

Abstract
Phase I clinical trials applying autologous progenitor cells to treat heart failure have yielded promising results; however, improvement in function is modest, indicating a need to enhance cardiac stem cell reparative capacity. Notch signaling plays a crucial role in cardiac development, guiding cell fate decisions that underlie myocyte and vessel differentiation. The Notch pathway is retained in the adult cardiac stem cell niche, where level and duration of Notch signal influence proliferation and differentiation of cardiac progenitors. In this study, Notch signaling promotes growth, survival and differentiation of cardiac progenitor cells into smooth muscle lineages in vitro. Cardiac progenitor cells expressing tamoxifen-regulated intracellular Notchl (CPCeK) are significantly larger and proliferate more slowly than control cells, exhibit elevated mTORCl and Akt signaling, and are resistant to oxidative stress. Vascular smooth muscle and cardiomyocyte markers increase in CPCeK and are augmented further upon ligand-mediated induction of Notch signal. Paracrine signals indicative of growth, survival and differentiation increase with Notch activity, while markers of senescence are decreased. Adoptive transfer of CPCeK into infarcted mouse myocardium enhances preservation of cardiac function and reduces infarct size relative to hearts receiving control cells. Greater capillary density and proportion of vascular smooth muscle tissue in CPCeK-treated hearts indicate improved vascularization. Finally, we report a previously undescribed signaling mechanism whereby Notch activation stimulates CPC growth, survival and differentiation via mTORCl and paracrine factor expression. Taken together, these findings suggest that regulated Notch activation potentiates the reparative capacity of CPCs in the treatment of cardiac disease.
Keywords: Notch signaling, Cardiac progenitor, Differentiation, Survival, Paracrine
Introduction
Heart disease remains the premier cause of death in the United States. The discovery that resident cardiac progenitor cells (CPCs) participate in cardiac homeostasis and repair has overturned the traditional view of the heart as a non-regenerative organ. However, endogenous repair by CPCs remains limited in the face of strong pathological challenge such as infarction, necessitating further understanding of the molecular mechanisms underlying CPC proliferation, survival and differentiation. Phase l clinical trials utilizing adoptive transfer of autologous CPCs into cardiac patients are promising; however, CPCs from patients with advanced cardiac disease may have compromised regenerative capacity [3, 20, 36, 43, 51]. Therefore, enhancement of CPC reparative potential is a necessary advancement in cell-based therapy as a treatment for heart disease [41].
The Notch signaling pathway plays a central role in cardiac development [12, 21, 45] and more recently has been associated with survival and proliferative signaling in adult heart tissue following injury [6, 10, 18, 32, 42, 44, 46, 53]. Canonical activation of the Notch cascade ensues upon binding of the extracellular domains of ligands to corresponding receptors on adjacent cells, triggering proteolytic cleavage and release of the receptor intracellular domain. The Notch intracellular domain (NICD) translocates to the nucleus and binds to the CBF1/RBP-JK/Suppressor of Hairless/LAG-1 (CSL) transcription factor, activating transcription of downstream targets including the Hairy and Hairy-related family of transcriptional repressors.
Notch signaling persists in the adult cardiac stem cell niche, with Notch receptors identified on the signal receiving stem cells, and Jagged1 ligand localized to myocyte and fibroblast niche support cells [54]. Activation of the pathway in adult cardiac progenitors stimulates proliferation and expression of Nkx2.5, while in vivo pharmacological inhibition of Notch activity following infarction interferes with endogenous progenitor commitment and repair [4, 53].
Successful enhancement of CPC reparative capacity through overexpression of Pim1 kinase has been established in mouse models of myocardial infarction [13, 39, 40]. In this study, we exploit the cardiogenic potential of Notch signaling by engineering stable CPC lines overexpressing intracellular mouse Notch1 (CPCeK). CPCeK exhibit increased size and resistance to oxidative stress compared to CPCe controls, and enhanced expression of vascular smooth muscle lineage markers. Proliferation is blunted with Notch activity in both CPCeK and Jagged1 stimulated CPCe, while MTT assay measurements and Akt and rS6 activity are elevated in response to Notch signaling. Cardiac function is slightly but significantly improved and infarct size reduced in infarcted mouse hearts following administration of CPCeK versus CPCe treatment. The proportion of smooth muscle expressing tissue and capillary density are significantly higher in CPCeK versus CPCe-treated hearts, suggesting that CPCeK cell therapy promotes formation or protection of vasculature in injured myocardium. Engraftment of adoptively transferred cells is nominal in both treatment groups. Therefore, survival signaling and possible stimulation of endogenous vascularization through paracrine effects by CPCeK may contribute to the improved function measured in CPCeK-treated hearts.
Materials and methods
Notch cloning, generation of lentivirus
Mouse intracellular Notch1 was subcloned from constructs kindly provided by Dr. Raphael Kopan, Washington University, St. Louis, MO (KNIC). KNIC was fused in frame to a FLAG tag mutated estrogen receptor fusion construct kindly provided by Dr. Christopher Glembotski, SDSU, San Diego, CA, to confer tamoxifen-mediated regulation of Notch activity (KNICmER, Fig. 1a). KNIC-mER insert was subcloned into the MND-VSV multiple cloning site upstream of an IRES_GFP encoding sequence (Fig. 1b [13]).
Fig. 1.
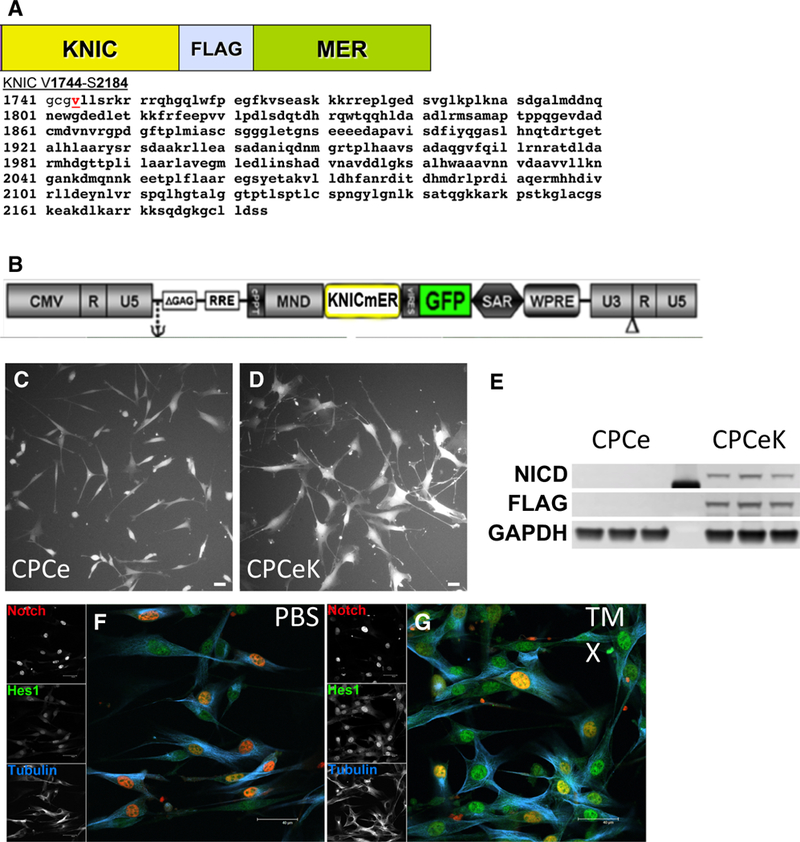
Constructs for creation of cell lines encoding intracellular Notch1. Schematic of construct encoding mouse intracellular Notch1 (KNIC) fused to the mutated estrogen receptor (mER) with protein sequence defining intracellular Notch as adapted from clones kindly provided by Professor Raphael Kopan of Washington University, St. Louis, MO. a Schematic representation of lentiviral construct encoding KNICmER used to generate stable CPCeK. b Live image of CPC transduced with control (c, CPCe) or Notch expressing virus (d CPCeK). Overexpression of KNICmER verified by immunoblot for activated Notch1 intracellular domain (NICD) and FLAG in CPCeK versus CPCe (e). Nuclear localization of overexpressed NICD (Notch1, red) in CPCeK treated with PBS (F) or 1 μM tamoxifen for 1 day (g). Increased expression of Hes1 (green) following activation of KNICmER by tamoxifen as shown by immunostain (f, g). Scale bar equals 40 μm in all images
Cardiac progenitor cell culture and creation of stable cell lines
Adult mouse cardiac progenitor cells were isolated as previously described [15]. Briefly, adult male FVB mouse hearts were digested and dissociated with collagenase, spun at low speed to pellet myocytes, and supernatant subjected to MACS selection for c-Kit expressing cells. CPCs were then transduced with either control IRES-GFP or KNIC-mER-IRES-GFP encoding lentivirus, passaged twice and subjected to clonal dilution by FACS sorting for GFP into 96-well plates. Clones were expanded (Fig. 1c, d), and KNICmER expression verified by immunoblot (Fig. 1e). Localization of KNICmER and elevation of Notch activity were verified by immunostaining using antibodies to Notch1 and downstream target Hes1 in CPCeK treated for 1 day with PBS or 1 iM tamoxifen (Fig. 1f, g). Live images were captured with a Leica DFC 340FX camera, DMIL fluorescence microscope, and FireCam 1.8.0 software. Diameters of dissociated cells were measured in ImageJ software using a hemocytometer to set the scale.
Immunostaining and immunoblotting
Hearts were arrested in diastole and retroperfused with formalin, then processed for paraffin embedding as previously described [18]. Paraffin sections were subjected to antigen retrieval and probed with antibodies to according to standard methods [18]. Fluorescent tyramide detection was used to enhance signal when necessary using the Perkin Elmer/NEN tyramide reagents. For immunofluorescent staining of cells, samples were fixed in 4 % buffered paraformaldehyde, washed in PBS, blocked in PBS with 10 % horse serum, and primary antibodies were applied in blocking buffer at 4 °C overnight. Fluorescent secondary antibodies were applied in blocking buffer. Fluorescent images were collected on a Leica SP2 or SP8 confocal laser scanning microscope and processed using Photoshop software. Area of isolectin B4 and sm22 staining was measured using NIH ImageJ software. Capillary density was measured by counting the number of capillaries per field of area in isolectin B4 stainings using NIH ImageJ software. Vessel length density (VLD) was measured as previously described with some modifications [49]. Briefly, paraffin sections were stained with isolectin B4 and smooth muscle actin to identify vascular endothelium and smooth muscle cells, respectively. Vessel length was measured in the remote region of the left ventricle in overlaid images captured using a Leica SP8 confocal microscope. VLD was measured in 3–5 fields from each of three hearts per group by tracing the total length of vessels in each field divided by the area of the field (mm of vessel length/mm2 of surface area). Analyses were performed using LASX software (Leica).
For immunoblotting, whole cell lysates were separated on Novex Bis/Tris gradient gels in MES running buffer and transferred to Immobilon-P PVDF membranes. Primary antibodies were incubated overnight in blocking buffer (8 % nonfat skim milk in Tris-buffered saline with 0.1 % Tween-20) at 4 °C, and secondary antibodies were incubated for 90 min at room temperature in blocking buffer. Secondary antibodies raised in donkey were purchased from Jackson Immuno or Life Technologies. Fluorescent signals were detected using a Typhoon Trio Imager and quantitated with Image Quant software. Primary antibodies and dilutions for immunostaining and immunoblotting are listed in Supplemental Table l.
Histology
Masson’s Trichrome staining was performed on paraffin sections using standard histological methods. Overview images were acquired with a Leica DMI6000 bright field microscope using the tiling function in LASX software. Length and area of infarct were measured using NIH ImageJ software.
Quantitative real-time PCR
Cells or tissues were harvested using the Zymo Research Quick-RNA MiniPrep kit (cat. #R1055). Reverse transcription to generate cDNA was performed using iScript according to manufacturer’s instructions and quantitative real-time PCR performed using iQ SYBR Green on a CFX96 Connect thermal cycler (all BioRad). Sequences of primers used to amplify mouse message targets are listed in Supplemental Table 2.
Proliferation and survival assays
Jaggedl was immobilized onto tissue culture plates as previously described [31]. Briefly, wells were treated with 25 μg/ml Goat anti-Human IgG (Fc specific, Sigma I2136) in PBS for 1 h at 37 °C, washed one time with PBS, blocked with 1 % BSA in PBS for 1 h at 37 °C, coated with either PBS or recombinant rat Jagged1-Fc chimera (RNDSystems 599-JG-100) 5 μg/ml in PBS for 1 h at 37 °C, and washed in PBS prior to plating. CPCe, CPCe grown on Jagged1-treated plates, or CPCeK were plated at 500 cells per well of a 96-well dish in growth media and assayed for cell number over 3 days using the Direct CyQuant method (Life Technologies) detected on a Tecan SpectraFluor Plus plate reader. Proliferation was plotted as fold change normalized to signal at day 0. Cell survival was assayed in CPCe, CPCe grown on Jagged1-treated plates or CPCeK plated at 50,000 cells per well of a 6-well dish in growth media. Media containing low serum (2.5 % FBS) was applied the following day for 24 h, then cells were treated with varying concentrations of stabilized hydrogen peroxide in low serum media. After 3–5 h when visible cell detachment became apparent, cells were detached and assayed for cell death by staining for AnnexinV (BD 550475, apoptosis) and 7-AAD (BD 51–6898E, necrosis). Cells positive for both AnnexinV and 7-AAD were chosen to define the percentage of cells undergoing cell death.
Surgery, echocardiography and in vivo hemodynamics
Myocardial infarction and intramyocardial injection of CPCs was performed in mice as previously described [13, 40]. Briefly, the left ascending coronary artery (LAD) was ligated and a total of 100,000 CPCe or CPCeK were injected into the border zone region adjacent to the infarct via five injections of 5 μl each. Cardiac function was monitored by echocardiography using a Visual Sonics Vevo 770, 707b probe at 1, 2 and 6 weeks following infarction. All animal procedures and treatments are approved by San Diego State University Institutional Animal Care and Use Committee.
Statistics
Statistical analysis was performed using GraphPad Prism 6.0 (Graphpad Software Inc; http://www.graphpad.com). Significance between two groups was measured using Student’s two-tailed t test. For comparison of more than two groups either one-way or two-way ANOVA statistical analysis was applied. A p value <0.05 was considered significant.
Results
CPCs engineered with tamoxifen-regulatable intracellular Notch1 express nuclear Notch and respond to ligand-mediated induction of Notch activity
The intracellular domain of mouse Notch1 was cloned in frame with a FLAG epitope tagged mutated estrogen receptor fusion protein (KNICmER, Fig. 1a). Mouse CPCs derived from male FVB hearts were transduced with lentivirus encoding KNICmER upstream of an IRES GFP construct (Fig. 1b) or IRES GFP only, then sorted for GFP expression by flow cytometry as single cells into 96-well plates to create clonal control (CPCe, Fig. 1c) and KNIC-mER expressing (CPCeK, Fig. 1d) cell lines. Overexpression of KNICmER was confirmed by immunoblot for FLAG tag and activated Notch1 (Fig. 1e) and immunolo-calization in a non-clonal CPCeK line treated with PBS or tamoxifen overnight to verify nuclear localization of intracellular Notch (Fig. 1f) and tamoxifen-mediated induction of Notch1 target Hes1 (Fig. 1g).
Augmented Notch activity increases area and roundness of cardiac progenitor cells in vitro
Notch signaling modulates proliferation, differentiation or cell cycle arrest in stem cells depending on cell type, context and signal dose [19]. CPCs were cultured on Jagged1-coated surfaces to activate endogenous Notch signaling. Jagged1-treated CPCs (CPCe JGD1, Fig. 2b) changed morphologically compared to BSA control-treated cells (CPCe BSA, Fig. 2a), increasing over twofold in area and roundness, and decreasing by half in length-to-width ratio within 4 days (Fig. 2d).
Fig. 2.
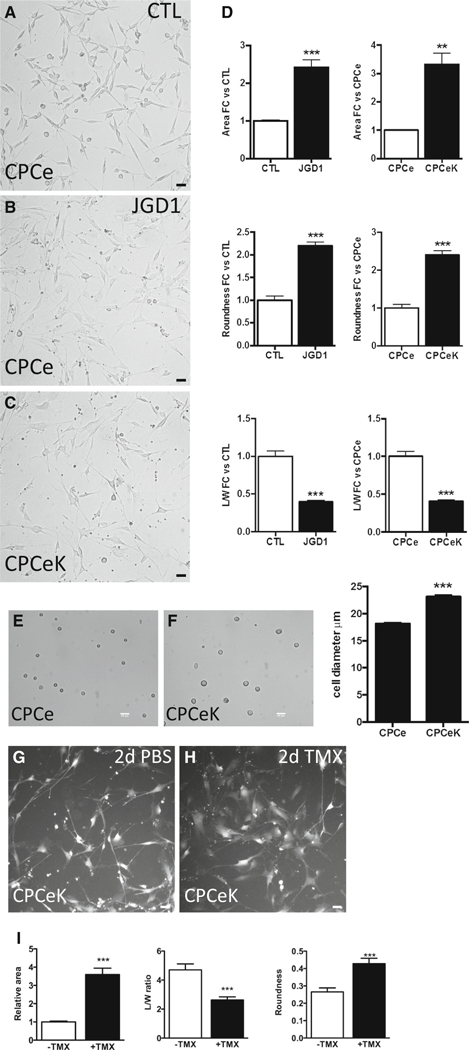
Notch activation increases area of cardiac progenitor cells. Live fluorescence images of CPCe cultured on control (a) or Jagged1 Fc-treated (b) plates for 4 days. CPCeK cultured on control-treated (c) plates. Quantitation of relative area, roundness and length-to-width (L/W) ratios as measured using NIH ImageJ (d), minimum three images per sample, at least ten measurements per image. Bright field images of resuspended CPCe (e) and CPCeK (f) with quantitation of cell diameter from multiple fields of view as measured in NIH ImageJ. Live fluorescence images of CPCeK treated with PBS (g) or 1 μM tamoxifen (h) for 2 days. Morphological measurements are shown in i. Scale bar 40 μm in all images. **p < 0.01, ***p < 0.001, significance determined using two-tailed t test
Similarly, stable CPC lines expressing KNICmER (CPCeK) exhibited morphological changes parallel to those observed after Notch activation by immobilized Jaggedl (Fig. 2c). Relative cell area increased more than threefold, roundness by more than twofold, and length-to-width ratio decreased by half in CPCeK versus CPCe (Fig. 2d). Average diameters of resuspended CPCe and CPCeK were 18 and 23 μm, respectively (Fig. 2e, f). The inner diameter of the needle used for intramyocardial injection is 147 μm, therefore resuspended CPCeK are sufficiently small for adoptive transfer. Acutely elevated Notch activity in CPCeK by tamoxifen treatment increased cell area further by nearly fourfold within 2 days compared to PBS-treated CPCeK (Fig. 2g–i). Similarly, length-to-width ratio was decreased and roundness augmented, indicative of CPC whole cell responsiveness to Notch activation. CPCe morphology was unchanged in response to tamoxifen treatment (Figure S1C). These results demonstrate that elevated Notch activity directs CPC morphology away from the spindle shape associated with proliferative stem cells to a larger, flatter appearance indicative of a more differentiated phenotype.
Elevated Notch activity decreases proliferation in CPCs in vitro
Stem cell proliferation and growth in response to Notch signaling varies depending on cell type, context and signal dose [1,4]. Proliferation rate was assessed in CPCs treated with immobilized Jagged1 (CPCe JGD1) or genetically engineered intracellular Notch1 (CPCeK) using the CyQuant assay. Proliferation of CPCe decreased by nearly half after 3 days of exposure to Jagged1 ligand compared to BSA controls (Fig. 3a) while CPCeK proliferation decreased by one-third at 3 days relative to CPCe (Fig. 3b). Boosting Notch activity in CPCeK by tamoxifen treatment further blunted proliferation (Figure S1B), but had no effect on CPCe proliferation rate (Figure S1A, C). Interestingly, assay of cell growth by MTT, which measures metabolic activity, gave inverse results to the Cyquant assay such that CPCe JGD1 and CPCeK readings were one-third higher than CPCe BSA and CPCe controls at 3 days, respectively (Fig. 3c, d). Taken together, these results indicate that Notch signaling slows CPC proliferation and may increase metabolic activity in conjunction with increased size.
Fig. 3.
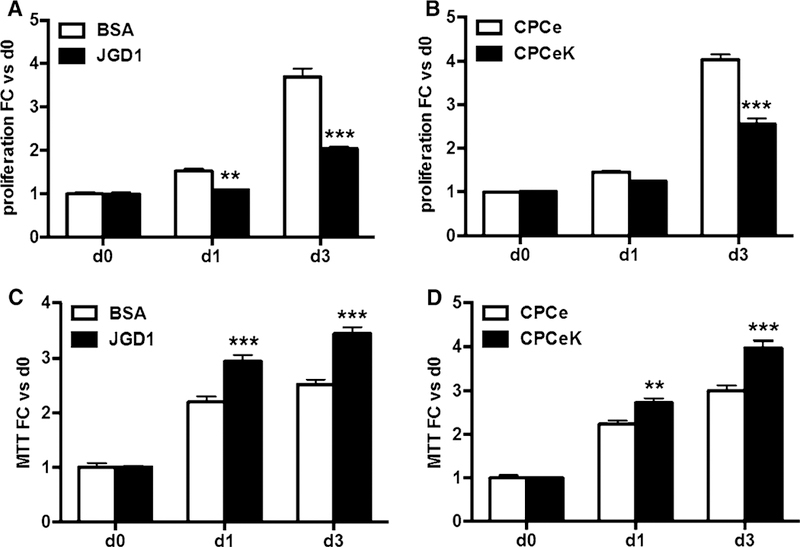
Notch activity decreases proliferation and increases metabolic activity in CPCs. Proliferation measured by CyQuant assay in CPCe cultured on BSA or Jagged1-coated surfaces (a) or in CPCe versus CPCeK (b). Metabolic activity in BSA versus JGD1-treated CPCe, and CPCe versus CPCeK as determined by MTT (c, d). Data combine at least three experiments, multiple replicates per data point. Significance determined by two-way ANOVA using Bonferroni’s multiple comparisons test. **p < 0.01, ***p < 0.001
Notch activity increases phosphorylation of mTORC1 and Akt in CPCs
The impact of Notch activity on CPC cell size and proliferation, and associated increase MTT values suggests that Notch upregulates growth signaling in CPCs. Therefore, levels of phosphorylated ribosomal S6 (rS6) and Akt were measured in CPCe JGD1 and CPCeK in low serum conditions. Jagged1-mediated induction of Notch signaling in CPCe resulted in a nearly eightfold increase in phospho-rS6 levels over BSA-treated controls and a modest but significant increase of one-third of phospho-Akt S473 (Fig. 4a). Levels of phospho-rS6 and phospho-Akt S473 were similarly elevated in CPCeK over CPCe (Fig. 4b). The effect persisted in full serum media, with a threefold increase in 70 % higher phosphor-Akt S473 in CPCeK versus CPCe (Figure S2A). Furthermore, increases in phospho-S6 kinase and phospho-4EBP1, but not phoshpo-ERK 1/2 (Figure S2B, C), in CPCeK substantiate that mTORC1 signaling is activated by Notch signaling in these cells and may be the growth signal driving CPCeK size increase. Notch signaling has been shown to activate protective Akt signaling in cardiomyocytes [18], therefore the impact of Notch activation on cell survival was assessed in CPC subjected to oxidative stress. The percentage of cells in late apoptosis coincident for apoptosis (Annexin V) and cell death (7-AAD) was measured by flow cytometry in CPCe, CPCe JGD1 and CPCeK treated with increasing doses of H2O2. Elevated Notch activity significantly suppressed cell death in both CPCe JGD1 and CPCeK; however, the protective effect was stronger in CPCeK (Fig. 4c, d). Collectively, these results demonstrate that Notch activation in CPCs stimulates growth and survival signaling, and mitigates cell death induced by oxidative stress.
Fig. 4.
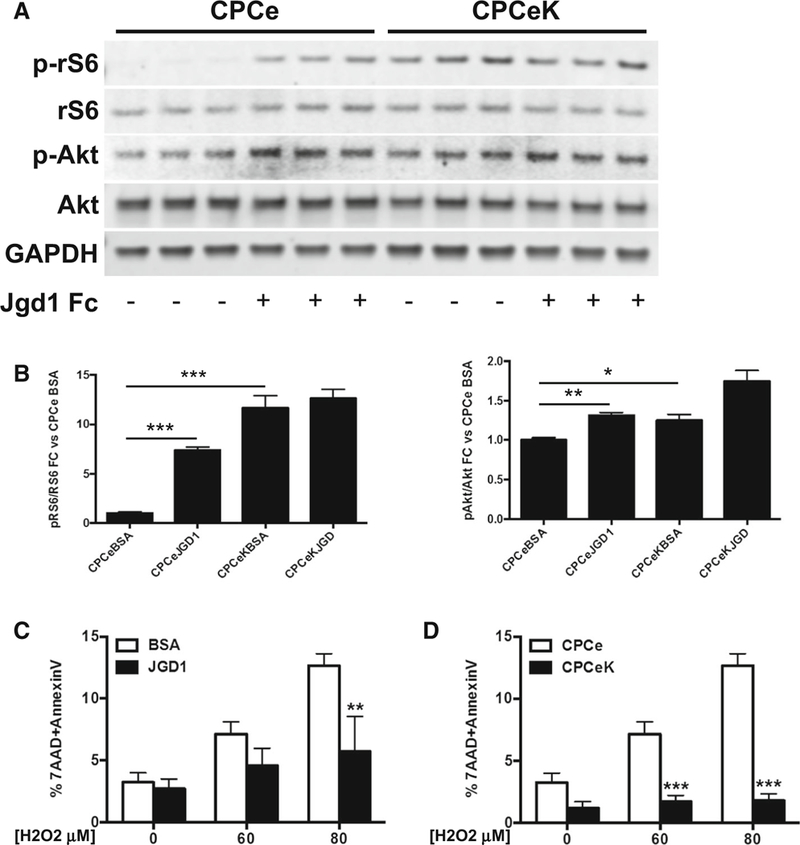
Notch signaling increases ribosomal S6 and Akt activity, and promotes resistance to oxidative stress in CPCs. Levels of phosphorylated ribosomal S6 (p-rS6) and Akt (p-Akt) in CPCe cultured on BSA versus Jaggedl (JGD1)-coated surfaces quantitated by immunoblot (a). Levels of p-rS6 and p-Akt in CPCe versus CPCeK quantitated by immunoblot (b). Cell death in response to H2O2 treatment at concentrations indicated was measured by flow cytometry analysis of AnnexinV and 7AAD signal in BSA versus JGDl-treated CPCe, and CPCe versus CPCeK (c, d). Significance for a and b determined by two-tailed t test. Significance for c and d determined by two-way ANOVA with Bonferroni’s multiple comparisons test. *p < 0.05, **p < 0.01, ***p < 0.001
Notch activation promotes expression of smooth muscle differentiation markers in CPCs in vitro
Notch signaling plays an established role in cardiovascular development [22, 27]. Given the impact of Notch activation on CPC size, proliferation and survival, we hypothesized that Notch signaling drives differentiation in CPCs. We therefore measured mRNA levels of known markers of cardiac differentiation in CPCe and CPCeK, with and without Jagged1 or tamoxifen treatment by qPCR analysis. Increased expression of hey1, hey2 and Jagged1, known targets of canonical Notch signaling, validated augmentation of Notch activity in CPCeK and in response to Jagged1 treatment (Figure S3A-C). Smooth muscle actin (sma), sm22 and myosin heavy chain 11 (MyHC11), all markers of vascular smooth muscle and early myocyte commitment, were significantly upregulated at the message level in CPCeK and Jagged1 stimulated CPCs versus controls, and tamoxifen induction in CPCeK further augmented expression of all three vascular smooth muscle genes (Fig. 5a–c). Interestingly, message levels of the Notch ligand Jagged1 also increased in response to Notch activity (Figure S3C), substantiating Jagged1 as a target of Notch involved in formation of vascular smooth muscle [38]. Likewise, expression of vascular smooth muscle protein calponin was clearly localized in CPCeK (Fig. 5e, white arrows), but not CPCe (Fig. 5d) following coculture with neonatal rat cardiomyocytes (NRCMs) as shown by immunostaining. Smooth muscle actin (SMA) was detected by immunoblot in CPCeK but not CPCe, and sarcomeric actinin levels were 45 % higher in Notch expressing cells (Fig. 5f). Expression of myocyte lineage markers GATA4 and MEF2C decreased with elevated Notch activity, as previously reported [4, 5]; however, cardiac troponin T mRNA increased significantly with elevated Notch signaling. Interestingly, vascular endothelial protein von Willebrand’s factor (vWF) levels were markedly decreased in CPCeK, as shown by immunoblot (Figure S4A–D). Paracrine survival signals contribute to protective effects following adoptive transfer of various progenitor cell types into damaged myocardium [16, 17, 25, 29, 47, 48]. Therefore, expression of several cytokines was measured by qPCR and immunoblot in Notch activated CPCs. Levels of IL-6, TGF1ß, and VEGF, paracrine factors associated with vascular smooth muscle, are higher with Notch activation (Fig. 6a–d). HGF mRNA expression was significantly downregulated in CPCe JGD1 and CPCeK (Figure S5A), consistent with suppression of HGF following Jagged1-mediated Notch signaling in COS-7 and MRC-5 cell lines [57]. Interestingly, expression of Wnt11 mRNA was dramatically upregulated in CPCeK following acute activation with tamoxifen. Wnt11, a noncanonical Wnt signaling protein, plays an important role in cardiomyogenesis during development, and supports cardiac lineage commitment in bone marrow mesenchymal stem cells [14]. Protein levels of cell cycle inhibitors p16 and p53 were significantly lower in CPCeK (Fig. 6e), suggesting that the increased size and decreased rate of proliferation observed in these cells are not consequences of cellular senescence. Collectively these data support the hypothesis that Notch activity drives differentiation of CPCs toward a vascular smooth muscle/early myocyte lineage in a dose-dependent manner, and that increased size and reduced proliferation rate are not due to senescence.
Fig. 5.
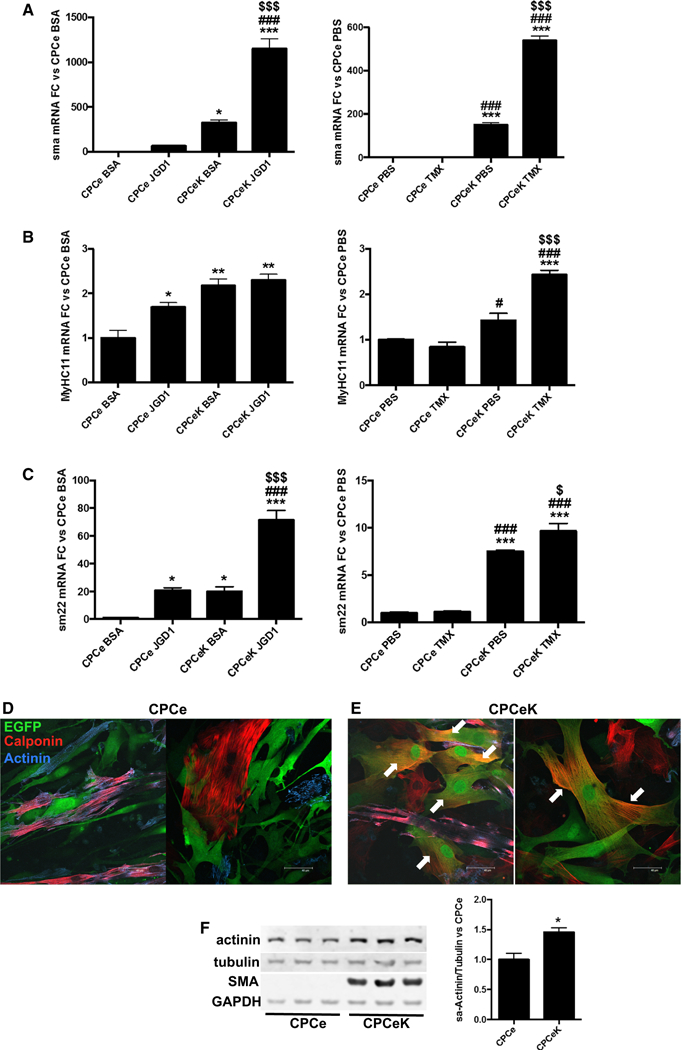
Smooth muscle differentiation markers are increased in CPCeK. Message levels of smooth muscle actin (sma, a), myosin heavy chain 11 (MyHC11, b) and sm22 (c) in response to Notch activity induced by Jaggedl treatment (CPCe JGD1) or overexpression of intracellular Notch (CPCeK PBS or CPCeK TMX) measured by qPCR. CPCeK treated with 1 μM tamoxifen (CPCeK TMX) display acute induction of Notch activity in CPCeK. Calponin (red) localizes to CPCeK (green, e) but not CPCe (green only, d) following coculture with NRCMs as detected by immunolabeling. Myocytes are labeled with alpha sarcomeric actinin (blue) and CPCe or CPCeK are identified by EGFP (green). Colocalization of EGFP and Calponin appears orange (white arrows). Protein levels of sarcomeric actinin and smooth muscle actin (SMA) in CPCe versus CPCeK, as measured by immunoblot (f). Significance in a-c determined by ordinary one-way ANOVA using Tukey’s multiple comparisons test. Significance in f determined by two-tailed t test. *p < 0.05, **p < 0.01, ***p < 0.001 versus CPCe BSA or CPCe PBS. #p < 0.05, ##p < 0.01, ###p < 0.001 versus CPCe JDG1 or CPCe TMX. $p < 0.05, $$p < 0.01, $$$p < 0.001 versus CPCeK BSA or CPCeK PBS. Scale bar 40 μM
Fig. 6.
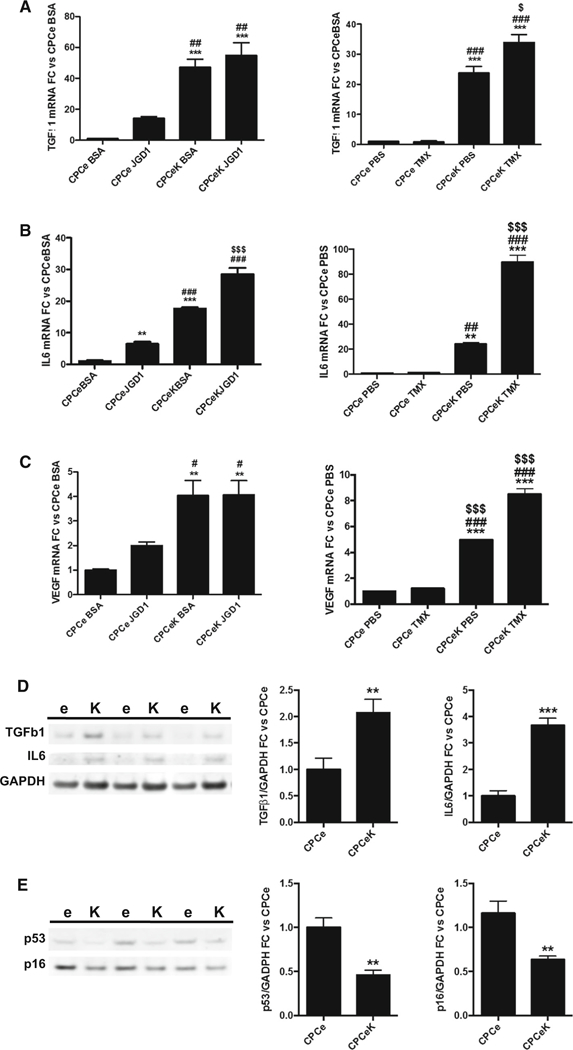
Notch activation induces expression of TGFßl, IL-6 and VEGF, but not p16 or p53. Message levels of TGFßl (a), IL6 (b) and VEGF (c) measured by qPCR in CPCe and CPCeK treated with Jagged1 or tamoxifen. Protein levels of TGFß1 and IL6 (d) and p53 and p16 (e) in CPCe and CPCeK quantitated by immunoblot. e CPCe, K CPCeK. Significance in a-c determined by ordinary one-way ANOVA using Tukey’s multiple comparisons test. **p < 0.01, ***p < 0.001 versus CPCe BSA or CPCe PBS. #p < 0.05, ##p < 0.01, ###p < 0.001 versus CPCe JDG1 or CPCe TMX. $p < 0.05, $ $ $p < 0.001 versus CPCeK BSA or CPCeK PBS. Significance for d and be determined by two-tailed t test. **p < 0.01, ***p < 0.001
Adoptive transfer of CPCeK into infarcted myocardium blunts loss of cardiac function
Given that CPCeK display enhanced growth, survival and differentiation over CPCe, adoptive transfer experiments were performed in infarcted mouse hearts comparing the regenerative potential of CPCe and CPCeK in a cell therapy application. Hearts receiving CPCeK at the time of infarction exhibited significantly decreased infarct length and area over hearts receiving CPCe, as measured from Trichrome staining (Fig. 7a). Heart weight-to-body weight ratios were also lower in CPCeK-treated hearts (Fig. 7b). Functional measurements by echocardiography (Fig. 7c) and in vivo hemodynamics (Fig. 7d) revealed significantly better function in CPCeK versus CPCe-treated hearts 6 weeks post-infarction. Furthermore, capillary density, as measured by isolectin B4 staining fluorescence and capillary numbers per field of view, were 35 % higher in CPCeK versus CPCe-treated hearts (Fig. 7e), while the percentage of tissue area positive for smooth muscle protein was 40 % higher with CPCeK treatment. Vessel length density (VLD) was 9 % higher in the remote region of CPCeK-treated hearts (Fig. 7f). Immunostaining for GFP did not reveal convincing evidence for engraftment of either CPCe or CPCeK (data not shown), perhaps due in part to low cell retention following injection [23]. These data indicate that the moderate but significantly increased protective effects of CPCeK may be mediated by enhanced growth and survival signaling following adoptive transfer, and possible stimulation of endogenous vascular repair mechanisms in CPCeK recipient hearts.
Fig. 7.
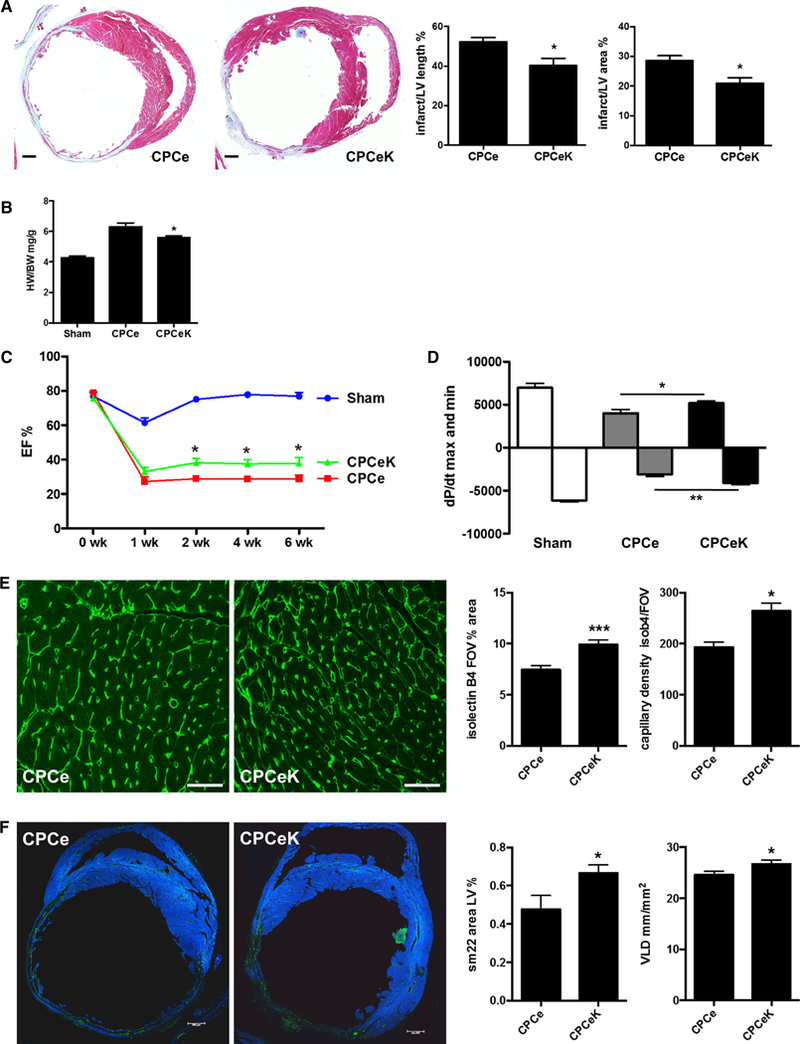
CPCeK provide enhanced protection to infarcted myocardium. Percentage of infarct length and area relative to total left ventricle (LV) in hearts receiving CPCe or CPCeK as measured by Masson’s Trichrome staining (a). Heart weight-to-body weight ratio in sham, CPCe and CPCeK-treated hearts (b). Cardiac function in CPCe and CPCeK-treated hearts during 6 weeks following infarction as measured by echocardiography (c) and in vivo hemodynamics (d). Capillary density in CPCe and CPCeK-treated hearts quantitated from isolectin B4 staining (e green, scale bar 40 μm). Area of smooth muscle in left ventricle as measured by sm22 staining (green, scale bar 500 μm) analyzed in NIH ImageJ, and vessel length density (VLD) as measured in isolectin B4 staining colocalized to smooth muscle actin analyzed in LASX (f). Significance between two groups was determined by two-tailed t test analysis. Significance between two or more groups over time was determined by two-way ANOVA. *p < 0.05, **p < 0.01. n = 4 for Sham and 9 for CPCe and CPCeK-treated groups for functional analyses
Discussion
Cardiac regeneration in adult mammals is restricted by the limited response of endogenous progenitor cells and proliferative cardiomyocytes to pathologic challenge. Recent clinical trials, in which heart failure patients showed functional improvement after receiving an intracoronary infusion of autologous cardiac stem cells, demonstrate feasibility and safety of cell-based therapy for the treatment of heart disease [3, 9, 36, 37]. However, cells derived from elderly or chronically ill patients may have compromised function, underscoring the need to enhance the reparative capacity of autologous adult stem cells. Modification of mouse and human CPCs with Pim1 survival kinase improves survival, engraftment and differentiation following adoptive transfer into infarcted mouse hearts, corroborating this approach for cell-based therapy [13, 40]. Within the cardiac stem cell niche, Notch/Jagged1 signaling mediates communication between niche support and stem cells to maintain a balance between self-renewal, proliferation and differentiation [4, 53]. In this study, CPCs engineered with activated Notch display morphological and molecular changes indicative of cardiac differentiation, and mitigate infarction damage, possibly through paracrine and survival signaling stimulated by elevated Notch activity.
Notch has been engineered into various stem cell types to drive proliferation, cardiac differentiation, or both. Forced expression of Notch4 receptor in mouse embryonic stem cells directs hemangioblasts to a cardiac cell fate, and transient overexpression of intracellular Notch in cardio-sphere-derived cells (CDCs) facilitates smooth muscle cell differentiation in vitro and in vivo [7, 8]. Likewise, overexpression of intracellular Notch1 in cardiac stem cells (CSCs) isolated from newborn mice stimulates differentiation into a transient amplifying pool of early myocytes [53]. Bone marrow from mice with a global hemizygous deletion in Notch1 (N1+/−) fails to decrease injury following transfer into infarcted wild-type hearts, while wild-type bone marrow transfer lessens injury in N1+/− infarcted myocardium [34]. Results presented here agree and conflict with these reports, such that Notch activation in adult CPCs decreases proliferation but drives differentiation toward a vascular smooth muscle and, to a lesser extent, myocyte phenotype in vitro. Cell origin and method of culture may explain these discrepancies. The CPCs described in this study are clonal stable lines created from adult mouse c-Kit+ selected cardiac progenitor cells. CPCeK lines exhibited a level of Notch activity consistent with cells subjected to ligand-mediated Jagged1 activation, driving morphological change while permitting proliferation. Deletion of Notch1 from fibroblasts has been reported to increase proliferation, while overexpression reverses the effect via induction of Wnt11 and downstream WISP1 [35]. Consistent with these findings, acute augmentation of Notch activity in CPCeK led to dramatic arrest of proliferation, increased cell size, and upregulation of Wnt11 expression, underscoring the sensitivity of these cells to Notch signaling levels and the potency of the Notch signal in progenitor cell differentiation and commitment.
Crosstalk between Notch and multiple signaling pathways plays a central role in stem cell proliferation, differentiation and survival [26]. Endogenous survival signaling in CPCs has been demonstrated in vivo [55] and in CPCs subjected to serum starvation, oxidative stress and hypoxic preconditioning [24, 28, 30, 50]. IGF-1-mediated Akt activation in CPCs isolated from patients undergoing cardiac bypass surgery was found to be one predictive indicator of postoperative cardiac remodeling [11]. Like-wise, we find here that elevated Notch activity enhances survival and growth signaling in CPCeK and CPCe JGD1, suggesting a role for Notch-mediated protection in adult CPCs. Moreover, adoptive transfer of CPCeK cells possessing enhanced growth and survival signaling may contribute to the improved cardiac function in recipient hearts. The paracrine profile for CPCeK is consistent with a vascular smooth muscle phenotype. Notch and TGFβ1 have been reported to promote differentiation of human mesenchymal stem cells (MSCs) into smooth muscle cell (SMCs) through induction of Notch ligand Jagged1 by TGFp [33]. IL-6 signaling through STAT3 decreases proliferation and promotes satellite stem cell myogenic differentiation [52]. Interestingly, results here indicate that Notch activation upregulates TGFβ1 and IL-6 message and protein levels, suggesting crosstalk among these signaling pathways during the CPC to SMC transition.
Combinatorial cell therapy is emerging as a promising strategy for boosting cardiac repair and regeneration by exploiting the regenerative potential of diverse, defined stem cell types [2, 56]. The improvement in cardiac function and vascular content of CPCeK-treated hearts does not appear to result from dramatically enhanced engraftment or differentiation of CPCeK at 6 weeks post-injection. Future studies investigating earlier time points after infarction and cell treatment will be required to assess the mechanistic role of Notch signaling on adoptive cell retention, proliferation and differentiation in vivo. However, the paracrine profile of CPCeK and resistance to oxidative stress suggest that CPCeK serve to protect recipient myocardium and stimulate endogenous cardiac repair mechanisms following injury. These properties, in addition to the predisposition toward a vascular smooth muscle phenotype, complement those of other modified cells such as CPCs expressing Pim1 kinase (CPCeP), which exhibit enhanced proliferation, endothelial commitment, engraftment and differentiation into cardiac myocytes. Concomitant administration of CPCeP and CPCeK would be expected to confer enhanced protection and repair beyond that observed with either cell type alone as a therapeutic treatment for cardiac injury.
Supplementary Material
Acknowledgments
Thank you to all members of the Sussman laboratory for critical reading of the manuscript. Thank you to Brett Collins and Daniela Michel for the excellent stewardship of the mouse colony. Thank you to Cameron Smurthwaite for assistance with flow cytometry performed in the SDSU FACS Core Facility. This study is supported by Grants of the National Institute of Health to Mark Sussman (R37HL091102, R01HL105759, R01HL067245, R01HL113647, R01HL117163, P01HL085577, R01HL122525), and to Pearl Quijada (F31HL117623). Grants of the American Heart Association support Natalie Gude (14BGIA187300511), Nirmala Hariharan (12P0ST12060191) and Haruhiro Toko (11P0ST7610164). Veronica Sacchi is funded by the Swiss National Science Foundation Fellowship (P2BSP3_155252). Mirko Voelkers was supported by the Deutsche Forschungsgemeinschaft DFG (1669/1–1). M. Villanueva is a SDSU/IMSD/MBRS scholar and CIRM undergraduate intern.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1007/s00395–015-0488–3) contains supplementary material, which is available to authorized users.
Conflict of interest
None.
References
- 1.Androutsellis-Theotokis A, Leker RR, Soldner F, Hoeppner DJ, Ravin R, Poser SW, Rueger MA, Bae SK, Kittappa R, McKay RD (2006) Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 442:823–826. doi: 10.1038/nature04940 [DOI] [PubMed] [Google Scholar]
- 2.Avolio E, Meloni M, Spencer HL, Riu F, Katare R, Mangialardi G, Oikawa A, Rodriguez-Arabaolaza I, Dang Z, Mitchell K, Reni C, Alvino VV, Rowlinson JM, Livi U, Cesselli D, Angelini G, Emanueli C, Beltrami AP, Madeddu PR (2015) Combined intramyocardial delivery of human pericytes and cardiac stem cells additively improves the healing of mouse infarcted hearts through stimulation of vascular and muscular repair. Circ Res. doi: 10.1161/CIRCRESAHA.115.306146 [DOI] [PubMed] [Google Scholar]
- 3. Bolli R, Chugh AR, D’Amario D, Loughran JH, Stoddard MF, Ikram S, Beache GM, Wagner SG, Leri A, Hosoda T, Sanada F, Elmore JB, Goichberg P, Cappetta D, Solankhi NK, Fahsah I, Rokosh DG, Slaughter MS, Kajstura J, Anversa P (2011) Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPI0): initial results of a randomised phase 1 trial. Lancet 378:1847–1857. doi: 10.1016/S0140-6736(11)61590-0 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 4.Boni A, Urbanek K, Nascimbene A, Hosoda T, Zheng H, Delucchi F, Amano K, Gonzalez A, Vitale S, Ojaimi C, Rizzi R, Bolli R, Yutzey KE, Rota M, Kajstura J, Anversa P, Leri A (2008) Notch1 regulates the fate of cardiac progenitor cells. Proc Natl Acad Sci USA 105:15529–15534. doi: 10.1073/pnas.0808357105 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Buas MF, Kabak S, Kadesch T (2010) The Notch effector Hey1 associates with myogenic target genes to repress myogenesis. J Biol Chem 285:1249–1258. doi: 10.1074/jbc.M109.046441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campa VM, Gutierrez-Lanza R, Cerignoli F, Diaz-Trelles R, Nelson B, Tsuji T, Barcova M, Jiang W, Mercola M (2008) Notch activates cell cycle reentry and progression in quiescent cardiomyocytes. J Cell Biol 183:129–141. doi: 10.1083/jcb.200806104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen L, Ashraf M, Wang Y, Zhou M, Zhang J, Qin G, Rubinstein J, Weintraub NL, Tang Y (2012) The role of Notch 1 activation in cardiosphere derived cell differentiation. Stem Cells Dev 21:2122–2129. doi: 10.1089/scd.2011.0463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen VC, Stull R, Joo D, Cheng X, Keller G (2008) Notch signaling respecifies the hemangioblast to a cardiac fate. Nat Biotechnol 26:1169–1178. doi: 10.1038/nbt.1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chugh AR, Beache GM, Loughran JH, Mewton N, Elmore JB, Kajstura J, Pappas P, Tatooles A, Stoddard MF, Lima JA, Slaughter MS, Anversa P, Bolli R (2012) Administration of cardiac stem cells in patients with ischemic cardiomyopathy: the SCIPIO trial: surgical aspects and interim analysis of myocardial function and viability by magnetic resonance. Circulation 126:S54–S64. doi: 10.1161/CIRCULATI0NAHA.112.092627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Croquelois A, Domenighetti AA, Nemir M, Lepore M, Rosen-blatt-Velin N, Radtke F, Pedrazzini T (2008) Control of the adaptive response of the heart to stress via the Notch1 receptor pathway. J Exp Med 205:3173–3185. doi: 10.1084/jem.20081427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D’Amario D, Leone AM, Iaconelli A, Luciani N, Gaudino M, Kannappan R, Manchi M, Severino A, Shin SH, Graziani F, Biasillo G, Macchione A, Smaldone C, De Maria GL, Cellini C, Siracusano A, Ottaviani L, Massetti M, Goichberg P, Leri A, Anversa P, Crea F (2014) Growth properties of cardiac stem cells are a novel biomarker of patients’ outcome after coronary bypass surgery. Circulation 129:157–172. doi: 10.1161/CIRCULATI0NAHA.113.006591 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.de la Pompa JL, Epstein JA (2012) Coordinating tissue interactions: Notch signaling in cardiac development and disease. Dev Cell 22:244–254. doi: 10.1016/j.devcel.2012.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischer KM, Cottage CT, Wu W, Din S, Gude NA, Avitabile D, Quijada P, Collins BL, Fransioli J, Sussman MA (2009) Enhancement of myocardial regeneration through genetic engineering of cardiac progenitor cells expressing Pim-1 kinase. Circulation 120:2077–2087. doi: 10.1161/CIRCULATI0NAHA.109.884403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flaherty MP, Kamerzell TJ, Dawn B (2012) Wnt signaling and cardiac differentiation. Prog Mol Biol Transl Sci 111:153–174. doi: 10.1016/B978-0-12-398459-3.00007-1 [DOI] [PubMed] [Google Scholar]
- 15.Fransioli J, Bailey B, Gude NA, Cottage CT, Muraski JA, Emmanuel G, Wu W, Alvarez R, Rubio M, Ottolenghi S, Schaefer E, Sussman MA (2008) Evolution of the c-kit-positive cell response to pathological challenge in the myocardium. Stem Cells 26:1315–1324. doi: 10.1634/stemcells.2007-0751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gnecchi M, He H, Liang OD, Melo LG, Morello F, Mu H, Noiseux N, Zhang L, Pratt RE, Ingwall JS, Dzau VJ (2005) Paracrine action accounts for marked protection of ischemic heart by Akt-modified mesenchymal stem cells. Nat Med 11:367–368. doi: 10.1038/nm0405-367 [DOI] [PubMed] [Google Scholar]
- 17.Gnecchi M, Zhang Z, Ni A, Dzau VJ (2008) Paracrine mechanisms in adult stem cell signaling and therapy. Circ Res 103:1204–1219. doi: 10.1161/CIRCRESAHA.108.176826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gude NA, Emmanuel G, Wu W, Cottage CT, Fischer K, Quijada P, Muraski JA, Alvarez R, Rubio M, Schaefer E, Sussman MA (2008) Activation of Notch-mediated protective signaling in the myocardium. Circ Res 102:1025–1035. doi: 10.1161/CIRCRESAHA.107.164749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guentchev M, McKay RD (2006) Notch controls proliferation and differentiation of stem cells in a dose-dependent manner. Eur J Neurosci 23:2289–2296. doi: 10.1111/j.1460-9568.2006.04766.x [DOI] [PubMed] [Google Scholar]
- 20.Heusch G (2011) SCIPIO brings new momentum to cardiac cell therapy. Lancet 378:1827–1828. doi: 10.1016/S0140-6736(11)61648-6 [DOI] [PubMed] [Google Scholar]
- 21.High FA, Epstein JA (2008) The multifaceted role of Notch in cardiac development and disease. Nat Rev Genet 9:49–61. doi: 10.1038/nrg2279 [DOI] [PubMed] [Google Scholar]
- 22.High FA, Lu MM, Pear WS, Loomes KM, Kaestner KH, Epstein JA (2008) Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc Natl Acad Sci USA 105:1955–1959. doi: 10.1073/pnas.0709663105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong KU, Li QH, Guo Y, Patton NS, Moktar A, Bhatnagar A, Bolli R (2013) A highly sensitive and accurate method to quantify absolute numbers of c-kit+ cardiac stem cells following transplantation in mice. Basic Res Cardiol 108:346. doi: 10.1007/s00395-013-0346-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu S, Yan G, Xu H, He W, Liu Z, Ma G (2014) Hypoxic pre-conditioning increases survival of cardiac progenitor cells via the pim-1 kinase-mediated anti-apoptotic effect. Circ J 78:724–731 [DOI] [PubMed] [Google Scholar]
- 25.Huang C, Gu H, Yu Q, Manukyan MC, Poynter JA, Wang M (2011) Sca-1 + cardiac stem cells mediate acute cardioprotection via paracrine factor SDF-1 following myocardial ischemia/reperfusion. PLoS One 6:e29246. doi: 10.1371/journal.pone.0029246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hurlbut GD, Kankel MW, Lake RJ, Artavanis-Tsakonas S (2007) Crossing paths with Notch in the hyper-network. Curr Opin Cell Biol 19:166–175. doi: 10.1016/j.ceb.2007.02.012 [DOI] [PubMed] [Google Scholar]
- 27.Jain R, Rentschler S, Epstein JA (2010) Notch and cardiac out-flow tract development. Ann N Y Acad Sci 1188:184–190. doi: 10.1111/j.1749-6632.2009.05099.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson AM, Kartha CC (2014) Proliferation of murine c-kit(pos) cardiac stem cells stimulated with IGF-1 is associated with Akt-1 mediated phosphorylation and nuclear export of FoxO3a and its effect on downstream cell cycle regulators. Growth Factors 32:53–62. doi: 10.3109/08977194.2014.889694 [DOI] [PubMed] [Google Scholar]
- 29.Kishore R, Verma SK, Mackie AR, Vaughan EE, Abramova TV, Aiko I, Krishnamurthy P (2013) Bone marrow progenitor cell therapy-mediated paracrine regulation of cardiac miRNA-155 modulates fibrotic response in diabetic hearts. PLoS One 8:e60161. doi: 10.1371/journal.pone.0060161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Konstandin MH, Toko H, Gastelum GM, Quijada P, De La Torre A, Quintana M, Collins B, Din S, Avitabile D, Volkers M, Gude N, Fassler R, Sussman MA (2013) Fibronectin is essential for reparative cardiac progenitor cell response after myocardial infarction. Circ Res 113:115–125. doi: 10.1161/CIRCRESAHA.113.301152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koyanagi M, Bushoven P, Iwasaki M, Urbich C, Zeiher AM, Dimmeler S (2007) Notch signaling contributes to the expression of cardiac markers in human circulating progenitor cells. Circ Res 101:1139–1145. doi: 10.1161/CIRCRESAHA.107.151381 [DOI] [PubMed] [Google Scholar]
- 32.Kratsios P, Catela C, Salimova E, Huth M, Berno V, Rosenthal N, Mourkioti F (2010) Distinct roles for cell-autonomous Notch signaling in cardiomyocytes of the embryonic and adult heart. Circ Res 106:559–572. doi: 10.1161/CIRCRESAHA.109.203034 [DOI] [PubMed] [Google Scholar]
- 33.Kurpinski K, Lam H, Chu J, Wang A, Kim A, Tsay E, Agrawal S, Schaffer DV, Li S (2010) Transforming growth factor-beta and Notch signaling mediate stem cell differentiation into smooth muscle cells. Stem Cells 28:734–742. doi: 10.1002/stem.319 [DOI] [PubMed] [Google Scholar]
- 34.Li Y, Hiroi Y, Ngoy S, Okamoto R, Noma K, Wang CY, Wang HW, Zhou Q, Radtke F, Liao R, Liao JK (2011) Notch1 in bone marrow-derived cells mediates cardiac repair after myocardial infarction. Circulation 123:866–876. doi: 10.1161/CIRCULATI0NAHA.110.947531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu ZJ, Li Y, Tan Y, Xiao M, Zhang J, Radtke F, Velazquez OC (2012) Inhibition of fibroblast growth by Notch1 signaling is mediated by induction of Wnt11-dependent WISP-1. PLoS One 7:e38811. doi: 10.1371/journal.pone.0038811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Makkar RR, Smith RR, Cheng K, Malliaras K, Thomson LE, Berman D, Czer LS, Marban L, Mendizabal A, Johnston PV, Russell SD, Schuleri KH, Lardo AC, Gerstenblith G, Marban E (2012) Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet 379:895–904. doi: 10.1016/S0140-6736(12)60195-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malliaras K, Makkar RR, Smith RR, Cheng K, Wu E, Bonow RO, Marban L, Mendizabal A, Cingolani E, Johnston PV, Ger-stenblith G, Schuleri KH, Lardo AC, Marban E (2014) Intra-coronary cardiosphere-derived cells after myocardial infarction: evidence of therapeutic regeneration in the final 1-year results of the CADUCEUS trial (CArdiosphere-Derived aUtologous stem CElls to reverse ventricUlar dySfunction). J Am Coll Cardiol 63:110–122. doi: 10.1016/j.jacc.2013.08.724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manderfield LJ, High FA, Engleka KA, Liu F, Li L, Rentschler S, Epstein JA (2012) Notch activation of Jagged1 contributes to the assembly of the arterial wall. Circulation 125:314–323. doi: 10.1161/CIRCULATI0NAHA.111.047159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mohsin S, Khan M, Nguyen J, Alkatib M, Siddiqi S, Hariharan N, Wallach K, Monsanto M, Gude N, Dembitsky W, Sussman MA (2013) Rejuvenation of human cardiac progenitor cells with Pim-1 kinase. Circ Res 113:1169–1179. doi: 10.1161/CIRCRESAHA.113.302302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mohsin S, Khan M, Toko H, Bailey B, Cottage CT, Wallach K, Nag D, Lee A, Siddiqi S, Lan F, Fischer KM, Gude N, Quijada P, Avitabile D, Truffa S, Collins B, Dembitsky W, Wu JC, Sussman MA (2012) Human cardiac progenitor cells engineered with Pim-I kinase enhance myocardial repair. J Am Coll Cardiol 60:1278–1287. doi: 10.1016/j.jacc.2012.04.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mohsin S, Siddiqi S, Collins B, Sussman MA (2011) Empowering adult stem cells for myocardial regeneration. Circ Res 109:1415–1428. doi: 10.1161/CIRCRESAHA.111.243071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nemir M, Metrich M, Plaisance I, Lepore M, Cruchet S, Berthonneche C, Sarre A, Radtke F, Pedrazzini T (2012) The Notch pathway controls fibrotic and regenerative repair in the adult heart. Eur Heart J. doi: 10.1093/eurheartj/ehs269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nurzynska D, Di Meglio F, Romano V, Miraglia R, Sacco AM, Latino F, Bancone C, Della Corte A, Maiello C, Amarelli C, Montagnani S, Castaldo C (2013) Cardiac primitive cells become committed to a cardiac fate in adult human heart with chronic ischemic disease but fail to acquire mature phenotype: genetic and phenotypic study. Basic Res Cardiol 108:320. doi: 10.1007/s00395-012-0320-2 [DOI] [PubMed] [Google Scholar]
- 44.Oie E, Sandberg WJ, Ahmed MS, Yndestad A, Laerum OD, Attramadal H, Aukrust P, Eiken HG (2010) Activation of Notch signaling in cardiomyocytes during post-infarction remodeling. Scand Cardiovasc J 44:359–366. doi: 10.3109/14017431.2010.511256 [DOI] [PubMed] [Google Scholar]
- 45.Pedrazzini T (2007) Control of cardiogenesis by the notch pathway. Trends Cardiovasc Med 17:83–90. doi: 10.1016/j.tcm.2007.01.003 [DOI] [PubMed] [Google Scholar]
- 46.Pei H, Yu Q, Xue Q, Guo Y, Sun L, Hong Z, Han H, Gao E, Qu Y, Tao L (2013) Notch1 cardioprotection in myocardial ischemia/reperfusion involves reduction of oxidative/nitrative stress. Basic Res Cardiol 108:373. doi: 10.1007/s00395-013-0373-x [DOI] [PubMed] [Google Scholar]
- 47.Quijada P, Sussman MA (2014) Making it stick: chasing the optimal stem cells for cardiac regeneration. Expert Rev Cardio-vasc Ther 12:1275–1288. doi: 10.1586/14779072.2014.972941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rogers TB, Pati S, Gaa S, Riley D, Khakoo AY, Patel S, Wardlow RD, 2nd Frederick CA, Hall G, He LP, Lederer WJ(2011) Mesenchymal stem cells stimulate protective genetic re-programming of injured cardiac ventricular myocytes. J Mol Cell Cardiol 50:346–356. doi: 10.1016/j.yjmcc.2010.09.001 [DOI] [PubMed] [Google Scholar]
- 49.Sacchi V, Mittermayr R, Hartinger J, Martino MM, Lorentz KM, Wolbank S, Hofmann A, Largo RA, Marschall JS, Groppa E, Gianni-Barrera R, Ehrbar M, Hubbell JA, Redl H, Banfi A (2014) Long-lasting fibrin matrices ensure stable and functional angio-genesis by highly tunable, sustained delivery of recombinant VEGF164. Proc Natl Acad Sci USA 111:6952–6957. doi: 10.1073/pnas.1404605111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sanada F, Kim J, Czarna A, Chan NY, Signore S, Ogorek B, Isobe K, Wybieralska E, Borghetti G, Pesapane A, Sorrentino A, Mangano E, Cappetta D, Mangiaracina C, Ricciardi M, Cimini M, Ifedigbo E, Perrella MA, Goichberg P, Choi AM, Kajstura J, Hosoda T, Rota M, Anversa P, Leri A (2014) c-Kit-positive cardiac stem cells nested in hypoxic niches are activated by stem cell factor reversing the aging myopathy. Circ Res 114:41–55. doi: 10.1161/CIRCRESAHA.114.302500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Siddiqi S, Sussman MA (2013) Cell and gene therapy for severe heart failure patients: the time and place for Pim-1 kinase. Expert Rev Cardiovasc Ther 11:949–957. doi: 10.1586/14779072.2013.814830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tierney MT, Aydogdu T, Sala D, Malecova B, Gatto S, Puri PL, Latella L, Sacco A (2014) STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat Med 20:1182–1186. doi: 10.1038/nm.3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Urbanek K, Cabral-da-Silva MC, Ide-Iwata N, Maestroni S, Delucchi F, Zheng H, Ferreira-Martins J, Ogorek B, D’Amario D, Bauer M, Zerbini G, Rota M, Hosoda T, Liao R, Anversa P, Kajstura J, Leri A (2010) Inhibition of notch1-dependent cardiomyogenesis leads to a dilated myopathy in the neonatal heart. Circ Res 107:429–441. doi: 10.1161/CIRCRESAHA.110.218487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Urbanek K, Cesselli D, Rota M, Nascimbene A, De Angelis A, Hosoda T, Bearzi C, Boni A, Bolli R, Kajstura J, Anversa P, Leri A (2006) Stem cell niches in the adult mouse heart. Proc Natl Acad Sci USA 103:9226–9231. doi: 10.1073/pnas.0600635103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Urbanek K, Rota M, Cascapera S, Bearzi C, Nascimbene A, De Angelis A, Hosoda T, Chimenti S, Baker M, Limana F, Nurzynska D, Torella D, Rotatori F, Rastaldo R, Musso E, Quaini F, Leri A, Kajstura J, Anversa P (2005) Cardiac stem cells possess growth factor-receptor systems that after activation regenerate the infarcted myocardium, improving ventricular function and long-term survival. Circ Res 97:663–673. doi: 10.1161/01.RES.0000183733.53101.11 [DOI] [PubMed] [Google Scholar]
- 56.Williams AR, Hatzistergos KE, Addicott B, McCall F, Carvalho D, Suncion V, Morales AR, Da Silva J, Sussman MA, Heldman AW, Hare JM (2013) Enhanced effect of combining human cardiac stem cells and bone marrow mesenchymal stem cells to reduce infarct size and to restore cardiac function after myocardial infarction. Circulation 127:213–223. doi: 10.1161/CIRCULATI0NAHA.112.131110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yuan ZR, Kobayashi N, Kohsaka T (2006) Human Jagged 1 mutants cause liver defect in Alagille syndrome by overexpression of hepatocyte growth factor. J Mol Biol 356:559–568. doi: 10.1016/j.jmb.2005.11.097 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.