

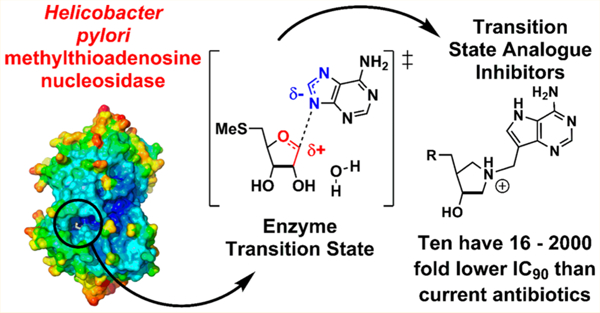
Abstract
Helicobacter pylori is a Gram-negative bacterium that colonizes the gut of over 50% of the world’s population. It is responsible for most peptic ulcers and is an important risk factor for gastric cancer. Antibiotic treatment for H. pylori infections is challenging as drug resistance has developed to antibiotics with traditional mechanisms of action. H. pylori uses an unusual pathway for menaquinone biosynthesis with 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase (MTAN) catalyzing an essential step. We validated MTAN as a target with a transition-state analogue of the enzyme [Wang, S.; Haapalainen, A. M.; Yan, F.; et al. Biochemistry 2012, 51, 6892−6894]. MTAN inhibitors will only be useful drug candidates if they can both include tight binding to the MTAN target and have the ability to penetrate the complex cell membrane found in Gram-negative H. pylori. Here we explore structural scaffolds for MTAN inhibition and for growth inhibition of cultured H. pylori. Sixteen analogues reported here are transition-state analogues of H. pylori MTAN with dissociation constants of 50 pM or below. Ten of these prevent growth of the H. pylori with IC90 values below 0.01 μg/mL. These remarkable compounds meet the criteria for potent inhibition and cell penetration. As a consequence, 10 new H. pylori antibiotic candidates are identified, all of which prevent H. pylori growth at concentrations 16−2000-fold lower than the five antibiotics, amoxicillin, metronidazole, levofloxacin, tetracyclin, and clarithromycin, commonly used to treat H. pylori infections. X-ray crystal structures of MTAN cocrystallized with several inhibitors show them to bind in the active site making interactions consistent with transition-state analogues.
Graphical Abstract
INTRODUCTION
The bacterial 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidases (MTANs) are dual substrate enzymes hydrolyzing 5′-methylthioadenosine (MTA) and S-adenosylhomo-cysteine (SAH) to produce 5-methylthioribose or S-ribosylho-mocysteine, and adenine. In most bacteria, MTAN is involved in methionine recycling, polyamine synthesis, and quorum sensing, but is not essential for cell growth or survival.1–3 Recently, Helicobacter pylori (Hp) and Campylobacter jeujeuni (Cj) were reported to have an unusual biosynthetic pathway for menaquinone in which the respective MTANs play essential roles.4,5 This pathway utilizes 6-amino-6-deoxyfutalosine which is also hydrolyzed by the Hp and Cj MTANs (Figure 1). Menaquinone is essential for electron transport in most Gram-positive and anaerobic Gram-negative bacteria and a block imposed on this pathway by inhibition of the MTANs would likely be lethal to the organisms.6–8
Figure 1.
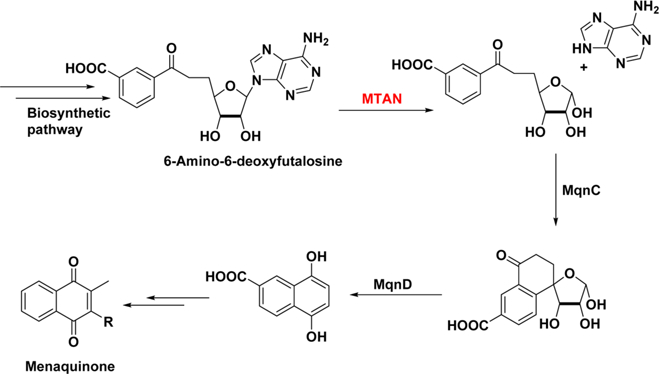
Role of MTAN in the biosynthesis of menaquinone.
H. pylori is the most common worldwide bacterial infection. It is related to 85% of gastric and 95% of duodenal ulcers and is an important risk factor for gastric malignancies.9,10 Antibiotic treatment for H. pylori infections is becoming more challenging, with even triple and quadruple therapies being often unsuccessful as drug resistance has developed.11 In addition, the combination of proton pump inhibitors and multiple antibiotics disrupt the normal gut microbiome and this treatment is associated with the development of Clostridium difficile associated diarrhea, a disorder linked with 29 000 annual deaths in the United States in 2011.12,13 New antibiotics are needed for H. pylori infections with novel targets and mechanisms of action. Transition-state-analogue inhibitors of the biosynthesis of menaquinone provide a unique approach to the design of new antibiotics for H. pylori.
Transition-state analogues capturing important features of an enzyme transition state can bind tightly with the potential for exceptionally potent inhibitors.14–16 Knowledge of the transition-state structure provides a blueprint for inhibitor design. The transition states of several bacterial MTANs have been analyzed and shown to have strong ribocationic character with weak involvement of the water nucleophile (Figure 2).17–20 A number of transition-state-analogue inhibitors of these MTANs have been developed, some with dissociation constants in the picomolar to femtomolar range.20–24 The HpMTAN has been compared to other bacterial MTANs and has been shown to be dissociative in character with a relatively early transition state.20 The inhibitors 1 and 2 were used in the comparison, with 1 better mimicking an early transition state and 2 a late transition state (Figure 3). Subsequently, 3 was shown to be a 36 pM inhibitor of the HpMTAN and additionally to have an IC90 of <8 ng/mL for the growth of H. pylori without arresting growth of other common bacterial species.25 Targeting the futalosine pathway has demonstrated the potential utility of HpMTAN transition-state-analogue inhibitors as new, and H. pylori specific, antibiotics.
Figure 2.
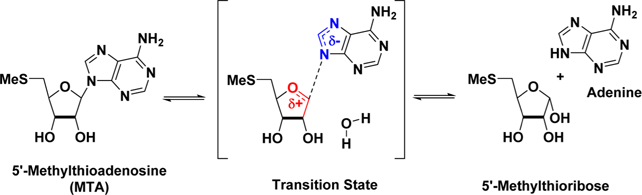
Hydrolysis of MTA by bacterial MTANs is characterized by dissociative transition states. Similar transition states are assumed for the S-adenosylhomocysteine and 6-amino-6-deoxyfutalosine substrates.
Figure 3.
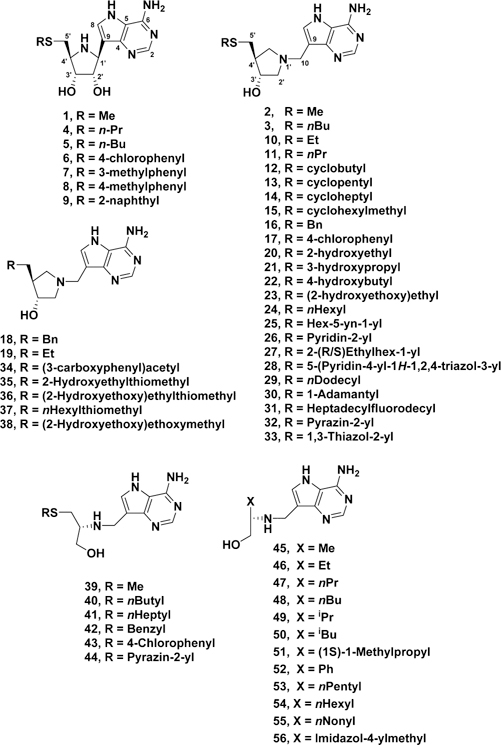
Compounds explored as transition state analogues of HpMTAN.
Here we present the HpMTAN inhibition data and IC90 values for transition-state analogues including novel and simplified structures. Sixteen analogues have dissociation constants of 50 pM or below. Ten of these prevent growth of the H. pylori at concentrations 16−2000-fold lower than the five antibiotics in current use to treat H. pylori infections. Additionally, the X-ray crystal structures of HpMTAN containing bound inhibitors are presented showing that all bind in the active site making interactions with the protein consistent with transition-state analogues.
RESULTS AND DISCUSSION
Synthesis of New Transition-State Analogues.
Detail of the synthesis of new H. pylori MTAN inhibitors is presented in the schemes and experimental procedures in the Supporting Information.
Biological Results.
Enzymes achieve the catalytic rate enhancement of reactions by lowering the activation energy for the transition states.26 When the electronic and spatial characteristics of an enzyme transition state are known, they provide a template for the design of transition-state analogues which can be powerful inhibitors. Kinetic isotope effects have been used to determine enzyme transition states leading to the design and synthesis of exceptionally potent inhibitors.14–32 The transition states of several bacterial MTANs have been shown to be dissociative in character, with the HpMTAN displaying characteristics of an early dissociative transition state.17–20 Transition-state-analogue inhibitors such as 1 mimic an early transition state with a short distance between C-1′ and N-9, whereas inhibitors such as 2 resemble a late transition state with a much longer distance between C-1′ and N-9. Consistent with transition state recognition, 1 is marginally a more potent inhibitor than 2 of the HpMTAN. However, the hydroxypyrrolidine compounds (e.g., 2) in general have better overall affinity for the MTANs. This may be due to a closer ion pair formation with the water nucleophile, the higher pKa of the pyrrolidine, and the geometric freedom inherent in the methylene bridge between the leaving group and ribocation mimics.
Enzyme Inhibition Results.
The HpMTAN shows broad substrate specificity, affecting the hydrolytic cleavage of the adenine moiety from the substrates methylthioadenosine, S-adenosylhomocysteine, and 6-amino-6-deoxyfutalosine. These substrates differ only in their adenosyl 5′-substituent. When the previously described MTAN inhibitors 1−2322,23,27–29 were assayed against HpMTAN, the results (Table 1 and Supporting Information Table 1) confirmed that the enzyme accepts varied substituents at the 5′-position and that the hydroxypyrrolidine structures (e.g., 3) provided a number of powerful picomolar inhibitors. Some alkylthio- (3, 5, 11), cycloalkylthio- (12−15), and desthio- (19) analogues showed good potency as enzyme inhibitors. Compounds 20−23 were originally synthesized in response to the observation that polyethylene glycol was present in the 5′-alkylthio binding site of crystalline Salmonella enterica MTAN.29 The PEG was from the crystallization solvent. Cocrystallization suggested that the 5′-binding site might easily accommodate ethylene glycol-type substituents, and 23 was indeed a 5 pM inhibitor of the SeMTAN. The H. pylori and S. enterica MTANs are very similar at the active site. In both cases, the active site entrance (the 5′-binding site) is rather hydrophobic, containing Met11, Ile53, Leu105, Phe108, Pro116, Phe154, and Phe209 in HpMTAN. In SeMTAN, all of these residues are the same except Leu105, which is a valine. Thus, hydrophobic 5’-substituents create tight binding, for example, 23 is a 15 pM inhibitor of the HpMTAN. Results from the other 5′-thio analogues 24−27, 32, and 33 showed that it is possible to vary the substituent at this position while retaining good inhibitor potency. Comparing compounds 21 and 35 demonstrates that the position of the sulfur is not important (and even can be removed as in 19) whereas replacing S with O (36 vs 38) demonstrated that hydrophobic groups (S and CH2) are preferred over oxygen.
Table 1.
Data of Selected Compounds for the Inhibition of H. pylori MTAN and IC90 values for H. pylori Growth on Blood Agar
|
H. pylori MTAN inhibition (nM) |
|||
|---|---|---|---|
| compd | Ki | Ki* | inhibition of H. pylori growth IC90 (ng/mL) |
| 1 | 0.16 ± 0.07a | 0.04 ± 0.02a | 80 |
| 2 | 0.19 ± 0.03 | 0.089 ± 0.019 | 6−12 |
| 3 | 0.79 ± 0.04 | 0.036 ± 0.002 | 6−8 |
| 4 | 0.79 ± 0.15 | 0.021 ± 0.004 | 16 |
| 11 | 0.058 ± 0.014 | 0.007 ± 0.002 | 10 |
| 12 | 0.27 ± 0.04 | 0.04 ± 0.01 | 16 |
| 13 | 0.78 ± 0.15 | 0.17 ± 0.01 | 7−14 |
| 15 | 0.56 ± 0.27 | 0.045 ± 0.004 | 18−35 |
| 19 | 0.17 ± 0.06 | 0.053 ± 0.007 | 16 |
| 20 | 0.43 ± 0.12 | 0.04 ± 0.01 | 40 |
| 22 | 0.34 ± 0.07 | 0.11 ± 0.04 | 9 |
| 23 | 0.96 ± 0.16 | 0.015 ± 0.004 | 35−70 |
| 24 | 0.21 ± 0.03 | 0.005 ± 0.002 | 4−8 |
| 25 | 0.5 ± 0.2 | 0.09 ± 0.02 | 4−8 |
| 26 | 0.32 ± 0.07 | 0.041 ± 0.002 | 40 |
| 27 | 0.16 ± 0.02 | 0.04 ± 0.01 | >80 |
| 32 | 0.043 ± 0.001 | 0.006 ± 0.001 | 8 |
| 33 | 0.24 ± 0.07 | 0.016 ± 0.005 | 20 |
| 44 | 0.10 ± 0.01 | N/Ob | 8 |
| 53 | 0.10 ± 0.01 | N/Ob | 8 |
| 54 | 0.030 ± 0.003 | N/Ob | 8 |
These errors were calculated based on the Km errors, to provide an upper limit to the errors to Ki based on the data from all other inhibitor determination.
No slow onset inhibition was observed.
Previous results with other N-ribosyltransferase enzymes have demonstrated that acyclic aminoalcohol derivatives can also act as transition-state analogues by acting as mimics of the ribocation moiety at the transition state.21,31–33 However, acyclic inhibitors of bacterial MTANs have been investigated without discovering potent inhibitors of interest.21 Here, for the HpMTAN, new substituted-thio analogues 40−44 all demonstrated improved potency over the methylthio compound 39, in particular 40, 41, and 44. The results from a number of desthio compounds 45−55 with hydrophobic side chains indicated that branched-chain compounds were not well tolerated and the pentyl derivative 54 was exceptional among these compounds with low picomolar potency.
Exploring the geometry and hydrophobicity of the 5′-substituents in a family of potential transition-state analogues for H. pylori MTAN has yielded 16 analogues with slow-onset inhibition properties and dissociation constants of 50 pM or below (Table 1). Included among these powerful inhibitors are three analogues with dissociation constants below 10 pM. This value is biologically significant, as it has been documented that transition-state analogues of 10 pM or below bind to their targets with inhibition times approaching or exceeding the lifetimes of cells.34 This inhibitory potential is significant for H. pylori, a bacterial target of the stomach, where exposure to oral drugs might be transitory. Given the inhibitory potential of these agents, we explored their ability to penetrate cells, inhibit the target enzyme, and prevent growth of cultured H. pylori.
Bacterial Growth Assays.
Candidate inhibitors were tested against H. pylori growth on 5% horse blood agar under conditions described previously (Table 1 and Supporting Information Table 1).25 Poor inhibitors had little or no effect on bacterial growth. Powerful inhibitors did not always show growth inhibition at low concentrations, an indication of biological access to the cell interior. Among the potent MTAN inhibitors, 10 gave H. pylori growth inhibition (IC90 values) ≤ 10 ng/mL (∼20 nM) while others required much higher concentrations.
H. pylori is a Gram-negative organism, the group of bacteria known to be difficult to achieve drug access because of the complex membrane and cell wall structure. We evaluated both the action of inhibitor on the MTAN target by analysis of kinetic constants, and biological access to the cell interior by measuring IC90 values (the inhibitor concentration reducing growth on blood agar plates by 90%). The hydroxypyrrolidine series of compounds with butylthio- 3, propylthio- 11, hexylthio- 24, hexynylthio- 25, and pyrazinylthio- 32 substituents demonstrated IC90 values ∼ 10 ng/mL, whereas in the acyclic aminoalcohol series those with pyrazinylthio- 44, and the desthio butyl- 53 and pentyl- 54 substituents showed similar potency. The pyrazinylthio substituent is common to both, perhaps again reflecting improved access to the cell interior. It is instructive to compare the IC90 values reported here with those of the current antibiotics used in multidrug therapy for H. pylori infections. Metronidazole (16 000 ng/mL), amoxicillin (125 ng/mL), clarithromycin (8000 ng/mL), levofloxacin (1000 ng/mL), tetracycline (500 ng/mL), and the new agent, moenomycin A (2000 ng/mL) are weaker than the 10 best MTAN inhibitors (∼8 ng/mL) by 16- (amoxicillin) to 2000-fold (metronidazole).35 We can conclude that the present compounds are promising leads with potent antibacterial activity against H. pylori.
Inhibitor-Bound HpMTAN Crystal Structure Comparisons.
HpMTAN was cocrystallized with the hydroxypyrrolidine compounds 2, 22, 32, and 36 and the acyclic aminoalcohol compound 54. All of the inhibitor-bound structures are homodimeric, with the asymmetric units containing between one and four monomer units (e.g., Figure 4). This oligomerization state is consistent with all other MTAN structures in the PDB from 15 other organisms. In PDB-reported MTAN structures, two of the active site residues strongly hydrogen bond to the adenine portion of the native substrates or inhibitors. The five inhibitors presented herein have the same interactions (Figure 5). The carboxylate of Asp199 (conserved in all deposited MTAN structures) hydrogen bonds to N-6 and N-7; and the backbone amide nitrogen and oxygen atoms of Val155 hydrogen bond to N-1 and N-6, respectively. (See numbering in Figure 4.) The tertiary amine (N-1′) in the inhibitors is protonated at neutral pH and mimics the ribocationic nature of the transition state. In the respective inhibitor-bound structures, the nearby nucleophilic water molecule that would act to hydrolyze the native substrates is highly coordinated, being hydrogen bonded to N-1′ of the inhibitor in addition to Arg195 and Glu13.
Figure 4.
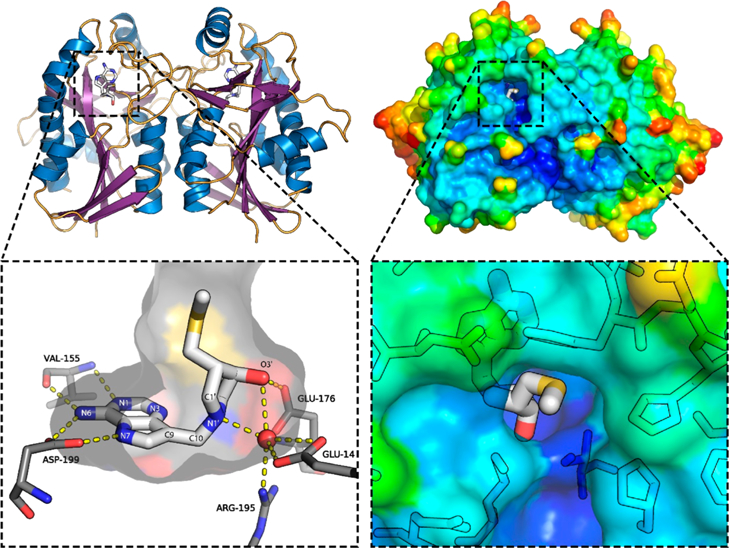
HpMTAN with 2 bound. Left: Ribbon diagram showing homodimeric structure. The expansion shows the binding pocket, with hydrogen bonding residues and “nucleophilic” water shown. Right: Surface representation showing the active site entrance, colored based on B-factors.
Figure 5.
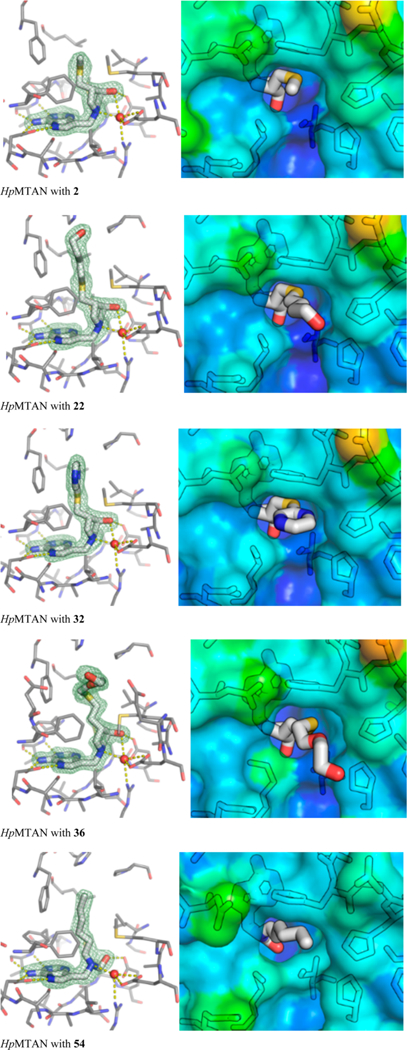
HpMTAN bound to various inhibitors. Left: electron densities greater than 1 RMSD around the ligand, in maps calculated with 2 Fobs − Fcalc coefficients. Right: Surface representation of protein residues at active site entrance (colored by B-factor), with respective inhibitors shown as sticks.
With inhibitors 2, 22, 32, and 36, there is an additional hydrogen bond to the 3′-hydroxy group, which is in turn hydrogen bonded to Glu176. In contrast, the hydroxymethyl group in the acyclic inhibitor 54 is hydrogen bonded only to Glu176. This acyclic inhibitor has greater conformational freedom and has one fewer carbon between N-1′ and O-3′ compared to the other inhibitors, and consequently is situated differently in the active site. This is exemplified by the torsion angle C9−C10−N1′−C1′, which is −115° in 54 and −75 ± 3° in inhibitors 2, 22, 32, and 36. The active site entrance (also the 5′-binding site) is rather hydrophobic (Met11, Ile53, Leu105, Phe108, Pro116, Phe154, Phe209) and spacious, and is able to accommodate a range of substrates, including MTA, SAH and aminofutalosine. As the 5′-substituent of 2 is small, there is extra room in the 5′-binding site for compounds with larger 5′-groups (Figures 4 and 5). Adenine-bound MTAN crystal structures sometimes have small molecules from crystallization buffer or cryoprotectant present in this cavity. For instance, PEG chains, glycerol, or ethylene glycol are observed near the ribose binding pocket or active site entrance in the S. enterica and E. coli MTAN structures, PDB codes 4F1W and 1Z5P, respectively. The synthesis of 36 was partly inspired by this observation. Given the largely hydrophobic nature of the active site, no substantial interactions are made to the 5′-alkylthio substituents, until the exterior of the protein is reached. At the protein exterior the hydroxybutyl and PEG fragment, in 32 and 36, respectively, are close enough to hydrogen bond to one of the imidazole nitrogen atoms of His110, although this is only observed in the hydroxybutyl inhibitor (N···O = 2.69 Å). Inhibitors with more extended 5’-substituents could also interact with Glu13. The HpMTAN structure with 54 bound also features well-ordered fusion tags. Two of the histidine residues from the fusion tag in one of the monomers, in addition to His42 from a second dimer and Glu211 from a third, form a tetrahedral coordination complex with a zinc ion (see the SI figure). The fusion tag from the second monomer does not form such an interaction, but is also well ordered.
CONCLUSIONS
A new antibiotic approach to H. pylori infections utilizes transition-state-analogue inhibitors of the H. pylori MTAN targeting the biosynthesis of menaquinone. The transition-state analogues include 16 with dissociation constants below 50 pM. Ten of these 16 powerful inhibitors also prevent growth of the H. pylori at ≤10 ng/mL, concentrations 16−2000-fold lower than the five antibiotics in current use to treat H. pylori infections.
Supplementary Material
ACKNOWLEDGMENTS
This work was financially supported by the New Zealand Foundation for Research Science and Technology Grant C08X0209 and by the National Institutes of Health Research Grant GM041916. Crystallographic data for this study were measured at beamline X29A of the National Synchrotron Light Source. Financial support comes principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the US Department of Energy, and from the National Center for Research Resources (P41RR012408) and the National Institute of General Medical Sciences (P41GM103473) of the National Institutes of Health. Herbert Wong and Yinrong Lu are thanked for high quality NMR and Mass Spec service.
Appendix
Figure.
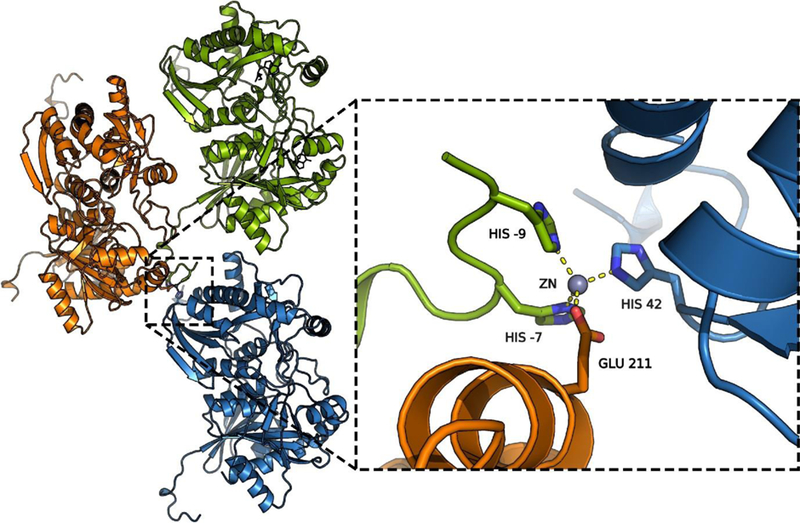
Three HpMTAN dimers with 54 bound, illustrating the tetrahedral zinc coordination complex formed by fusion tag histidine residues (His −7 and His −9) and main chain residues (His 42 and Glu 211) from neighboring dimers.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b06110.
Full experimental details including compound syntheses, characterization and NMR spectra, enzyme inhibition and cell culture assays, protein crystallization details, and diffraction data statistics (PDF)
Figure of three (symmetry-generated) HpMTAN dimers (PDF)
Crystallographic data (CIF)
The authors declare no competing financial interest.
REFERENCES
- (1).Miller MB; Bassler BL Annu. Rev. Microbiol 2001, 55, 165. [DOI] [PubMed] [Google Scholar]
- (2).Parveen N; Cornell KA Mol. Microbiol 2011, 79, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Gutierrez JA; Crowder T; Rinaldo-Matthis A; Ho M-C; Almo SC; Schramm VL Nat. Chem. Biol 2009, 5, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Li X; Apel D; Gaynor EC; Tanner ME J. Biol. Chem 2011, 286, 19392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Dairi TJ Antibiot 2009, 62, 347. [DOI] [PubMed] [Google Scholar]
- (6).Suttie JW Vitamin K in Health and Disease; CRC Press: Boca Raton, FL, 2009. [Google Scholar]
- (7).Popp JL; Berliner C; Bentley R Anal. Biochem 1989, 178, 306. [DOI] [PubMed] [Google Scholar]
- (8).Kurosu M; Begari E Molecules 2010, 15, 1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).De Falco M; Lucariello A; Iaquinto S; Esposito V; Guerra G; De Luca AJ Cell. Physiol 2015, 230, 1702. [DOI] [PubMed] [Google Scholar]
- (10).Kuipers EJ; Thijs JC; Feston HP Aliment Pharmacol. Ther 1995, 9, 59. [PubMed] [Google Scholar]
- (11).Selgrad M; Malfertheiner P Curr. Opin. Gastroenterol 2011, 27, 565. [DOI] [PubMed] [Google Scholar]
- (12).Dial S; Delaney JAC; Barkun AN; Suissa SJ Am. Med. Assoc 2005, 294, 2989. [DOI] [PubMed] [Google Scholar]
- (13).Lessa FC; Mu Y; Bamberg WM; Beldavs ZG; Dumyati GK; Dunn JR; Farley MM; Holzbauer SM; Meek JI; Phipps EC; Wilson LE; Winston LG; Cohen JA; Limbago BM; Fridkin SK; Gerding DN; McDonald LC N. Engl. J. Med 2015, 372, 825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wolfenden R Nature 1969, 223, 704. [DOI] [PubMed] [Google Scholar]
- (15).Wolfenden R Annu. Rev. Biophys. Bioeng 1976, 5, 271. [DOI] [PubMed] [Google Scholar]
- (16).Schramm VL Arch. Biochem. Biophys 2005, 433, 13. [DOI] [PubMed] [Google Scholar]
- (17).Singh V; Lee JE; Nunez S; Howell PL; Schramm VL Biochemistry 2005, 44, 11647. [DOI] [PubMed] [Google Scholar]
- (18).Singh V; Luo M; Brown RL; Norris GE; Schramm VL J. Am. Chem. Soc 2007, 129, 13831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Singh V; Schramm VL J. Am. Chem. Soc 2007, 129, 2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Gutierrez JA; Luo M; Singh V; Li L; Brown RL; Norris GE; Evans GB; Furneaux RH; Tyler PC; Painter GF; et al. ACS Chem. Biol 2007, 2, 725. [DOI] [PubMed] [Google Scholar]
- (21).Clinch K; Evans GB; Frohlich RFG; Gulab SA; Gutierrez JA; Mason JM; Schramm VL; Tyler PC; Woolhouse AD Bioorg. Med. Chem 2012, 20, 5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Longshaw AI; Adanitsch F; Gutierrez JA; Evans GB; Tyler PC; Schramm VL J. Med. Chem 2010, 53, 6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Singh V; Evans GB; Lenz DH; Mason JM; Clinch K; Mee S; Painter GF; Tyler PC; Furneaux RH; Lee JE; et al. J. Biol. Chem 2005, 280, 18265. [DOI] [PubMed] [Google Scholar]
- (24).Singh V; Shi W; Almo SC; Evans GB; Furneaux RH; Tyler PC; Painter GF; Lenz DH; Mee S; Zheng R; et al. Biochemistry 2006, 45, 12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wang S; Haapalainen AM; Yan F; Du Q; Tyler PC; Evans GB; Rinaldo-Matthis A; Brown RL; Norris GE; Almo SC; et al. Biochemistry 2012, 51, 6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Schramm VL Curr. Opin. Struct. Biol 2005, 15, 604. [DOI] [PubMed] [Google Scholar]
- (27).Evans GB; Furneaux RH; Lenz DH; Painter GF; Schramm VL; Singh V; Tyler PC J. Med. Chem 2005, 48, 4679. [DOI] [PubMed] [Google Scholar]
- (28).Evans GB; Furneaux RH; Schramm VL; Singh V; Tyler PC J. Med. Chem 2004, 47, 3275. [DOI] [PubMed] [Google Scholar]
- (29).Haapalainen AM; Thomas K; Tyler PC; Evans GB; Almo SC; Schramm VL Structure 2013, 21, 963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Schramm VL ACS Chem. Biol 2013, 8, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ho MC; Shi W; Rinaldo-Matthis A; Tyler PC; Evans GB; Clinch K; Almo SC; Schramm VL Proc. Natl. Acad. Sci. U. S. A 2010, 107, 4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Clinch K; Crump DR; Evans GB; Hazleton KZ; Mason JM; Schramm VL; Tyler PC Bioorg. Med. Chem 2013, 21, 5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Clinch K; Evans GB; Fröhlich RF; Furneaux RH; Kelly PM; Legentil L; Murkin AS; Li L; Schramm VL; Tyler PC; et al. J. Med. Chem 2009, 52, 1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lewandowicz A; Tyler PC; Evans GB; Furneaux RH; Schramm VL J. Biol. Chem 2003, 278, 31465. [DOI] [PubMed] [Google Scholar]
- (35).Tseng Y-Y; Liou J-M; Hsu T-L; Cheng W-C; Wu M-S; Wong C-H Bioorg. Med. Chem. Lett 2014, 24, 2412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.