

Bacteria have acquired multiple mechanisms to evade the lethal effects of current therapeutics, hindering treatment of bacterial infections, such as those caused by the pathogen Pseudomonas aeruginosa, which is responsible for nosocomial and cystic fibrosis lung infections. One resistance mechanism involves membrane-embedded multidrug efflux pumps that can effectively extrude an array of substrates, including common antibiotics, dyes, and biocides.
KEYWORDS: Pseudomonas aeruginosa, biocides, efflux pump inhibitors, membrane proteins, multidrug resistance, peptides, protein-protein interactions, small multidrug resistance proteins
ABSTRACT
Bacteria have acquired multiple mechanisms to evade the lethal effects of current therapeutics, hindering treatment of bacterial infections, such as those caused by the pathogen Pseudomonas aeruginosa, which is responsible for nosocomial and cystic fibrosis lung infections. One resistance mechanism involves membrane-embedded multidrug efflux pumps that can effectively extrude an array of substrates, including common antibiotics, dyes, and biocides. Among these is a small multidrug resistance (SMR) efflux protein, consisting of four transmembrane (TM) helices, that functions as an antiparallel dimer. TM helices 1 to 3 (TM1 to TM3) comprise the substrate binding pocket, while TM4 contains a GG7 heptad sequence motif that mediates the SMR TM4-TM4 dimerization. In the present work, we synthesized a series of peptides containing the residues centered on the TM4-TM4 binding interface found in the P. aeruginosa SMR (PAsmr), typified by Ac-Ala-(Sar)3-LLGIGLIIAGVLV-KKK-NH2 (helix-helix interaction residues are underlined). Here, the acetylated N-terminal sarcosine (N-methyl-Gly) tag [Ac-Ala-(Sar)3] promotes membrane penetration, while the C-terminal Lys tag promotes selectivity for the negatively charged bacterial membranes. This peptide was observed to competitively disrupt PAsmr-mediated efflux, as measured by efflux inhibition of the fluorescent dye ethidium bromide, while having no effect on cell membrane integrity. Alternatively, a corresponding peptide in which the TM4 binding motif is scrambled was inactive in this assay. In addition, when Escherichia coli cells expressing PAsmr were combined with sublethal concentrations of several biocides, growth was significantly inhibited when peptide was added, notably, by up to 95% with the disinfectant benzylalkonium chloride. These results demonstrate promise for an efflux pump inhibitor to address the increasing threat of antibiotic-resistant bacteria.
INTRODUCTION
Antibiotic-resistant bacteria are becoming a serious threat to public health, and yet, the development of new antibiotics is at a virtual standstill (1). The emergence of multidrug-resistant (MDR) strains has made treatment of numerous bacterial infections difficult, particularly in diseases like cystic fibrosis (CF) (2–4). Infection by Pseudomonas aeruginosa in the lungs of patients with CF is a primary factor in the onset of lung disease and an ultimate cause of mortality (5). P. aeruginosa is also commonly associated with nosocomial infections, making the disinfection of hospital surfaces and equipment a pressing challenge to stop the spread of MDR infections (2). Disinfectants and biocides are used heavily in the health care sector on both mucosal and inanimate surfaces, for the disinfection of wounds and as topical agents in the sterilization of medical equipment (6). However, these methods are becoming ineffective at sterilization of these surfaces, as increasing levels of resistance are being observed across various bacterial strains (7–9).
Membrane-embedded multidrug efflux pumps are a principal component underlying bacterial resistance, as these proteins can export a variety of toxic compounds. MDR efflux pumps are found ubiquitously across bacterial species, conferring their inherent resistance (10). Five families of these pumps have been characterized, among which the focus of the present study is on the small multidrug resistance (SMR) efflux pumps (11). The primary substrates of SMRs include quaternary ammonium compounds (QACs), a broad category that includes the fluorescent toxic molecule ethidium bromide (EtBr) and many biocides, including cetylpyridinium chloride (CP+), cetyltrimethylammonium bromide (CTAB), and benzalkonium chloride (BZK); the latter is a commonly used hospital disinfectant (12). Of particular relevance to the present study is the strong correlation observed between the presence of genes encoding SMR proteins in P. aeruginosa clinical isolates and increased resistance to common disinfectants, including benzalkonium chloride (13).
SMRs are present in the bacterial inner membrane and consist of approximately 110 residues, including four transmembrane (TM) helices, designated TM1 to TM4, of which TM1 to -3 make up the substrate-binding pocket and TM4 contains the binding motif to form the obligate, antiparallel homodimer required for substrate efflux (12, 14). When considering approaches to inhibit the action of SMRs, the binding pocket itself does provide an appealing drug target; however, given the promiscuity of this pocket in transporting a wide variety of substrates, we decided to focus on disrupting the dimerization motif in the TM4-TM4 helix-helix interaction (15). We have earlier shown that peptides designed to target and disrupt the membrane-buried protein-protein interactions that stabilize the functional SMR dimer effectively reduced efflux activity of the SMR from archaebacterium Halobacter salinarum (Hsmr) (16, 17). In the present work, we report the design of peptide-based efflux pump inhibitors against the SMR from P. aeruginosa (PAsmr) that target and disrupt its TM4-TM4 interface and, hence, act to reduce the SMR-mediated substrate efflux, as is highlighted by their ability to improve susceptibility to disinfectants.
RESULTS
Peptide inhibitor design.
We have previously shown that a synthesized transmembrane α-helical peptide of the Hsmr TM4 that includes the GG7 TM4-TM4 dimerization motif in its sequence significantly reduces Hsmr efflux activity (15, 16, 18). Here, we have synthesized a series of peptides designed to target the corresponding TM4 region of the SMR from the opportunistic pathogen P. aeruginosa (Table 1). As depicted schematically in Fig. 1, the synthetic peptide is anticipated to corkscrew into the membrane, N terminus first, align with the corresponding region of native TM4-TM4 interaction, and competitively disrupt SMR dimerization, thereby creating nonfunctional monomeric SMR species. The full-length PAsmr TM4 peptide (PAsmrFL) was synthesized, and since previous studies have identified the minimal sequence length needed for disruption of H. salinarum to be TM4 residues 88 to 100 (15, 18), the shortened PAsmrTM4 peptide corresponding to residues 88 to 100 was synthesized to compare its activity against that of the full-length peptide. Furthermore, to assess the specificity of the binding motif and advent of any nonspecific interactions, a motif-scrambled peptide was prepared (PAsmrScr), which contained the same sequence composition as PAsmrTM4 but whose GG7 motif was disrupted from residues 90 to 98. In addition, an all-d-enantiomer of PAsmrTM4 (PAsmrD) was used as a control, since this peptide should be unable to interact with the corresponding l-enantiomeric SMR sequence in the native protein. The C terminus of each shortened peptide was extended by three Lys residues and that of the full-length peptide extended by five to improve solubility and specificity, as the positive charges should direct the peptides selectively toward the negatively charged bacterial membrane (15, 19). The N terminus was tagged with an acetyl-Ala-(Sar)3 sequence (where the sarcosine [Sar] is N-methyl-glycine) to further improve solubility and provide the peptide with an uncharged terminus that could direct its partitioning of the peptide into the membrane (15, 19). A corresponding Trp-containing peptide (PAsmrW) was also prepared for quantification of other peptides and fluorescence studies (see below).
TABLE 1.
Peptide inhibitor sequences used in the present work and their MICs
| Peptide | Sequencea | MIC (μM)b |
|---|---|---|
| PAsmrFL | Ac-A-(Sar)3 -DPAALL GI G LI IA GV LVIQLFS-KKKKK-NH2 | >64 |
| PAsmrTM4 | Ac-A-(Sar)3 -LL GI G LI IA GV LV-KKK-NH2 | 16 |
| PAsmrDc | Ac-a-(Sar)3 -ll gi g li ia gv lv-kkk-NH2 | 32 |
| PAsmrScr | Ac-A-(Sar)3 -LLVLGAIGIIGLV-KKK-NH2 | >64 |
| PAsmrW | Ac-A-(Sar)3 -LL GI G LI WA GV LV-KKK-NH2 | >64 |
Sequences for full-length (FL) TM4 (residues 84 to 105) and shortened TM4 (residues 88 to 100) from PAsmr are shown, along with solubility tags, where Ac is an acetylated N terminus, NH2 is an amidated C terminus, and Sar (sarcosine) is N-methyl-glycine. The TM4-TM4 dimerization motif is underlined.
MICs determined against E. coli BL21 cells cloned with PAsmr.
Peptide is composed of d-enantiomeric amino acids, as indicated by the lowercase letters.
FIG 1.
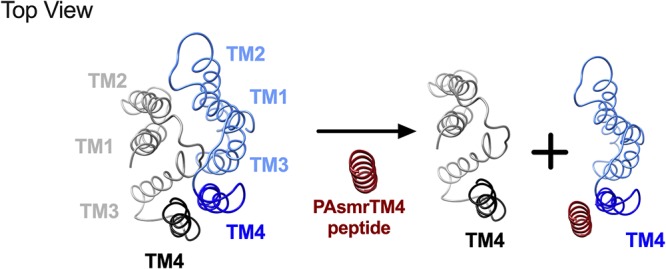
Schematic representation of peptide-based mechanism of SMR inhibition. PAsmr homolog EmrE is depicted in a top view in its functional, dimeric form (antiparallel monomers rendered in blue and black) (PDB code 3B5D [14]). Upon the addition of PAsmrTM4 peptide (red), peptide binds antiparallel to the SMR monomer (blue), leading to disruption of the six-helix substrate transport pathway and concomitant SMR inactivation.
Peptides display low antimicrobial activity.
As these peptides are inherently hydrophobic, it may be anticipated that they have some capacity to cause membrane disruption, despite being designed for a specific protein target. The antimicrobial activities of the designed peptides against Escherichia coli BL21 cells in which the PAsmr protein had been expressed (see Materials and Methods) were determined (Table 1). It is seen that the peptides exhibited relatively low levels of antimicrobial activity (largely >64 μM), with the lowest MIC (16 μM for PAsmrTM4) being well above the peptide concentrations used in the assays described herein.
Peptide inhibitors interact favorably with membranes.
To determine if the synthesized peptides properly fold and adopt an α-helix, as would be required for activity in the bacterial membrane environment, their secondary structures were assessed by circular dichroism spectroscopy. This was performed in both an aqueous phase and in the membrane-mimetic environment of sodium dodecyl sulfate (SDS) micelles (Fig. 2), noting that bacterial membranes and SDS are both anionic. All peptides with the shortened chain (residues 88 to 100) adopted a random coil structure in the aqueous environment, while the longer PAsmrFL peptide adopted a partial helix (Fig. 2A); in contrast, all peptides adopted an α-helix in the presence of SDS micelles (Fig. 2B), indicating their ability to partition into detergent particles. The full-length, shortened, and d-enantiomer TM4 peptides exhibited a strongly helical character in SDS, evidenced by large negative (or positive for the d-enantiomer) mean residue ellipticity (MRE) values at 208 nm and 222 nm. The PAsmrScr and PAsmrW peptides exhibited relatively reduced degrees of helicity in detergent, as the alteration of sequence from the wild type apparently limited helicity, likely resulting in helical fraying at the peptide termini.
FIG 2.
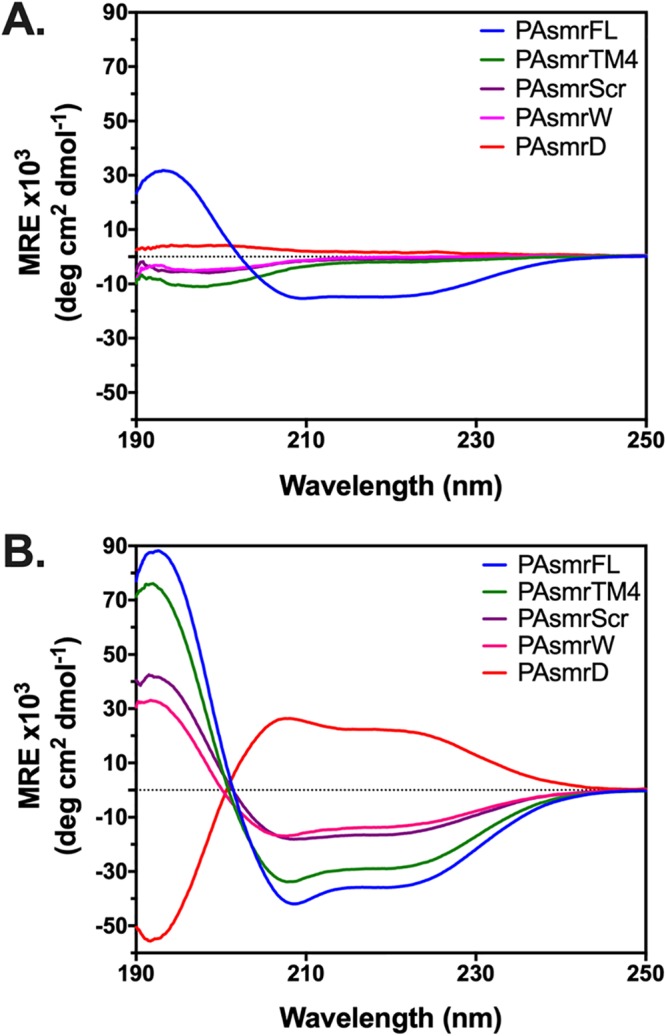
Circular dichroism spectra of PAsmr peptides. (A) Aqueous environment: 20 μM peptides in 10 mM Tris-Cl, 10 mM NaCl, pH 7.4. (B) Membrane environment: 20 μM peptide in 10 mM Tris-Cl, 10 mM NaCl, pH 7.4, supplemented with 20 mM SDS; 1:1,000 peptide/detergent ratio. Results are the average values from 3 independent trials in both panels A and B. Peptide notations are given in Table 1. Spectra are color coded as shown in the key in each panel.
Additionally, we assessed membrane interaction through measurement of the Trp fluorescence of the PAsmrW peptide in SDS micelles. The PAsmrW peptide displayed a significant blue shift in SDS micelles (18 nm), confirming that the peptide partitions into the detergent micelle (see Fig. S2 in the supplemental material).
Peptides mediate efflux inhibition of P. aeruginosa SMR.
To explore peptide-mediated SMR efflux inhibition, efflux rates of the fluorescent toxin ethidium bromide (EtBr) by E. coli BL21 cells expressing PAsmr were determined (Fig. 3A). Cells were first treated with carbonyl cyanide m-chlorophenylhydrazone (CCCP), an ionophore that disrupts the proton motive force needed for SMR pumping and therefore allows EtBr uptake into the cells. An increase in intracellular EtBr in the presence of CCCP, as evidenced by increased EtBr fluorescence, demonstrates that EtBr efflux is a result of SMR pumping rather than nonspecific diffusion. The baseline efflux rate of ethidium bromide by E. coli cells with endogenous SMR (EmrE) was then determined by measuring the slope of the fluorescence decay, followed by determining the efflux rate of E. coli cells expressing PAsmr. The difference in efflux rates between E. coli cells expressing PAsmr and E. coli cells that are not can be considered the zone of inhibition (Fig. 3B, gray region). PAsmr-expressing cells treated with the full-length PAsmr peptide (PAsmFL) displayed significant inhibition of EtBr efflux, with efflux rates comparable to those of E. coli cells not expressing PAsmr. Furthermore, an increase in the final EtBr plateau intensity suggests that a higher concentration of EtBr remains within the cell when efflux is disrupted. The shortened PAsmrTM4 peptide was also found to induce significant inhibition, increasing the amount of EtBr remaining in the cells, similar to the results for PAsmrFL, showing that activity was not lost by the TM4 peptide length reduction but had actually improved. Both TM4 peptides, despite inducing a higher efflux rate than E. coli BL21 cells not expressing the PAsmr protein (BL21 empty vector), exhibit higher final EtBr plateaus, a result that may be attributed to interactions of the TM peptides with the endogenous SMR (EmrE) in the E. coli cells.
FIG 3.

Peptide-mediated efflux inhibition of E. coli cells expressing PAsmr. E. coli cells expressing PAsmr were incubated with 1 μg/ml of EtBr, and fluorescence decay measured at 600 nm for 20 min. (A) Baseline efflux was measured for E. coli cells alone (gray curve) and for those expressing PAsmr (black). Cells were then incubated with 8 μM PAsmrFL (blue), PAsmrTM4 (green), PAsmrScr (purple), or PAsmrD (red). Efflux from cells treated with CCCP was also measured (yellow). AFU, arbitrary fluorescence units. (B) Efflux rates as obtained by curve fits; lowest values indicate lowest EtBr efflux rates. Gray region indicates zone of inhibition. Error bars show standard deviations [n = 4, except that for BL21 (Empty Vector), n = 3]. ****, P > 0.0001; ns, not significant.
The PAsmrScr and PAsmrD peptides were then used to assess nonspecific binding, as these peptides should not interact favorably with the PAsmr dimerization motif. The PAsmrScr peptide showed no inhibitory activity on the EtBr efflux rate or EtBr plateau. The PAsmrD peptide was also seen not to reduce efflux activity significantly, albeit the baseline EtBr content was increased. Both PAsmrFL and PAsmrTM4 peptides exhibited dose-dependent responses, as inhibition was incremental from 4 μM and 8 μM (Fig. S3). These results suggest that the PAsmrTM4 peptides are specifically interacting with the PAsmr protein, likely at the TM4-TM4 dimerization site, to result in reduced EtBr efflux activity.
Transmembrane peptides have minimal nonspecific membrane disruption.
To ensure that EtBr efflux could not be attributed to cell death as a result of nonspecific membrane disruption during the EtBr efflux assay, a propidium iodide (PI) assay was used to assess inner membrane integrity (Fig. 4). The experiment setup matched the conditions of the efflux assay but instead measured the uptake of PI, an EtBr analog that is impermeable to the bacterial inner membrane. If the inner membrane is intact, PI should not be able to enter the cells, resulting in low fluorescence of PI. However, if there is peptide-mediated disruption of the membrane, PI will enter the cells and produce an increase in fluorescence. This is demonstrated in cells treated with the pore-forming peptide melittin (20), where an increase in PI fluorescence is observed and corresponds to about 40% bacterial inner membrane disruption (Fig. 4). While the presence of inhibitor peptides actually resulted in a slightly lower fluorescence value than for cells alone, which may be attributed to cell growth in the presence of peptide during the time course of the experiment, these results demonstrate that there is no measurable nonspecific membrane disruption caused by the peptides at the concentrations tested.
FIG 4.
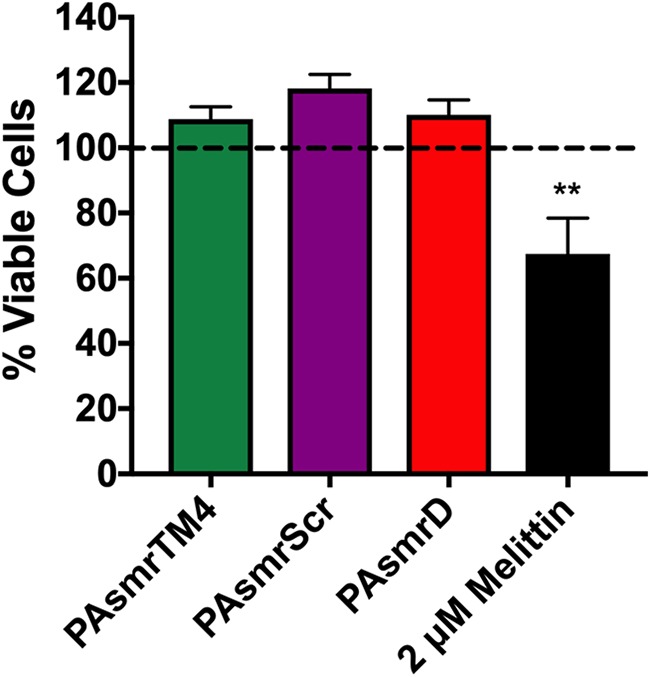
Propidium iodide uptake as a measure of membrane disruption in response to peptide addition. Overnight cultures of E. coli cells expressing PAsmr were resuspended in minimal medium A, representing viable cells. Cells were diluted to an OD600 of 0.1, and a standard curve was generated using the viable cells and cells 100% disrupted by 70% isopropanol. Diluted cells in minimal medium A were then incubated with 8 μM peptide or 2 μM melittin, followed by the addition of 5 μM PI. Fluorescence was measured, and using the equation of the standard curve, the relative amounts of viable cells were calculated. Error bars show standard deviations (n ≥ 3). **, P > 0.001 for comparison of melittin treatment to PAsmrTM4 and PAsmrScr treatments; no significant difference was noted among peptides.
Peptide-mediated resensitization to sublethal doses of biocides.
Since biocides are a primary substrate of SMRs, the biocides BZK, CP+, and CTAB were tested against E. coli cells expressing the PAsmr protein. We used a resensitization assay to determine whether the peptides’ efflux inhibitory capacities were sufficient to increase the toxicity of substrates and thereby reduce cell growth. In this assay, a sublethal (below-MIC) dose of biocide was determined (see Materials and Methods), at which the amount of toxicant entering the cell equilibrated with the corresponding amount being effluxed by SMRs out of the cells; under these conditions, cells maintain their growth. Cells were then treated with sublethal concentrations of biocide along with increasing amounts of PAsmrTM4, with the lowest dose of both peptide and biocide to cause resensitization being determined (Fig. 5A to C). The addition of peptide to each of the three biocides resulted in a significant level of resensitization and relative growth inhibition compared to the combination of biocide with the PAsmrScr peptide at the same concentration. PAsmrTM4 peptide in combination with biocides CP+ and CTAB displayed 38% and 22% growth inhibition, respectively. Importantly, essentially complete inhibition of growth was seen from the combination of PAsmrTM4 peptide with the common hospital disinfectant BZK, which resulted in ∼95% inhibition of cell growth. To ensure that the reduction of cell growth could not be attributed to detergent-like properties of peptide and/or biocide, the resensitization was performed with nonsubstrate detergents. In the presence of either PAsmrTM4 and n-octyl-β-d-glucopyranoside (OG) (Fig. 5D) or SDS (Fig. S4), there was no significant effect on cell growth, demonstrating the specificity of SMR substrates for the PAsmr protein.
FIG 5.
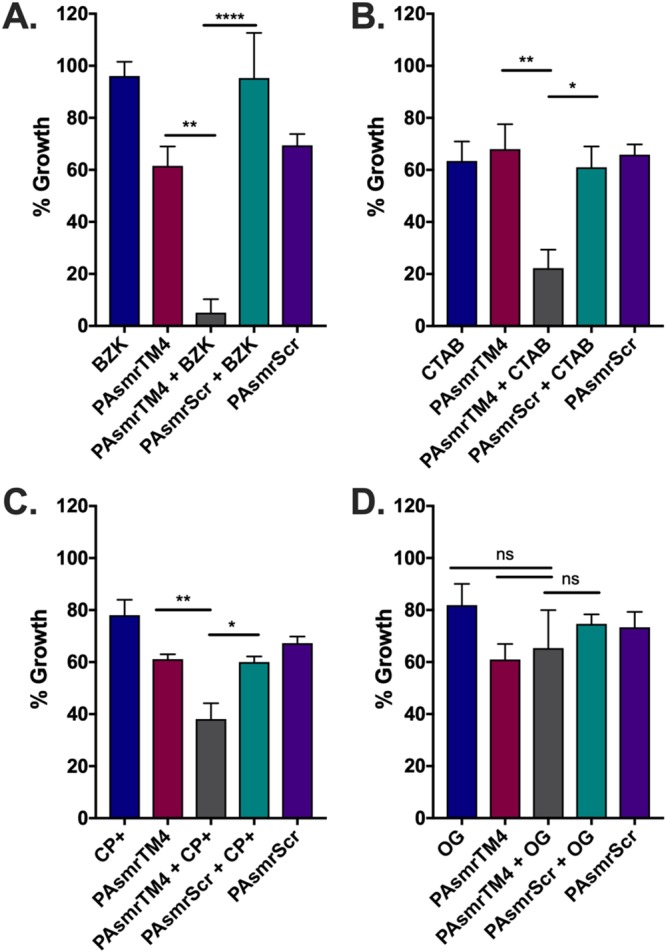
Resensitization of BL21 cells expressing PAsmr to biocide upon treatment with PAsmrTM4 peptide. Cells were treated with a sublethal dose of each biocide (A to C) or nonsubstrate detergent (D) as determined for E. coli cells expressing PAsmr (blue bar), as well for the PAsmrTM4 peptide alone (red) and the PAsmrScr peptide alone (purple). (A) Cells were plated with a sublethal dose of BZK (1.5 μg/ml) and 2 μM of either PAsmrTM4 (gray) or PAsmrScr (turquoise). (B) Cells were plated with 1 μg/ml CP+ and 1 μM of peptide as indicated. (C) Cells were plated with 4 μg/ml CTAB and 2 μM of peptide as indicated. (D) A sublethal concentration of 500 μg/ml n-octyl-β-d-glucopyranose (OG) was added to cells treated with 2 μM of peptide as indicated. After 20 h of incubation under all conditions shown, the OD600 was measured to determine cell growth. Error bars show standard deviations (n = 3). *, P < 0.05; **, P < 0.01; ****, P < 0.0001; ns, not significant.
DISCUSSION
There is now a strong need for the development of new classes of antibiotics and adjuvants targeting antibiotic resistance mechanisms in bacteria to improve the efficiency of current therapeutics (21–23). While small multidrug resistance proteins (SMRs) are not the only or major factor contributing to the effluxome, the importance of SMRs working together with other efflux pumps to efficiently extrude cationic dyes and biocides from the cell has been shown by their ability to export toxic compounds into the periplasm to be further exported by the resistance-nodulation-cell division (RND) pump (24–26). This, alongside the broad range of substrates (QAC compounds, disinfectants, antibiotics, etc.) that SMRs can extrude and their increasing prevalence on plasmids and integrons in clinical and multidrug-resistant strains of bacteria (13, 27–35), marks these proteins as important therapeutic targets.
In the work reported here, we have developed novel peptide efflux pump inhibitors targeting the P. aeruginosa SMR, PAsmr. The peptide design has a hydrophobic core containing the PAsmr transmembrane helix 4 (TM4) binding motif that is anticipated to bind at the PAsmr TM4-TM4 dimer interface. We demonstrated that these TM4 peptides are able to reduce efflux activity and increase EtBr retention in bacteria, which is consistent with inhibition of SMR pumping activity. Corresponding scrambled-motif and d-enantiomer peptides were largely inactive, confirming the specificity of the PAsmrTM4 inhibitor.
Inherent in the design of TM peptides is their intrinsic hydrophobicity, which, in turn, has the potential to nonspecifically disrupt bacterial membranes (36). For these reasons, the concentrations of peptides used in assays were below their respective MICs (Fig. 3). Minor variations in MICs of antimicrobial enantiomeric pairs have been suggested to be due to protease resistance, likely resulting in a higher persisting concentration of the nonnatural peptide enantiomer (37, 38). At the concentrations we used, no nonspecific interaction of the peptides with the bacterial inner membrane was observed, as demonstrated by the lack of measurable uptake of inner membrane-impermeable PI (Fig. 4). These observations, together with circular dichroism (CD) and Trp fluorescence data, demonstrate that the peptides are interacting with bacterial membranes but do not affect membrane integrity. The ability of the PAsmrTM4 peptide to reduce efflux activity, as seen in EtBr efflux assays, was further manifested in the reduced concentration of biocide substrate needed to inhibit cell growth. This effect was observed to various degrees for three common disinfectants (Fig. 5), demonstrating the ability of PAsmrTM4-based peptides to broadly reduce SMR activity and to increase bacterial susceptibility to biocides. If increased bacterial killing can be achieved by peptide-mediated resensitization of bacteria to biocides, such an approach could improve hospital disinfection and lower rates of nosocomial infections (6, 22, 23). The therapeutic application of these peptides could be further explored through testing against antibiotic-resistant strains of P. aeruginosa and optimization of reduced toxicity and improved stability as previously demonstrated by our laboratory (18).
Our overall results suggest that the approach, which targets and disrupts functional protein-protein interactions within membranes, can in principle be extended to SMR pumps found in other pathogenic bacteria. The present inhibitor design has been shown to be effective at reducing efflux activity against P. aeruginosa SMR here and against the respective SMRs in H. salinarum and E. coli previously (15, 18, 39). Sequence alignments of SMRs from different bacteria have shown that the GG7 dimerization motif in TM4 is highly conserved among EmrE homologs, including those of Mycobacterium tuberculosis and Acinetobacter baumannii, which have become highly resistant and have been described as among the top bacterial threats (1, 40, 41). These considerations raise the prospect that a designed TM4 peptide could be used as a broad-spectrum efflux pump inhibitor targeting the conserved dimerization motif in SMRs.
MATERIALS AND METHODS
Peptide synthesis and purification.
Peptides were synthesized as previously described (15), using an automated PS3 peptide synthesizer (Protein Technologies, Inc., AZ, USA). A 0.1 mmol-scale synthesis using Fmoc [N-9(-fluorenyl)methoxycarbonyl] chemistry was carried out using a low-load peptide amide linker-polyethylene glycol (PAL-PEG) resin. After the synthesis, the N terminus was acetylated using 10% acetic anhydride. Cleavage of the peptide from the resin, using a solution of 88% trifluoroacetic acid (TFA), 5% phenol, and 2% triisopropylsilane (TIPS), resulted in an amidated C terminus. Peptides were ether precipitated and purified using reverse-phase high-performance liquid chromatography (RP-HPLC) and a C4 column (Phenomenex, Canada). Peptides were eluted using a water-acetonitrile gradient, starting with 80% water, 20% acetonitrile, and 0.1% TFA and increasing the acetonitrile concentration by 1% per minute. Peptide purity was confirmed by mass spectrometry or to a single peak on an analytical column.
Peptide quantification.
Due to the hydrophobic nature of the peptides studied herein, they were treated with 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) to disrupt any aggregates, dried into a film, and dissolved in 2,2,2-trifluoroethanol (TFE) to be quantified (42). Since the wild-type TM4 peptides do not contain a native Trp residue, one PAsmrTM4 peptide was mutated by replacing Ile-95, in a nonconserved region, with a Trp residue (PAsmrW) (Table 1). This peptide was then used as a standard to determine the extinction coefficient at 215 nm, since this peptide is the same length as and maintains a degree of hydrophobicity similar to those of other TM4 peptides. A standard curve at 215 nm was generated, and using Beer’s law (A = εLc), the extinction coefficient was calculated to be 51,870 M−1cm−1, from which the concentrations of the remaining peptides were quantified. The PAsmrFL peptide was quantified using an extinction coefficient generated from a 31-residue TM peptide from the viral protein Vpu (39). Peptides were stored as lyophilized powders at −20°C and dissolved in dimethyl sulfoxide (DMSO) for further study.
MIC assay.
Peptide MICs were determined using previously described methods (43). Briefly, E. coli BL21(DE3) cells with the endogenous SMR EmrE were used to express a His-Myc-tagged PAsmr on a pT7-7 plasmid. Cells were not induced, as the T7 vector displays leaky expression. The presence of membrane-embedded PAsmr was confirmed by Western blotting (see Fig. S1 in the supplemental material). The cells were grown overnight in Mueller-Hinton broth (MHB). Two-fold serial dilutions of peptide were made from 0 to 64 μM peptide, and the contents were plated with 50,000 CFU/well. The plates were incubated at 37°C for 20 h, and the optical density at 600 nm (OD600) was measured to determine cell growth.
CD spectroscopy.
Circular dichroism (CD) spectra of peptides were collected on a Jasco J-720 circular dichroism spectropolarimeter. Peptide spectra were recorded in an aqueous environment (20 μM peptide, 10 mM Tris, 10 mM NaCl, pH 7.2) and in a membrane mimetic (20 μM peptide, 20 mM sodium dodecyl sulfate [SDS], 10 mM Tris, 10 mM NaCl, pH 7.2). The spectra shown in Fig. 2 represent the average results of three independent trials, which have had the background signal subtracted and have been converted to mean residue ellipticity (MRE) using standard formulas.
EtBr efflux assay.
Overnight cultures of BL21 cells expressing PAsmr in Luria-Bertani (LB) broth were diluted to an OD600 of 0.2 and incubated at 37°C. Once an OD600 of 0.4 was reached, cells were grown for an additional 2 h. Cells were spun down at 4,000 rpm for 10 min and resuspended in M9 minimal media to an OD600 of 0.1. For each run, 2 ml of cells was used. Carbonyl cyanide m-chlorophenylhydrazine (CCCP) (40 μM) was added and incubated with cells for 5 min. This was followed by the addition of 1 μg/ml ethidium bromide (EtBr), and for peptide samples, increasing concentrations of peptide were added at 2 μM, 4 μM, or 8 μM. Cells were incubated for 30 min and then spun down for 10 min at 4,000 rpm. Fluorescence was measured using a Photon Technology International fluorimeter, with an excitation wavelength of 530 nm and emission wavelength of 600 nm. Slit widths were set to 2 nm for excitation and 4 nm for emission. After cells were pelleted, supernatant was removed, cells were resuspended in fresh minimal medium A without CCCP, and measurements were immediately recorded. Samples were monitored for 20 min. Raw data were buffer subtracted and normalized to maximum fluorescence values. Slopes were determined using the program GraphPad Prism 7, fitting a line using the plateau feature followed by exponential decay, given by the following equation, where X0 is the time at which fluorescence decay begins, Y0 is the average fluorescence until time X0, “plateau” represents the fluorescence value reached at equilibrium, and K is the rate constant of fluorescence decay over time: Y = plateau + (Y0 − plateau)−K(X−X0).
PI assay.
Membrane disruption, if any, was determined by adapting methods previously described (44). Overnight cultures of E. coli cells expressing PAsmr were inoculated into 50 ml of LB and incubated overnight at 37°C. Cultures were spun down and the pellets resuspended in 1 ml minimal medium, a 500-μl amount was inoculated into 20 ml of minimal medium to achieve “viable” cells, and another 500 μl was added to 70% isopropanol to achieve 100% “disrupted” cells, as isopropanol permeabilizes membranes (44). Cells were incubated with shaking at room temperature for 1 h. Samples were diluted to an OD600 of 0.1 for consistency with the efflux assay, and a standard curve was generated. From the “viable” culture, dilutions had been made earlier and incubated for an hour with 8 μM TM peptide(s) and 2 μM melittin; the latter peptide was used as a reference as it causes membrane disruption through pore formation (20). Amounts of 100 μl of incubated cells were added to an opaque 96-well Greiner Microlon Fluotrac 200 plate (Sigma-Aldrich, MO, USA), and 50 μl of 15 μM propidium iodide (PI) was added to each well. Plates were covered to protect the contents from light and shaken for 15 min. Fluorescence was measured using a SpectraMax Gemini EM microplate reader at an excitation wavelength of 488 nm and recording emissions at 630 nm. After blanks were subtracted, standard curves were generated by interpolating a standard curve through the program GraphPad Prism 7. Using the slope of the line for the standard curve, the values for percentages of viable cells incubated with peptide were determined. The values presented represent the average results of at least three independent trials.
Biocide resensitization assay.
MICs for all three biocides were determined for E. coli cells expressing PAsmr, using methods described above for the MIC assays. Additionally, the MICs for two SMR nonsubstrates, SDS and n-octyl-β-d-glucopyranose (OG), were determined and compared to the MICs for E. coli cells not expressing PAsmr to confirm they were nonsubstrates. Sublethal concentrations (below the critical micelle concentration for detergents) of biocide and detergent were then plated with a 2-fold serial dilution of peptide from 0.25 μM to 8 μM. Cells were plated at 50,000 CFU/well. Plates were incubated at 37°C for 20 h, and the OD was measured at 600 nm to determine the extent of cell growth.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by a grant to C.M.D. from the Canadian Institutes of Health Research (CIHR project grant number 376666). C.JM. is the recipient of a RESTRACOMP research studentship award from the Hospital for Sick Children.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00730-19.
REFERENCES
- 1.Centers for Disease Control and Prevention. 2013. Antibiotic resistance threats in the United States, 2013. CDC, Atlanta, GA. [Google Scholar]
- 2.Aloush V, Navon-Venezia S, Seigman-Igra Y, Cabili S, Carmeli Y. 2006. Multidrug-resistant Pseudomonas aeruginosa: risk factors and clinical impact. Antimicrob Agents Chemother 50:43–48. doi: 10.1128/AAC.50.1.43-48.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Poole K. 2011. Pseudomonas aeruginosa: resistance to the max. Front Microbiol 2:65. doi: 10.3389/fmicb.2011.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hirsch EB, Tam VH. 2010. Impact of multidrug-resistant Pseudomonas aeruginosa infection on patient outcomes. Expert Rev Pharmacoecon Outcomes Res 10:441–451. doi: 10.1586/erp.10.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhagirath AY, Li Y, Somayajula D, Dadashi M, Badr S, Duan K. 2016. Cystic fibrosis lung environment and Pseudomonas aeruginosa infection. BMC Pulm Med 16:174. doi: 10.1186/s12890-016-0339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maillard J-Y. 2005. Antimicrobial biocides in the healthcare environment: efficacy, usage, policies, and perceived problems. Ther Clin Risk Manag 1:307–320. [PMC free article] [PubMed] [Google Scholar]
- 7.Poole K. 2002. Mechanisms of bacterial biocide and antibiotic resistance. J Appl Microbiol 92(Suppl):55S–64S. doi: 10.1046/j.1365-2672.92.5s1.8.x. [DOI] [PubMed] [Google Scholar]
- 8.Hegstad K, Langsrud S, Lunestad BT, Scheie AA, Sunde M, Yazdankhah SP. 2010. Does the wide use of quaternary ammonium compounds enhance the selection and spread of antimicrobial resistance and thus threaten our health? Microb Drug Resist 16:91–104. doi: 10.1089/mdr.2009.0120. [DOI] [PubMed] [Google Scholar]
- 9.Furi L, Ciusa ML, Knight D, Di Lorenzo V, Tocci N, Cirasola D, Aragones L, Coelho JR, Freitas AT, Marchi E, Moce L, Visa P, Northwood JB, Viti C, Borghi E, Orefici G, Morrissey I, Oggioni MR. 2013. Evaluation of reduced susceptibility to quaternary ammonium compounds and bisbiguanides in clinical isolates and laboratory-generated mutants of Staphylococcus aureus. Antimicrob Agents Chemother 57:3488–3497. doi: 10.1128/AAC.00498-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bay DC, Turner RJ. 2009. Diversity and evolution of the small multidrug resistance protein family. BMC Evol Biol 9:140. doi: 10.1186/1471-2148-9-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun J, Deng Z, Yan A. 2014. Bacterial multidrug efflux pumps: mechanisms, physiology and pharmacological exploitations. Biochem Biophys Res Commun 453:254–267. doi: 10.1016/j.bbrc.2014.05.090. [DOI] [PubMed] [Google Scholar]
- 12.Bay DC, Rommens KL, Turner RJ. 2008. Small multidrug resistance proteins: a multidrug transporter family that continues to grow. Biochim Biophys Acta 1778:1814–1838. doi: 10.1016/j.bbamem.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 13.Kadry AA, Serry FM, El-Ganiny AM, El-Baz AM, Kadry AA, Serry FM, El-Ganiny AM, Ahmed M. 2017. Integron occurrence is linked to reduced biocide susceptibility in multidrug resistant Pseudomonas aeruginosa. Br J Biomed Sci 74:78–84. doi: 10.1080/09674845.2017.1278884. [DOI] [PubMed] [Google Scholar]
- 14.Chen Y-J, Pornillos O, Lieu S, Ma C, Chen AP, Chang G. 2007. X-ray structure of EmrE supports dual topology model. Proc Natl Acad Sci U S A 104:18999–19004. doi: 10.1073/pnas.0709387104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poulsen BE, Deber CM. 2012. Drug efflux by a small multidrug resistance protein is inhibited by a transmembrane peptide. Antimicrob Agents Chemother 56:3911–3916. doi: 10.1128/AAC.00158-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poulsen BE, Rath A, Deber CM. 2009. The assembly motif of a bacterial small multi drug resistance protein. J Biol Chem 284:9870–9875. doi: 10.1074/jbc.M900182200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poulsen BE, Cunningham F, Lee KKY, Deber CM. 2011. Modulation of substrate efflux in bacterial small multidrug resistance proteins by mutations at the dimer interface. J Bacteriol 193:5929–2935. doi: 10.1128/JB.05846-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bellmann-Sickert K, Stone TA, Poulsen BE, Deber CM. 2015. Efflux by small multidrug resistance proteins is inhibited by membrane-interactive helix-stapled peptides. J Biol Chem 290:1752–1759. doi: 10.1074/jbc.M114.616185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melnyk RA, Partridge AW, Yip J, Wu Y, Goto NK, Deber CM. 2003. Polar residue tagging of transmembrane peptides. Biopolymers 71:675–685. doi: 10.1002/bip.10595. [DOI] [PubMed] [Google Scholar]
- 20.Van Den Bogaart G, Guzmán JV, Mika JT, Poolman B. 2008. On the mechanism of pore formation by melittin. J Biol Chem 283:33854–33857. doi: 10.1074/jbc.M805171200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Domalaon R, Idowu T, Zhanel GG, Schweizer F. 2018. Antibiotic hybrids: the next generation of agents and adjuvants against gram-negative pathogens? Clin Microbiol Rev 31:e00077-17. doi: 10.1128/CMR.00077-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wright GD. 2016. Antibiotic adjuvants: rescuing antibiotics from resistance. Trends Microbiol 24:862–871. doi: 10.1016/j.tim.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 23.Wright GD. 2012. Antibiotics: a new hope. Chem Biol 19:3–10. doi: 10.1016/j.chembiol.2011.10.019. [DOI] [PubMed] [Google Scholar]
- 24.Schuldiner S. 2018. The Escherichia coli effluxome. Res Microbiol 169:357–362. doi: 10.1016/j.resmic.2018.02.006. [DOI] [PubMed] [Google Scholar]
- 25.Li XZ, Poole K, Nikaido H. 2003. Contributions of MexAB-OprM and an EmrE homolog to intrinsic resistance of Pseudomonas aeruginosa to aminoglycosides and dyes. Antimicrob Agents Chemother 47:27–33. doi: 10.1128/aac.47.1.27-33.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tal N, Schuldiner S. 2009. A coordinated network of transporters with overlapping specificities provides a robust survival strategy. Proc Natl Acad Sci U S A 106:9051–9056. doi: 10.1073/pnas.0902400106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paulsen IT, Littlejohn TG, Rådström P, Sundström L, Sköld O, Swedberg G, Skurray RA. 1993. The 3′ conserved segment of integrons contains a gene associated with multidrug resistance to antiseptics and disinfectants. Antimicrob Agents Chemother 37:761–768. doi: 10.1128/aac.37.4.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kazama H, Hamashima H, Sasatsu M, Arai T. 1998. Distribution of the antiseptic-resistance gene qacE delta 1 in gram-positive bacteria. FEMS Microbiol Lett 165:295–299. doi: 10.1111/j.1574-6968.1998.tb13160.x. [DOI] [PubMed] [Google Scholar]
- 29.Poirel L, Lambert T, Turkoglu S, Ronco E, Gaillard J, Nordmann P. 2001. Characterization of class 1 integrons from Pseudomonas aeruginosa that contain the bla(VIM-2) carbapenem-hydrolyzing beta-lactamase gene and of two novel aminoglycoside resistance gene cassettes. Antimicrob Agents Chemother 45:546–552. doi: 10.1128/AAC.45.2.546-552.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahzounieh M, Khoshnood S, Ebrahimi A, Habibian S, Yaghoubian M. 2014. Detection of antiseptic-resistance genes in Pseudomonas and Acinetobacter spp. isolated from burn patients. Jundishapur J Nat Pharm Prod 9:e15402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Babaei MR, Sulong A, Hamat RA, Nordin SA, Neela VK. 2015. Extremely high prevalence of antiseptic resistant quaternary ammonium compound E gene among clinical isolates of multiple drug resistant Acinetobacter baumannii in Malaysia. Ann Clin Microbiol Antimicrob 14:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu WJ, Fu L, Huang M, Zhang JP, Wu Y, Zhou YS, Zeng J, Wang GX. 2017. Frequency of antiseptic resistance genes and reduced susceptibility to biocides in carbapenem-resistant Acinetobacter baumannii. J Med Microbiol 66:13–17. doi: 10.1099/jmm.0.000403. [DOI] [PubMed] [Google Scholar]
- 33.Romao C, Miranda CA, Silva J, Mandetta Clementino M, de Filippis I, Asensi M. 2011. Presence of qacEDelta1 gene and susceptibility to a hospital biocide in clinical isolates of Pseudomonas aeruginosa resistant to antibiotics. Curr Microbiol 63:16–21. doi: 10.1007/s00284-011-9934-0. [DOI] [PubMed] [Google Scholar]
- 34.Helal ZH, Khan MI. 2015. QacE and QacEdelta1 genes and their correlation to antibiotics and biocides resistance Pseudomonas aeruginosa. Am J Biomed Sci 7:52–62. doi: 10.5099/aj150200052. [DOI] [Google Scholar]
- 35.Shafaati M, Boroumand M, Nowroozi J, Amiri P, Kazemian H. 2016. Correlation between qacE and qacEΔ1 efflux pump genes, antibiotic and disinfectant resistant among clinical isolates of E. coli. Recent Pat Antiinfect Drug Discov 11:189–195. doi: 10.2174/1574891X11666160815094718. [DOI] [PubMed] [Google Scholar]
- 36.Glukhov E, Burrows LL, Deber CM. 2008. Membrane interactions of designed cationic antimicrobial peptides: the two thresholds. Biopolymers 89:360–371. doi: 10.1002/bip.20917. [DOI] [PubMed] [Google Scholar]
- 37.Chen Y, Vasil AI, Rehaume L, Mant CT, Burns JL, Vasil ML, Hancock REW, Hodges RS. 2006. Comparison of biophysical and biologic properties of α-helical enantiomeric antimicrobial peptides. Chem Biol Drug Design 67:162–173. doi: 10.1111/j.1747-0285.2006.00349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hunter HN, Jing W, Schibli DJ, Trinh T, Park IY, Kim SC, Vogel HJ. 2005. The interactions of antimicrobial peptides derived from lysozyme with model membrane systems. Biochim Biophys Acta 1668:175–189. doi: 10.1016/j.bbamem.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 39.Ovchinnikov V, Stone TA, Deber CM, Karplus M. 2018. Structure of the EmrE multidrug transporter and its use for inhibitor peptide design. Proc Natl Acad Sci U S A 115:E7932–E7941. doi: 10.1073/pnas.1802177115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brill S, Sade-Falk O, Elbaz-Alon Y, Schuldiner S. 2015. Specificity determinants in small multidrug transporters. J Mol Biol 427:468–477. doi: 10.1016/j.jmb.2014.11.015. [DOI] [PubMed] [Google Scholar]
- 41.WHO. 2017. Antibacterial agents in clinical development: an analysis of the antibacterial clinical development pipeline, including tuberculosis. WHO, Geneva, Switzerland. [Google Scholar]
- 42.Duarte AMS, De Jong ER, Koehorst RBM, Hemminga MA. 2010. Conformational studies of peptides representing a segment of TM7 from H+-VO-ATPase in SDS micelles. Eur Biophys J 39:639–646. doi: 10.1007/s00249-009-0522-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stone TA, Cole GB, Nguyen HQ, Sharpe S, Deber CM. 2018. Influence of hydrocarbon-stapling on membrane interactions of synthetic antimicrobial peptides. Bioorganic Med Chem 26:1189–1196. doi: 10.1016/j.bmc.2017.10.020. [DOI] [PubMed] [Google Scholar]
- 44.Stiefel P, Schmidt-Emrich S, Maniura-Weber K, Ren Q. 2015. Critical aspects of using bacterial cell viability assays with the fluorophores SYTO9 and propidium iodide. BMC Microbiol 15:36. doi: 10.1186/s12866-015-0376-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.