

Abstract
An important aspect of the interaction between the opportunistic bacterial pathogen Streptococcus pneumoniae and its human host is its ability to harvest host glycans. The pneumococcus can degrade a variety of complex glycans, including N- and O-linked glycans, glycosaminoglycans, and carbohydrate antigens, an ability that is tightly linked to the virulence of S. pneumoniae. Although S. pneumoniae is known to use a sophisticated enzyme machinery to attack the human glycome, how it copes with fucosylated glycans, which are primarily histo-blood group antigens, is largely unknown. Here, we identified two pneumococcal enzymes, SpGH29C and SpGH95C, that target α-(1→3/4) and α-(1→2) fucosidic linkages, respectively. X-ray crystallography studies combined with functional assays revealed that SpGH29C is specific for the LewisA and LewisX antigen motifs and that SpGH95C is specific for the H(O)-antigen motif. Together, these enzymes could defucosylate LewisY and LewisB antigens in a complementary fashion. In vitro reconstruction of glycan degradation cascades disclosed that the individual or combined activities of these enzymes expose the underlying glycan structure, promoting the complete deconstruction of a glycan that would otherwise be resistant to pneumococcal enzymes. These experiments expand our understanding of the extensive capacity of S. pneumoniae to process host glycans and the likely roles of α-fucosidases in this. Overall, given the importance of enzymes that initiate glycan breakdown in pneumococcal virulence, such as the neuraminidase NanA and the mannosidase SpGH92, we anticipate that the α-fucosidases identified here will be important factors in developing more refined models of the S. pneumoniae–host interaction.
Keywords: Streptococcus, glycoside hydrolase, host-pathogen interaction, structure-function, X-ray crystallography
Introduction
The structural repertoire of glycans present in the human glycome is diverse, with the glycoconjugates that carry these glycans also being highly abundant. Approximately 1% of the human genome is dedicated to the synthesis and modification of glycans, and most human proteins are thought to be glycosylated (1, 2). Common human glycans include N- and O-linked glycans, histo-blood group antigens, glycosaminoglycans, glycogen, and the glycan families attached to glycosphingolipids (3). These glycans, both secreted and conjugated, have numerous important and varied functions. These include cell–cell interactions and cellular signaling; glycans also influence folding, stability, and function of glycoproteins (4). Commensurate with the importance and abundance of the human glycome, many commensal and pathogenic organisms have evolved strategies to degrade, transport, and process human glycans (e.g. Refs. 5–7).
One bacterium that is particularly adept at harvesting human glycans is Streptococcus pneumoniae (8–10). This commensal bacterium frequently inhabits the human nasopharynx and upper respiratory tract; however, it is also an important etiological agent of a number of serious and potentially life-threatening diseases such as pneumonia, bacteremia, and meningitis (11). Among respiratory pathogens, S. pneumoniae is unique in its capacity to degrade, transport, and metabolize a wide range of complex carbohydrates, most of which are host-derived (9, 12). This ability to break down and transport human glycans has been identified as a key virulence mechanism within this bacterium (13–15), contributing to nutrient acquisition, the uncovering of host receptors required for adherence and invasion, and immune modulation through the deglycosylation of host glycoproteins (16–18). The human respiratory tract is rich in functionally important glycoconjugates that bear a range of glycans, including complex, high-mannose, and hybrid N-linked glycans; core 1– and core 2–type O-linked glycans; and histo-blood group capping motifs (19). These glycan structures and those present on other glycoconjugates, such as glycosphingolipids, are also found disseminated throughout the human body and at sites of invasive pneumococcal disease, for example on the surface of erythrocytes and immune cells, and in the brain (20). Together, these glycans contain more than 20 different linkages among the monosaccharides N-acetylneuraminic acid (sialic acid), d-galactose, N-acetyl-d-glucosamine (GlcNAc),3 N-acetyl-d-galactosamine (GalNAc), d-mannose, d-glucose, and l-fucose. The S. pneumoniae genome encodes for more than 40 known or predicted proteins that break glycosidic bonds, the majority of which are glycoside hydrolases (GHs) (10). Many of these GHs have now been functionally characterized and found to cleave one or more of the above linkages as well as contribute directly to the virulence of this pathogen (10). Therefore, a comprehensive picture of human glycan degradation by S. pneumoniae is now emerging.
The 14 human glycan-specific pneumococcal GHs that have been functionally characterized to date include exo-α-sialidases (21, 22), exo- and endo-β-galactosidases (16, 23, 24), exo-α-mannosidases (25, 26), exo- and endo-β-N-acetylglucosaminidases (27, 28), an endo-β-N-acetylgalactosaminidase (29), and a general exo-β-N-acetylhexosaminidase (30) that participate in the degradation of N-glycans, O-glycans, histo-blood group antigens, and glycosphingolipids (10). α-Linked fucose is a core component of histo-blood group antigens and a frequent “capping” residue found on other human glycans (20, 31, 32); however, a pneumococcal α-fucosidase has yet to be functionally identified.
Fucose is attached to human glycans via α-(1→2), α-(1→3), α-(1→4), and α-(1→6) linkages, with the latter found as decorations on the core GlcNAc of N-glycans (33). Fucose residues attached via linkages other than α-(1→6) are typically found in the histo-blood group antigens, which comprise the A, B, and O antigens, and four Lewis antigens, LewisA, LewisB, LewisX, and LewisY. ABO and Lewis antigens are commonly observed as capping motifs on the arms of N- and O-glycans, as well as glycosphingolipids, in a wide variety of tissues (19, 34). As the majority of pneumococcal GHs are exoglycosidases, the presence of fucose as a capping residue on these glycans would likely necessitate the deployment of an enzyme, or enzymes, that can remove fucose residues, thereby allowing other pneumococcal GHs to access the main glycan.
We have recently identified a highly conserved carbohydrate-processing locus in S. pneumoniae (26). Located within this locus is SP_2146 (TIGR4 locus tag), a gene encoding for a putative α-fucosidase belonging to GH family 29. This gene (and its protein product, herein referred to as SpGH29C; superscript “C” for belonging to the core genome) has been identified as a putative virulence factor in multiple signature-tagged mutagenesis studies of pneumococcal disease and is a component of the core pneumococcal genome (13, 14, 35). A second putative α-fucosidase belonging to GH family 95 is also encoded by the core genome (locus tag SP_1654, herein referred to as SpGH95C). Like SpGH29C, SpGH95C has been identified as a putative virulence factor in multiple signature-tagged mutagenesis studies (13, 37). SpGH95C does not reside within an operon or functional locus; the only protein predicted to be functionally associated with SpGH95C via STRING analysis with a score of 0.95 (38) is SpGH29C. Given the classification of SpGH29C and SpGH95C into GH families 29 and 95, respectively (39), and their predicted functional association, we hypothesized that these two enzymes are α-fucosidases with complementary linkage specificities. Here, we show that SpGH29C and SpGH95C are indeed α-fucosidases with differing linkage specificities, that they are active against histo-blood group antigens, and that together they act as keystone enzymes to “uncap” fucosylated human glycans, enabling complete depolymerization by other enzymes. By recapitulating pneumococcal glycan degradation pathways in vitro, we also demonstrate the competence of S. pneumoniae to degrade a wide range of human glycans.
Results
Pneumococcal α-fucosidases active on histo-blood group antigens
To test our hypothesis that SpGH29C and SpGH95C are α-fucosidases with complementary linkage specificities, we produced the proteins recombinantly in Escherichia coli. Following purification, neither enzyme exhibited activity against 4-nitrophenyl α-l-fucopyranoside with substrate concentrations in the mm range (data not shown); however, a screen against histo-blood group antigens and other α-fucosylated glycans by TLC revealed that SpGH29C and SpGH95C have α-fucosidase activity (Table 1 and Fig. S1). SpGH29C displayed activity against substrates containing α-(1→3)– and α-(1→4)–linked fucose units, including 3-fucosyllactose and the four Lewis antigens; however, it was unable to cleave fucose from substrates smaller than a trisaccharide. In contrast, SpGH95C exhibited exclusive activity against substrates containing α-(1→2)–linked fucose units (namely 2-fucosyllactose, blood group H-antigens, LewisY, and LewisB), and it was able to cleave a disaccharide substrate. Activity on the H-disaccharide (Fuc-α-(1→2)-Gal) but lack of activity on 4-nitrophenyl α-l-fucopyranoside suggests quite strict accommodation of the residue preceding the terminal fucose. Likewise, despite the presence of α-(1→2)–linked fucose units, the blood group A- and B-antigens were resistant to SpGH95C activity (Table 1 and Fig. S1).
Table 1.
Activity of SpGH29C and SpGH95C against fucose-containing glycans
ND, not determined; N/A, not applicable.
| Substrate |
SpGH29C |
SpGH95C |
||
|---|---|---|---|---|
| Activity by TLCa | kcat/Km ± S.E. | Activity by TLCa | kcat/Km ± S.E. | |
| min−1 mm−1 | min−1 mm−1 | |||
| Fuc-α-(1→3)-GlcNAc | − | N/A | − | N/A |
| Fuc-α-(1→4)-GlcNAc | − | N/A | − | N/A |
| Fuc-α-(1→6)-GlcNAc | − | N/A | − | N/A |
| 2-fucosyllactose Gal-β-(1→4)-[Fuc-α-(1→2)]-Glc | − | N/A | + | 67.3 ± 1.9 |
| 3-fucosyllactose Gal-β-(1→4)-[Fuc-α-(1→3)]-Glc] | + | 8.9 ± 0.1 | − | N/A |
| Type II A-tetrasaccharide GalNAc-α-(1→3)-[Fuc-α-(1→2)]-Gal-β-(1→4)-GlcNAc | ND | N/A | − | N/A |
| Type II B-tetrasaccharide Gal-α-(1→3)-[Fuc-α-(1→2)]-Gal-β-(1→4)-GlcNAc | ND | N/A | − | N/A |
| H-disaccharide Fuc-α-(1→2)-Gal | − | N/A | + | 10.0 ± 0.2 |
| Type I H-trisaccharide Fuc-α-(1→2)-Gal-β-(1→3)-GlcNAc | ND | N/A | ND | 23.5 ± 0.5 |
| Type II H-trisaccharide Fuc-α-(1→2)-Gal-β-(1→4)-GlcNAc | − | N/A | + | 128.5 ± 3.9 |
| Type IV H-tetrasaccharide Fuc-α-(1→2)-Gal-β-(1→3)-GalNAc-β-(1→3)-Gal | ND | N/A | ND | 40.2 ± 0.6 |
| LewisA trisaccharide Gal-β-(1→3)-[Fuc-α-(1→4)]-GlcNAc | + | 16.3 ± 0.1 | − | N/A |
| LewisB tetrasaccharide Fuc-α-(1→2)-Gal-β-(1→3)-[Fuc-α-(1→4)]-GlcNAc | + | 14.3 ± 0.2 | + | 8.1 ± 0.1 |
| LewisX trisaccharide Gal-β-(1→4)-[Fuc-α-(1→3)]-GlcNAc | + | 19.3 ± 0.6 | − | N/A |
| LewisY tetrasaccharide Fuc-α-(1→2)-Gal-β-(1→4)-[Fuc-α-(1→3)]-GlcNAc | + | 21.1 ± 0.6 | + | 7.5 ± 0.1 |
a See Fig. S1 for TLC images. +/− indicate presence/absence of activity.
We followed up this initial activity screen by determining the kinetic parameters of SpGH29C and SpGH95C against relevant α-fucosylated substrates using an enzyme-coupled fucose detection assay (40) (see “Experimental procedures” for details). Both SpGH29C and SpGH95C exhibited a linear increase in initial velocity with increasing substrate concentration for a number of substrates; therefore, precise Km values could not be determined. However, kcat/Km values for each substrate–enzyme combination were determined (Table 1). SpGH29C exhibited very similar kcat/Km values for all four Lewis antigens; therefore, it demonstrated no significant preference for glycan size (trisaccharide or tetrasaccharide) or fucose linkage (α-(1→3) or α-(1→4)). Conversely, SpGH95C exhibited kcat/Km values that varied by up to 17-fold among substrates depending on the size and configuration of the glycan. All of the substrates for SpGH95C contained the same core H-motif. The H-disaccharide acted as a substrate for SpGH95C with a kcat/Km of 10.0 ± 0.2 min−1 mm−1 (±S.E.; Table 1). The linkage of a glucose or GlcNAc unit to the galactose on this H-disaccharide motif resulted in a 2–10-fold increase in kcat/Km. Specifically, addition of a GlcNAc residue via a β-(1→3) linkage (H-trisaccharide type I) resulted in an ∼2-fold increase in kcat/Km, whereas addition of this same residue via a β-(1→4) linkage (H-trisaccharide type II) resulted in a >10-fold increase in catalytic efficiency. SpGH95C also exhibited higher catalytic efficiency when the H-disaccharide was modified by the addition of a GalNAc-β-(1→3)-Gal disaccharide to the galactose via a β-(1→3) linkage (H-tetrasaccharide type IV). Despite the fact that the LewisB and LewisY tetrasaccharides contain the H-trisaccharide type I and II antigens, respectively, the catalytic efficiency of SpGH95C against these substrates was similar to that observed with the H-disaccharide (Table 1).
Structural analysis of SpGH29C
The activity of SpGH29C reveals it to be of the “B” group of GH29 fucosidases, which are defined as having little/no activity on pNP-α-l-fucopyranoside (where pNP is p-nitrophenyl) and specificity for terminal α-(1,3/4)-fucosidic linkages (40). Furthermore, SpGH29C displays an absolute requirement for a more complex glycan substrate than a simple disaccharide, which is similar to the GH29 BiAfcB enzyme from Bifidobacterium longum subsp. infantis (41). We used X-ray crystallography to probe the molecular basis for the ability of SpGH29C to recognize complex glycans and, specifically, accommodate substrates with both type I and type II core motifs (e.g. LewisX versus LewisA). Initially, a single crystal of SpGH29C was obtained, but subsequent trials failed to reproduce the crystals. This crystal yielded a good diffraction data set to a resolution of 1.72 Å, allowing the structure to be solved by molecular replacement.
The final refined structure comprised two molecules per asymmetric unit with each polypeptide chain unexpectedly terminating at residue 452 (of 559 expected residues). This C-terminally truncated form of the protein, which was presumably generated by degradation during the crystallization experiment, had an overall fold containing two domains that is typical of several GH29 enzymes (Fig. 1). The C-terminal domain is a β-sandwich domain made up of three and five antiparallel β-strands arranged in β-sheets (Fig. 1). The N-terminal (α/β)8-barrel domain houses the catalytic machinery, which on the basis of similarity to other GH29 enzymes can be identified as Asp-171 for the nucleophile and Glu-215 for the acid/base (Fig. 1).
Figure 1.

Overall structure of SpGH29C. The X-ray crystal structure of SpGH29C is represented as a cartoon colored from blue (N terminus (Nter)) to red (C terminus (Cter)). Both catalytic residues and a Bis-Tris molecule observed to be bound in the active site are shown as gray sticks. The numbering of helices and β-strands comprising the (α/β)8 catalytic module is indicated. The strand numbering of the ancillary module is also indicated.
To enable reproducible crystallization of SpGH29C, we used the native structure to inform the generation of a shorter construct (amino acids 1–451; SpGH29CT) into which we also introduced a D171N/E215Q double mutation to catalytically inactivate the enzyme. This protein crystallized easily and showed no hydrolytic activity, allowing us to determine the structure of the protein in complex with intact LewisA, LewisX, and LewisY antigen substrates. In all three cases, clear electron density for the complete glycans in the active site was present, allowing us to model these substrates (Fig. S2).
SpGH29CT interacts with the LewisA antigen in a manner that is largely indistinguishable from the interaction of BiAfcB with the same antigen structure (Fig. 2A) (41). The terminal fucose residue, which is in a standard 1C4 chair conformation, sits in the −1 subsite, making a series of hydrogen-bonding interactions and a classical CH–π interaction with Trp-264. This poise for the fucose residue places the oxygen of its glycosidic bond in proximity to Gln-215, which in the unmutated enzyme would be a glutamate residue, thus indicating the appropriate positioning of this residue to act as the catalytic acid/base (Fig. 2A). Asn-171, which in the unmutated enzyme would be an aspartate residue, is placed ∼3.5 Å beneath C1 of the fucose, consistent with its role as a nucleophile in the active enzyme (Fig. 2A).
Figure 2.

The SpGH29CT D171N/E215Q catalytic pocket in complex with histo-blood group antigens. A, structure of SpGH29T D171N/E215Q (green sticks) in complex with LewisA antigen. The insets focus on the +2′ galactose-binding subsite (top right inset), the −1 fucose-binding subsite (bottom right inset), and +1* GlcNAc pseudo-subsite (left inset). B, structural overlay of BiAfcB D172A/E217A (PDB code 3EUT; orange sticks) in complex with the LewisA antigen (gray lines) with the LewisA antigen from the complex with SpGH29T D171N/E215Q. The active-site side chains of SpGH29T D171N/E215Q were omitted as they are completely conserved with those of BiAfcB. SpGH29T residue numbering is shown in black, and that of BiAfcB is shown in gray. C and D, SpGH29T D171N/E215Q in complex with the LewisX and LewisY antigens, respectively. Fucose, galactose, and GlcNAc are shown in red, yellow, and blue sticks, respectively. The water molecule (wat) is represented by a purple sphere. Dashed lines denote hydrogen bonds. Subsites are indicated in red.
The GlcNAc residue that precedes the fucose residue and is in the type I motif of the LewisA antigen does not appear to make any interactions with the enzyme active site, and thus we cannot structurally define a distinct +1 subsite. However, this residue must be present in the minimal trisaccharide substrate of the enzyme, so we consider this as a pseudo +1 subsite (referred to as +1*). The terminal galactose residue of the antigen, however, sits in a subsite, which we refer to as a +2′ subsite, where the plane of C3–C4–C5 packs against Trp-211 and the C6, C3, and, notably, C4 hydroxyl groups make a series of hydrogen bonds with the active site. This particular constellation of interactions thereby provides specificity for galactose in this subsite.
The structures of SpGH29CT D171N/E215Q in complex with the LewisX (Fig. 2B) and LewisY (Fig. 2C) antigens revealed the molecular basis for accommodation of the type II core motif as well as the recognition of the additional α-(1,2)–linked terminal fucose residue in the LewisY antigen (Figs. 2C and S2). In both complexes, the fucose and galactose residues in the −1 and +2′ subsites, respectively, employ an identical set of interactions to those described for the LewisA complex. Likewise, the GlcNAc is positioned in the +1* subsite; however, revealing the plasticity of this pseudo-subsite, the GlcNAc is flipped 180° in accordance with accommodating the altered linkages to the fucose and galactose residues. The terminal α-(1→2)–linked fucose of the LewisY antigen makes only a water-mediated hydrogen bond and thus is largely just accommodated by the active site of the enzyme rather than appearing to act as a key recognition determinant. Presumably, the terminal α-(1→2)–linked fucose of the LewisB antigen, with its type I core motif, would be accepted in a similar manner.
Overall, therefore, the specificity of SpGH29C is determined by the unique spatial arrangement of the −1 and +2′ subsites and the occupation of these subsites by the appropriately positioned fucose and galactose residues, respectively, in the nonsialylated series of Lewis antigens. The accommodation of both the type I and II motifs in these antigens is enabled by the lack of specific interactions between the +1* subsite and the GlcNAc residue in these glycans. Notably, this distinctive set of interactions legislates against recognition and hydrolysis of α-(1→2)-fucosidic bonds, necessitating the presence of SpGH95C to process glycans with this modification.
SpGH29C and SpGH95C initiate a cascade of histo-blood group degradation
SpGH29C and SpGH95C are α-fucosidases with differing linkage specificities and therefore have the potential to uncap fucosylated glycans to expose potential substrates for other pneumococcal exoglycosidases. We tested the ability of SpGH29C and SpGH95C to initiate complete degradation of human histo-blood group antigens into monosaccharides by other known pneumococcal GHs using fluorophore-assisted carbohydrate electrophoresis (FACE). This is illustrated, as an example, by the sequential depolymerization of the type IV H-tetrasaccharide (Fig. 3). This glycan is resistant to depolymerization by pneumococcal enzymes unless first treated with SpGH95C. Uncapping of this glycan by SpGH95C exposes a terminal Gal-β-(1→3)-GalNAc motif, which could be hydrolyzed by the exo-β-(1→3)-galactosidase BgaC (23) to release galactose. The sequential action of SpGH95C and BgaC then allowed GH20C, a known exo-β-N-acetylhexosaminidase (30), to cleave the remaining GalNAc-β-(1→3)-Gal disaccharide. This general approach was used to examine the depolymerization of a wider range of glycans.
Figure 3.
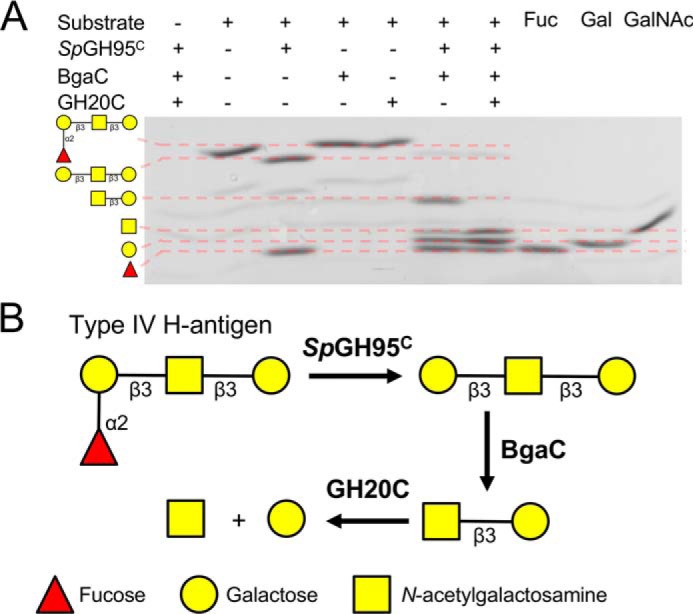
Example of sequential degradation of a human glycan by pneumococcal GHs. A, the substrate type IV H-tetrasaccharide was incubated with enzyme(s) overnight, and the products were labeled and visualized by fluorophore-assisted carbohydrate electrophoresis. ± above the gel indicates the presence/absence of substrate or enzyme. B, schematic depiction of the sequential breakdown of type IV H-tetrasaccharide by pneumococcal GHs. GHs are indicated in bold next to the arrow for the reaction they catalyze.
The lacto-N-biose (Gal-β-(1→3)-GlcNAc) and LacNAc (Gal-β-(1→4)-GlcNAc), which are found in type I H-trisaccharide/LewisA/LewisB and type II H-trisaccharide/LewisX/LewisY, respectively, are known targets for the characterized pneumococcal exoglycosidases BgaC (23) and BgaA (16, 42). In the absence of SpGH95C, these β-galactosidases are unable to degrade the H-trisaccharides (Fig. S3, A and B). SpGH29C is required to uncap the LewisA and LewisX antigens (Figs. 4 and S3, C and D). Both SpGH95C and SpGH29C are required to remove the capping fucose residues from LewisB and LewisY and to allow degradation by BgaC or BgaA, respectively (Figs. 4 and S3, E and F). We observed that either fucosidase is able to initiate the degradation of these glycans (Figs. 4 and S3, E and F). Degradation of LewisY can proceed either via SpGH29C, which generates the type II H-trisaccharide, or via SpGH95C, which generates LewisX. These trisaccharides are then acted on by the complementary fucosidase and converge at LacNAc, a substrate for BgaA. A parallel degradation pathway takes place for LewisB, with type I H-trisaccharide, LewisA, and lacto-N-biose acting as intermediates, followed by BgaC activity. LewisX and LewisA are sometimes sialylated; therefore, we also determined the order of enzymatic degradation of sialyl-LewisX and sialyl-LewisA (Figs. 4 and S3, G and H). The presence of the α-(2→3)–linked sialic acid on both antigens influenced the activity of SpGH29C by abrogating it on sialyl-LewisA and limiting activity on sialyl-LewisX (Fig. S3, E and F). However, desialylation of sialyl-LewisX or sialyl-LewisA by the exo-α-sialidase NanA (21) allowed the activity of SpGH29C and the other pneumococcal GHs to proceed to full depolymerization of the glycans.
Figure 4.
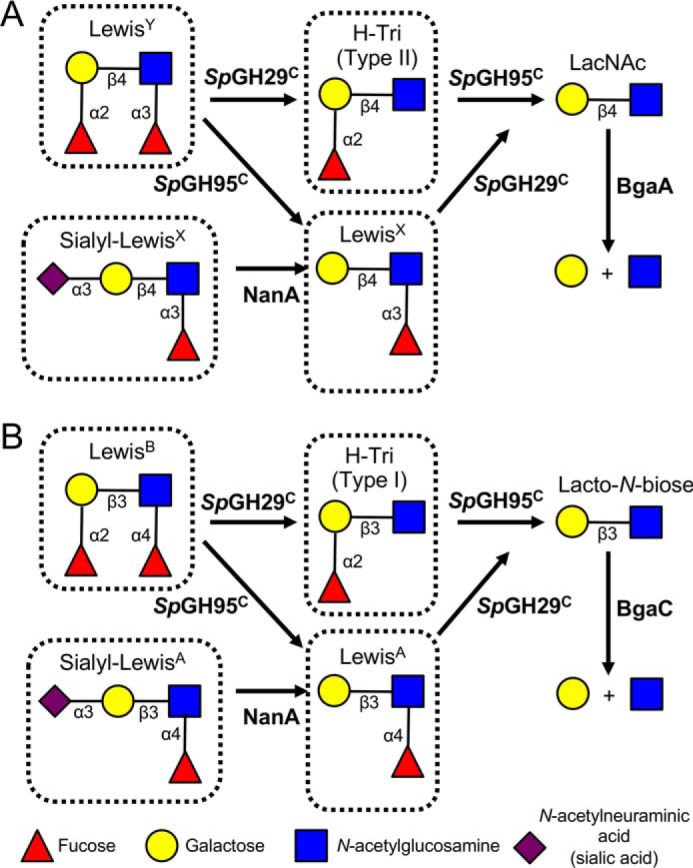
Schematic depiction of the sequential degradation of histo-blood group antigens by pneumococcal GHs. GHs are indicated in bold next to the arrow for the reaction they catalyze. A, degradation of LewisY can be initiated either by SpGH29C, which yields the type II H-trisaccharide (H-Tri), or by SpGH95C, which yields LewisX. The complementary α-fucosidase then acts to produce N-acetyllactosamine (LacNAc), which is cleaved into its constituent monosaccharides by BgaA. Sialyl-LewisX must be desialylated by NanA prior to SpGH29C activity. B, degradation of LewisB can be initiated either by SpGH29C, which yields the type I H-trisaccharide, or by SpGH95C, which yields LewisA. The complementary α-fucosidase then acts to produce lacto-N-biose, which is cleaved into its constituent monosaccharides by BgaC. Sialyl-LewisA must be desialylated by NanA prior to SpGH29C activity. See Fig. S3 for experimental validation for the sequential depolymerization of each of the boxed species in this figure.
Cellular localization of SpGH29C
Neither SpGH29C nor SpGH95C possesses an LPXTG cell wall–anchoring motif, and protein localization prediction software (43) did not identify any signal peptides. However, S. pneumoniae is known to export many of its carbohydrate-active enzymes, both classically and nonclassically, either into the supernatant or to be associated with the cell wall (10, 44). Given the initiating role SpGH29C and SpGH95C play in the degradation of fucosylated glycans and the fact that BgaA, BgaC, and GH20C are all known or strongly suspected to be exported (23, 30, 45, 46), we hypothesized that SpGH29C and SpGH95C function extracellularly. To experimentally test our hypothesis, we assayed isolated cellular fractions of S. pneumoniae TIGR4 for SpGH29C activity. Exposure of LewisX to the cell wall–associated fraction (CWF) and total soluble fraction (TSF) resulted in loss or significant reduction of the band corresponding to LewisX on a FACE gel, indicating processing of the glycan (Fig. 5A). Production of bands corresponding to monosaccharides could also be seen in the TSF-treated sample, but the CWF-treated sample contained a contaminating species that migrated the same distance as the monosaccharides, which prevented conclusive visualization of monosaccharides in this sample. Notably, neither the TSF-treated sample nor the CWF-treated samples displayed the presence of a LacNAc intermediate, as seen in the sample of LewisX treated with recombinant SpGH29C. LacNAc is the substrate of BgaA, which is cell wall–associated via its N-terminal signal peptide and C-terminal LPXTG motif (46). The absence of LacNAc in the CWF-treated sample, therefore, most likely indicates that SpGH29C and BgaA are localized together in this fraction. To confirm that the degradative activity against LewisX observed with the CWF and TSF was initiated by SpGH29C, we repeated this experiment with a deletion mutant of SpGH29C (Δspgh29C; Fig. 5B). In this experiment, none of the cellular fractions exhibited activity against LewisX, and no band corresponding to fucose was observed in the TSF-treated sample. These results are most consistent with SpGH29C being associated with the bacterial cell wall, placing it as another likely example of a nonclassically secreted pneumococcal protein.
Figure 5.

Cellular localization of SpGH29C. A and B, activity of different cellular fractions of WT TIGR4 (A) and ΔspGH29C (B) against LewisX as detected by fluorophore-assisted carbohydrate electrophoresis. The activities of recombinant SpGH29C and BgaA against LewisX are shown as controls, and fucose is shown as a standard. C, background labeling of the different cellular fractions in the absence of LewisX. LewisX and LewisX treated with total soluble protein are shown for comparison. LeX, LewisX; EF, extracellular fraction; CF, cytoplasmic fraction; MF, membrane fraction. The 8-aminonaphthalene-1,3,6-trisulfonic acid (ANTS) lane indicates background labeling due to the fluorophore alone. Due to the background labeling of the cell wall–associated fraction, SpGH29C activity can be observed as a disappearance of LewisX rather than an appearance of fucose.
Similar attempts were made to determine the localization of SpGH95C by testing cellular fractions for activity against the type II H-trisaccharide (the substrate against which SpGH95C exhibited the highest kcat/Km; Table 1). However, no degradative activity was observed in any of the fractions, including the TSF (data not shown). Therefore, we suggest that SpGH95C is not expressed under typical laboratory growth conditions.
The ability of pneumococcal GHs to degrade important human glycans
We have demonstrated the ability of SpGH29C and SpGH95C, together with other pneumococcal GHs, to completely degrade H- and Lewis blood group antigens into their monosaccharide constituents. As previously mentioned, these antigens are frequently observed as capping motifs on more complex glycans (19, 34). Therefore, we set out to assess the overall ability of the S. pneumoniae glycan-processing machinery to depolymerize important human glycans. Trifucosyllacto-N-hexaose (TFLNH) is a human milk oligosaccharide, but it mimics a complex O-glycan containing many of the linkages and motifs that S. pneumoniae likely encounters during colonization and infection, including terminal LewisX and LewisB motifs as well as an internal lacto-N-tetraose motif (Gal-β-(1→3)-GlcNAc-β-(1→3)-Gal-β-(1→4)-Glc), which forms the core of the lacto series of glycosphingolipids (20). Thus, this complex glycan makes an excellent model glycan, and therefore we used it as a substrate to demonstrate the capacity of the pneumococcal GH arsenal to depolymerize a highly modified glycan (Fig. 6). Using FACE analysis, we observed the ability of pneumococcal GHs to cleave all eight different linkages present in TFLNH and the fundamental dependence on SpGH29C and SpGH95C for initiation of this process (Figs. 6 and S4). SpGH29C was able to remove both the α-(1→3)–linked fucose residue from the arm bearing a LewisX motif and the α-(1→4)–linked fucose from the LewisB arm of TFLNH without prior action of SpGH95C. In contrast, SpGH95C exhibited only partial activity against TFLNH, and the presence of SpGH29C was required to facilitate complete removal of the α-(1→2)–linked fucose. In the absence of SpGH95C, SpGH29C was able to initiate degradation of the LewisX arm of TFLNH by BgaA and GH20C; however, the LewisB arm could not be degraded. Therefore, both fucosidases were required to uncap the two arms of TFLNH. The complete degradation of TFLNH by BgaA, BgaC, and GH20C following defucosylation is consistent with their published activities (23, 30, 42).
Figure 6.

Schematic depiction of the sequential degradation of TFLNH by pneumococcal GHs. GHs are indicated in bold next to the arrow for the reaction they catalyze; numbers in green refer to the gel lane in Fig. S4. SpGH95C and SpGH29C are required to remove the capping fucose residues from TFLNH and allow access to the oligosaccharide by other GHs. Treatment of TFLNH with SpGH29C results in removal of the α-(1→3)– and α-(1→4)–linked fucose units and allows BgaA and GH20C to degrade the arm proximal to the reducing end; however, without SpGH95C, the distal arm cannot be degraded. Treatment of TFLNH with SpGH95C results in partial removal of the α-(1→2)–linked fucose unit and a difucosylated oligosaccharide that cannot be acted upon by other GHs (except SpGH29C). If the α-(1→3)– and α-(1→4)–linked fucose units are removed by SpGH29C first, SpGH95C is able to fully remove the α-(1→2)–linked fucose unit from the distal arm to produce lacto-N-hexaose. This hexasaccharide can then be fully degraded into galactose and glucose by the combined actions of BgaA, BgaC, and GH20C. See Fig. S4 for experimental validation of this pathway.
Discussion
S. pneumoniae is considered an accomplished degrader of human glycans, with known capacity to depolymerize complex and high-mannose N-linked glycans as well as some O-linked glycans (e.g. Refs. 20, 24, 27, 28, and 46). The bacterium also has the ability to metabolize the glycosaminoglycan hyaluronan (e.g. Refs. 47 and 48) and glycogen (e.g. Refs. 49 and 50). The activities of some of the pneumococcal enzymes are also consistent with depolymerization of glycosphingolipid glycans (23, 30). Here, we have focused on the previously uncharacterized capacity of S. pneumoniae to degrade the full complement of fucosylated blood group H- and Lewis antigens and the underlying glycans that can bear these motifs.
Fucose is an important and common monosaccharide that often decorates, and more frequently terminates, a number of human glycans (52). We have previously reported that all sequenced strains of S. pneumoniae carry one of two types of fucose utilization operon (24, 53, 54). Both operon types encode for a set of intracellular enzymes dedicated to processing free fucose to dihydroxyacetone phosphate and lactaldehyde, whereas the transporter systems and GHs that process the glycans vary between the operons (14, 55). The type 1 operon is found in the majority of pneumococcal strains, including TIGR4, and encodes for a member of GH family 98, Sp4GH98, which is an extracellular endo-β-galactosidase that cleaves the type II linkage of LewisY. This action releases a free H-disaccharide, whereas the GlcNAc and α-(1→3)–linked fucose remain attached to the glycoconjugate. The released H-disaccharide is thought to be imported by a phosphotransferase system transporter and then degraded by a putative intracellular GH95 (encoded by a gene distinct from the gene encoding SpGH95C). The type 2 operon, which was originally identified in a serotype 3 strain of S. pneumoniae, also encodes for a GH98 endo-β-galactosidase, Sp3GH98, that cleaves type II linkages; however, this enzyme is specific for blood group A- and B-antigens. Sp3GH98 releases soluble A/B-trisaccharides, which are then imported by an ABC transporter into the cytoplasm where they are degraded by a putative GH29 (encoded by a gene distinct from the gene encoding SpGH29C) and two putative GH family 36 members (10). Thus, there is evidence that S. pneumoniae can harvest fucosylated glycans from host tissues. Indeed, in TIGR4, the presence of the type 1 fucose operon is strongly linked to the full virulence of the microbe (56). However, by virtue of the well-characterized endo-acting enzymes that initiate LewisY or A/B-antigen harvesting, the models of the pathways encoded by these operons indicate highly specific glycan targets, which do not include LewisA, LewisB, LewisX, or H-antigens.
The presence of SpGH29C and SpGH95C as part of the core arsenal of GHs deployed by S. pneumoniae indicates that all strains of this bacterium likely have an innate capacity to target the H(O)-blood group antigen and all Lewis antigens, again suggesting the importance of fucosylated glycan degradation to the host-adapted lifestyle of S. pneumoniae. However, it also reveals potential redundancy, and even competition, between the functions of the “core” fucosidases and the fucose utilization pathways. For example, the processing of LewisY by SpGH29C or SpGH95C would prevent the action of Sp4GH98, which is unable to cleave the type II H-trisaccharide or LewisX products, respectively, that would be left by exo-α-fucosidase activity (24). Conversely, the cleavage of LewisY by Sp4GH98 leaves a glycoconjugate terminating in Fuc-α-(1→3)-GlcNAc, which is not a substrate for any of the known pneumococcal enzymes. Therefore, unless these enzymes are competing for substrates, they are likely expressed under different conditions in vivo, which are yet to be uncovered.
Although SpGH95C was able to cleave α-(1→2)–linked fucose residues found on histo-blood group antigens, the blood group A- and B-antigens were resistant to defucosylation by this enzyme. We were unable to obtain the X-ray crystal structure of SpGH95C; however, this enzyme is clearly unable to accommodate the additional terminal GalNAc/galactose residue found on blood group A/B-antigens. The lack of this activity is consistent with the well-characterized GH95 enzyme from Bifidobacterium bifidum (57). One potential mechanism for the degradation of the A/B-antigens could involve removal of the terminal GalNAc/galactose by an exo-α-N-acetylgalactosaminidase/galactosidase, which would allow degradation of the resulting H-antigen by SpGH95C and additional enzymes, depending on the glycan core type. The core S. pneumoniae genome, however, encodes for only a single GH having this possible activity, Aga, which is a member of GH36. This enzyme exhibits α-(1→6)-galactosidase activity against the plant oligosaccharide raffinose (14, 58). Furthermore, in direct tests, we failed to find activity for Aga on blood group A/B-antigens (not shown). Thus, deconstruction of the H(O)-blood group antigen and all Lewis antigens is a conserved feature in the glycan-degrading capacity of all S. pneumoniae strains, but targeting the A/B-antigens is not. Nevertheless, the type 2 fucose utilization operon found in some strains of S. pneumoniae is specific for the blood group A/B-antigens; therefore, at least a subset of strains has the ability to target these glycans. As we have inferred previously, the apparent differential ability of particular S. pneumoniae strains to degrade A/B-antigens may have implications for host susceptibility to infection (24).
A key distinction between the fucosylated glycan degradation pathways described here and those encoded by the type 1/2 operons is the cellular location in which defucosylation occurs. S. pneumoniae is able to import galactose and GlcNAc, which would be released from histo-blood group antigens extracellularly by BgaA and BgaC, and use them as a carbon source for growth (12, 59, 60); however, it is unable to grow on exogenous fucose (54, 56). Both type 1 and 2 fucose utilization operons are known or predicted to import fucosylated glycans and utilize intracellular α-fucosidases. Therefore, the released fucose can then be processed by the other components of the operon and feed into central metabolism (54). In contrast, we have shown that SpGH29C is cell wall–associated. Likewise, based on its uncapping function and the cellular location of the enzymes that act after it, we predict that SpGH95C is also extracellular. Therefore, the fucosylated glycan degradation pathways described here would release free fucose that apparently cannot be utilized by S. pneumoniae. As such, the bacterium may view fucose as a capping residue that has to be removed for the pneumococcus to release other monosaccharides that it can import. This apparent “waste” of fucose may point more importantly toward the functional significance of fucose in the context of human glycoconjugates and the importance of defucosylation to other aspects of the host–pneumococcus interaction rather than simple nutrition.
SpGH29C and SpGH95C possess complementary linkage specificities that, together, allow them to expose a wide range of human glycans to the action of other pneumococcal GHs. It is common for deletion mutants of pneumococcal initiating enzymes, such as NanA and the high-mannose N-glycan degradation initiator SpGH92, to display strong virulence phenotypes (10). Therefore, it is consistent that SpGH29C and SpGH95C have been identified as putative virulence factors in multiple animal models of disease (13, 35, 37). Given the known role of the type 1 operon in pneumococcal virulence (56) and the uncapping function of SpGH29C and SpGH95C, we hypothesize that directed studies into the contributions of these fucosidases to the host–pathogen interaction would confirm their roles as important virulence factors.
During our exploration of glycan degradation by the enzymes of S. pneumoniae, we unexpectedly uncovered a previously unknown activity for BgaC. Jeong et al. (23) previously reported that BgaC is unable to cleave the Gal-β-(1→3)-GalNAc motif in the context of the ganglioside GA1 (Gal-β-(1→3)-GalNAc-β-(1→4)-Gal-β-(1→4)-Glc; also known as asialo GM1). However, we observed that BgaC cleaved the terminal Gal-β-(1→3)-GalNAc motif in the type IV H-tetrasaccharide after it was uncapped by SpGH95C. This suggests that the substrate repertoire for BgaC is broader than previously suspected, which is notable because this linkage also occurs in the core of O-linked glycans as well as the globoside series of glycosphingolipids.
S. pneumoniae possesses a considerable ability to degrade distinct linkages found in human glycans. Of the >20 linkages commonly found in N-glycans, O-glycans, histo-blood group antigens, and glycosphingolipids, many are now associated with the activity of a characterized pneumococcal GH. Overall, our characterization of two complementary α-fucosidases and the in vitro recapitulation of glycan degradation pathways employed by S. pneumoniae expands the known capacity of this pathogen to degrade human glycans and highlights the comprehensive nature of its ability to target the human glycome.
Experimental procedures
Materials
Fucosyllactose, Lewis antigens, H-disaccharide, and type II H-trisaccharide were purchased from Carbosynth Ltd. (Berkshire, UK). Type I H-trisaccharide, type IV H-tetrasaccharide, type II A- and B-tetrasaccharides, and lacto-N-tetraose were obtained from Elicityl (Crolles, France). TFLNH was purchased from ProZyme (Hayward, CA). All other materials were from Millipore-Sigma unless otherwise stated.
Cloning and mutagenesis
The gene encoding for full-length SpGH29C (amino acids 1–559) from TIGR4 (locus tag SP_2146) was amplified by PCR with the primers GH29-F and GH29-R (Table S1) and cloned into pET28a between the NdeI and SalI sites to produce pET28a-SpGH29C. A truncated version of SpGH29C (amino acids 1–451) was also cloned into pET28a using the primers GH29-F and GH29T-R to produce pET28a-SpGH29CT. The gene encoding for full-length SpGH95C (locus tag SP_1654) was codon-optimized for expression in E. coli and synthesized by GenScript (Piscataway, NJ). This synthetic gene was then cloned into pET28a between the NdeI and XhoI sites to produce pET28a-SpGH95C. BgaC (locus tag SP_0060) and the catalytic domain of NanA (amino acids 303–777; locus tag SP_1693) were amplified using the primers BgaC-F, BgaC-R, NanA-F, and NanA-R and cloned into pET28a between the NheI and NotI or NdeI and XhoI sites to produce pET28a-BgaC and pET28a-NanA, respectively. Cloning of BgaA and GH20C has been reported previously (16, 30). Mutagenesis of pET28a-SpGH29CT to generate the SpGH29CT D171N/E215Q double mutation was performed using the “megaprimer” PCR method (61). All mutagenic primers are listed in Table S1. The integrity of all constructs was confirmed by bidirectional sequencing.
Protein expression and purification
Protein expression constructs were transformed into BL21(DE3) (or TunerTM(DE3) for expression of β-galactosidases). Expression of SpGH29C, SpGH29CT, and BgaC was performed in LB broth with 0.5 mm isopropyl β-d-1-thiogalactopyranoside induction at 16 °C for 18 h; SpGH95C and NanA were expressed in autoinduction medium at 16 °C for 4 days. Expression of BgaA and GH20C has been reported previously (16, 30). Standard procedures, as detailed previously (62), were used to lyse cells and purify the released proteins by immobilized metal-affinity chromatography and size-exclusion chromatography using either an S200 or S300 HiPrep 16/60 Sephacryl column (GE Healthcare) as appropriate. Protein purity was judged by SDS-PAGE analysis, and protein concentrations were determined using extinction coefficients calculated by ProtParam on the ExPASy server (63).
α-Fucosidase assays
The activity of SpGH29C and SpGH95C on α-fucosylated glycans was assayed by TLC and the detection of liberated fucose using an l-fucose assay kit that contains an NADP+-dependent fucose dehydrogenase (Megazyme Inc., Chicago, IL). TLC reactions contained 5 mm substrate and 1 μm enzyme in 20 mm Tris, pH 8.0, and were incubated at 37 °C for 1 h. Reactions were spotted onto precoated POLYGRAM SIL G/UV254 TLC sheets (Thermo Fisher Scientific, Waltham, MA), separated in a solvent of 7:2:1 propanol:H2O:ethanol, and visualized with 5% (v/v) H2SO4 in ethanol followed by heating at 90 °C. For the determination of kinetic parameters, the fucose detection kit method was adapted to allow both the α-fucosidase and fucose dehydrogenase reactions to occur simultaneously. Conditions were optimized to ensure that neither the fucose dehydrogenase nor NADP+ were limiting. Reactions (100 μl) contained 5 μl of substrate (at varying concentrations), 4 μl of NADP+ (kit supply), 2 μl of fucose dehydrogenase, and 1 μm α-fucosidase in 100 mm Tris, 50 mm NaCl, pH 8.0. Reactions (in triplicate) were incubated at 37 °C in a SpectraMax M5 plate reader (Molecular Devices, San Jose, CA), and the absorbance at 340 nm was read every 5 s. Slopes for each substrate concentration were converted into NADPH concentrations using an extinction coefficient of 6220 m−1 cm−1. The kcat/Km for each substrate-enzyme combination was calculated by linear regression of the initial velocities versus substrate concentration using GraphPad Prism 6.0.7.
General crystallography procedures
Crystals were obtained using sitting-drop vapor diffusion for screening and hanging-drop vapor diffusion for optimization at 18 °C. Prior to data collection, single crystals were flash-cooled with liquid nitrogen in crystallization solution supplemented with 20% (v/v) ethylene glycol as cryoprotectant. Diffraction data were collected either on beamline 9-2 or 11-1 at the Stanford Linear Accelerator Center (SLAC, Stanford Synchrotron Radiation Lightsource (SSRL), CA) or beamline 08B1-1 at the Canadian Light Source (CLS, Saskatoon, Saskatchewan, Canada) as indicated in Table 2. All diffraction data were processed using MOSFLM and SCALA (64–66). Data collection and processing statistics are shown in Table 2. For all structures, manual model building was performed with Coot (67), and refinement of atomic coordinates was performed with REFMAC (68). Water molecules were added in Coot with Find Waters and manually checked after refinement. In all data sets, refinement procedures were monitored by flagging 5% of all observations as “free” (69). Model validation was performed with MolProbity (70).
Table 2.
X-ray data collection and structure statistics
Values for the highest-resolution shells are shown in parentheses. r.m.s.d., root mean square deviation; Le, Lewis; EDO, 1,2-ethanediol; BTB, 2-[bis(2-hydroxyethyl)amino]-2-hydroxymethylpropane-1,3-diol; CA, calcium ion; SSRL, Stanford Synchrotron Radiation Lightsource; CLS, Canadian Light Source.
| SpGH29C native | SpGH29CT D171N/E215Q LewisX | SpGH29CT D171N/E215Q LewisY | SpGH29CT D171N/E215Q LewisA | |
|---|---|---|---|---|
| Data collection | ||||
| Beamline | SSRL BL9-2 | SSRL BL11-1 | SSRL BL11-1 | CLS 08B1-1 |
| Wavelength (Å) | 0.97946 | 0.97945 | 0.97945 | 0.9795 |
| Space group | P21 | P21 | P21 | P1 |
| Cell dimensions | ||||
| a, b, c (Å) | 60.9, 117.2, 64.9; β = 90.0° | 70.0, 99.0, 79.1; β = 97.6° | 69.8, 98.4, 79.5; β = 97.3° | 50.0, 68.8, 72.7, α = 76.6°; β = 73.2°, γ = 73.6° |
| Resolution (Å) | 41.50–1.72 (1.82–1.72) | 50.0–1.70 (1.79–1.70) | 27.8–1.62 (1.71–1.62) | 46.54–2.10 (2.16–2.10) |
| Rmerge | 0.082 (0.425) | 0.068 (0.307) | 0.050 (0.337) | 0.062 (0.344) |
| Rpim | 0.031 (0.162) | 0.035 (0.165) | 0.026 (0.183) | 0.062 (0.344) |
| CC 1/2 | 0.998 (0.940) | 0.997 (0.919) | 0.998 (0.882) | 0.998 (0.895) |
| 〈I/σI〉 | 16.1 (4.8) | 13.4 (4.2) | 17.0 (4.7) | 14.1 (3.5) |
| Completeness (%) | 98.0 (96.6) | 96.7 (97.1) | 97.2 (98.4) | 98.0 (96.9) |
| Redundancy | 7.8 (7.8) | 4.4 (4.3) | 4.5 (4.5) | 3.9 (4.0) |
| No. of reflections | 735,056 | 498,568 | 587,623 | 198,020 |
| No. unique | 93,880 | 113,304 | 131,486 | 50,203 |
| Refinement | ||||
| Resolution (Å) | 1.72 | 1.70 | 1.62 | 2.10 |
| Rwork/Rfree | 0.17/0.20 | 0.17/0.21 | 0.17/0.20 | 0.17/0.23 |
| No. of atoms | ||||
| Protein | 3,627 (A), 3,629 (B) | 3,656 (A), 3,630 (B) | 3,713 (A), 3,650 (B) | 3,605 (A), 3,582 (B) |
| Ligand | 2 (CA), 28 (BTB), 32 (EDO) | 36 (LeX-A), 36 (LeX-B), 24 (EDO) | 46 (LeY-A), 46 (LeY-B), 56 (EDO) | 36 (LeA-A), 36 (LeA-B) |
| Water | 913 | 1071 | 1032 | 673 |
| B-factors | ||||
| Protein | 19.9 (A), 19.8 (B) | 16.0 (A), 18.5 (B) | 18.2 (A), 23.0 (B) | 22.6 (A), 22.7 (B) |
| Ligand | 24.9 (CA), 29.4 (BTB), 35.9 (EDO) | 12.5 (LeX-A), 13.9 (LeX-B), 31.0 (EDO) | 16.7 (LeY-A), 19.8 (LeY-B), 39.7 (EDO) | 34.3 (LeA-A), 29.5 (LeA-B) |
| Water | 29.5 | 27.5 | 31.2 | 27.3 |
| r.m.s.d. | ||||
| Bond lengths (Å) | 0.012 | 0.008 | 0.007 | 0.010 |
| Bond angles (°) | 1.637 | 1.433 | 1.423 | 1.374 |
| Ramachandran (%) | ||||
| Preferred | 96.8 | 97.2 | 97.3 | 96.8 |
| Allowed | 3.2 | 2.8 | 2.7 | 3.2 |
| Disallowed | 0.0 | 0.0 | 0.0 | 0.0 |
SpGH29C and SpGH29CT D171N/E215Q structure determinations
A unique crystal of SpGH29C (25 mg ml−1) was obtained in 16% (w/v) polyethylene glycol (PEG) 3350, 0.15 m potassium chloride, 1 mm DTT, 0.1 m Bis-Tris, pH 6.0. This crystal was flash frozen in liquid nitrogen using the crystallization solution supplemented with 20% (v/v) ethylene glycol. After data collection, initial phases for SpGH29C were determined by molecular replacement using Phaser (71) and the structure of an α-l-fucosidase from Bacteroides thetaiotamicron as the search model (Protein Data Bank (PDB) code 3EYP). An initial model of SpGH29C was generated by automatic model building using the program Buccaneer (72). SpGH29CT D171N/E215Q (35 mg ml−1) was cocrystallized in the presence of an excess of LewisX or LewisY in 21–23% (w/v) PEG 4000, 0.22 m sodium acetate, 1 mm DTT, 0.1 m Tris, pH 8.5. Cocrystals of SpGH29CT D171N/E215Q with LewisA were obtained in 20–24% (w/v) PEG 3350, 0.18–0.22 m sodium chloride, 1 mm DTT, 0.1 m Tris, pH 8.5. All complexes were solved by molecular replacement using Phaser and the SpGH29C crystal structure.
Generation of SpGH29C deletion mutant
A PCR ligation technique was used to replace sp_2146 with a chloramphenicol resistance cassette as described previously (30). Briefly, the chloramphenicol resistance cassette was amplified with the primers CAM-F and CAM-R (Table S1), which introduced a 5′ NheI site and a 3′ XhoI site. Up- and downstream regions flanking sp_2146 were amplified using the primers Upstream-F, Upstream-R, Downstream-F, and Downstream-R, which introduced a 3′ NheI site into the upstream flank and a 5′ XhoI site into the downstream flank. Following digestion, all three amplicons were ligated together, and this ligation mixture was used to transform S. pneumoniae TIGR4 as described previously (30). The presence and location of the chloramphenicol resistance cassette and absence of sp_2146 were confirmed by multiple PCR analyses and bidirectional DNA sequencing.
Localization of SpGH29C
S. pneumoniae TIGR4 and ΔspGH29 were grown in 50 ml of AGCH medium (73) with 1% glucose at 37 °C in a candle jar to an OD600 of 0.6, then pelleted, and resuspended in AGCH medium containing no carbohydrate for a further 30 min (in an attempt to induce expression of GHs). The cells were then pelleted again, and the supernatant was retained as the extracellular fraction and concentrated 100-fold using an Amicon ultrafiltration cell fitted with a 10-kDa molecular-mass-cutoff membrane. The pelleted cells were split into two samples: one was used to produce protoplasts and obtain the cell wall, cytoplasmic, and membrane fractions, whereas the other was resuspended in 50 mm Tris-HCl, pH 7.5; sonicated on ice; and centrifuged to obtain the total protein fraction. The cell pellet intended for protoplast production was washed with 50 mm Tris-HCl, pH 7.5; resuspended in cell wall digestion buffer (74); and incubated at 37 °C with gentle shaking for 2 h. The protoplasts were then pelleted, and the supernatant was retained as the cell wall fraction. The cytoplasmic fraction was obtained by gently washing the protoplasts with 50 mm Tris-HCl, pH 7.5, 30% sucrose; resuspending and lysing them in 50 mm Tris-HCl, pH 7.5; pelleting the protoplast membranes at 20,000 rpm for 30 min; and retaining the supernatant. Finally, the membrane fraction was obtained by solubilizing the membranes in 50 mm Tris-HCl, pH 7.5, 0.05% Triton as described previously (75). The different fractions were kept on ice, and 5 μl of each was used to set up reactions with LewisX. Reactions were incubated at 37 °C for 48 h and then processed for fluorophore-assisted carbohydrate electrophoresis as described below.
FACE
FACE reactions contained 10 μg of glycan substrate and 1 μm enzyme in 50 mm sodium phosphate buffer, pH 6.5, 45 mm β-mercaptoethanol and were incubated at 37 °C for 20 h. Reactions were stopped by the addition of ethanol, dried in a SpeedVac for 4 h, and then labeled overnight with 5 μl of 0.2 m 8-aminonaphthalene-1,3,6-trisulfonic acid (Thermo Fisher Scientific) and 5 μl of 1 m sodium cyanoborohydride at 37 °C as described previously (36). Labeled reaction products were separated on a 35% polyacrylamide gel, and labeled glycans were visualized under UV light.
Author contributions
J. K. H., B. P., M. R., S. P. S., and A. B. B. formal analysis; J. K. H., B. P., M. R., and S. P. S. investigation; J. K. H., B. P., M. R., and S. P. S. methodology; J. K. H., B. P., and A. B. B. writing-original draft; J. K. H., B. P., and A. B. B. writing-review and editing; B. P. validation; A. B. B. conceptualization; A. B. B. supervision; A. B. B. funding acquisition; A. B. B. project administration.
Supplementary Material
Acknowledgments
We thank the beamline staff at the Stanford Synchrotron Research Laboratory (SSRL). SSRL is a Directorate of Stanford Linear Accelerator Center (SLAC) National Accelerator Laboratory and an Office of Science User Facility operated for the United States Department of Energy (DOE) Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program (Grant P41RR001209), and the National Institute of General Medical Sciences. Research described in this paper was performed using beamline 08B1-1 at the Canadian Light Source, which is supported by the Canada Foundation for Innovation, Natural Sciences and Engineering Research Council of Canada, the University of Saskatchewan, the Government of Saskatchewan, Western Economic Diversification Canada, the National Research Council Canada, and the Canadian Institutes of Health Research.
This work was supported by Canadian Institutes of Health Research Operating Grant PJT 159786. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4 and Table S1.
The atomic coordinates and structure factors (codes 6ORG, 6ORF, 6ORH, and 6OR4) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- GlcNAc
- N-acetyl-d-glucosamine
- GH
- glycoside hydrolase
- GalNAc
- N-acetyl-d-galactosamine
- Fuc
- fucose
- Gal
- galactose
- Glc
- glucose
- LacNAc
- N-acetyllactosamine
- FACE
- fluorophore-assisted carbohydrate electrophoresis
- CWF
- cell wall–associated fraction
- TSF
- total soluble fraction
- TFLNH
- trifucosyllacto-N-hexaose
- Sp
- S. pneumoniae
- Bi
- B. longum subsp. infantis
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Varki A., and Marth J. (1995) Oligosaccharides in vertebrate development. Semin. Dev. Biol. 6, 127–138 10.1016/S1044-5781(06)80022-8 [DOI] [Google Scholar]
- 2. Khoury G. A., Baliban R. C., and Floudas C. A. (2011) Proteome-wide post-translational modification statistics: frequency analysis and curation of the Swiss-Prot database. Sci. Rep. 1, srep00090 10.1038/srep00090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gagneux P., Aebi M., and Varki A. (2015) Evolution of glycan diversity, in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., and Seeberger P. H., eds) 3rd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 4. Reitsma S., Slaaf D. W., Vink H., van Zandvoort M. A., and oude Egbrink M. G. (2007) The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. 454, 345–359 10.1007/s00424-007-0212-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Unione L., Gimeno A., Valverde P., Calloni I., Coelho H., Mirabella S., Poveda A., Arda A., and Jimenez-Barbero J. (2017) Glycans in infectious diseases. A molecular recognition perspective. Curr. Med. Chem. 24, 4057–4080 10.2174/0929867324666170217093702 [DOI] [PubMed] [Google Scholar]
- 6. Tailford L. E., Crost E. H., Kavanaugh D., and Juge N. (2015) Mucin glycan foraging in the human gut microbiome. Front. Genet. 6, 81 10.3389/fgene.2015.00081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Koropatkin N. M., Cameron E. A., and Martens E. C. (2012) How glycan metabolism shapes the human gut microbiota. Nat. Rev. Microbiol. 10, 323–335 10.1038/nrmicro2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. King S. J. (2010) Pneumococcal modification of host sugars: a major contributor to colonization of the human airway? Mol. Oral Microbiol. 25, 15–24 10.1111/j.2041-1014.2009.00564.x [DOI] [PubMed] [Google Scholar]
- 9. Buckwalter C. M., and King S. J. (2012) Pneumococcal carbohydrate transport: food for thought. Trends Microbiol. 20, 517–522 10.1016/j.tim.2012.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hobbs J. K., Pluvinage B., and Boraston A. B. (2018) Glycan-metabolizing enzymes in microbe-host interactions: the Streptococcus pneumoniae paradigm. FEBS Lett. 592, 3865–3897 10.1002/1873-3468.13045 [DOI] [PubMed] [Google Scholar]
- 11. Weiser J. N., Ferreira D. M., and Paton J. C. (2018) Streptococcus pneumoniae: transmission, colonization and invasion. Nat. Rev. Microbiol. 16, 355–367 10.1038/s41579-018-0001-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bidossi A., Mulas L., Decorosi F., Colomba L., Ricci S., Pozzi G., Deutscher J., Viti C., and Oggioni M. R. (2012) A functional genomics approach to establish the complement of carbohydrate transporters in Streptococcus pneumoniae. PLoS One 7, e33320 10.1371/journal.pone.0033320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hava D. L., and Camilli A. (2002) Large-scale identification of serotype 4 Streptococcus pneumoniae virulence factors. Mol. Microbiol. 45, 1389–1406 10.1046/j.1365-2958.2002.03106.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Obert C., Sublett J., Kaushal D., Hinojosa E., Barton T., Tuomanen E. I., and Orihuela C. J. (2006) Identification of a candidate Streptococcus pneumoniae core genome and regions of diversity correlated with invasive pneumococcal disease. Infect. Immun. 74, 4766–4777 10.1128/IAI.00316-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen H., Ma Y., Yang J., O'Brien C. J., Lee S. L., Mazurkiewicz J. E., Haataja S., Yan J.-H., Gao G. F., and Zhang J.-R. (2007) Genetic requirement for pneumococcal ear infection. PLoS One 3, e2950 10.1371/journal.pone.0002950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Singh A. K., Pluvinage B., Higgins M. A., Dalia A. B., Woodiga S. A., Flynn M., Lloyd A. R., Weiser J. N., Stubbs K. A., Boraston A. B., and King S. J. (2014) Unravelling the multiple functions of the architecturally intricate Streptococcus pneumoniae β-galactosidase, BgaA. PLoS Pathog. 10, e1004364 10.1371/journal.ppat.1004364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pluvinage B., Higgins M. A., Abbott D. W., Robb C., Dalia A. B., Deng L., Weiser J. N., Parsons T. B., Fairbanks A. J., Vocadlo D. J., and Boraston A. B. (2011) Inhibition of the pneumococcal virulence factor StrH and molecular insights into N-glycan recognition and hydrolysis. Structure 19, 1603–1614 10.1016/j.str.2011.08.011 [DOI] [PubMed] [Google Scholar]
- 18. Dalia A. B., Standish A. J., and Weiser J. N. (2010) Three surface exoglycosidases from Streptococcus pneumoniae, NanA, BgaA, and StrH, promote resistance to opsonophagocytic killing by human neutrophils. Infect. Immun. 78, 2108–2116 10.1128/IAI.01125-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Walther T., Karamanska R., Chan R. W., Chan M. C., Jia N., Air G., Hopton C., Wong M. P., Dell A., Malik Peiris J. S., Haslam S. M., and Nicholls J. M. (2013) Glycomic analysis of human respiratory tract tissues and correlation with influenza virus infection. PLoS Pathog. 9, e1003223 10.1371/journal.ppat.1003223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schnaar R. L., and Kinoshita T. (2015) Glycosphingolipids, in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., and Seeberger P. H., eds) 3rd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 21. Xu G., Kiefel M. J., Wilson J. C., Andrew P. W., Oggioni M. R., and Taylor G. L. (2011) Three Streptococcus pneumoniae sialidases: three different products. J. Am. Chem. Soc. 133, 1718–1721 10.1021/ja110733q [DOI] [PubMed] [Google Scholar]
- 22. King S. J., Hippe K. R., and Weiser J. N. (2006) Deglycosylation of human glycoconjugates by the sequential activities of exoglycosidases expressed by Streptococcus pneumoniae. Mol. Microbiol. 59, 961–974 10.1111/j.1365-2958.2005.04984.x [DOI] [PubMed] [Google Scholar]
- 23. Jeong J. K., Kwon O., Lee Y. M., Oh D.-B., Lee J. M., Kim S., Kim E.-H., Le T. N., Rhee D.-K., and Kang H. A. (2009) Characterization of the Streptococcus pneumoniae BgaC protein as a novel surface β-galactosidase with specific hydrolysis activity for the Galβ1–3GlcNAc moiety of oligosaccharides. J. Bacteriol. 191, 3011–3023 10.1128/JB.01601-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Higgins M. A., Whitworth G. E., El Warry N., Randriantsoa M., Samain E., Burke R. D., Vocadlo D. J., and Boraston A. B. (2009) Differential recognition and hydrolysis of host carbohydrate antigens by Streptococcus pneumoniae family 98 glycoside hydrolases. J. Biol. Chem. 284, 26161–26173 10.1074/jbc.M109.024067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gregg K. J., Zandberg W. F., Hehemann J.-H., Whitworth G. E., Deng L., Vocadlo D. J., and Boraston A. B. (2011) Analysis of a new family of widely distributed metal-independent α-mannosidases provides unique insight into the processing of N-linked glycans. J. Biol. Chem. 286, 15586–15596 10.1074/jbc.M111.223172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Robb M., Hobbs J. K., Woodiga S. A., Shapiro-Ward S., Suits M. D., McGregor N., Brumer H., Yesilkaya H., King S. J., and Boraston A. B. (2017) Molecular characterization of N-glycan degradation and transport in Streptococcus pneumoniae and its contribution to virulence. PLoS Pathog. 13, e1006090 10.1371/journal.ppat.1006090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Muramatsu H., Tachikui H., Ushida H., Song X., Qiu Y., Yamamoto S., and Muramatsu T. (2001) Molecular cloning and expression of endo-β-N-acetylglucosaminidase D, which acts on the core structure of complex type asparagine-linked oligosaccharides. J. Biochem. 129, 923–928 10.1093/oxfordjournals.jbchem.a002938 [DOI] [PubMed] [Google Scholar]
- 28. Clarke V. A., Platt N., and Butters T. D. (1995) Cloning and expression of the β -N-acetylglucosaminidase gene from Streptococcus pneumoniae. Generation of truncated enzymes with modified aglycon specificity. J. Biol. Chem. 270, 8805–8814 10.1074/jbc.270.15.8805 [DOI] [PubMed] [Google Scholar]
- 29. Marion C., Limoli D. H., Bobulsky G. S., Abraham J. L., Burnaugh A. M., and King S. J. (2009) Identification of a pneumococcal glycosidase that modifies O-linked glycans. Infect. Immun. 77, 1389–1396 10.1128/IAI.01215-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Robb M., Robb C. S., Higgins M. A., Hobbs J. K., Paton J. C., and Boraston A. B. (2015) A second β-hexosaminidase encoded in the Streptococcus pneumoniae genome provides an expanded biochemical ability to degrade host glycans. J. Biol. Chem. 290, 30888–30900 10.1074/jbc.M115.688630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brockhausen I., and Stanley P. (2015) O-GalNAc glycans, in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., and Seeberger P. H., eds) 3rd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 32. Flynn J. M., Niccum D., Dunitz J. M., and Hunter R. C. (2016) Evidence and role for bacterial mucin degradation in cystic fibrosis airway disease. PLoS Pathog. 12, e1005846 10.1371/journal.ppat.1005846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stanley P., Taniguchi N., and Aebi M. (2015) N-Glycans, in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., and Seeberger P. H., eds) 3rd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 34. Stanley P., and Cummings R. D. (2015) Structures common to different glycans, in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., and Seeberger P. H., eds) 3rd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 35. Polissi A., Pontiggia A., Feger G., Altieri M., Mottl H., Ferrari L., and Simon D. (1998) Large-scale identification of virulence genes from Streptococcus pneumoniae. Infect. Immun. 66, 5620–5629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Robb M., Hobbs J. K., and Boraston A. B. (2017) Separation and visualization of glycans by fluorophore-assisted carbohydrate electrophoresis. Methods Mol. Biol. 1588, 215–221 10.1007/978-1-4939-6899-2_17 [DOI] [PubMed] [Google Scholar]
- 37. Lau G. W., Haataja S., Lonetto M., Kensit S. E., Marra A., Bryant A. P., McDevitt D., Morrison D. A., and Holden D. W. (2001) A functional genomic analysis of type 3 Streptococcus pneumoniae virulence. Mol. Microbiol. 40, 555–571 10.1046/j.1365-2958.2001.02335.x [DOI] [PubMed] [Google Scholar]
- 38. Szklarczyk D., Gable A. L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N. T., Morris J. H., Bork P., Jensen L. J., and Mering C. V. (2019) STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 10.1093/nar/gky1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., and Henrissat B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 10.1093/nar/gkt1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ashida H., Miyake A., Kiyohara M., Wada J., Yoshida E., Kumagai H., Katayama T., and Yamamoto K. (2009) Two distinct α-l-fucosidases from Bifidobacterium bifidum are essential for the utilization of fucosylated milk oligosaccharides and glycoconjugates. Glycobiology 19, 1010–1017 10.1093/glycob/cwp082 [DOI] [PubMed] [Google Scholar]
- 41. Sakurama H., Fushinobu S., Hidaka M., Yoshida E., Honda Y., Ashida H., Kitaoka M., Kumagai H., Yamamoto K., and Katayama T. (2012) 1,3–1,4-α-l-fucosynthase that specifically introduces Lewis a/x antigens into type-1/2 chains. J. Biol. Chem. 287, 16709–16719 10.1074/jbc.M111.333781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zähner D., and Hakenbeck R. (2000) The Streptococcus pneumoniae beta-galactosidase is a surface protein. J. Bacteriol. 182, 5919–5921 10.1128/JB.182.20.5919-5921.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Almagro Armenteros J. J., Tsirigos K. D., Sønderby C. K., Petersen T. N., Winther O., Brunak S., von Heijne G., and Nielsen H. (2019) SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423 10.1038/s41587-019-0036-z [DOI] [PubMed] [Google Scholar]
- 44. Pérez-Dorado I., Galan-Bartual S., and Hermoso J. A. (2012) Pneumococcal surface proteins: when the whole is greater than the sum of its parts. Mol. Oral Microbiol. 27, 221–245 10.1111/j.2041-1014.2012.00655.x [DOI] [PubMed] [Google Scholar]
- 45. Terra V. S., Homer K. A., Rao S. G., Andrew P. W., and Yesilkaya H. (2010) Characterization of novel β-galactosidase activity that contributes to glycoprotein degradation and virulence in Streptococcus pneumoniae. Infect. Immun. 78, 348–357 10.1128/IAI.00721-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kharat A. S., and Tomasz A. (2003) Inactivation of the srtA gene affects localization of surface proteins and decreases adhesion of Streptococcus pneumoniae to human pharyngeal cells in vitro. Infect. Immun. 71, 2758–2765 10.1128/IAI.71.5.2758-2765.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hughes R. C., and Jeanloz R. W. (1964) The extracellular glycosidases of Diplococcus pneumoniae. I. Purification and properties of a neuraminidase and a β-galactosidase. Action on the α-1-acid glycoprotein of human plasma. Biochemistry 3, 1535–1543 10.1021/bi00898a025 [DOI] [PubMed] [Google Scholar]
- 48. Berry A. M., Lock R. A., Thomas S. M., Rajan D. P., Hansman D., and Paton J. C. (1994) Cloning and nucleotide sequence of the Streptococcus pneumoniae hyaluronidase gene and purification of the enzyme from recombinant Escherichia coli. Infect. Immun. 62, 1101–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maruyama Y., Nakamichi Y., Itoh T., Mikami B., Hashimoto W., and Murata K. (2009) Substrate specificity of streptococcal unsaturated glucuronyl hydrolases for sulfated glycosaminoglycan. J. Biol. Chem. 284, 18059–18069 10.1074/jbc.M109.005660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bongaerts R. J., Heinz H. P., Hadding U., and Zysk G. (2000) Antigenicity, expression, and molecular characterization of surface-located pullulanase of Streptococcus pneumoniae. Infect. Immun. 68, 7141–7143 10.1128/IAI.68.12.7141-7143.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Abbott D. W., Higgins M. A., Hyrnuik S., Pluvinage B., Lammerts van Bueren A., and Boraston A. B. (2010) The molecular basis of glycogen breakdown and transport in Streptococcus pneumoniae. Mol. Microbiol. 77, 183–199 10.1111/j.1365-2958.2010.07199.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Becker D. J., and Lowe J. B. (2003) Fucose: biosynthesis and biological function in mammals. Glycobiology 13, 41R–53R 10.1093/glycob/cwg054 [DOI] [PubMed] [Google Scholar]
- 53. Higgins M. A., Abbott D. W., Boulanger M. J., and Boraston A. B. (2009) Blood group antigen recognition by a solute-binding protein from a serotype 3 strain of Streptococcus pneumoniae. J. Mol. Biol. 388, 299–309 10.1016/j.jmb.2009.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Higgins M. A., Suits M. D., Marsters C., and Boraston A. B. (2014) Structural and functional analysis of fucose-processing enzymes from Streptococcus pneumoniae. J. Mol. Biol. 426, 1469–1482 10.1016/j.jmb.2013.12.006 [DOI] [PubMed] [Google Scholar]
- 55. Blomberg C., Dagerhamn J., Dahlberg S., Browall S., Fernebro J., Albiger B., Morfeldt E., Normark S., and Henriques-Normark B. (2009) Pattern of accessory regions and invasive disease potential in Streptococcus pneumoniae. J. Infect. Dis. 199, 1032–1042 10.1086/597205 [DOI] [PubMed] [Google Scholar]
- 56. Embry A., Hinojosa E., and Orihuela C. J. (2007) Regions of diversity 8, 9 and 13 contribute to Streptococcus pneumoniae virulence. BMC Microbiol. 7, 80 10.1186/1471-2180-7-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Katayama T., Sakuma A., Kimura T., Makimura Y., Hiratake J., Sakata K., Yamanoi T., Kumagai H., and Yamamoto K. (2004) Molecular cloning and characterization of Bifidobacterium bifidum 1,2-l-fucosidase (AfcA), a novel inverting glycosidase (glycoside hydrolase family 95). J. Bacteriol. 186, 4885–4893 10.1128/JB.186.15.4885-4893.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rosenow C., Maniar M., and Trias J. (1999) Regulation of the alpha-galactosidase activity in Streptococcus pneumoniae: characterization of the raffinose utilization system. Genome Res. 9, 1189–1197 10.1101/gr.9.12.1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Paixão L., Oliveira J., Veríssimo A., Vinga S., Lourenço E. C., Ventura M. R., Kjos M., Veening J.-W., Fernandes V. E., Andrew P. W., Yesilkaya H., and Neves A. R. (2015) Host glycan sugar-specific pathways in Streptococcus pneumoniae: galactose as a key sugar in colonisation and infection. PLoS One 10, e0121042 10.1371/journal.pone.0121042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Afzal M., Shafeeq S., Manzoor I., Henriques-Normark B., and Kuipers O. P. (2016) N-Acetylglucosamine-mediated expression of nagA and nagB in Streptococcus pneumoniae. Front. Cell. Infect. Microbiol. 6, 158 10.3389/fcimb.2016.00158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Barik S. (1996) Site-directed mutagenesis in vitro by megaprimer PCR. Methods Mol. Biol. 57, 203–215 10.1385/0-89603-332-5:203 [DOI] [PubMed] [Google Scholar]
- 62. McLean R., Hobbs J. K., Suits M. D., Tuomivaara S. T., Jones D. R., Boraston A. B., and Abbott D. W. (2015) Functional analyses of resurrected and contemporary enzymes illuminate an evolutionary path for the emergence of exolysis in polysaccharide lyase family 2. J. Biol. Chem. 290, 21231–21243 10.1074/jbc.M115.664847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., and Bairoch A. (2005) Protein identification and analysis tools on the ExPASy server, in The Proteomics Protocols Handbook (Walker John M., ed), pp. 571–607, Humana Press, New York [Google Scholar]
- 64. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., and Leslie A. G. W. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 10.1107/S0907444910048675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 10.1107/S0907444905036693 [DOI] [PubMed] [Google Scholar]
- 67. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 10.1107/S0907444911001314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Brünger A. T. (1992) Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475 10.1038/355472a0 [DOI] [PubMed] [Google Scholar]
- 70. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cowtan K. (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 10.1107/S0907444906022116 [DOI] [PubMed] [Google Scholar]
- 73. Lacks S. (1968) Genetic regulation of maltosaccharide utilization in pneumococcus. Genetics 60, 685–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Greene N. G., Narciso A. R., Filipe S. R., and Camilli A. (2015) Peptidoglycan branched stem peptides contribute to Streptococcus pneumoniae virulence by inhibiting pneumolysin release. PLoS Pathog. 11, e1004996 10.1371/journal.ppat.1004996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Vijayakumar M. N., and Morrison D. A. (1986) Localization of competence-induced proteins in Streptococcus pneumoniae. J. Bacteriol. 165, 689–695 10.1128/jb.165.3.689-695.1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.