

Abstract
The gut microbiota influences both local and systemic inflammation. Inflammation contributes to development, progression, and treatment of cancer, but it remains unclear whether commensal bacteria affect inflammation in the sterile tumor microenvironment. Here, we show that disruption of the microbiota impairs the response of subcutaneous tumors to CpG-oligonucleotide immunotherapy and platinum chemotherapy. In antibiotics-treated or germ-free mice, tumor-infiltrating myeloid-derived cells responded poorly to therapy, resulting in lower cytokine production and tumor necrosis after CpG-oligonucleotide treatment and deficient production of reactive oxygen species and cytotoxicity after chemotherapy. Thus, optimal responses to cancer therapy require an intact commensal microbiota that mediates its effects by modulating myeloid-derived cell functions in the tumor microenvironment. These findings underscore the importance of the microbiota in the outcome of disease treatment.
The commensal microbiota influences metabolism, tissue development, inflammation, and immunity (1). The gut microbiota affects inflammation and immunity not only locally at the mucosal level but also systemically (2–5), raising the question of whether the microbiota affects inflammatory processes that contribute to cancer and its therapy. Tumor-associated inflammatory cells promote cancer cell proliferation and suppress antitumor immunity (6). Anticancer treatments alter this microenvironment and may evoke tumor-destructive immune responses (7–9), but whether commensal organisms affect this process is unexplored.
To investigate whether commensal bacteria alter inflammation in the tumor microenvironment, mice received an antibiotic cocktail (ABX) of vancomycin, imipenem, and neomycin in drinking water starting 3 weeks before tumor inoculation and continuing throughout the experiment. To analyze tumors of comparable stages of progression in both wild-type (WT) and genetically altered mice, three C57Bl/6 mouse transplantable tumors were selected based on susceptibility to therapy: EL4 lymphoma, MC38 colon carcinoma, and B16 melanoma. Tumor cells were injected subcutaneously, and mice were analyzed or treated when the tumors reached a diameter of ≥4 mm. Gene expression analysis of pretherapy MC38 and EL4 tumors revealed down-regulation of genes related to inflammation, phagocytosis, antigen presentation, and adaptive immune responses in ABX-treated mice but up-regulation of genes related to tissue development, cancer, and metabolism (fig. S1, A and B). ABX lowered the frequency of monocyte-derived Ly6C+ MHC class II+ cells in EL4 and MC38 tumors and the spleen of MC38 tumor-bearing mice. ABX also decreased Ly6Ghigh neutrophil frequency in MC38 tumors (fig. S2).
To determine whether ABX affect tumor immunotherapy, MC38 and B16 tumor-bearing mice were treated with a combination of intratumoral CpG-oligodeoxynucleotides (ODN), a ligand of Toll-like receptor 9 (TLR9), and inhibitory interleukin-10 (IL-10) receptor antibodies (anti-IL-10R) (10). This immunotherapy retards tumor growth and prolongs survival by rapidly inducing hemorrhagic necrosis dependent on tumor necrosis factor (TNF) production by tumor-associated myeloid cells followed by a CD8 T cell response required for tumor eradication (11). ABX impaired therapy efficacy to retard tumor growth and lengthen survival (Fig. 1, A and B, and fig. S3A). Anti-IL-10R/CpG-ODN treatment induced MC38 tumor necrosis in control mice that was greatly reduced both macroscopically and histologically in ABX-treated animals (Fig. 1, C and D). Similar observations in immunodeficient Ragl−/− mice demonstrated that ABX regulate the response to anti-IL-10R/CpG-ODN independently of adaptive immunity (Fig. 1D). However, in Tnf−/− mice, anti-IL-10R/CpG-ODN induced minimal necrosis and had no effect on tumor growth in either control or ABX-treated mice (Fig. 1, C and D, and fig. S3B).
Fig. 1. Oral administration of anti biotics impai rs CpG-ODN-based immunotherapy.
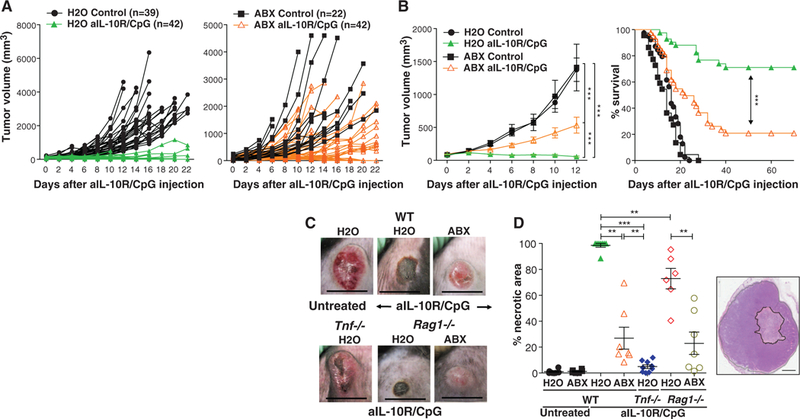
(A) MC38 tumor growth kinetics in H2O-drinking (left) or ABX-treated animals (right) treated once with intratumoral injection of CpG-ODN in combination with anti-IL-10R antibody injected intraperitoneally a day earlier (anti-IL10R/CpG-ODN) or left untreated (control). Data show individual mice combined from multiple independent experiments. (B) Combined tumor volume data from the mice shown in (A) (left; means ± SEM) and corresponding survival data (right). (C) Macroscopic appearance of MC38 tumors in WT, Tnf−/−, and Rag1−/− mice, drinking H2O or ABX, 72 hours after anti-IL10R/CpG-ODN treatment. Scale bars, 1 cm. (D) Quantification of necrotic area in tumors from WT, Tnf−/−, and Ragl−/− mice 48 hours after anti-IL10R/CpG-ODN treatment analyzed in hematoxylin and eosin stained sections. Data are means±SEM from two experiments combined. The right inset shows an an example of the histological appearance, with the area of necrosis outlined in black. Scale bar, 200 μm. *p < 0.05, **p < 0.01, and ***p < 0.001.
ABX significantly impaired CpG-ODN–induced Tnfexpression and frequencies of TNF-positive leukocytes in MC38 tumors at 3 hours (Fig. 2, A and B). ABX decreased the frequencies of TNF-producing cells and the amount of cytokine per cell in Ly6C+MHC-IT monocytes, Ly6C-F4/80hi macrophages, CD11chiMHC-IIhi dendritic cells (DC), and Ly6C+F4/80intMHC-II+ monocyte-derived cells (fig. S4, A to C). ABX also decreased CD86 expression and IL-12B production in tumor-associated DC (fig. S4, D and E). After anti-IL-10R/CpG-ODN, tumors from ABX-treated mice also displayed diminished expression of pro-inflammatory Il1a, Il1b, Il12b, and Cxcl10, but not of anti-inflammatory Il10, Il1rn, and Socs3 or Nos2 genes (fig. S5). To determine whether the ABX effect was dependent on decreased bacteria load, germ-free (GF) or specific pathogen-free (SPF) mice bearing MC38 tumors received anti-IL-10R/CpG-ODN treatment. Tumors of GF as compared to SPF control mice produced significantly lower amounts of TNF and IL-12B (Fig. 2, C to E). Thus, commensal microbiota primes tumor-associated innate myeloid cells for inflammatory cytokine production in response to anti-IL-10R/CpG-ODN, and ABX or the GF status of the mice attenuates this response and the TNF-dependent early tumor necrosis.
Fig. 2. Commensal bacteria and TLR4 are necessary for optimal TNF production by tumor myeloid cells after anti-IL-10R/CpG treatment.

(A) Tnf mRNA (Nanostring nCounter) in MC38 tumors from H2O− or ABX-treatedmice exposed or not to anti-IL-10R/CpG-ODN therapy. (B) TNF-producing MC38 tumor infiltrating CD45+ cells in H2O− or ABX-treated mice. (C to E) Tnf mRNA [real-time polymerase chain reaction (RT-PCR)] (C) and percentage TNF− (D) or IL-12p40− (E) producing CD45+ cells (flow cytometry) in MC38 tumors from SPF or GF mice. (F) MC38 tumor Tnf mRNA in WT mice or in Tlr4−/− mice orally gavaged or not with LPS (25 mg per kg of weight, three times per week, 2 weeks before and 1 week after tumor injection). (G) MC38 tumor growth in H2O− or ABX-treated WT mice or Tlr4−/− mice treated or not with anti-IL-10R/CpG-ODN. Data show individual mice [(A) to (F)] or means T SEM (G). *P < 0.05, **P < 0.01, and ***P < 0.001.
Gavage administration thrice weekly of bacterial lipopolysaccharide (LPS) largely restored Tnf expression and frequency of TNF-producing leukocytes in tumors of ABX-treated WT mice but not Tlr4−/−mice [lacking the Toll-like receptor 4 (TLR4) receptor for LPS] (Fig. 2F and fig. S6A). Conversely, Tlr4 deficiency led to lower induction of Tnf and reduced tumor regression after immunotherapy (Fig. 2, F and G), whereas Tlr2 deficiency had no effect (fig. S6B). Overall, these findings suggest that bacterial products, in part through TLR4 activation, directly or indirectly prime tumor-associated myeloid cells for TLR9-dependent response to CpG-ODN. The ability of oral ABX treatment to reduce gastrointestinal but not skin bacterial load, combined with an absence of bacterial DNA in tumors and the restorative effect of gastrointestinal LPS administration, suggest that intestinal bacteria regulate the inflammatory response in the tumor microenvironment.
To determine how individual bacterial species affect the antitumor response, we correlated fecal microbiota composition with TNF production. Principal-component analysis of natural variation in microbiota composition versus TNF production suggested a codependence (fig. S7). Two experimental microbiota perturbations were used: (i) three-week ABX treatment followed by a 1-to 4-week recovery, and (ii) three-week singleantibiotic treatments. Bacterial abundance was restored to pretreatment levels 1 week after ABX cessation (Fig. 3A); bacterial diversity (fig. S8) and composition gradually increased, although neither reached the pre-ABX condition (Fig. 3, B and C, and figs. S9 and S10). Tnf expression did not recover until week 4 post-ABX (Fig. 3D). All the single antibiotics tested increased variability in Tnf expression, whereas only imipenem consistently reduced total bacterial abundance (fig. S11, A to D, and fig. S12). The different perturbations resulted in different bacteria being present. Thus, partially overlapping genera were identified that positively or negatively correlated with Tnf expression (figs. S13 and S14A). Among the positive correlates were Gram-negative genera [e.g., Alistipes (figs. S13 and S14A)], consistent with a role for TLR4 in priming TNF production. However, Gram-positive genera also showed positive correlation, suggesting that recognition pathways other than TLR4 were also involved. Identification of the same genera in different data sets increased the confidence in the validity of the correlation. Particularly, one genus (Lactobacillus) negatively and one (Ruminococcus) positively correlated with TNF expression in all three analyses (fig. S13). The Lactobacillus genus included L. murinum, L. intestinalis, and L. fermentum, the latter previously identified to have anti-inflammatory effects (12, 13). The Ruminococcus genus was represented by a single unclassified operational taxonomic unit (OTU) and the Alistipes genus by the single A. shahii species. Lactobacillus species recovered quickly after ABX withdrawal, whereas recovery of Ruminococcus and Alistipes species was delayed and coincided with restoration of the TNF response (fig. S14B). Administration of cultured A. shahii by gavage to mice preexposed to ABX reconstituted the ability of tumor-associated myeloid cells to produce TNF (Fig. 3E and fig. S15A). Conversely, administration of L. fermentum to intact mice attenuated the response (fig. S15B). These association experiments validate the results of the correlation analysis and confirm the ability of these species to influence the tumor response to CpG-ODN.
Fig. 3. Commensal microbiota composition correlates with tumor TNF response.
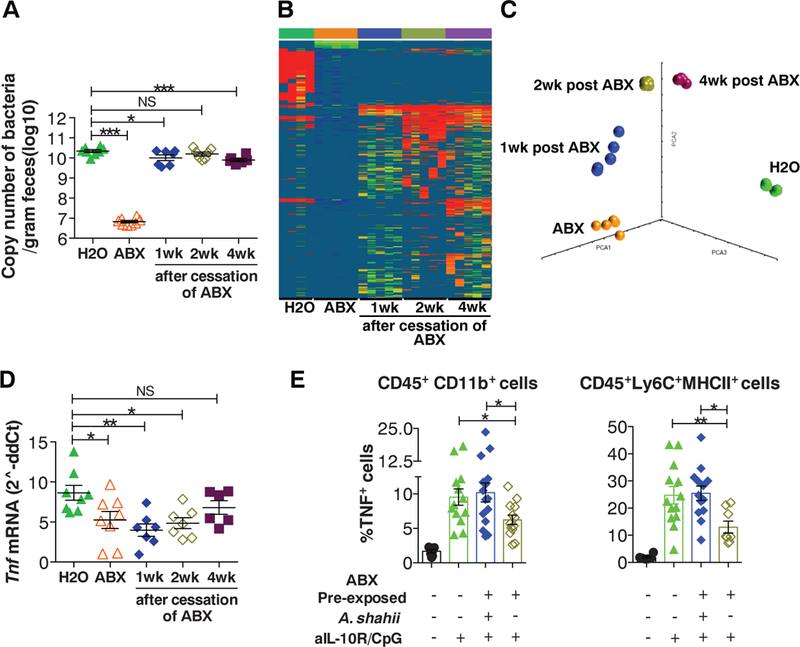
(A and B) MC38 tumors were subcutaneously implanted 1, 2, or 4 weeks after the cessation of a 3-week ABX treatment. Control mi ce continuously drinking H2O or ABX were included. (A) Eubacteria 16S ribosomal RNA gene copy number in feces was determined by RT-PCR. (B) 16S-sequence frequencies were analyzed by pyrosequencing in fecal microbi ota. Data are shown as heat map of OTUs (97% similarity) normalized to copy number of 16S per gram of feces (OTUs represented by <0.1% of total reads were removed from the analysi s). (C) Principal-component analysis of unweighted Unifrac di stances. (D) Tumor Tnf mRNA expression 3 hours after anti-IL-10R/CpG-ODN treatment was determined by RT-PCR. (E) Control H2O-drinking mice or 1 week after cessation of ABX treatment were exposed to anti-IL-10R/CpG-ODN therapy. A group of ABX pre-exposed mice was subjected to oral gavage with A. shahii. Mice were killed 3 hours after CpG-ODN treatment, and intracellular TNF was measured in the indicated tumor-associated myeloid cell subsets. Data show individual mice and means ± SEM from one representative experiment out of two performed [(A) and (D)] or combined data from two experiments (E).
To determine whether the microbiota also affects cancer treatments not considered to be directly dependent on inflammation processes, we next examined the response to agents used in human cancer chemotherapy. The platinum compounds oxaliplatin and cisplatin cause tumor cytotoxicity by forming platinum DNA adducts and intrastrand cross-links (14). Reactive oxygen species (ROS) are important for DNA damage and apoptosis in response to platinum compounds (15). Oxaliplatin, unlike cisplatin, also induces immunogenic cell death that drives antitumor T cell immunity (9,16). Oxaliplatin eradicated most subcutaneous EL4 tumors and prolonged survival in control mice, but ABX-treated animals displayed significantly reduced tumor regression and survival (Fig. 4A and fig. S16A). GF animals similarly failed to respond to oxaliplatin treatment (fig. S16B). The therapeutic effects of oxaliplatin on a different tumor (MC38) and of cisplatin on EL4 tumors were also significantly reduced in ABX-treated mice (fig. S16, C and D).
Fig. 4. Commensal bacteria control oxaliplatin therapy response by modulating ROS production.

(A) Subcutaneous EL4 tumor-bearing H2O− or ABX-treated mice were treated with oxaliplatin (10 mg per kg of weight); tumor growth (top) and survival (bottom) are shown. (B) Global gene expression (q < 0.1, >2-fold change compared to time 0, two-way analysis of variance analysis) in tumors from H2O− and ABX-treated mice before (0 hours) and after (6 and 18 hours) oxaliplatin treatment. (C and D) ROS production 24 hours after oxaliplatin injection in subcutaneous EL4 tumors from H2O− or ABX-treated WT mice and Cybb−/− mice was measured in vivo by bioluminescence (probe L-012) [inset in (C) shows a representative mouse with tumor area marked in red] and ex vivo in tumor-infiltrating cells (D, fluorescent probe, flow cytometry). (E and F) EL4-bearing WT and Cybb−/− mice (E) and Gr-1 antibody− or immunoglobulin G isotype-treated mice (F) were treated with oxaliplatin. (A), (C), and (E) show data combined from several independent experiments; (D) and (F) show a representative experiment out of two.
The effect of ABX was observed as early as day 2 after treatment with oxaliplatin (Fig. 4A), suggesting suppression of early cytotoxicity rather than of inflammation/immunity after immunogenic cell death. Indeed, ABX attenuated most gene expression changes observed in EL4 tumors 18 hours after oxaliplatin treatment (Fig. 4B). The genes inhibited by ABX were related to monocyte differentiation, activation, and function, whereas the genes that were increased in ABX-treated mice were related to cellular functions such as metabolism, transcription, translation, and DNA replication (fig. S17). Gene expression analysis using the Nanostring nCounter technology confirmed the expression changes of selected genes fig. S18).
Gene expression analysis revealed that after oxaliplatin treatment the induction of pro-inflammatory genes was decreased in the absence of microbiota, suggesting that inflammation was important for the antitumor effect of the drug. However, unlike anti-IL-10R/CpG-ODN, oxaliplatin antitumor effects were independent of TNF (fig. S19A). The observation that ABX treatment attenuated the induction of Nox1 and Cybb encoding ROS-generating NADPH (reduced nicotinamide adenine dinucleotide phosphate) oxidase 2 (NOX2) and of the ROS-responsive Nos2, Sod1, and Sod2 after oxaliplatin led us to examine oxaliplatin-induced ROS production. Oxaliplatin induced ROS in tumors from control but not ABX-treated mice, as detected in vivo by bioluminescence (Fig. 4C). Induction of ROS was dependent on NOX2 encoded by the Cybb gene (Fig. 4C). Because NOX2 is expressed in phagocytic cells (17), we examined ROS production in tumor-infiltrating hematopoietic cells ex vivo. The fluorescence intensity of the ROS probe in tumor-infiltrating CD11b+Gr-1hi neutrophils and in F4/80+ Gr-1int macrophage-like cells increased after oxaliplatin treatment in control mice, but this response was impaired in ABX-treated or Cybb−/− animals (Fig. 4D). ROS inhibition by the antioxidant N-acetyl cysteine (NAC) was previously shown to block oxaliplatin-induced apoptosis of cancer cell lines in vitro and inhibit the effectiveness of oxaliplatin therapy of mouse tumor cell lines in vivo (18,19). We found that inhibition of ROS by either Cybb deficiency or NAC impaired oxaliplatin-induced EL4 tumor regression as early as day 2 and decreased overall survival at an extent comparable to that of ABX (Fig. 4E and fig. S19B). Depletion of myeloid cells by Gr-1 antibody impaired the ability of oxaliplatin to induce tumor regression and to increase survival (Fig. 4F and fig. S19C). These data suggest that the reduced effect of oxaliplatin in ABX-treated or GF mice is partially due to reduced myeloid-cell ROS production. To investigate whether gut commensals prime these cells by release of TLR agonists, we studied Tlr4−/− and Myd88−/− mice. (Myd88 is the adaptor protein that signals downstream of IL-1 family receptors and most TLRs.) Myd88, but not Tlr4, deficiency impaired the early antitumor effect of oxaliplatin, although Tlr4 deficiency had a partial effect on long-term tumor growth and survival (fig. S19D). Il1r or Il18r deficiency did not impair the response (fig. S19D), suggesting that the commensal effect was related to microbial product sensing.
The levels of tumor DNA-bound platinum were similar in control and ABX-treated mice, although tumors from ABX-treated mice showed reduced DNA damage (fig. S20). These data suggest that ABX prevent oxaliplatin-induced DNA damage and apoptosis after DNA adduct formation in part by decreasing ROS production (15,18,19). In ABX-treated mice, the decreased induction of the DNA damage response gene Atr and of the p53 downstream genes Bax, Fas, Cdkn1a, and Rb1 is compatible with reduced tumor DNA damage and p53-dependent activation of apoptosis. Although tumor-cell-produced ROS were previously reported to be required for the cytotoxic effect of platinum compounds in vitro, our data indicate that the ROS required for oxaliplatin genotoxicity in vivo are mostly derived from tumor-associated inflammatory cells. Because ABX treatment affected tumor cytotoxicity shortly after chemotherapy, and this effect was observed using both oxaliplatin and its relative cisplatin that is unable to trigger immunogenic cell death, the data suggest that commensal bacteria modulate the genotoxicity of platinum compounds independently of immunity elicited by immunogenic cell death. However, DC activation and adaptive antitumor immune responses after oxaliplatin may also require a commensal-dependent inflammatory response.
Besides platinum complexes, drugs such as anthracyclines, alkylating agents, podophyllotoxins, and camptothecine induce ROS as part of their anticancer activity and may be similarly regulated (15, 20–22). Mechanisms by which the gut microbiota affects the systemic inflammatory tone are only now being uncovered (23, 24). In a different setting, total body irradiation was shown to increase the efficacy of adoptively transferred tumor-specific CD8+ T cells in part by inducing microbial translocation (25). Our study indicates that commensal bacteria differentially affect the type of inflammatory tone required for response to different therapeutic protocols and highlights the potential to improve cancer treatment by manipulating human gut microbiota.
Supplementary Material
Acknowledgments:
We greatly appreciate the support of D. Trageser-Cessler and A. Cesar in the NIAID germ-free facility; H. Zhou in the NIH Center for Human Immunology; T. Plona and K. Pike in the Laboratory of Molecular Technology, Leidos Biomedical Research, Inc.; and R. Winkler-Pickett, M. Karwan, and D. Zhao in the Cancer and Inflammation Program, CCR/NCI. We thank D. Klinman and A. Hurwitz for advice and R. Germain for critically reviewing the manuscript. The data presented in this manuscript are tabulated in the main paper and in the supplementary materials. This research was supported by the Intramural Research Program of the NIH (NCI, Center for Cancer Research, and NIAID) and the Japanese Society for Promotion of Science (JSPS) Research Fellowship for Japanese Biomedical and Behavioral Researchers at NIH. The 16S sequences were deposited in the Sequence Read Archive (SRA) under accession PRJNA221649. Array data were deposited at the Gene Expression Omnibus (GEO) with accession no. GSE51414.
Footnotes
Supplementary Materials
www.sciencemag.org/content/342/6161/967/suppl/DC1 Materials and Methods
References and Notes
- 1.Lee YK, Mazmanian SK, Science 330, 1768–1773 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clemente JC, Ursell LK, Parfrey LW, Knight R, Cell 148, 1258–1270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McAleer JP, Kolls JK, Immunity 37, 10–12 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Abt MC et al. , Immunity 37, 158–170 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ichinohe T et al. , Proc. Natl. Acad. Sci. U.S.A. 108, 5354–5359 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grivennikov SI, Greten FR, Karin M, Cell 140, 883–899 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shirota Y, Shirota H, Klinman DM, J. Immunol. 188, 1592–1599 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zitvogel L, o. Kepp, G. Kroemer, Cell 140, 798–804 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Apetoh L et al. , Nat. Med. 13, 1050–1059 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Vicari AP et al. , J. Exp. Med. 196, 541–549 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guiducci C, Vicari AP, Sangaletti S, Trinchieri G, Colombo MP, Cancer Res. 65, 3437–3446 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Jenq RR et al. , J. Exp. Med. 209, 903–911 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peran L et al. , Br. J. Nutr. 97, 96–103 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Siddik ZH, Oncogene 22, 7265–7279 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Ozben T, J. Pharm. Sci. 96, 2181–2196 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Ghiringhelli F et al. , Nat. Med. 15, 1170–1178 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Drummond GR, Selemidis S, Griendling KK, Sobey CG, Nat. Rev. Drug Discov. 10, 453–471 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim S, Lee TJ, Park JW, Kwon TK, J. Cell. Biochem. 105, 971–979 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Laurent A et al. , Cancer Res. 65, 948–956 (2005). [PubMed] [Google Scholar]
- 20.Conklin KA, Integr. Cancer Ther. 3, 294–300 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Fang J, Nakamura H, Iyer AK, J. Drug Target. 15, 475–486 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Simizu S, Takada M, Umezawa K, Imoto M, J. Biol. Chem. 273, 26900–26907 (1998). [DOI] [PubMed] [Google Scholar]
- 23.Ganal SC et al. , Immunity 37, 171–186 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Huang Y et al. , Genes Immun. 13, 311–320 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Paulos CM et al. , J. Clin. Invest. 117, 2197–2204 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.