

Abstract
Late-onset ataxia is common, often idiopathic, and can result from cerebellar, proprioceptive, or vestibular impairment; when in combination, it is also termed cerebellar ataxia, neuropathy, vestibular areflexia syndrome (CANVAS). We used non-parametric linkage analysis and genome sequencing to identify a biallelic intronic AAGGG repeat expansion in the replication facsstor C subunit 1 (RFC1) gene as the cause of familial CANVAS and a frequent cause of late-onset ataxia, particularly if sensory neuronopathy and bilateral vestibular areflexia coexist. The expansion, which occurs in the poly(A) tail of an AluSx3 element and differs in both size and nucleotide sequence from the reference (AAAAG)11 allele, does not affect RFC1 expression in patient peripheral and brain tissue, suggesting no overt loss of function. These data, along with an expansion carrier frequency of 0.7% in Europeans, implies that biallelic AAGGG expansion in RFC1 is a frequent cause of late-onset ataxia.
Late-onset ataxia, postural imbalance, and falls are a frequent reason for neurological consultation. Physiologically, motor coordination is achieved under visual control thanks to the cerebellar integration of proprioceptive information conveyed by large-fiber sensory neurons and vestibular inputs. Failure of any or a combination of these systems can result in ataxia1–6. Both acquired and genetic causes are known, but a large proportion remains idiopathic.
Previous studies suggested that there is a spectrum of clinical signs, from pure idiopathic late-onset cerebellar degeneration (ILOCA) through to the combined degeneration of the cerebellum and its vestibular and sensory afferents, which has been named CANVAS (Fig. 1a)7. CANVAS is an adult-onset, slowly progressive neurological disorder characterized by imbalance, sensory neuropathy (neuronopathy), bilateral vestibulopathy8, chronic cough, and occasionally autonomic dysfunction9. Typically, sensory action potentials and somatosensory potentials are absent throughout, brain magnetic resonance imaging (MRI) shows cerebellar atrophy, and vestibular testing is consistent with impaired vestibular function bilaterally9–17. Late-onset ataxia and CANVAS are usually sporadic, but occasionally occur in siblings, raising the possibility of recessive transmission. However, initial attempts to identify the underlying genetic defect by whole-exome sequencing were unsuccessful.
Fig. 1 |. Clinical spectrum and pedigrees of late-onset ataxia.
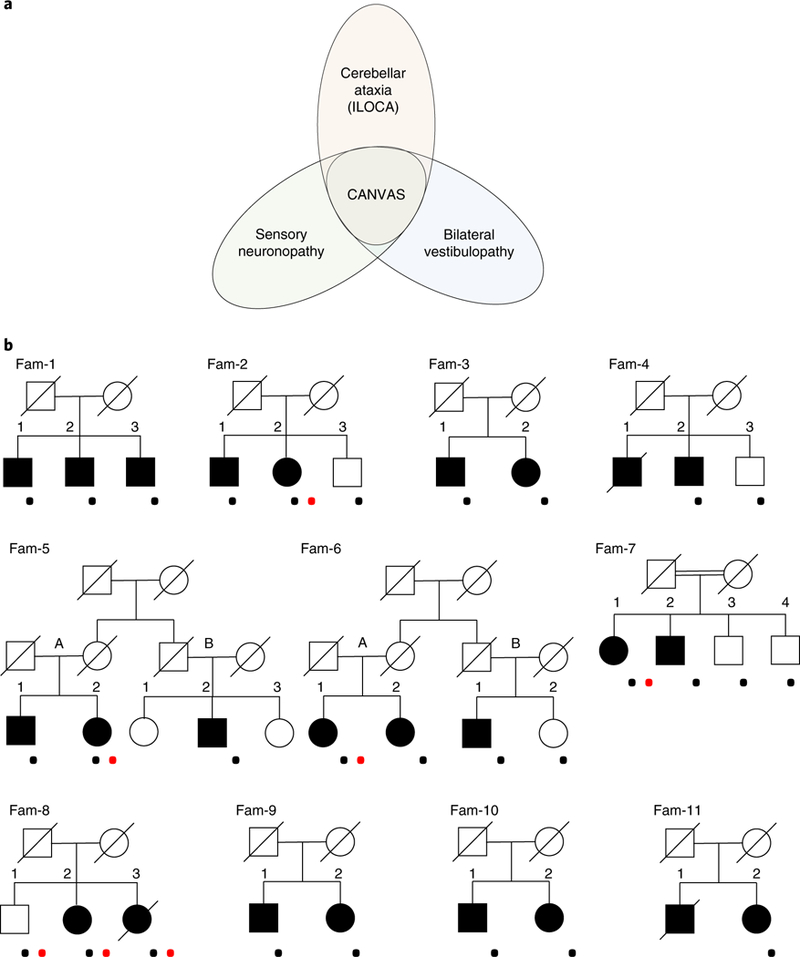
a, Clinical spectrum of idiopathic late-onset ataxia from isolated cerebellar, vestibular, and sensory variants to full-blown CANVAS. b, Pedigrees of CANVAS families. The squares indicate males and the circles females. The diagonal lines are used for deceased individuals. CANVAS patients are indicated with filled shapes. The black dots indicate genotyped individuals. The red dots indicate patients enrolled for WGS study.
Using non-parametric linkage analysis and whole-genome sequencing (WGS), we identified a recessive intronic AAGGG repeat expansion in the RFC1 gene as a cause of familial CANVAS. The expansion occurs in the poly(A) tail of an AluSx3 element and differs in both size and nucleotide sequence from the reference (AAAAG)11 allele. Screening of additional sporadic cases with late- onset ataxia confirmed the presence of the mutated AAGGG repeat expansion in 22% of them, and in higher percentages if sensory neuronopathy and/or bilateral vestibular areflexia coexisted, suggesting that it represents a frequent and underrecognized cause of late-onset ataxia.
Results
Genetic study.
We genotyped 29 individuals (23 affected and 6 unaffected) from 11 families (Fig. 1b). The majority of the families consisted of affected sibships, except for two first-degree cousins from non-consanguineous families (Fam 5b-2 and Fam 6b-1). None of the families had convincing evidence of vertical disease transmission.
Assuming a recessive mode of inheritance, non-parametric linkage analysis identified a single peak at position 4p14 with a cumulative maximum heterogeneity logarithm of the odds (HLOD) score of 5.8 (Fig. 2a). Haplotype analysis defined a 1.7-Mb region between markers rs6814637 and rs10008483 (chr4:38977921–40712231) where, within single families, affected siblings shared the same maternal and paternal alleles as opposed to unaffected brothers and sisters, who had at most one of them (Fig. 2b). The region contains 21 known HUGO Gene Nomenclature Committee genes (Supplementary Table 1). Homozygosity mapping in consanguineous family Fam 7 showed that the previously identified 1.7-Mb region is encompassed in a larger run of homozygosity of 12 Mb shared by the affected siblings (Supplementary Fig. 1). Of interest, inside the 1.7-Mb region, four single-nucleotide polymorphisms (SNPs; rs2066790, rs11096992, rs17584703, and rs6844176, bold highlighted) mapping inside a region encompassing all exons of RFC1 and the last exon of the WD repeat domain 19 (WDR19) gene were shared by all affected individuals from different families except for individual Fam 5b-2, raising the possibility of a founder haplotype (Fig. 2c,d).
Fig. 2 |. Identification of CANVAS locus.
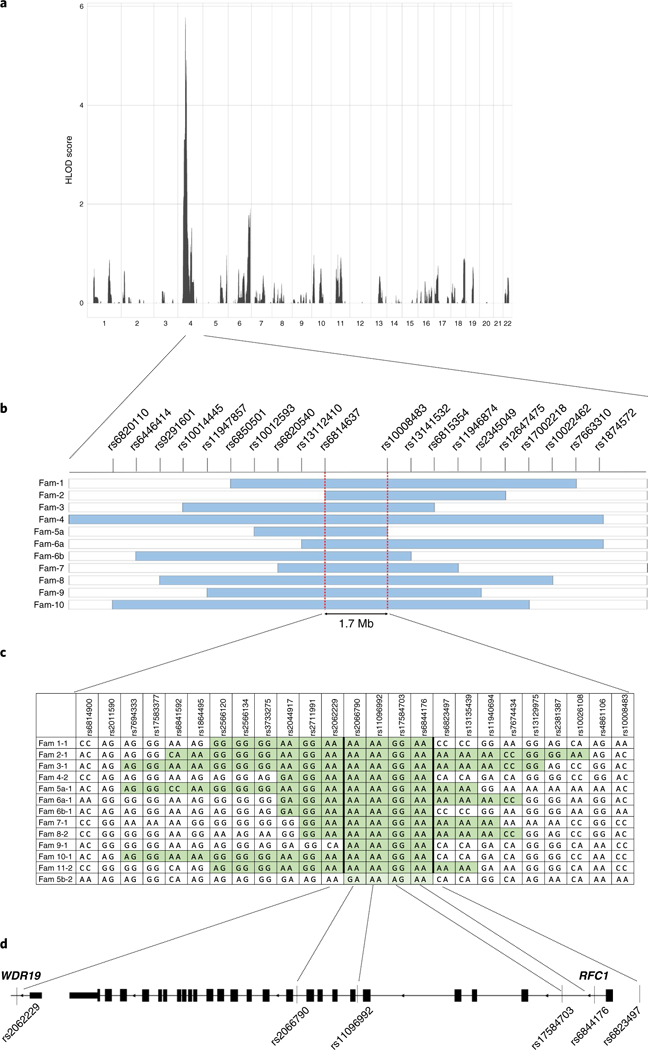
a, Non-parametric multipoint linkage analysis identifies a unique locus associated with the disease in chromosomal region 4p14 with a maximal HLOD score of 5.8. b, Schematic representation of shared haplotypes within single families. The light blue bars indicate a genomic region shared by affected siblings in a family and for which unaffected siblings are discordant. Two red dashed lines define a 1.7-Mb region common to the different families. SNPs defining the haplotypes are represented on the top line. c, Fine mapping inside the 1.7-Mb region identifies a recessive haplotype shared by all distinct families (highlighted in green), except for individual Fam 5b-2, who probably shares only one allele (highlighted in light green). d, Schematic representation of the candidate region encompassing all 24 exons and flanking regions of RFC1 and the last exon and flanking intron of WDR19.
Whole-exome sequencing was previously performed in seven individuals (Fam 1–1, Fam 1–2, Fam 1–3, Fam 3–1, Fam 3–2, Fam 4–2, and Fam 4–3) from three unrelated families (Fam1, Fam3, Fam4), but did not identify recurrent non-synonymous variants within the coding regions of the genes encompassed in the 1.7-Mb region (data not shown). We next performed WGS in an additional six affected individuals (Fam 2–2, Fam 5a-2, Fam 6a-1, Fam 7–1, Fam 8–2, and Fam 8–3), one unaffected individual (Fam 8–1) from four unrelated families, and one sporadic case (s9). The analysis of non-synonymous and copy number variants did not reveal changes recurring in the affected families. By visually inspecting the aligned paired reads inside the 1.7-Mb region, we noted in all CANVAS patients a reduced read depth in a region encompassing a simple tandem (AAAAG)11 repeat at position chr4:39350045–39350103 (hg19, Fig. 3a). Inside the microsatellite region, the reference (AAAAG)11 repeat was replaced in patients by a variable number of AAGGG repeated units, which were detected on the reads mapped to either side of the short tandem repeat (STR). However, none of the reads spanned across the microsatellite region from one side to the other, suggesting the presence of a biallelic expansion of the AAGGG repeat unit (Fig. 3b). WGS from an unaffected sibling (Fam 8–1) showed an equal distribution of interrupted reads containing the mutated AAGGG repeated unit change as well as reads containing the AAAAG repeat.
Fig. 3 |. Recessive expansion of a mutated AAGGG repeated unit in intron 2 of RFC1 causes CANVAS and late-onset ataxia in familial and sporadic cases.
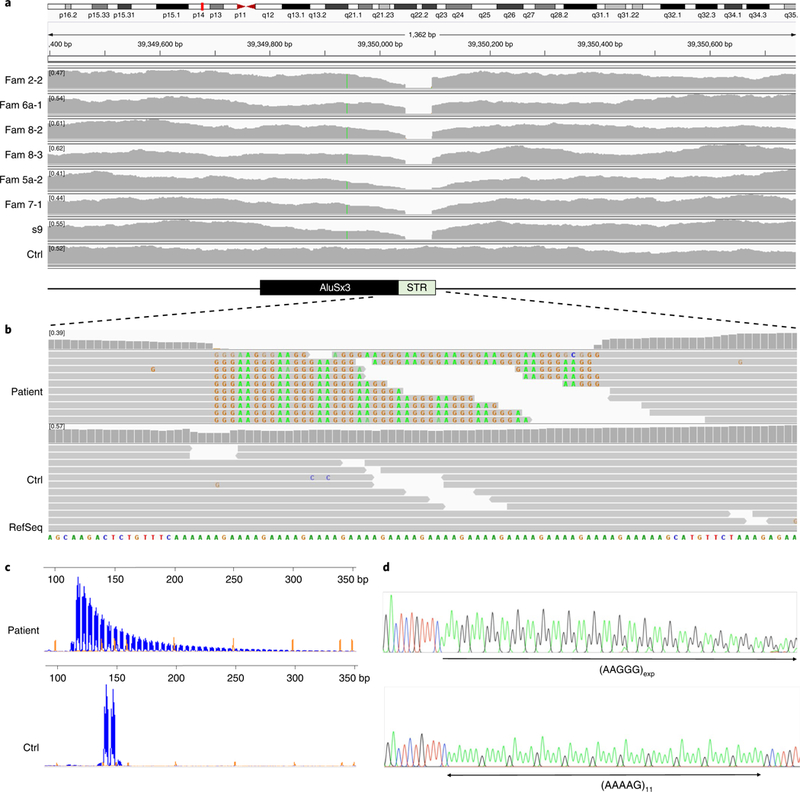
a, A reduced read depth of WGS is observed in CANVAS patients (n = 6) in a region corresponding to a short tandem AAAAG repeat in intron 2 of RFC1. b, Visualization on IGV of reads aligned to the short repeat and flanking region shows in patients (n = 6) the presence of a mutated AAGGG repeat unit (representative image). Reads from both sides are interrupted and are unable to cover the entire length of the microsatellite region. Note that, per IGV default setting, AAGGG repeated units that do not map to the (AAAAG)11 reference sequence are soft-clipped and do not contribute to the coverage of the STR in a, which is virtually absent. However, ≥20 reads containing the AAGGG repeated unit could be observed in each patient if soft-clipped reads were shown. c, RP-PCR targeting the mutated AAGGG repeated unit. Fluorescein amidite-labeled PCR products were separated on an ABI 3730 DNA Analyzer. Electropherograms were visualized on GeneMapper at 2,000 relative fluorescence units. The representative plots from a patient carrying the AAGGG repeat expansion and one non-carrier are shown. RP-PCR experiments were repeated independently twice with similar results. d, Sanger sequencing of long-range PCR reactions confirms the AAAAG to AAGGG nucleotide change of the repeated unit in patients.
We then performed repeat-primed PCR (RP-PCR) with primers targeting the mutant AAGGG pentanucleotide unit and confirmed the presence of an AAGGG repeat expansion in all affected members from 11 families, as well as in unaffected carriers (Fig. 3c). Flanking PCR using standard conditions failed to amplify the region in all patients, suggesting the presence of a large expansion on both alleles, as opposed to their unaffected siblings for whom at least one allele could be amplified by PCR (data not shown).
We next screened a cohort of 150 patients diagnosed with sporadic late-onset ataxia and identified an additional 33 (22%) sporadic cases carrying the recessive AAGGG repeat expansion, as defined by a positive RP-PCR for AAGGG repeat unit and the absence of PCR-amplifiable products by standard flanking PCR. The percentage of positive cases increased to 63% (32 out of 51) if cases with late-onset cerebellar ataxia and sensory neuronopathy were considered and to 92% (11 out of 12) in cases will full CANVAS syndrome. Taking advantage of two informative SNPs (rs11096992 and rs2066790), using PCR and direct sequencing, we observed that all additional sporadic cases except for individual s23 shared the same haplotype as familial CANVAS cases.
Using long-range PCR, we amplified and confirmed by Sanger sequencing the presence of the AAGGG expansion in all patients (Fig. 3d). However, long-range PCR did not allow sizing of the repeat expansion, since PCR is error-prone and contraction of repeated regions during PCR cycling has been demonstrated previously18. Therefore, Southern blots were conducted in 34 cases; they confirmed the presence of biallelic large expansions in all 34 cases. Biallelic expansions could be visualized as two distinct bands in individuals carrying expansions of different sizes, or one thick band if the expanded alleles had a similar size (Supplementary Fig. 2). Four unaffected siblings from four families were also included, and they all carried one expanded and one normal allele. Although the expansion size varied across different families, ranging from around 400 to 2,000 repeats, in the majority of cases approximately 1,000 repeats were observed. Repeat size was relatively stable in siblings within single families. There was no association between age at onset and the number of AAGGG repeat units on either the smaller or larger allele (n = 34; r = +0.007, P = 0.97 and r = −0.04, P = 0.81, respectively).
Polymorphic conformations and allelic distribution of the STR locus in the normal population.
Recessive AAGGG expansion, as defined by the combination of positive RP-PCR targeting the AAGGG repeat and the absence of a PCR-amplifiable product on flanking PCR, were not observed in 304 healthy controls screened. RP-PCR analysis targeting the AAGGG repeat showed that 0.7% (4 out of 608 chromosomes tested) carried an AAGGG expansion in the heterozygous state. Southern blot analysis was performed and confirmed the presence of an expanded allele in all of them. The chr4:39350045–39350103 locus, where the expansion resides, was shown to be highly polymorphic in the normal population and, besides the rare AAGGG expansion allele (AAGGG)exp, three other conformations were observed: (AAAAG)11; (AAAAG)exp; and (AAAGG)exp (Fig. 4a). The (AAAGG)exp often showed interruptions and nucleotide changes of the expanded sequence. By a combinatory approach of flanking PCR, RP-PCR targeting one of the three possible nucleotide sequences, as well as Southern blot and Sanger sequencing in selected cases, we observed an allelic distribution of 75.5% (n = 459) for the (AAAAG)11 allele, 13.0% (n = 79) for the (AAAAG)exp allele, 7.9% (n = 48) for the (AAAGG)exp allele, and 0.7% (n = 4) for the (AAGGG)exp allele (Fig. 4b). The average size of (AAAAG)exp ranged from 15 to 200 repeats (mean ± s.d. 72 ± s.d. 43), and (AAAGG)exp ranged from 40 to 1,000 (mean ± s.d. 173 ± s.d. 232) (Fig. 4c).
Fig. 4 |. Polymorphic configurations of the repeat expansion locus and allelic distribution in healthy controls.
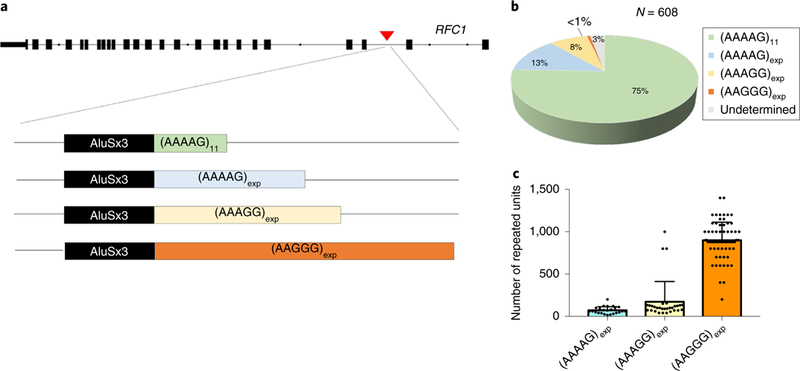
a, Schematic representation of the repeat expansion locus in intron 2 of RFC1 and its main allelic variants. b, Estimated allelic frequencies in 608 chromosomes from 304 healthy controls. c, Average size and s.d. of (AAAAG)exp (n = 24) and (AAAGG)exp (n = 30) expansions in healthy controls and (AAGGG)exp (n = 72) in controls and CANVAS patients.
Eight healthy individuals had biallelic repeat expansions of a distinct repeated unit: (AAAAG)exp/(AAGGG)exp in one case; (AAAGG)exp/(AAGGG)exp in one case; and (AAAAG)exp / (AAAGG)exp in six cases. Twenty-two cases probably had two expansions of the repeated AAAAG unit and nine of the repeated AAAGG unit, as defined by a positive RP-PCR for the target repeat and two distinct bands on the Southern blot, although we cannot exclude that one of the two alleles may be characterized by a distinct nucleotide sequence, which was not considered in the present study. Indeed, nine additional individuals had no PCR-amplifiable product on flanking PCR and were negative for RP-PCR targeting the AAAAG, AAAGG, or AAGGG repeated units, suggesting the potential existence of other possible allelic conformations in 3% (n = 18) of tested chromosomes. Southern blot analysis could not be performed because of insufficient amounts of DNA in these cases.
The haplotype associated in most patients with the AAGGG repeat expansion has an allelic carrier frequency in the 1000 Genomes Project control population of 18%. Based on rs11096992 and rs2066790 genotyping, the disease-associated haplotype rs2066790 (AA), rs11096992 (AA) was absent in the recessive state from healthy individuals who carried two (AAAAG)11 alleles, two (AAAAG)exp alleles, or a compound (AAAAG)11/ (AAAAG)exp genotype, but was observed in three out of nine carriers of two (AAAGG)exp alleles and one healthy individual with the (AAGGG)exp/(AAAGG)exp alleles, suggesting its possible stronger association with both (AAGGG)exp and (AAAGG)exp configurations of the repeated unit.
Clinical features of patients carrying the recessive AAGGG repeat expansion.
The clinical features of 56 cases carrying the recessive intronic AAGGG repeat expansion, including 23 familial and 33 sporadic cases, are summarized in Table 1 and detailed in Supplementary Table 2. All cases were of European ancestry. Apart from a higher frequency of vestibular areflexia in familial CANVAS, clinical features were otherwise similar in familial and sporadic cases; hence, data are presented together. Mean age of onset was 54 ± 9 (35–73) years, and mean disease duration at examination was 11 ± 7 (1–30) years. The most common complaint at disease onset was unsteadiness, which was reported by 84% of patients, and frequently described as being worse in the dark; 37% of patients complained of chronic cough, which in some cases could precede by decades the onset of the walking difficulties. Neurological examination invariably showed signs in keeping with a large-fiber sensory neuropathy; 80% of patients had signs of cerebellar involvement and overall 54% had evidence of bilateral vestibular areflexia. Twenty-three percent of patients had concurrent autonomic nervous system involvement, particularly affecting micturition and defecation. Nerve conduction studies confirmed the presence of a non-length- dependent sensory neuropathy in all cases tested, as opposed to an entirely normal motor conduction study in most patients. Cerebellar atrophy was identified in 35 (83%) of 42 cases who underwent an MRI or CT scan.
Table 1 |.
Clinical features of patients with familial or sporadic late-onset ataxia carrying the recessive AAGGG repeat expansion in RFC1
| Familial cases (n = 23) | Sporadic cases (n = 33) | All cases (n = 56) | P | |
|---|---|---|---|---|
| Male | 12 (52%) | 15 (45%) | 27 (48%) | NS |
| Age at onset | 53±8 | 54±10 | 54±9 | NS |
| Disease duration at examination | 13±9 | 10±6 | 11±7 | NS |
| Sensory neuropathy | 23 (100%) | 33 (100%) | 56 (100%) | NS |
| Cerebellar syndrome | 18 (78%) | 27 (82%) | 45 (80%) | NS |
| Bilateral vestibular impairment | 17 (74%) | 13 (39%) | 30 (53%) | 0.01 |
| Dysautonomia | 4 (17%) | 9 (27%) | 13 (23%) | NS |
| Cough | 7 (30%) | 14 (42%) | 21 (37%) | NS |
| SAP upper limbs | ||||
| Reduced | 6/21 (29%) | 4/31 (13%) | 10/52 (19%) | NS |
| Absent | 15/21 (71%) | 27/31 (87%) | 42/52 (81%) | NS |
| SAP lower limbs | ||||
| Reduced | 2/21 (10%) | 1/31 (3%) | 3/52 (6%) | NS |
| Absent | 19/21 (90%) | 30/31 (97%) | 49/52 (94%) | NS |
| Normal motor conduction | 19/21 (90%) | 26/31 (84%) | 45/52 (87%) | NS |
| Cerebellar atrophy at CT/MRI scan | 14/17 (82%) | 21/25 (84%) | 35/42 (83%) | NS |
| Full-blown CANVAS syndrome | 15 (65%) | 11 (33%) | 26 (46%) | 0.02 |
NS, not significant; SAP, sensory action potential.
Neuropathological examination.
The neuropathological examination was conducted in a patient with CANVAS who carried the biallelic AAGGG repeat expansion, compared with a patient with genetically confirmed Friedreich’s ataxia (FRDA), a patient with spinocerebellar ataxia type 17 (SCA17), and one case with C9orf72-related frontotemporal dementia (FTD), as well as control brains (Fig. 5). The patient with CANVAS showed severe, widespread depletion of Purkinje cells with associated prominent Bergmann gliosis, while cell density in the granule cell layer was well preserved. Loss of Purkinje cells was also observed in FRDA, SCA17 and, to a much lesser extent, in C9orf72-related FTD, but not in control brains. Similarly to FRDA and control brains, and as opposed to SCA17 and a C9orf72-related FTD, which were tested as positive controls, p62 immunostaining showed no pathological cytoplasmic or intranuclear inclusions in the cerebellar cortex of the patient with CANVAS. Examination of the brain, in addition to prominent cerebellar atrophy, revealed age-related changes in the form of neurofibrillary tangle tau pathology and amyloid-β pathology (Supplementary Fig. 3).
Fig. 5 |. Pathology of cerebellar degeneration in a patient with CANVAS carrying the recessive AAGGG repeat expansion.
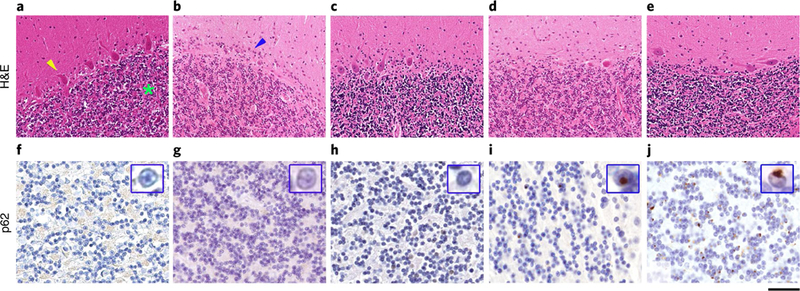
a-j, Hematoxylin and eosin (H&E)-stained sections (a-e) and sections immunostained for p62 (f-j). In a control brain (a), age-matched for the patient with CANVAS syndrome, there is well preserved density of Purkinje cells (yellow arrowhead); the granule cell layer is densely populated with small neurocytes (green asterisk). b, In CANVAS syndrome, there is severe, widespread depletion of Purkinje cells with associated prominent Bergmann gliosis (blue arrowhead), while cell density in the granule cell layer is well preserved. c, In a patient with genetically confirmed FRDA, there is patchy depletion of Purkinje cells associated with Bergmann gliosis and unremarkable appearance of the granule cell layer. d, In a patient with genetically confirmed SCA17, there is widespread Purkinje cell loss with only occasional Purkinje cells remaining; also, in this patient, the granule cell layer is densely populated with small neurocytes. e, In a patient with FTD due to C9orf72 expansion, Purkinje cell loss is patchy and the granule cell layer is unremarkable. f-h, Immunostaining for p62 shows no pathological cytoplasmic or intranuclear inclusions in the cerebellar cortex in the control patient (f), the patient with CANVAS syndrome (g), and also in the patient with FRDA (h). i, In the SCA17 patient, there are scattered discrete intranuclear p62 immunoreactive inclusions in the small neurons within the granule cell layer (high-power view of a representative intranuclear inclusion is demonstrated in the inset within i). j, In the patient with the C9orf72 expansion, there are frequent characteristic perinuclear p62 positive inclusions in the granule cell layer (high-power view of a representative inclusion is shown in the inset within j). Scale bar, 100 μm in a-e, 30 μm in f-j, and 5μm in the insets in f-j. Staining was carried out once on patient samples with appropriate controls according to standard practice and histopathology procedures in an ISO 15189-accredited laboratory.
Eight nerve biopsies and ten muscle biopsies were also available for assessment from patients carrying the biallelic AAGGG repeat expansion. In all nerve biopsies, there was prominent widespread depletion of myelinated fibers; the muscle biopsies confirmed chronic denervation with reinnervation (Supplementary Fig. 4).
Fluorescence in situ hybridization using sense (AAGGG)5 and antisense (TTCCC)5 repeat-specific oligonucleotides was performed on vermis postmortem tissue from one CANVAS patient and disease and healthy controls. As opposed to SH-SY5Y cells transfected with pcDNA3.1/CT-green fluorescent protein-TOPO vector containing either (TTCCC)94 or (AAGGG)54, where intranuclear and cytoplasmic inclusion were clearly detectable, we did not observe the presence of endogenous RNA foci in any of the samples examined (Supplementary Fig. 5).
RNA sequencing (RNA-seq).
We performed whole transcriptome analysis to assess the presence of changes in RFC1 expression, as well as the cis and trans effects at more distant genomic regions. RNA-seq data showed that RFC1 messenger RNA (mRNA) was unchanged in CANVAS (n = 3) and control (n = 4) fibroblasts (P=0.42) and in CANVAS (n = 2) and control (n = 3) lymphoblasts (P=0.45; data not shown). We also performed RNA-seq from the frontal cortex and cerebellar vermis of autopsied brains from one CANVAS patient, FRDA cases (n = 2), and controls without evidence of neurological disease (n = 3). In the single CANVAS patient, RFC1 appeared to be unchanged in both cortex and cerebellum, as compared to the other samples (Fig. 6a). However, the frataxin (FXN) gene was clearly downregulated in the frontal cortex and cerebellum of the FRDA cases compared to the controls (cerebellum P = 0.007; log2 fold change = −1.2; frontal cortex P = 0.0003; log2 fold change = −1.3) (Fig. 6a). The single CANVAS sample resembled the controls for FXN expression.
Fig. 6 |. RFC1 expression is not affected by the AAGGG repeat expansion.
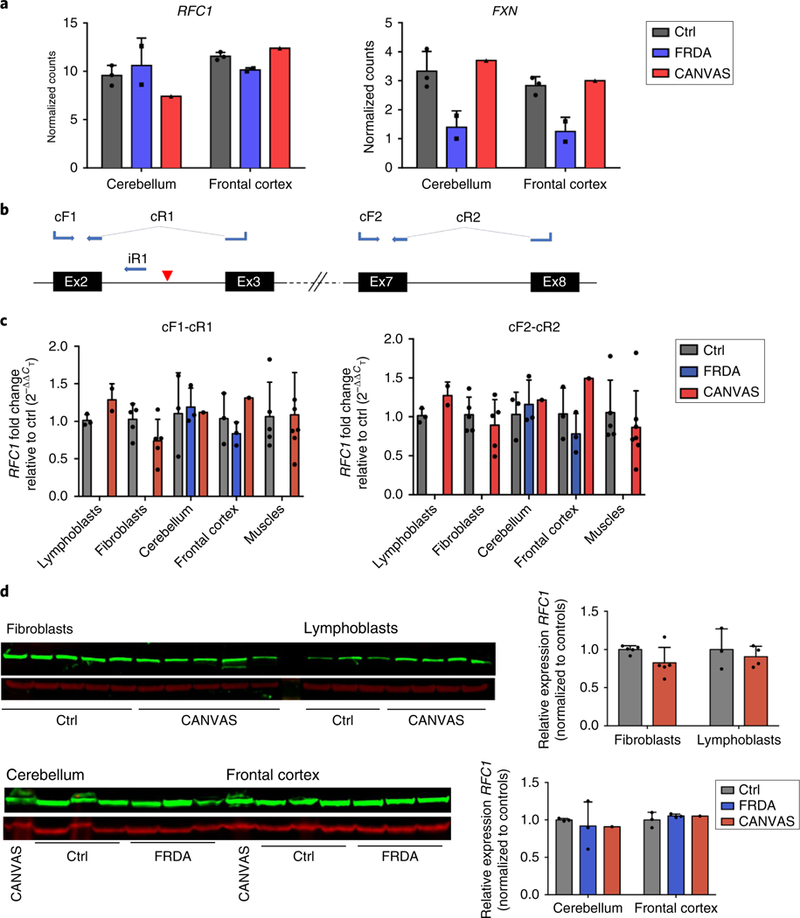
a, Plots showing the expression levels of RFC1 and FXN in controls (n = 3), patients with FRDA (n = 2), and one CANVAS patient in postmortem cerebellum and frontal cortex. b, Mapping on RFC1 transcript 1 of the primers used for assessment by qRT-PCR of RFC1 mRNA (cF1-cR1 and cF2-cR2) and pre-mRNA (cF1/iR1) expression. The blue arrows indicate the primers mapping to the exonic and intronic regions of the canonical RFC1 transcript. Primers spanning across exonic junctions are connected by dotted lines. A red triangle indicates the site of the AAGGG repeat expansion. c, Expression levels of the canonical coding RFC1 mRNA as measured by qRT-PCR using two separate sets of primers, cF1-cR1 and cF2-cR2, in control (n = 3) and CANVAS (n = 2) lymphoblasts, control (n = 5) and CANVAS (n = 5) fibroblasts, control (n = 3), FRDA (n = 3), and CANVAS (n = 1) cerebellum and frontal cortex, and control (n = 5) and CANVAS muscles (n = 7). d, RFC1-encoded protein levels as measured by western blotting using the polyclonal antibody GTX129291 and normalized to β-actin in control (n = 5) and CANVAS (n = 5) fibroblasts, control (n = 3) and CANVAS (n = 4) lymphoblasts, and control (n = 3), FRDA (n = 3), and CANVAS (n = 1) postmortem cerebellum and frontal cortex. The bar graphs show the mean±s.d. and data distribution (black dots). A two-tailed t-test was performed to compare RFC1 transcript and encoded protein expression in patients versus healthy or disease controls. All experiments were repeated independently twice with similar results.
There were no differentially expressed genes between patient and control fibroblasts, whereas 132 differentially expressed genes were identified between patient and control lymphoblasts. Gene Ontology analysis showed enrichment for immune terms, whose relevance to the disease warrants further work. Notably, only eight differentially expressed genes were located on chromosome 4 and were all separated by at least 25 Mb from the locus of the repeat expansion. Analysis of differentially expressed genes in the frontal cortex and vermis was not possible because of the limited number of CANVAS samples (n = 1).
Splicing analysis was performed in lymphoblasts. We identified 145 exons in 108 genes that had evidence of differential exon usage in CANVAS patients compared to healthy controls. Motif analysis of the alternatively spliced exons showed enrichment of motifs targeted by serine/arginine-rich splicing factor proteins, in particular serine/arginine-rich splicing factor 3. RFC1 did not show aberrant splicing of its coding exons in mature mRNA. Also, no reads containing the AAGGG or TTCCC repeated unit mapping to intron 2 of the RFC1 pre-mRNA transcript were detected; no antisense or non-coding transcript was observed at the RFC1 locus in any of the tissues examined. Gene Ontology analysis of alternatively spliced genes indicated enrichment for focal adhesion and non-specific cellular response terms. Lists of differentially expressed genes and exons in lymphoblasts, their normalized count values in brain samples, and motif analysis for the alternatively spliced exons are provided in the Supplementary Data.
RFC1 expression in patient tissues.
Quantitative reverse transcriptionPCR (qRT-PCR) was performed using two sets of primers(Fig. 6b); concordantly with RNA-seq data, it did not show any significant decrease of RFC1 mRNA (RefSeq NM_002913) level in patient fibroblasts (n = 5), lymphoblasts (n = 2), muscle (n = 7), frontal cortex, and cerebellar vermis (n = 1), as compared to healthy controls or FRDA cases (Fig. 6c). Exons 2 and 3 were correctly spliced in the mature RFC1 mRNA as shown by RNA-seq, qRT-PCR, and sequencing. However, assessment of pre-mRNA expression by qRT-PCR showed a consistent increase of intron 2 retention in patient lymphoblasts (n = 2), muscle (n = 7) (P = 0.0077), cerebellum, and frontal cortex (n = 1), as compared to healthy controls (Supplementary Fig. 6). The low level of RFC1 expression in fibroblasts prevented the assessment of pre-mRNA processing.
The western blot analysis showed that RFC1 protein (isoform 1, UniProt identifier P35251–1) was not decreased in patient fibroblasts (n = 5), lymphoblasts (n = 4), or brain (n = 1), as compared to healthy controls or FRDA cases (Fig. 6d and Supplementary Fig. 7). Assessment of RFC1 protein expression in muscle could not be performed because of limited tissue availability.
Since RFC1 plays a key role in DNA damage recognition and recruitment of DNA-repair enzymes, we assessed whether patient- derived fibroblasts have an impaired response to DNA damage. Patient-fibroblasts did not show an increased susceptibility to DNA damage; their treatment with double-stranded DNA-break-inducing agents, ultraviolet light, and methyl methanesulfonate triggered a grossly normal response to DNA damage (Supplementary Fig. 8).
Discussion
We identified a recessive repeat expansion in intron 2 of RFC1 as a cause of CANVAS and late-onset ataxia. Twenty-three cases from 11 families and 33 sporadic cases carried the biallelic AAGGG repeat expansion. Notably, out of 150 cases from a single center diagnosed with late-onset ataxia, 22% tested positive for the biallelic AAGGG repeat expansion; the percentage was higher if only patients with sensory neuronopathy and cerebellar involvement (62%), CANVAS disease (92%), and familial CANVAS disease (100%) were considered, highlighting that a higher diagnostic can be achieved in cases with well-defined clinical features and a positive family history. Not since the discovery two decades ago of the most common genes causing ataxia19–22 and Charcot-Marie-Tooth disease23–26 has a novel gene explained percentages above 10% of genetically undetermined cases27,28.
We determined that the allelic carrier frequency of the AAGGG repeat expansion in healthy controls was 0.7%, which is similar to the allelic carrier frequency of the GAA expansion in FXN, which ranges from 0.9 to 1.6% and, in the biallelic state, which causes the most common recessive ataxia, FRDA. Together, these data suggest that the recessive AAGGG expansion in RFC1 may represent a frequent cause of late-onset ataxia in the general population, with an estimated prevalence at birth of the recessive trait of approximately 1 in 20,000.
The expansion resides at the 3′ end of a deep intronic AluSx3 element, and it increases the size of the poly(A) tail from 11 to over 400 repeated units, but also alters its sequence. Of interest, expansions in terminal and mid A-stretches of Alu elements have been previously identified to cause FRDA19, spinocerebellar ataxia type 37 (SCA37; see Seixas et al.29), more recently benign adult familial myoclonic epilepsy (BAFME)30, and now CANVAS and late-onset ataxia. Together, these observations suggest that variations and expansion of these highly polymorphic regions of Alu elements represent a common mechanism underlying different inherited neurological disorders. Notably, both SCA37 and BAFME are characterized by expansion of a mutated repeated unit, ATTTC and TTTCA, respectively29,30. In this study, as well as in BAFME and SCA37, the presence in the normal population of large expansions of the reference repeated unit suggests that nucleotide change rather than expansion size may be the driving pathogenic mechanism.
Alu elements are repetitive elements about 300 base pairs (bp) long and are highly conserved within primate genomes. The 3′ end of an Alu element has a longer A-rich region that plays a critical role in its amplification mechanism31. Active elements degrade rapidly on an evolutionary timescale by poly(A) tail shortening or heterogeneous base interruptions accumulating in the poly(A) tail, such as G insertions. We hypothesize that the mutation of the AAGGG repeated unit occurred as part of the inactivation process by G interruption of the poly(A) tail of the retrotransposon AluSx3. Repetitive DNA motifs, particularly G-rich regions, can form secondary or tertiary nucleotide structures such as hairpins, parallel and antiparallel G-quadruplexes, and, if transcribed, DNA-RNA hybrids also known as R loops. These structures have been shown to increase the exposure of single-stranded DNA to damaging environmental agents; they can initiate repeat expansion and perpetrate genomic instability across meiotic and mitotic divisions or after DNA damage32.
Since the same ancestral haplotype is shared by the majority of familial and positive cases, as well as some healthy carriers of two (AAAGG)exp alleles, we speculate that the nucleotide change from AAAAG to AAAGG or AAGGG may represent an ancestral founder event, which was followed by the pathological expansion of the repeated unit, whose size seems to correlate positively with its guanine-cytosine content. However, the identification of two patients (Fam 5b-2 and s23) with a recessive AAGGG repeat expansion who share only one allele of the common haplotype implies that repeat expansions of the mutated AAGGG unit can also occur on a different genetic background. Interestingly, Fam 5b-2 was also found to carry the largest repeat expansion (10 kb or 2,000 repeats) in the cohort of patients tested.
In the majority of patients, the expansion encompassed 1,000 repeats; however, as few as 400 AAGGG repeats were shown to be sufficient to cause disease. The size of the expanded alleles was relatively stable in siblings within single families, but no parent of the affected patients was available to assess whether this also applied across generations. We did not observe a correlation between age at onset of the neuropathy and the size of the repeat expansion, although the disease course was very slowly progressive, and initial symptoms might have been neglected in some patients but reported by others.
So far, approximately 40 neurological or neuromuscular genetic disorders have been associated with nucleotide repeat expansions. Two of them are known to be inherited in a recessive mode, namely FRDA and myoclonic epilepsy type 1; both are associated with loss of function of the repeat-hosting gene33–35.
A remarkable aspect of the recessive expansion described in this study is that our data do not suggest a direct mechanism of loss of function for RFC1. We did not observe a reduced level of RFC1 expression at either the transcript or the protein level in CANVAS patients, although as a known loss-of-function control, we detected a significant reduction of FXN transcript in postmortem brain from patients with FRDA. Also, RNA-seq data did not show a clear effect on the expression of neighboring or distant genes. We cannot exclude that the repeat expansion may cause more subtle tissue-specific alterations of RFC1 transcript and protein or alter the structural organization of chromatin.
RFC1 encodes the large subunit of replication factor C, a five subunit DNA polymerase accessory protein. It loads proliferating cell nuclear antigen onto DNA and activates DNA polymerases 8 and e to promote the coordinated synthesis of both strands during replication or after DNA damage36–38. It is interesting to note that mutations in many of the genes involved in DNA repair have been already associated with degenerative neurological disorders39, including ataxia-telangiectasia, xeroderma pigmentosum, Cockayne syndrome, and ataxia with oculomotor apraxia types 1 and 2. Interestingly, ataxia and neuropathy are common clinical features to all of them, suggesting a particular susceptibility of the cerebellum and peripheral nerves to DNA damage. However, our preliminary study did not show an impaired response to DNA damage in patient-derived fibroblasts.
In fact, late-onset Mendelian disorders represent a unique interpretative challenge, since risk variants may exert subtle effects, rather than a clear loss of function of the mutated gene, that are compatible with normal development until adult or old age40. In this regard, although unusual in the context of a recessive mode of inheritance, other mechanisms, including the production of toxic RNA containing the expanded repeat, or the translation of a repeat-encoded polypeptide, should be considered41. We did not observe in patient brains the presence of RNA foci of either the sense or antisense repeated unit. However, we detected a consistent increase across different tissues of the retention of intron 2 in RFC1 pre-mRNA. Retention of the repeat-hosting intron was recently identified as a common event associated with other disease-causing guanine-cytosine-rich intronic expansions, such as in myotonic dystrophy type 2 and C9orf72-amyotrophic lateral sclerosis/FTD but not AT-rich repeat expansions such as in FRDA42. Intron retention and abnormal pre-mRNA processing bear potential effects on nuclear retention and nucleocytoplasmic transport of the pre-mRNA, which, if efficiently exported to the cytoplasm, would be accessible to the translationary machinery.
Notwithstanding the enormous progress in Mendelian gene identification during the last decade, up to 40% of patients with ataxia and inherited neuropathy remain genetically undiagnosed, and the percentage can rise up to 80–90% in particular subtypes, such as late-onset ataxia2,5,43 and hereditary sensory neuropathies27,28. Our study, together with other studies from recent years30,44–46, provides evidence that the combined use of WGS and classical genetic investigations, such as linkage analysis, can provide a powerful tool to unravel a part of the missing heritability hidden in non-coding regions of the human genome.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of code and data availability and associated accession codes are available at https://doi.org/10.1038/s41588–019-0372–4.
Methods
Patients.
For the initial linkage study, we enrolled 29 individuals (23 affected and 6 unaffected) from 11 families with a clinical diagnosis of CANVAS across four centers: the National Hospital for Neurology and Neurosurgery (London, UK); C. Mondino National Neurological Institute (Pavia, Italy); C. Besta Neurological Institute (Milan, Italy); and the Department of Neurology, School of Medicine at Ribeirão Preto (Brazil).
An additional 150 patients with sporadic CANVAS or late-onset ataxia (onset after 35 years of age) were identified from the neurogenetic database of the National Hospital for Neurology and Neurosurgery. For the experimental procedures, patient samples are generally referred to as CANVAS samples; no distinction between samples from patients with full-blown CANVAS or other more limited variants of late-onset ataxia is made. A skin biopsy was performed in five (Fam 1–3, Fam 2–2, Fam 5a-2, Fam 5b-2, and Fam 6b-1) genetically confirmed individuals and six age- and sex-matched controls. Fibroblast cultures were maintained according to standard procedures47. EBV-transformed lymphoblast cultures from four patients (Fam 6–1, Fam 8–2, Fam 8–3, and Fam 11–2) were generated and maintained. EBV-transformed lymphoblast cultures from three age- and sex-matched healthy controls were provided by the European Collection of Authenticated Cell Cultures.
Paraffin-embedded and snap-frozen cerebellum (vermis) and frontal cortex from postmortem brain from one sporadic CANVAS patient carrying the biallelic AAGGG repeat expansion (s16), three patients with genetically confirmed FRDA, one patient with genetically confirmed SCA17, one patient with genetically confirmed C9orf72--related FTD, and three neurologically healthy controls were obtained from the Queen Square Brain Bank for Neurological Disorders.
Eight nerve biopsies and ten muscle biopsies were obtained from patients carrying the homozygous AAGGG repeat expansion and healthy controls for pathological examination. Muscle biopsy tissue from seven patients and five controls was also used for qRT-PCR.
The study was approved by the UCL Institute of Neurology institutional review board, and all participants gave written informed consent to participate. The study complied with all relevant ethical regulations.
SNP genotyping and linkage analysis.
Genotype calls were generated by the UCL Genomics genotyping facility using Infinium Core Exome arrays (Illumina). Raw data were processed and quality-checked using the GenomeStudio software (Illumina). All individuals passed the 99% call rate threshold and were included in the subsequent analysis using the PLINK 1.9 software48. Uninformative markers or markers with missing genotypes > 10% were removed, and the resulting dataset was further pruned to remove markers in high linkage equilibrium. Finally, the dataset was thinned to include 1-cM-spaced markers covering all autosomes. In total, 3,476 markers were included. For fine-mapping analyses, all available informative markers were included.
Parametric linkage analysis was performed using Merlin49 assuming a highly penetrant recessive model of inheritance and disease allele frequency < 1:10,000 The Merlin software was also used to obtain the most likely haplotypes in the candidate region. All genotyped individuals were included for haplotype analysis.
The rs11096992 and rs2066790 SNPs were genotyped in sporadic CANVAS patients and unaffected individuals using PCR followed by Sanger sequencing. Primers sequences, concentrations, and PCR thermocycling conditions are provided in Supplementary Table 3.
WGS.
WGS was performed by deCODE genetics. Paired-end sequencing reads (100 bp) were generated using a HiSeq 4000 system (Illumina) and aligned to GRCh37 using the Burrows-Wheeler Aligner50. The mean coverage per sample was 35×. Variants were called according to the Genome Analysis Toolkit UnifiedGenotyper51 workflow and annotated with ANNOVAR52. Variants were prioritized based on segregation, minor allele frequency (<0.001 in the 1000 Genomes Project53, National Heart, Lung, and Blood Institute (NHLBI) GO Exome Sequencing Project (ESP) (Exome Variant Server, NHLBI GO ESP, (http://evs.gs.washington.edu/EVS/), or the Genome Aggregation Database54), evolutionary conservation, and in silico prediction of pathogenicity for coding variants. Copy number analysis was performed with LUMPY55 using its default parameters. The candidate region on chromosome 4 was also visually inspected for any copy number or structural variants using the Integrative Genomics Viewer (IGV)56.
RP-PCR.
RP-PCR was performed to provide qualitative assessment of the presence of an expanded AAGGG repeat as well as expansions of the reference AAAAG allele or AAAGG variant. The RP-PCR was designed so that the reverse primers bind at different points within the repeat expansion to produce multiple amplicons of incremental size; 25–27 nucleotides flanking the repeat were added to increase binding affinity of the reverse primer to the polymorphic (A/AA/-) 3′ end of the microsatellite and flanking region and give preferential amplification of the larger PCR product, thus allowing sizing of the expansion in some cases. Primer sequences, concentrations, and PCR thermocycling conditions are provided in Supplementary Table 3.
Reverse primers were used in equimolar concentrations. Fragment length analysis was performed on an ABI 3730xl Genetic Analyzer (Applied Biosystems), and data were analyzed with the GeneMapper software (v. 4.0, Applied Biosystems). Expansions with a characteristic ‘sawtooth’ pattern were identified and put forward for Southern blotting where sufficient DNA allowed this.
Southern blot.
Five micrograms of genomic DNA (gDNA) was digested for 3 h with EcoRI (10 U) before electrophoresis. DNA was transferred to a positively charged nylon membrane (Roche Applied Science) by capillary blotting and was cross-linked by exposure to ultraviolet light. Digoxygenin (DIG)-labeled probes were prepared by PCR amplification of a genomic fragment cloned into a pGEM-T Easy Vector (Promega) by using the PCR DIG Probe Synthesis Kit (Roche Applied Science). Primer pairs used for the cloning of the gDNA fragment, PCR amplification of the DIG-labeled probe, and the PCR conditions are shown in Supplementary Table 3. Filter hybridization was undertaken as recommended in the DIG Application Manual (Roche Diagnostics) except for the supplementation of DIG Easy Hyb buffer with 100 mg ml−1 denatured fragmented salmon sperm DNA. After pre-hybridization at 46 °C for 3 h, hybridization was allowed to proceed at 46 °C overnight. A total of 600 μl of PCR products containing the labeled oligonucleotide probe was used in 30 ml of hybridization solution. Membranes were washed initially in 23 standard sodium citrate (SSC) and 0.1% sodium dodecyl sulfate (SDS), while the oven was being ramped from 48 to 65 °C and then washed three times in fresh solution at 65 °C for 15 min. Detection of the hybridized probe DNA was carried out as recommended in the DIG Application Manual with CSPD ready-to-use (Roche Applied Science) as a chemiluminescent substrate. Signals were visualized on Fluorescent Detection Film (Roche Diagnostics) after 1 h. All samples were electrophoresed against the DIG-labeled DNA molecular weight markers II and III (Roche Diagnostics). The pentanucleotide repeat number was estimated after subtraction of the wild-type allele fragment size (5,037 bp). The sizes of the detected bands were recorded for each individual and the number of expanded repeated units was estimated using the following formula: repeated pentanucleotide unit = (size of the expanded band in bp — 5,000 bp)/5.
Neuropathological examination.
The formalin-fixed cerebellar tissue was embedded in paraffin wax; 5-μm-thick sections were cut for routine hematoxylin and eosin staining and immunohistochemistry. The sections were immunostained with anti-SQSTM1/p62 antibody (1:500; catalog no. ab56416, Abcam), anti-TDP-43/TARDBP antibody (1:500; catalog no. 2E2-D3, Novus Biologicals), anti-α-synuclein antibody (1:1,000; catalog no. 4D6, Abcam), anti-phospho-Tau (Ser202, Thr205) antibody (1:100; catalog no. AT-8, Innogenetics), and anti-βA4 (1:50; 6F/3D clone, Dako). Immunostaining, together with appropriate controls, was performed on a Ventana Discovery automated staining platform (Ventana Medical Systems) following the manufacturer’s guidelines, using biotinylated secondary antibodies and streptavidin-conjugated horseradish peroxidase (HRP) and 3,3’-diaminobenzidine as the chromogen. Assessment of neuronal density in the cerebellar cortex was performed semiquantitatively. Nerve and muscle biopsy specimens were obtained and analyzed according to standard procedures57,58. In brief, all nerve biopsies were examined after processing for paraffin histology (immunostaining for neurofilaments was performed with the SMI 31 antibody (1:5,000; Sternberger) and in resin blocks (semi-thin resin sections were stained with methylene blue azure-basic fuchsin). The muscle biopsies were examined with routine histochemical stains after freezing in isopentane cooled in liquid nitrogen.
qRT-PCR.
Total RNA was extracted from fibroblasts, lymphoblasts, and brain regions using 1 ml Qiazol (Qiagen) and 200 μl chloroform. The aqueous phase was loaded and purified on columns using the RNeasy Lipid Tissue Mini Kit (Qiagen) and treated with RNAse-free DNase I (Qiagen). Complementary DNA (cDNA) was synthesized using 500 ng total RNA for all samples, with a SuperScript III First Strand cDNA Synthesis Kit (Invitrogen) and an equimolar mixture of oligo (dT)18 and random hexamer primers. Real-time qRT-PCR was carried out using the Power SYBR Green Master Mix (Applied Biosystems) and measured with a QuantStudio 7 Flex Real-Time PCR System (Applied Biosystems). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the housekeeping gene to normalize across different samples. Amplified transcripts were quantified using the comparative CT method and presented as normalized fold expression change (2ΔΔCT). Oligonucleotide sequences and thermocycling conditions are provided in Supplementary Table 3.
Western blot.
Cells and tissues were lysed in radioimmunoprecipitation assay (RIPA) buffer supplemented with a complete EDTA-free protease inhibitor cocktail (Roche Diagnostics). Brain lysates were homogenized on ice using a tissue ruptor with disposable probes (Qiagen). Protein lysate concentrations were measured with a DC protein assay (Bio-Rad). After adding 5 μl of sample buffer (Bio-Rad) and 2 μl of NuPAGE reducing agent (Invitrogen) and boiling at 95 °C for 5 min, 15–30 μg of protein for each sample were separated on 4–12% SDS-polyacrylamide gel (Bio-Rad) in 2-(N-morpholino)ethanesulfonic acid buffer and transferred onto nitrocellulose membranes (GE Healthcare) using a Turbo Transfer Pack (Bio-Rad). After blocking in 5% milk, immunoblotting was performed by incubating overnight at 4 °C with the following primary antibodies: anti-RFC1 (1:1,000; catalog no. GTX129291, GeneTex); anti-β-actin (1:2,000; catalog no. A2228, Sigma-Aldrich). Secondary antibodies were as follows: IRDye 800CW goat anti-rabbit IgG (LI-COR catalog no. 926–32211); IRDye 680RD donkey anti-mouse IgG (LI-COR catalog no. 926–68072); and IgG (LI-COR). Signals of RFC1 bands were normalized to those of the corresponding β-actin bands as internal controls. Signals were digitally acquired with an Odyssey Fc infrared scanner (LI-COR) and quantified with the Image Studio v. 5.2 software (LI-COR Bioscience).
RNA-seq.
Reads were aligned to the hg38 human genome build using STAR (version 2.4.2a)59. BAM files were sorted and duplicate reads flagged using NovoSort (version 1.03.09; Novocraft). The aligned reads overlapping the human exons (Ensembl 82) were counted with HTSeq (version 0.1)60. For each gene and each sample, the fragments per kilobase of exon per million mapped reads (FPKM) was calculated. Any gene with a mean FPKM across all samples in a dataset < 1 was discarded from further analysis. Differential gene expression was assessed with DESeq2 (version 1.8.2)61 and differential splicing was assessed with DEXSeq62 running on R (version 3.3.2). The significance thresholds for differential expression and splicing were set at a Benjamini-Hochberg false discovery rate of 10%. Quality control reports were collated with MultiQC63. Gene ontology enrichment testing was done using g:Profiler64 with both the Gene Ontology and Kyoto Encyclopedia of Genes and Genomes ontologies, with a minimum term size of five genes and all P values Bonferroni-corrected for multiple testing. Motif analysis was conducted on 49 alternatively spliced exons in lymphoblasts identified by unambiguous sequences with known strands using RBPmap65. The prediction of non-coding RNA sequences in intron 2 of RFC1 was tested with Rfam66.
Statistical analyses.
Clinical variables were compared between familial and sporadic cases with a two-tailed Student’s t-test (continuous variables) and chi-squared test2 (categorical variables). The correlation between repeat expansion size and age at onset of neuropathy was calculated using the Pearson’s correlation coefficient. The FPKM of FXN and RFC1 was compared using a two-tailed Student’s t-test. The relative expression of the RFC1 transcript 1 versus GAPDH as measured by qRT-PCR was compared with a two-tailed Student’s t-test. The statistical analysis of the results of the western blot was performed with a two-tailed Student’s t-test after confirmation of equality of variances. P < 0.05 was considered significant.
Cloning of the RFC1 repeat expansion locus.
The RFC1 locus containing the AAGGG repeat expansion was amplified with long-range PCR from the gDNA of a CANVAS patient carrying the biallelic AAGGG repeat expansion and a healthy control carrying two (AAAAG)11 alleles. PCR products were cloned into the pcDNA3.1/TOPO vector (Invitrogen), according to the manufacturer’s instructions. Primers and thermocycling conditions are provided in Supplementary Table 3. The size of the insert was determined by digestion with BstXI (New England Biolabs). The integrity of repeats and their orientation were confirmed by DNA sequencing (Eurofins Genomics), which revealed uninterrupted 94 × (CCCTT) and 54 × (AAGGG) repeats in mutant clones, as well as 11 × (CTTTT) and 11 × (AAAAG) repeat sequences in the wild-type clone. Once confirmed, the four clones used for the experimental procedures were amplified using a Maxi-prep plasmid purification system (Qiagen).
RNA in situ hybridization.
Paraffin-embedded, formalin-fixed postmortem vermis sections from a CANVAS case, two healthy, and two cerebellar degeneration age-matched controls were deparaffinized in xylene twice for 10 min, then rehydrated in 100, 90, and 70% ethanol, then in PBS. Approximately 105 SH-SY5Y cells were seeded on coverslips in 24-well plates and transfected using Lipofectamine 3000 (Thermo Fisher Scientific) with plasmids expressing wild-type sense (TTTTC)11, wild-type antisense (AAAAG)11, mutant sense (TTCCQ94, or mutant antisense (AAGGG)54 repeat sequences and were analyzed after 24 h. Cells were fixed in 4% methanol-free paraformaldehyde (Pierce) for 10 min at room temperature, dehydrated in a graded series of alcohols, air-dried and rehydrated in PBS, permeabilized for 10 min in 0.1% Triton X-100 in PBS, briefly washed in 2× SSC, and incubated for 30 min in pre-hybridization solution (40% formamide, 2× SSC, 1 mg ml−1 transfer RNA (tRNA), 1 mg ml−1 salmon sperm DNA, 0.2% BSA, 10% dextran sulfate, and 2 mM ribonucleoside vanadyl complex) at 57 °C. Hybridization solution (40% formamide, 2× SSC, 1 mg ml−1 tRNA, 1 mg ml−1 salmon sperm DNA, 0.2% BSA, 10% dextran sulfate, 2 mM ribonucleoside vanadyl complex, 0.2 ng μl−1 (AAGGG)5 or (CCCTT)5 locked nucleic acid probe, 5’-TYE563-labeled (Exiqon)) was heated at 95 °C for 10 min before incubation with sections for 1 h at 57 °C. Cells were washed for 30 min at 57 °C with high-stringency buffer (2× SSC, 0.2% Triton X-100, 40% formamide) and then for 20 min each, in 0.2× SSC buffer. Nuclei were stained with 4,6-diamidino-2-phenylindole. Coverslips were then dehydrated in 70 then 100% ethanol and mounted onto slides in VECTASHIELD Antifade Mounting Medium (Vector Laboratories). Images were acquired using an LSM 710 confocal microscope (ZEISS) using a planapochromat ×63 oil immersion objective.
Response to DNA damage.
Fibroblasts were grown in 10-cm dishes in Dulbecco’s Modified Eagle’s medium supplemented with 10% fetal bovine serum. Asynchronous cell cultures were grown to approximately 80% confluency and treated with ultraviolet light or methyl methanesulfonate, or left untreated. For ultraviolet light irradiation, cells were washed with PBS and exposed to 30 or 120 J m−2 ultraviolet light (254 nm) using a Stratalinker UV crosslinker (Stratagene). For genotoxin treatment, methyl methanesulfonate (Sigma-Aldrich) was added to the culture medium to give a final concentration of 1 mM, and cells were exposed for 8 h. After ultraviolet irradiation or genotoxin treatment, cells were allowed to recover for 24 h before analysis.
Cells were homogenized in RIPA buffer containing 50 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, and protease inhibitor. Samples were sonicated and centrifuged before protein levels were quantified using a BCA assay (Thermo Fisher Scientific). For the western blot analysis, protein (5 μg) was size-separated using SDS-PAGE, transferred to nitrocellulose membranes, and subjected to standard immunoblotting procedures using antibodies to the following: γH2AX (1:1,000; ab11174, Abcam) and β-actin (1:1,000; A1978, Sigma-Aldrich). γH2AX has been extensively used as a marker for double-strand DNA breaks67,68. It is one of the initial markers of double-strand DNA breaking common to all DNA-repair pathways. Secondary HRP-conjugated antibodies were purchased from Proteintech and used at a 1:2,000 concentration. Antibody staining was detected by enhanced chemiluminescence (Pierce ECL Western Blotting Substrate; Thermo Fisher Scientific) and visualized by X-ray film.
Cell viability was assessed with a CellTiter-Glo Luminescent Cell Viability Assay (Promega) according to the manufacturer’s protocol. To assess cell viability, 20,000 cells per well were seeded in 96-well plates before treatment and were treated as described previously.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
A.C. is funded by the Inherited Neuropathies Consortium (INC), which is part of the National Institutes of Health Rare Diseases Clinical Research Network (RDCRN) (grant no. U54NS065712) and the Wellcome Trust (grant no. 204841/Z/16/Z). A.M.R. is funded by a Wellcome Trust Postdoctoral Fellowship for Clinicians (no. 110043/Z/15/Z). H.H. is supported by the Rosetree Trust, Ataxia UK, MSA Trust, Brain Research UK, Muscular Dystrophy UK, Muscular Dystrophy Association, Higher Education Commission of Pakistan and Wellcome Trust (Synaptopathies Strategic Award, 165908). M.M.R. is grateful to the Medical Research Council (MRC), MRC Centre grant (G0601943) and to the National Institutes of Neurological Diseases and Stroke and Office of Rare Diseases (U54NS065712) for their support. The INC (grant no. U54NS065712) is part of the National Center for Advancing Translational Sciences (NCATS) RDCRN. The RDCRN is an initiative of the NCATS Office of Rare Diseases Research and is funded through a collaboration between NCATS and the National Institute of Neurological Disorders and Stroke. S.Z. thanks the National Institutes of Health (NIH; grant no. 4R01NS075764) for its support. This research was also supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre (176718). Neuromuscular and brain tissue samples were obtained from University College London Hospitals NHS Foundation Trust as part of the UK Brain Archive Information Network (BRAIN UK), which is funded by the Medical Research Council and Brain Tumour Research and the NIH-funded NeuroBioBank. We also thank F. Launchbury from the UCL IQPath laboratory for her technical assistance in histology slide preparation. We would also like to thank Muscular Dystrophy UK and Muscular Dystrophy Association USA (award 171011). P.F. is funded by an MRC/MNDA CSF (MR/M008606/1).
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41588–019-0372–4.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Data availability
The genotyping microarray and sequence data obtained by WGS and RNA-seq are available upon request from the corresponding authors (A.C., H.H.). They are not publicly available because some of the study participants did not give their full consent to release data publicly. Since WGS data are protected by the Personal Information Protection Law, availability of these data is under the regulation of the institutional review board. The data obtained from RNA-seq have been deposited on the NCBI Sequence Read Archive under accession no. SRP186868.
Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Harding AE “Idiopathic” late onset cerebellar ataxia. A clinical and genetic study of 36 cases. J. Neurol. Sci. 51, 259–271 (1981). [DOI] [PubMed] [Google Scholar]
- 2.Muzaimi MB et al. Population based study of late onset cerebellar ataxia in south east Wales. J. Neurol. Neurosurg. Psychiatry 75, 1129–1134 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sghirlanzoni A, Pareyson D & Lauria G Sensory neuron diseases. Lancet Neurol. 4, 349–361 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Strupp M, Feil K, Dieterich M & Brandt T Bilateral vestibulopathy. Handb. Clin. Neurol. 137, 235–240 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Abele M et al. The aetiology of sporadic adult-onset ataxia. Brain 125(Pt. 5), 961–968 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Kirchner H et al. Clinical, electrophysiological, and MRI findings in patients with cerebellar ataxia and a bilaterally pathological head-impulse test. Ann. N. Y. Acad. Sci. 1233, 127–138 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Migliaccio AA, Halmagyi GM, McGarvie LA & Cremer PD Cerebellar ataxia with bilateral vestibulopathy: description of a syndrome and its characteristic clinical sign. Brain 127(Pt. 2), 280–293 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Szmulewicz DJ et al. Proposed diagnostic criteria for cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS). Neurol. Clin. Pract. 6, 61–68 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu TY et al. Autonomic dysfunction is a major feature of cerebellar ataxia, neuropathy, vestibular areflexia ‘CANVAS’ syndrome. Brain 137(Pt. 10), 2649–2656 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Szmulewicz DJ, Merchant SN & Halmagyi GM Cerebellar ataxia with neuropathy and bilateral vestibular areflexia syndrome: a histopathologic case report. Otol. Neurotol. 32, e63–e65 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szmulewicz DJ et al. Dorsal root ganglionopathy is responsible for the sensory impairment in CANVAS. Neurology 82, 1410–1415 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cazzato D, Bella ED, Dacci P, Mariotti C & Lauria G Cerebellar ataxia, neuropathy, and vestibular areflexia syndrome: a slowly progressive disorder with stereotypical presentation. J. Neurol. 263, 245–249 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Rust H et al. VEMPs in a patient with cerebellar ataxia, neuropathy and vestibular areflexia (CANVAS). J. Neurol. Sci. 378, 9–11 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Pelosi L et al. Peripheral nerve ultrasound in cerebellar ataxia neuropathy vestibular areflexia syndrome (CANVAS). Muscle Nerve. 56, 160–162 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Pelosi L et al. Peripheral nerves are pathologically small in cerebellar ataxia neuropathy vestibular areflexia syndrome: a controlled ultrasound study. Eur. J. Neurol. 25, 659–665 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Taki M et al. Cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS). Auris Nasus Larynx 45, 866–870 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Infante J et al. Cerebellar ataxia, neuropathy, vestibular areflexia syndrome (CANVAS) with chronic cough and preserved muscle stretch reflexes: evidence for selective sparing of afferent Ia fibres. J. Neurol. 265, 1454–1462 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Hommelsheim CM, Frantzeskakis L, Huang M & Ülker B PCR amplification of repetitive DNA: a limitation to genome editing technologies and many other applications. Sci. Rep. 4, 5052 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Campuzano V et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271, 1423–1427 (1996). [DOI] [PubMed] [Google Scholar]
- 20.Orr HT et al. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat. Genet. 4, 221–226 (1993). [DOI] [PubMed] [Google Scholar]
- 21.Pulst SM et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat. Genet. 14, 269–276 (1996). [DOI] [PubMed] [Google Scholar]
- 22.Kawaguchi Y et al. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet. 8, 221–228 (1994). [DOI] [PubMed] [Google Scholar]
- 23.Lupski JR et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 66, 219–232 (1991). [DOI] [PubMed] [Google Scholar]
- 24.Hayasaka K et al. Charcot-Marie-Tooth neuropathy type 1B is associated with mutations of the myelin P0 gene. Nat. Genet. 5, 31–34 (1993). [DOI] [PubMed] [Google Scholar]
- 25.Bergoffen J et al. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science 262, 2039–2042 (1993). [DOI] [PubMed] [Google Scholar]
- 26.Züchner S et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 36, 449–451 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Fridman V et al. CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis. J. Neurol. Neurosurg. Psychiatry 86, 873–878 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murphy SM et al. Charcot-Marie-Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J. Neurol. Neurosurg. Psychiatry 83, 706–710 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seixas AI et al. A pentanucleotide ATTTC repeat insertion in the non-coding region of DAB1, mapping to SCA37, causes spinocerebellar ataxia. Am. J. Hum. Genet. 101, 87–103 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishiura H et al. Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat. Genet. 50, 581–590 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Deininger P Alu elements: know the SINEs. Genome Biol. 12, 236 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haeusler AR, Donnelly CJ & Rothstein JD The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat. Rev. Neurosci. 17, 383–395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dürr A et al. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N. Engl. J. Med. 335, 1169–1175 (1996). [DOI] [PubMed] [Google Scholar]
- 34.Lazaropoulos M et al. Frataxin levels in peripheral tissue in Friedreich ataxia. Ann. Clin. Transl. Neurol. 2, 831–842 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paulson H Repeat expansion diseases. Handb. Clin. Neurol. 147, 105–123 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Majka J & Burgers PMJ The PCNA-RFC families of DNA clamps and clamp loaders. Prog. Nucleic Acid Res. Mol. Biol. 78, 227–260 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Tomida J et al. DNA damage-induced ubiquitylation of RFC2 subunit of replication factor C complex. J. Biol. Chem. 283, 9071–9079 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Overmeer RM et al. Replication factor C recruits DNA polymerase delta to sites of nucleotide excision repair but is not required for PCNA recruitment. Mol. Cell. Biol. 30, 4828–4839 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKinnon PJ Maintaining genome stability in the nervous system. Nat. Neurosci. 16, 1523–1529 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Higuchi Y et al. Mutations in MME cause an autosomal-recessive Charcot-Marie-Tooth disease type 2. Ann. Neurol. 79, 659–672 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.La Spada AR & Taylor JP Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat. Rev. Genet. 11, 247–258 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sznajder ŁJ et al. Intron retention induced by microsatellite expansions as a disease biomarker. Proc. Natl Acad. Sci. USA 115, 4234–4239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gebus O et al. Deciphering the causes of sporadic late-onset cerebellar ataxias: a prospective study with implications for diagnostic work. J. Neurol. 264, 1118–1126 (2017). [DOI] [PubMed] [Google Scholar]
- 44.DeJesus-Hernandez M et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Renton AE et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aneichyk T et al. Dissecting the causal mechanism of X-linked dystonia-parkinsonism by integrating genome and transcriptome assembly. Cell 172, 897–909.e21 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manole A et al. Clinical, pathological and functional characterization of riboflavin-responsive neuropathy. Brain 140, 2820–2837 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Purcell S et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abecasis GR, Cherny SS, Cookson WO & Cardon LR Merlin: rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 30, 97–101 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Li H & Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McKenna A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang K, Li M & Hakonarson H ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Auton A et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Layer RM, Chiang C, Quinlan AR & Hall IM LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 15, R84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robinson JT et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weis J, Brandner S, Lammens M, Sommer C & Vallat J-M Processing of nerve biopsies: a practical guide for neuropathologists. Clin. Neuropathol. 31, 7–23 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dubowitz V, Sewry CA & Oldfors A Muscle Biopsy: a Practical Approach (Elsevier, 2013). [Google Scholar]
- 59.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Anders S, Pyl PT & Huber W HTSeq: a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Anders S, Reyes A & Huber W Detecting differential usage of exons from RNA-seq data. Genome Res. 22, 2008–2017 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ewels P, Magnusson M, Lundin S & Käller M MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reimand J et al. g:Profiler: a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 44, W83–W89 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paz I, Kosti I, Ares M Jr., Cline M & Mandel-Gutfreund Y RBPmap: a web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 42, W361–W367 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Griffiths-Jones S, Bateman A, Marshall M, Khanna A & Eddy SR Rfam: an RNA family database. Nucleic Acids Res. 31, 439–441 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Podhorecka M, Skladanowski A & Bozko P H2AX phosphorylation: its role in DNA damage response and cancer therapy. J. Nucleic Acids 2010, 920161 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sharma A, Singh K & Almasan A Histone H2AX phosphorylation: a marker for DNA damage. Methods Mol. Biol. 920, 613–626 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.