

Abstract
High throughput tandem mass spectrometry has enabled the detection and identification of over 75% of all proteins predicted to result in translated gene products in the human genome. In fact, the galloping rate of data acquisition and sharing of mass spectrometry data has led to the current availability of many tens of terabytes of public data in thousands of human datasets. The systematic reanalysis of these public datasets has been used to build a community-scale spectral library of 2.1 million precursors for over 1 million unique sequences from over 19,000 proteins (including spectra of synthetic peptides). However, it has remained challenging to find and inspect spectra of peptides covering functional protein regions or matching to novel proteins. ProteinExplorer addresses these challenges with an intuitive interface mapping tens of millions of identifications to functional sites on nearly all human proteins while maintaining provenance for every identification back to the original dataset and data file. Additionally, ProteinExplorer facilitates the selection and inspection of HPP-compliant peptides whose spectra can be matched to spectra of synthetic peptides and already includes HPP-compliant evidence for 107 missing (PE 2, PE3 and PE4) and 23 dubious (PE5) proteins. Finally, ProteinExplorer allows users to rate spectra and to contribute to a community library of peptides entitled PrEdict (Protein Existance dictionary) mapping to novel proteins but whose preliminary identities have not yet been fully established with community-scale false discovery rates and synthetic peptide spectra. ProteinExplorer can be now be accessed at https://massive.ucsd.edu/ProteoSAFe/protein_explorer_splash.jsp.
Keywords: tandem mass spectrometry, proteomics, human proteome, missing proteins, big data, community-scale science, peptide-spectrum match, spectral library, computational analysis, user submission
Introduction
Sustained improvements to tandem mass spectrometry (MS/MS) instruments and their application to the analysis of a broad range of protein samples have resulted in the generation of a large volume of mass spectrometry data12. But while this increased capacity has allowed for the community-wide exploration of the protein content of many types of samples, it has also led to new challenges where the significance of an identification (e.g., peptide or protein) made in the context of a single sample or dataset may not hold when considering aggregate identifications from hundreds of datasets taken to express what is known by the community as a whole. Since the vast majority of the true matches coincide across most datasets (i.e., most human samples share many proteins, which are typically identified mostly by the same peptides) but false matches are much more likely to be unique to each dataset (or at least less consistent across datasets), this leads to a situation where the naïve union of discoveries across many datasets would result in an uncontrolled increase in false discovery rates (FDR) – a problem that affects any question referring to community-scale identifications, as is certainly the case for the HUPO Human Proteome Project’s (HPP3) quest for reliable detection of translated protein products.
To address this issue, our MassIVE Knowledge Base (MassIVE-KB4) spectral library applied strict spectrum, peptide and protein level FDR controls at the aggregate level for all search results included in its construction. As such, its reanalysis of over 31 TB of human HCD data resulted in the largest HCD spectral library to date, with 2.1+ million spectra of >1 million unique peptide sequences mapped to >16,000 proteins and covering over 50% of all amino acid content in the human proteome. But while MassIVE-KB’s identifications offer an FDR-controlled, repository-scale route towards analysis of protein detection in datasets from many sources, it remains cumbersome and time consuming to i) to explore the genomic or functional considerations associated with different identifications and ii) inspect the evidence in support of the detection of missing or dubious proteins whose translated expression has not been confirmed by mass spectrometry data. Our ProteinExplorer application addresses these issues by offering integrated and intuitive access to exon and functional information mapped to peptide and protein identifications, as well as integrating with synthetic peptide5/protein6 expression resources and applying official HPP criteria for detection of novel proteins3. In addition, ProteinExplorer provides access to detailed provenance records for every single identification, thereby enabling seamless direct access to the public datasets from which identifications were derived, as well as to the standardized search jobs that were used to generate the original identifications from the public mass spectrometry data – an aspect that is often overlooked when aggregating results from multiple datasets (i.e., access to the full set of search parameters and original search results). In particular, the hundreds of public datasets in the current release of ProteinExplorer reported here already include identifications from systematic reanalysis of multiple datasets that were incorporated into previous HPP releases (PXD000529/MSV0000802557, PXD000533/MSV0000802547, PXD000561/MSV0000795148, PXD000612/MSV0000807019, PXD000865/MSV00007952610, PXD003947/MSV00008082611), thereby allowing for open access to inspect whether reanalysis confirms the original reports after considering repository-scale FDR corrections.
To further support the validation of peptide and protein identifications beyond what is supported in existing resources, ProteinExplorer facilitates the comparison of experimental spectra identified from public datasets to spectra of synthetic peptides (from the ProteomeTools5 project) or proteins (from the BioPlex6 project) by including these in separate spectral libraries (altogether covering over 19,000 proteins) and by providing a simple, one-click access to the generation of interactive spectrum images for matching peptides in multiple libraries. Finally, since there are multiple ways in which identifications may not be currently eligible to fully claim detection of novel proteins (e.g., protein identifications with only one peptide or absence of corroborating spectra of synthetic peptides), ProteinExplorer further incorporates a user library entitled PrEdict (Protein Existence dictionary) where users can submit spectra in partial support of the detection of novel proteins, and thus iteratively progress towards collaborative detection of proteins across datasets. Illustrating how this feature supports community inspection of evidence of detection and supports convergence towards a common consensus, the quality and interpretation of these identifications has also been rated and sometimes commented in records associated with each entry in the user library.
Methods
ProteinExplorer is built on the MassIVE Knowledge Base4 (MassIVE-KB), a repository-scale spectral library resulting from the reanalysis of 658 million MS/MS HCD spectra from 27,404 LC/MS runs in 227 datasets. In brief, spectra were searched with MS-GF+12 database search against the UniProt human reference proteome with isoforms as well as contaminants, such as porcine trypsin. Variable modifications included in the searches were oxidation, N-term acetylation, N-term Carbamylation, Pyro-glu, and deamidation were considered as variable modifications and carbamidomethylation on Cysteine as a fixed modification. For the purpose of the analysis reported here, only matches identified to canonical proteins, and not matches to contaminants, isoforms, or TrEMBL were retained. Spectrum identifications were considered to be ambiguous and removed from consideration if there were two or more peptides matching the spectrum with scores passing FDR thresholds. FDR was applied at the spectrum level (0.1% library-level FDR), length-specific peptide level (1% local FDR), and 1% protein-level FDR for all proteins matched by at least one unique peptide13,14, which corresponds to 0.013% protein-level FDR for proteins matched by 2+ peptides15 (i.e., expect two false positive protein identifications). In addition to the MassIVE-KB spectral library constructed from public datasets of natural sequences, spectral libraries of synthetic peptide spectra were also constructed using the same process and used to assess the correlation of fragmentation patterns with MassIVE-KB spectra.
Protein metadata and functional information was integrated into ProteinExplorer from both PhosphoSitePlus16 and Uniprot17, with the current release based on downloads on March 26, 2018. Protein Existence (PE) classifications were downloaded from the neXtProt18 ftp (the 2018–01-17 release) and incorporated into ProteinExplorer as well (rare discrepancies between UniProtKB and neXtProt due to lags in synchronization between database versions were conservatively assigned a PE of 0 to remove those matches from consideration). In brief, protein existence tiers are defined as PE1 if there is evidence at protein level, PE2 if evidence at transcript level, PE3 if inferred from homology, PE4 if predicted and PE5 if uncertain; proteins are also referred to as “missing” if classified as PE2, PE3 or PE4 and “dubious” if classified as PE518. To further assess the potential biological significance of peptides, all peptides were mapped onto genomic exons included in reference human transcripts. Using a previously described approach16,17, we mapped all peptides in MassIVE-KB to exons in ENSEMBL 8919,20 and annotated whether the peptide maps uniquely to an exon, covers a splice junction, or is mapped to an exon at all (some peptides are not mapped due to differences between UniProt and ENSEMBL sequences). The algorithms used to determine whether proteins were matched by two or more HPP-compliant peptides are described in Supplementary Materials.
ProteinExplorer is developed as a community hub for examining the human proteome and is built as a Java application with a JavaScript front-end and a RESTful API built with Tomcat to fit in the ecosystem of the MassIVE repository and the ProteoSAFe parallel workflow engine; ProteinExplorer can be accessed at https://massive.ucsd.edu/ProteoSAFe/protein_explorer_splash.jsp. The underlying database is built in MySQL and contains tables for libraries, proteins, peptides, representatives (spectral library PSMs selected to best characterize a modified peptide at a given precursor charge out of the set of all PSMs which pass FDR), provenance spectra (the PSMs that pass FDR for each precursor), and comments. In particular, PSMs are stored in a way that is independent of libraries, so when updates are made to a library, or even representatives, it is unlikely that most PSMs will need to be updated. An overview of the ProteinExplorer functionality and data sources is provided in Figure 1 and the full schema for the database is provided in Figure S1.
Figure 1.
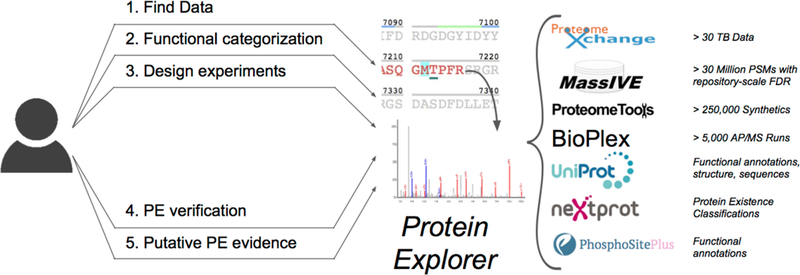
ProteinExplorer overview including use cases and major sources of information.
ProteinExplorer was designed to facilitate the productive exploration of repository-scale identification of tens of millions of peptide spectrum matches (PSMs) for over 1 million distinct peptide sequences identified from 30+ TB of public mass spectrometry data.
The JavaScript user interface provides two views for exploring the proteome. The first is a proteome-wide view with information about all proteins, which we call the proteome page, and the second is a protein-centric view that contains amino-acid level coverage information from libraries as well as metadata from community resources, we which title the protein page. The proteome page consists of two panels. The first is a series of filters for protein accession identifiers, as well as other common fields such as the Uniprot protein description, protein existence (PE) information from neXtProt, and options to consider only specific public datasets. Under these filter boxes is a table that displays all proteins that satisfy the search criteria specified in the filter boxes. The table also provides a one-click option to separate protein information per dataset, including dataset specific expression and peptide-uniqueness; the table is also downloadable for offline processing (see supplementary materials for examples of how the proteome view can be utilized).
The protein page provides a detailed view of each protein, including sequence coverage, representatives, and provenance, as well as filters for library expression and functional information overlays. This page is specific per protein and can also be filtered to show only combinations of libraries (e.g. only the natural peptides). The coverage map superimposes sequence-level expression for the protein with functional information from UniProt and PhosphoSitePlus, and is interactive allowing for users to hover over amino acids for more information about metadata as well as click on individual amino acids to filter the view to just peptides covering that amino acid. As the interface displays ample information in a relatively small space, distinctions are made between different libraries by using different colors, which are fully customizable to be more accessible to all users, using a color picker21. When two libraries overlap, we blend the colors by simply averaging the RGB values for each color, allowing for an easy distinction between red, blue, and their mix (purple). The final two sections of the protein page provide interactive and downloadable tables for the representative peptides for the library and for all the supporting PSM provenance information for the protein. The representatives table includes information about representative length, number of proteins that each peptide maps onto when considering single amino acid variants, number of exons matched by each peptide, and the coordinates of the peptide on the protein. The provenance table contains all information necessary to track the peptide back to its original experiment as well as search task, including the filename, scan, charge, search algorithm, and link to the search. These tables are all filterable, and any filter made to any table is applied to the entire view, facilitating discovery by eliminating redundant clicks.
Results
The high diversity of peptide sequences and expression patterns observed across many datasets in repository-scale searches open up the possibility of investigating protein biology in more detail by exploring patterns of protein expression across datasets (see Supplementary Materials) as well as comprehensive coverage of genomic exons and functional sites. As shown in Figure 2, multiple features are mapped onto a sequence coverage view of the protein sequence and it is possible to highlight different aspects of the coverage by selecting between different types of rendering of the protein sequence, with “Flat coverage” highlighting all amino acids covered by at least one PSM, “Spectrum Coverage” highlighting amino acids on a color scale based on how many PSMs covered it and “Peptide Coverage” and “Variants Coverage” highlighting amino acids based on how many unique peptides or modified peptide variants cover each site (respectively). This coverage view is then extended to facilitate the analysis of exon matches and functional sites.
Figure 2.
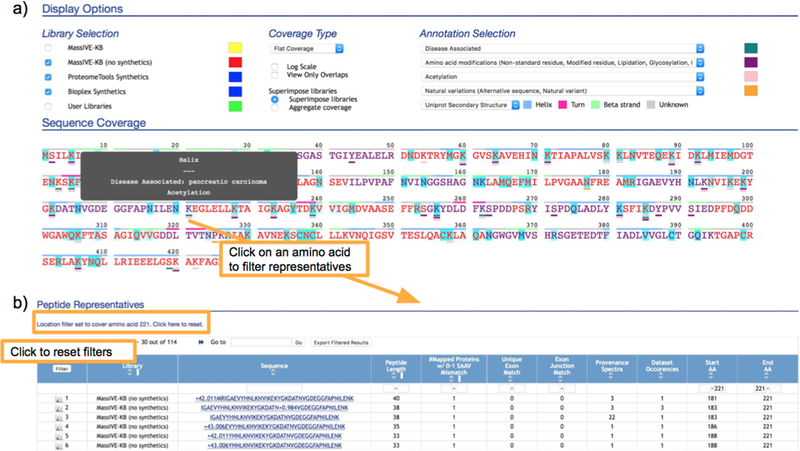
Protein page
(a) For each protein, the sequence coverage display provides an easy view to explore the spectrum, peptide and modified variants coverage with superimposed metadata from UniProt and PhosphoSitePlus. In this example, we highlight UniProt amino acid modifications and disease associated sites from PhosphoSitePlus as dashes below their associated site, with secondary structure also shown above the protein sequence. (b) Modified peptide variants covering a site that is disease-associated (from PhosphoSitePlus) and also a known modification (from UniProt), as well as covered by modified peptide variants in MassIVE-KB.
Peptide sequences are mapped to genomic coordinates22,23 on ENSEMBL 8919,20 to i) determine whether the mapping is unique (and thus indicative of exon presence/absence) and ii) whether the peptide is fully contained within an exon or it’s a junction peptide spanning one or more exons. As such, filtering by “Unique Exon Match” or by “Exon Junction Match” in the “Peptide Representatives” table (i.e., by requiring a minimum value of one on either of these columns) would select only for peptides matching the corresponding category and update the sequence coverage to highlight only the locations covered by the sequences of the selected peptides. Sorting by “Start AA” or “End AA” in the “Peptide Representatives” table further facilitates inspection of overlapping sequences and modification variants covering the same protein regions.
Functional knowledge of modified sites was integrated from UniProt17 and PhosphoSitePlus16 are shown on the protein coverage view as underlines, with additional details shows by hovering over annotated sites. These be further explored to examine if any of the peptides contain modified variants of a site or whether modification or expression patterns vary across a range of datasets. Clicking on the amino acid location in the protein coverage panel filters the page for only sequences covering the site of interest. Upon filtering, the views renormalize the expression levels to examine specific sites rather than global expression.
Beyond the sequence level understanding in the coverage view, the “Peptide representatives” table then provides additional information on these peptides including the diversity of peptide sequences and modification variants covering the site and the MS/MS spectrum, as well as their corresponding expression patterns in terms of both number of PSM identifications (“Provenance Spectra” column) and number of datasets in which each peptide variant was detected (“Dataset Occurrences” column), thereby facilitating the estimation of which peptides are most representative of the protein (e.g., identified in the highest number of datasets), rather than just peptides with large number of PSMs (which might also be explained by dataset-specific protocols strongly selecting for some protein regions over others).
We illustrate this functionality by considering a disease-associated site annotated by PhosphoSitePlus on protein P06733 (Figure 2) and examining the overlapping variants at position 221. As all views are filtered by clicking on this site, we see that there are 4058 PSMs in 114 variants that cover this site. Further filtering for acetylated peptide sequences by entering “+42.011” in the filter box for the “Sequence” field in the “Peptide Representatives” table reveals that, even though the original searches did not consider Lysine acetylation as a possible modification, there were still 9 peptide variants covering this site and identified as modified with N-terminal acetylation. Out of these, 3 variants account for over 90% of all observed PSMs and are the most commonly observed across multiple datasets. Interestingly, selecting for Asparagine deamidation (by filtering for “N+0.984”) also reveals separate deamidation on each of the three Asparagines on the most abundant peptide sequence covering the site (DATNVGDEGGFAPNILENK), each of which is clearly supported by site-localizing peaks, as can be readily seen by clicking on the spectrum icon on the leftmost column of the peptide representatives table.
ProteinExplorer also facilitates the design of targeted experiments. The standard course of action in a targeted experiment is to first find a few unique precursors to the protein and then find transitions, (i.e. precursor and ion pairs) that are frequently observed in a reproducible manner. In the protein view it is possible to click on a site to see all covering peptides which can then be further filtered to emphasize particular characteristics, e.g. selecting for a particular dataset that might be illustrative of experimental conditions. Once a peptide is selected, one can then rank PSMs and compare them side by side to pick transitions (see Supplementary Materials).
Detection of PE2–4 and PE5 missing proteins
The inspection of evidence provided in support of the detection of PE2–4 and PE5 proteins (missing and uncertain/dubious proteins) is a core need of the Human Proteome Project’s (HPP) goal of confirming the in vivo translation of proteins predicted from the human genome. Establishing the detection of these proteins requires that the supporting identifications meet a well-defined set of criteria for extraordinary detection claims24, including verification of the identifications using spectra of synthetic peptides, all of which are addressed in ProteinExplorer as follows.
First, the requirements for proper estimation of false discovery rates at the spectrum, peptide and protein levels are all guaranteed by the spectral library construction processes used to assemble the MassIVE-KB spectral library from over 190 million PSMs identified from over 30 TB of human higher-energy collisional dissociation (HCD) data4. Second, the additional requirements for peptide length of at least nine amino acids, unique protein matches even with one single amino acid variation (SAAV), and detection of at least two non-contained peptides were addressed in ProteinExplorer and are reflected in the protein page in the “# Matched Proteins w/0–1 SAAV Mismatch”. As such, filtering on this column for peptides matching exactly one protein selects for all candidates potentially supporting the detection of missing proteins.
The final step in the confirmation of peptide identifications (matching fragmentation to spectra of synthetic peptides) is implemented in ProteinExplorer by separately listing representative PSMs with matching sequences but from different libraries (the “Library” field in the “Peptide Representatives” table). Further, clicking on the “View Only Overlaps” option under the “Coverage Type” options (as shown in Figure 3) refines the protein coverage to render only peptides matched by two or more libraries, thereby facilitating the visual selection of sequences. Also in this view, it is possible to track every single PSM back to the scan number, raw file and public dataset where the spectral data came from (data provenance), as well as trace every PSM back to the search workflow, parameters, job and results from which the PSM was extracted (analytical provenance). Both of these elements are critical to the evaluation of detection of missing proteins and likely should be added as formal requirements for all HPP submissions.
Figure 3.
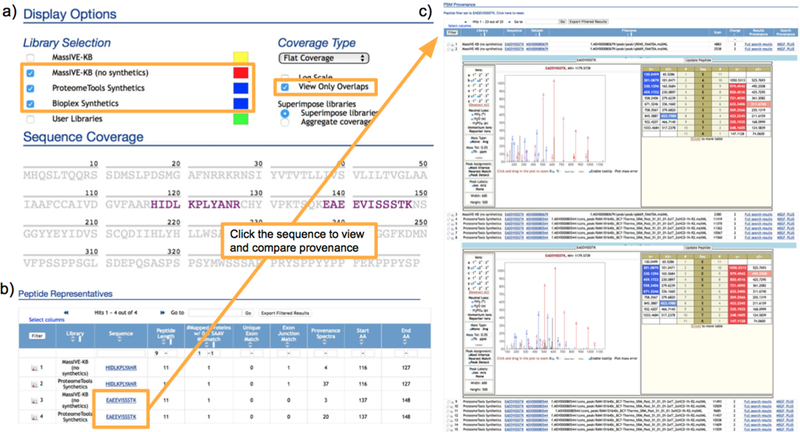
Matching spectra from different libraries.
(a) ProteinExplorer facilitates comparison of spectra from different libraries (e.g., for comparison of spectra from synthetic and natural sources) by providing an option to automatically select only peptides with the same sequence in the two libraries; e.g., selecting ProteomeTools and natural MassIVE-KB to allow for inspection of full compliance with HPP criteria. (b) List of selected peptides with entries in multiple libraries, also displaying whether the peptides are sufficiently long and do not match more than one protein even when allowing for single amino acid variations (SAAVs). (c) Interactive Lorikeet panels render annotated PSMs for assessment of matches to theoretical ions masses, as well as for inspection of correlated fragmentation between spectra from synthetic and natural sources.
Using all of these ProteinExplorer features to analyze identifications in the current release of MassIVE-KB, we were able to detect 296 PE2–4 and 55 PE5 proteins with at least one HPP-compliant peptide, with 107 missing (PE2, PE3 and PE4) and 23 dubious (PE5) proteins of these identified with two or more peptides meeting all HPP criteria for peptide identifications (see Figure 4a). We include PE5 proteins as we have found ample evidence for the detection of many but continue to report them separately from the missing proteins. Out of these PE2–4 proteins, 60 (3 for PE5) had two or more peptides with matching spectra from synthetic peptides and 21 (4 for PE5) proteins had only one of the two peptides for which spectra of synthetic peptides were available (see Table S2 which is split into two sheets for both missing and dubious proteins and is further sortable by number of matching synthetics on the “HPP Non-overlapping Matching Synthetics” column as well as containing chromosome location and a link to the ProteinExplorer page where all MS/MS spectra for the peptides can be examined). The distribution of cosines between MassIVE-KB representative spectra (identified from public datasets) and corresponding spectra of synthetic peptides is shown in Figure 4b; spectra and rationale for approval are provided in supplementary materials as well as can be browsed online for all matches with a cosine less than 0.6 for further examination (see Supplementary Materials and Figure S5). While spectra of synthetic peptides were not available to validate all 107 PE2–4 and 23 PE5 protein identifications with 2+ HPP-compliant peptides, we note that out of 60*2+21=141 PE2–4 and 3*2+4=10 PE5 cases where matching sequences were present in both MassIVE-KB and synthetic peptide collections, all spectrum matches confirmed the identifications – thereby strongly suggesting that the vast majority of proteins currently detected with 2+ HPP-compliant peptides will also be confirmed as soon as spectra of corresponding synthetic peptides become available.
Figure 4.
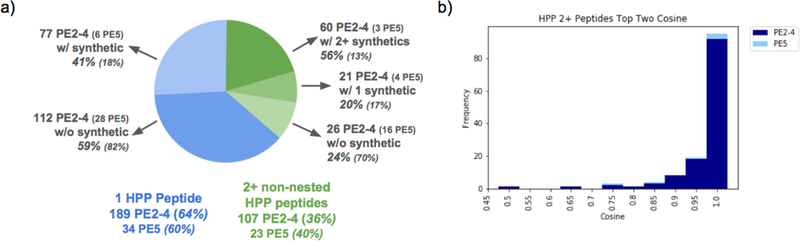
Detection of PE2–4 and PE5 proteins.
(a) MasslVE-KB identifications with repository-scaLe FDR detect 365 protein annotated by neXtProt at the level of protein existence 2 or higher (PE2+), out of which we find that 63 PE2+ proteins were identified with 2+ non-nested peptides whose spectra matched to spectra of synthetic peptides, (b) Cosine distribution for synthetic/natural matches for all peptides supporting the detection of PE2+ proteins; all cases with cosine below 0.6 were manually inspected and are shown and discussed in supplementary materials.
In addition to facilitating the confirmation of HPP criteria for detection of missing proteins, ProteinExplorer also helps ascertain whether proteins are likely to ever generate enough peptides meeting the current criteria. For example, protein H3BUK9 is currently identified in MassIVE-KB by 26 distinct peptide variants (and further matched an additional 34 shared peptides) and is matched by over 200 variants in ProteomeTools and Bioplex synthetics combined, altogether accumulating 15 peptide sequences (of length 9 or longer) uniquely matching to this protein – yet not a single one of these peptides is a unique match after considering a single amino acid variation (see Figure S2).
Updating libraries with user-added spectra
Since many proteins in the human proteome have not yet been detected or have been matched only to a degree that does not meet HPP guidelines, ProteinExplorer also supports contributions to a user library entitled PrEdict (Protein Existence dictionary) constituting a community resource of exploratory `peptide hints` that may eventually accumulate to the point of fully supporting the detection of previously unobserved proteins. Submissions to the PrEdict library can come from complete dataset submissions or from reanalyses of public datasets, even if these have not yet been aggregated into repository-scale spectral libraries (or otherwise guaranteed to meet repository-scale FDR thresholds, as is the case with MassIVE-KB spectral library construction workflows)). That said, ProteinExplorer retains the full provenance of every PSM by linking it back to the dataset and specific context from where it was contributed (e.g., a dataset reanalysis), thereby providing a route for direct inspection of the quality of search producing the PSM.
To demonstrate the potential utility of the ProteinExplorer PrEdict library, we added CID spectra from the CPTAC colorectal dataset25 (MSV000079852) in support of an additional 25 missing proteins, each matched by 2 non-overlapping peptides matching uniquely to the protein sequence even when considering SAAVs (see Table S4). These PSMs were originally either submitted with the dataset or identified by subsequent CCMS reanalysis using MS-GF+ database search. As an example of the utility of manual revision of tentative spectra, we added a peptide ARHSEAEATRAR that uniquely maps (including SAAV matches) to the protein A6NNA2, a protein with 8 synthetic peptides (125 PSMs) but only a limited amount of observations in natural data (1 peptide with 1 PSM). While the spectrum initially appears to have a fair amount of unexplained ion current, it turns out that the highest intensity peaks can be explained as doubly-charged ions for neutral losses from the unfragmented precursor. As such, the spectrum was marked as a 4 star match and a comment was entered (and is directly attached to the spectrum for immediate access by anyone inspecting it further) describing the reasons for the rating. All spectra in all ProteinExplorer libraries can be rated and annotated in the same way, thereby empowering the community to review all matches submitted in support of the detection of missing proteins.
Discussion
The identification of over 1.4 million modified human peptide variants from systematic reanalysis of public mass spectrometry data has created new opportunities for understanding the human proteome, but also brings new challenges for the meaningful and efficient exploration of such a large number of identifications from a volume of data so large that would not be accessible to the vast majority of proteomics labs. ProteinExplorer thus expands beyond other resources in i) its ability to enable interactive exploration of multiple libraries in dynamically updated views displaying expression and functional metadata and ii) by supporting the submission and curation of spectrum identifications for missing/dubious proteins, thereby empowering the community to add spectra to the PrEdict user library, as well as curate (i.e., rate and comment on) other spectra which were also submitted as hints.
As a key example of the utility of ProteinExplorer to assist with the exploration of the human proteome, we have also described how its features enable the inspection of evidence submitted in support of the detection of novel proteins, and show how these select identifications for dozens of proteins that are fully compliant with HPP guidelines, as well as detect many more proteins whose peptide identifications match HPP guidelines but for which there are currently no spectra of the same synthetic peptide sequences. Since it is thus expected that it will be common for evidence in support of the detection of novel proteins to be accumulated over time as new data becomes available (including data for spectra of synthetic peptides), ProteinExplorer supports this iterative and collaborative process by allowing for the submission of PSMs to the PrEdict user library that is shared by the whole community. Finally, progressing towards community-wide consensus of which identifications to accept requires full transparency in complete provenance records (from data, tools and search procedures, all the way to identifications) but should also be supported by an interactive platform where (dis)agreements can be recorded and used to eventually converge on a community-curated collection of reliably identified spectra, especially for cases of high biological relevance (e.g., binding regions in monoclonal antibodies) or for evidence supposed to support the detection of novel proteins.
Supplementary Material
Supplementary Text. Includes sections on algorithms, synthetics, the proteome view, and the ability to update libraries with user-added spectra.
Figure S1 - Database Schema. Complete schema of the MySQL database of ProteinExplorer.
Figure S2 - Non-unique but ample coverage for H3BUK9. H3BUK9 is a protein that has many natural peptide matches in ProteinExplorer, but they are all shared with other proteins.
Figure S3 - Main user interfaces. (a) The ProteinExplorer main page allows users to search for proteins by accession, gene, neXtprot PE, and UniProt description. (b) The protein list panel shows all proteins meeting the filtration criteria, either at the level of the whole repository or per dataset. (c) The dataset option allows users to see and filter by dataset accessions to find dataset-specific protein information, including unique peptide and exon counts.
Figure S4 - User libraries. ProteinExplorer allows for user submission to shared community libraries. While user libraries are not FDR-controlled in the same way as MassIVE-KB libraries, the flexibility to share spectra of any PSMs facilitates the exchange of information and could help converge towards collaborative accumulation of evidence supporting the detection of novel proteins. (a) All public datasets are potential sources for submission of spectra to user libraries. (b) Dataset results or reanalyses allow for inspection of PSMs and submission to the user libraries. (c) The spectrum submission form curates the PSM for resolution of modifications (if needed) before final submission to the library. (d) Submitted PSMs are mapped onto the proteome and shown in both the coverage map and in the (e) representative and provenance sections. (f) Spectra in the user library (and in all other libraries) can be rated and commented on by the community to assess and promote consensus around the proposed identifications.
Figure S5 - Low Cosine Match Between Synthetic and Natural PSM. These are two spectra from the same variant of a PE2 protein Q4KMG9 that has 2+ matching HPP peptides, but the cosine match for this example is lower than expected at 0.461. However, this score can be explained due to a high intensity contaminant peak, though otherwise the y-series y4+ through y9+ matches and many other fragmentations peaks correlate. A link is provided for further examination.
Table S1 - Datasets. All the datasets used in MassIVE-KB and ProteinExplorer.
Table S2 - PE2–4 and PE5 Proteins with matched peptides. PE2–4 and PE5 (as defined by neXtProt) proteins that have uniquely mapped peptides in ProteinExplorer and also synthetic evidence (when it exists). The table is divided into two sheets, missing proteins (PE2–4) and dubious proteins (PE5).
Table S3 - Exon Mapping. All peptides and exon/protein mappings in ProteinExplorer.
Table S4 - CPTAC PE2–4 and PE5 Proteins. PE2–4 and PE5 proteins with 2 HPP-compliant peptides in CPTAC that are not in MassIVE-KB without synthetics.
ACKNOWLEDGMENT
This work was supported by the US National Institutes of Health (grant 2 P41 GM103484–06A1 from the National Institute of General Medical Sciences to BSP, JW, JC and NB; NB is an Alfred P. Sloan Research Fellow. NB was a co-founder, had an equity interest and received income from Digital Proteomics, LLC through 2017. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. Digital Proteomics was not involved in the research presented here.
Footnotes
SUPPORTING INFORMATION
The following supporting information is available free of charge at ACS website http://pubs.acs.org.
REFERENCES
- (1).Zhang Y; Fonslow BR; Shan B; Baek M-C; Yates JR Protein Analysis by Shotgun/Bottom-up Proteomics. Chem. Rev. 2013, 113 (4), 2343–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bensimon A; Heck AJR; Aebersold R Mass Spectrometry-Based Proteomics and Network Biology. Annu. Rev. Biochem. 2012, 81, 379–405. [DOI] [PubMed] [Google Scholar]
- (3).Omenn GS; Lane L; Lundberg EK; Overall CM; Deutsch EW Progress on the HUPO Draft Human Proteome: 2017 Metrics of the Human Proteome Project. J. Proteome Res. 2017, 16 (12), 4281–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Wang M; Wang J; Carver J; Pullman BS; Cha SW; Bandeira N Assembling the Community-Scale Discoverable Human Proteome. Cell Syst. 2018, 7, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Zolg DP; Wilhelm M; Schnatbaum K; Zerweck J; Knaute T; Delanghe B; Bailey DJ; Gessulat S; Ehrlich H-C; Weininger M; et al. Building ProteomeTools Based on a Complete Synthetic Human Proteome. Nat. Methods 2017, 14 (3), 259–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Huttlin EL; Bruckner RJ; Paulo JA; Cannon JR; Ting L; Baltier K; Colby G; Gebreab F; Gygi MP; Parzen H; et al. Architecture of the Human Interactome Defines Protein Communities and Disease Networks. Nature 2017, 545 (7655), 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Liu Y; Ying W; Ren Z; Gu W; Zhang Y; Yan G; Yang P; Liu Y; Yin X; Chang C; et al. Chromosome-8-Coded Proteome of Chinese Chromosome Proteome Data Set (CCPD) 2.0 with Partial Immunohistochemical Verifications. J. Proteome Res. 2014, 13 (1), 126–136. [DOI] [PubMed] [Google Scholar]
- (8).Kim M-S; Pinto SM; Getnet D; Nirujogi RS; Manda SS; Chaerkady R; Madugundu AK; Kelkar DS; Isserlin R; Jain S; et al. A Draft Map of the Human Proteome. Nature 2014, 509 (7502), 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sharma K; D’Souza RCJ; Tyanova S; Schaab C; Wiśniewski JR; Cox J; Mann M Ultradeep Human Phosphoproteome Reveals a Distinct Regulatory Nature of Tyr and Ser/Thr-Based Signaling. Cell Rep. 2014, 8 (5), 1583–1594. [DOI] [PubMed] [Google Scholar]
- (10).Wilhelm M; Schlegl J; Hahne H; Gholami AM; Lieberenz M; Savitski MM; Ziegler E; Butzmann L; Gessulat S; Marx H; et al. Mass-Spectrometry-Based Draft of the Human Proteome. Nature 2014, 509, 582. [DOI] [PubMed] [Google Scholar]
- (11).Vandenbrouck Y; Lane L; Carapito C; Duek P; Rondel K; Bruley C; Macron C; Gonzalez de Peredo A; Coute Y; Chaoui K; et al. Looking for Missing Proteins in the Proteome of Human Spermatozoa: An Update. J. Proteome Res. 2016, 15 (11), 3998–4019. [DOI] [PubMed] [Google Scholar]
- (12).Kim S; Pevzner PA MS-GF+ Makes Progress towards a Universal Database Search Tool for Proteomics. Nat. Commun. 2014, 5, 5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Elias JE; Gygi SP Target-Decoy Search Strategy for Increased Confidence in Large-Scale Protein Identifications by Mass Spectrometry. Nat. Methods 2007, 4 (3), 207–214. [DOI] [PubMed] [Google Scholar]
- (14).Gupta N; Pevzner PA False Discovery Rates of Protein Identifications: A Strike against the Two-Peptide Rule. J. Proteome Res. 2009, 8 (9), 4173–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Savitski MM; Wilhelm M; Hahne H; Kuster B; Bantscheff M A Scalable Approach for Protein False Discovery Rate Estimation in Large Proteomic Data Sets. Mol. Cell. Proteomics 2015, 14 (9), 2394–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hornbeck PV; Zhang B; Murray B; Kornhauser JM; Latham V; Skrzypek E PhosphoSitePlus, 2014: Mutations, PTMs and Recalibrations. Nucleic Acids Res. 2015, 43 (Database issue), D512–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).The UniProt Consortium. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2017, 45 (D1), D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Gaudet P; Michel P-A; Zahn-Zabal M; Britan A; Cusin I; Domagalski M; Duek PD; Gateau A; Gleizes A; Hinard V; et al. The NeXtProt Knowledgebase on Human Proteins: 2017 Update. Nucleic Acids Res. 2017, 45 (D1), D177–D182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Aken BL; Achuthan P; Akanni W; Amode MR; Bernsdorff F; Bhai J; Billis K; Carvalho-Silva D; Cummins C; Clapham P; et al. Ensembl 2017. Nucleic Acids Res. 2017, 45 (D1), D635–D642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zerbino DR; Achuthan P; Akanni W; Amode MR; Barrell D; Bhai J; Billis K; Cummins C; Gall A; Girón CG; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46 (D1), D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Odvárko J jscolor.
- (22).Woo S; Cha SW; Na S; Guest C; Liu T; Smith RD; Rodland KD; Payne S; Bafna V Proteogenomic Strategies for Identification of Aberrant Cancer Peptides Using Large-Scale next-Generation Sequencing Data. Proteomics 2014, 14 (23–24), 2719–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Woo S; Cha SW; Bonissone S; Na S; Tabb DL; Pevzner PA; Bafna V Advanced Proteogenomic Analysis Reveals Multiple Peptide Mutations and Complex Immunoglobulin Peptides in Colon Cancer. J. Proteome Res. 2015, 14 (9), 3555–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Deutsch EW; Overall CM; Van Eyk JE; Baker MS; Paik Y-K; Weintraub ST; Lane L; Martens L; Vandenbrouck Y; Kusebauch U; et al. Human Proteome Project Mass Spectrometry Data Interpretation Guidelines 2.1. J. Proteome Res. 2016, 15 (11), 3961–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Rudnick PA; Markey SP; Roth J; Mirokhin Y; Yan X; Tchekhovskoi DV; Edwards NJ; Thangudu RR; Ketchum KA; Kinsinger CR; et al. A Description of the Clinical Proteomic Tumor Analysis Consortium (CPTAC) Common Data Analysis Pipeline. J. Proteome Res. 2016, 15 (3), 1023–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Text. Includes sections on algorithms, synthetics, the proteome view, and the ability to update libraries with user-added spectra.
Figure S1 - Database Schema. Complete schema of the MySQL database of ProteinExplorer.
Figure S2 - Non-unique but ample coverage for H3BUK9. H3BUK9 is a protein that has many natural peptide matches in ProteinExplorer, but they are all shared with other proteins.
Figure S3 - Main user interfaces. (a) The ProteinExplorer main page allows users to search for proteins by accession, gene, neXtprot PE, and UniProt description. (b) The protein list panel shows all proteins meeting the filtration criteria, either at the level of the whole repository or per dataset. (c) The dataset option allows users to see and filter by dataset accessions to find dataset-specific protein information, including unique peptide and exon counts.
Figure S4 - User libraries. ProteinExplorer allows for user submission to shared community libraries. While user libraries are not FDR-controlled in the same way as MassIVE-KB libraries, the flexibility to share spectra of any PSMs facilitates the exchange of information and could help converge towards collaborative accumulation of evidence supporting the detection of novel proteins. (a) All public datasets are potential sources for submission of spectra to user libraries. (b) Dataset results or reanalyses allow for inspection of PSMs and submission to the user libraries. (c) The spectrum submission form curates the PSM for resolution of modifications (if needed) before final submission to the library. (d) Submitted PSMs are mapped onto the proteome and shown in both the coverage map and in the (e) representative and provenance sections. (f) Spectra in the user library (and in all other libraries) can be rated and commented on by the community to assess and promote consensus around the proposed identifications.
Figure S5 - Low Cosine Match Between Synthetic and Natural PSM. These are two spectra from the same variant of a PE2 protein Q4KMG9 that has 2+ matching HPP peptides, but the cosine match for this example is lower than expected at 0.461. However, this score can be explained due to a high intensity contaminant peak, though otherwise the y-series y4+ through y9+ matches and many other fragmentations peaks correlate. A link is provided for further examination.
Table S1 - Datasets. All the datasets used in MassIVE-KB and ProteinExplorer.
Table S2 - PE2–4 and PE5 Proteins with matched peptides. PE2–4 and PE5 (as defined by neXtProt) proteins that have uniquely mapped peptides in ProteinExplorer and also synthetic evidence (when it exists). The table is divided into two sheets, missing proteins (PE2–4) and dubious proteins (PE5).
Table S3 - Exon Mapping. All peptides and exon/protein mappings in ProteinExplorer.
Table S4 - CPTAC PE2–4 and PE5 Proteins. PE2–4 and PE5 proteins with 2 HPP-compliant peptides in CPTAC that are not in MassIVE-KB without synthetics.