannii isolates. J AOAC Int 100:152–158. doi: 10.5740/jaoacint.16-0139. [DOI] [PubMed] [Google Scholar]
- 10.Antunes LCS, Imperi F, Minandri F, Visca P. 2012. In vitro and in vivo antimicrobial activities of gallium nitrate against multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 56:5961–5970. doi: 10.1128/AAC.01519-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Runci F, Bonchi C, Frangipani E, Visaggio D, Visca P. 2017. Acinetobacter baumannii biofilm formation in human serum and disruption by gallium. Antimicrob Agents Chemother 61:e01563-16. doi: 10.1128/AAC.01563-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Léséleuc L, Harris G, KuoLee R, Chen W. 2012. In vitro and in vivo biological activity of iron chelators and gallium nitrate against Acinetobacter baumannii. Antimicrob Agents Chemother 56:5397–5400. doi: 10.1128/AAC.00778-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chitambar CR. 2010. Medical applications and toxicities of gallium compounds. Int J Environ Res Public Health 7:2337–2361. doi: 10.3390/ijerph7052337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holbein BE, Feng M, Huber AL, Kidby DK. 2011. Polymeric metal chelating compositions and methods of preparing same for controlling growth and activities of living cells and organisms. International patent WO2012167368 A1.
- 15.Ang MTC, Gumbau-Brisa R, Allan DS, McDonald R, Ferguson MJ, Holbein BE, Bierenstiel M. 2018. DIBI, a 3-hydroxypyridin-4-one chelator iron-binding polymer with enhanced antimicrobial activity. Medchemcomm 9:1206–1212. doi: 10.1039/c8md00192h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parquet MC, Savage KA, Allan DS, Davidson RJ, Holbein BE. 2018. Novel iron-chelator DIBI inhibits Staphylococcus aureus growth, suppresses experimental MRSA infection in mice and enhances the activities of diverse antibiotics in vitro. Front Microbiol 9:1811. doi: 10.3389/fmicb.2018.01811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Savage KA, Parquet MC, Allan DS, Davidson RJ, Holbein BE, Lilly EA, Fidel PL Jr. 2018. Iron restriction to clinical isolates of Candida albicans by the novel chelator DIBI inhibits growth and increases sensitivity to azoles in vitro and in vivo in a murine model of experimental vaginitis. Antimicrob Agents Chemother 62:e02576-17. doi: 10.1128/AAC.02576-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Power Coombs MR, Grant T, Greenshields AL, Arsenault DJ, Holbein BE, Hoskin DW. 2015. Inhibitory effect of iron withdrawal by chelation on the growth of human and murine mammary carcinoma and fibrosarcoma cells. Exp Mol Pathol 99:262–270. doi: 10.1016/j.yexmp.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 19.Harris G, Kuo Lee R, Lam CK, Kanzaki G, Patel GB, Xu HH, Chen W. 2013. A mouse model of Acinetobacter baumannii-associated pneumonia using a clinically isolated hypervirulent strain. Antimicrob Agents Chemother 57:3601–3613. doi: 10.1128/AAC.00944-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Faassen H, KuoLee R, Harris G, Zhao X, Conlan JW, Chen W. 2007. Neutrophils play an important role in host resistance to respiratory infection with Acinetobacter baumannii in mice. Infect Immun 75:5597–5608. doi: 10.1128/IAI.00762-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ou HY, Kuang SN, He X, Molgora BM, Ewing PJ, Deng Z, Osby M, Chen W, Xu HH. 2015. Complete genome sequence of hypervirulent and outbreak-associated Acinetobacter baumannii strain LAC-4: epidemiology, resistance genetic determinants and potential virulence factors. Sci Rep 5:8643. doi: 10.1038/srep08643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Antunes LCS, Imperi F, Towner KJ, Visca P. 2011. Genome-assisted identification of putative iron-utilization genes in Acinetobacter baumannii and their distribution among a genotypically diverse collection of clinical isolates. Res Microbiol 162:279–284. doi: 10.1016/j.resmic.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 23.de Léséleuc L, Harris G, KuoLee R, Xu HH, Chen W. 2014. Serum resistance, gallium nitrate tolerance, and extrapulmonary dissemination are linked to heme consumption in a bacteremic strain of Acinetobacter baumannii. Int J Med Microbiol 304:360–369. doi: 10.1016/j.ijmm.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 24.Eijkelkamp BA, Hassan KA, Paulsen IT, Brown MH. 2011. Investigation of the human pathogen Acinetobacter baumannii under iron limiting conditions. BMC Genomics 12:126. doi: 10.1186/1471-2164-12-126. [DOI] [PMC free article] [PubMed] [Google Scholar]