

SUMMARY
Ras GTPases are mutated at codons 12, 13, and 61, with different frequencies in KRas, HRas, and NRas and in a cancer-specific manner. The G13D mutant appears in 25% of KRas-driven colorectal cancers, while observed only rarely in HRas or NRas. Structures of Ras G13D in the three isoforms show an open active site, with adjustments to the D13 backbone torsion angles and with disconnected switch regions. KRas G13D has unique features that destabilize the nucleotide-binding pocket. In KRas G13D bound to GDP, A59 is placed in the Mg2+ binding site, as in the HRas-SOS complex. Structure and biochemistry are consistent with an intermediate level of KRas G13D bound to GTP, relative to wild-type and KRas G12D, observed in genetically engineered mouse models. The results explain in part the elevated frequency of the G13D mutant in KRas over the other isoforms of Ras.
Graphical Abstract
In Brief
Johnson et al. show that conformational states and biochemical properties of the KRas G13D oncogenic mutant in the context of isoform-specific residues unique to KRas lead to destabilization of the active site, consistent with its intermediate phenotype between wild-type KRas and KRas G12D in genetically engineered mice.
INTRODUCTION
Cancer is the second leading cause of death in the United States and the world (Siegel et al., 2018). Oncogenic alleles of KRAS, HRAS, and NRAS are found in ~20% of cancers, with mutant KRAS alleles observed at frequencies of 95%, 40%, and 35% in pancreatic, colorectal, and lung cancers, respectively (Haigis, 2017; Prior et al., 2012). In general, particular mutations have different frequencies of occurrence in the three RAS isoforms and are associated with different survival outcomes (De Roock et al., 2010; Imamura et al., 2012; Messner et al., 2013). Furthermore, in vivo mouse studies have demonstrated that similar codon mutations produce different biological outcomes, depending on their isoform background (Haigis et al., 2008; Wang et al., 2013). However, there is little understanding of the factors leading to the isoform-specific preferences associated with Ras oncogenic mutants, and, in spite of great need, there are no US Food and Drug Administration (FDA)-approved inhibitors of Ras available in the clinic (Haigis, 2017; Prior et al., 2012).
Oncogenic alleles of Ras cluster predominantly at codons 12, 13, and 61. These mutations result in increased levels of Ras bound to guanosine triphosphate (GTP), promoting a hyper-proliferative state by the activation of, among others, Mek/Erk and Akt/mTOR (mammalian target of rapamycin) signaling pathways via the effector protein Raf and phosphatidylinositol 3-kinase (PI3K) (Haigis, 2017). Increase in the levels of Ras-GTP can occur by the impairment of intrinsic and GTPase-activating protein (GAP)-catalyzed GTP hydrolysis (e.g., G12V, Q61L) (Buhrman et al., 2011; Scheffzek et al., 1997) or by increased intrinsic and guanine nucleotide exchange factor (GEF)-catalyzed nucleotide exchange activity (e.g., G13D, A146T) (Hunter et al., 2015; Smith et al., 2013). The G13D mutant is particularly interesting because it is the most common mutation at codon 13 and it appears significantly more frequently in KRas than in either the NRas or HRas isoforms (Forbes et al., 2017). KRas G13D represents ~25% of KRas mutations in colorectal cancer (CRC)—more than in any other type of cancer—and is associated with a better prognosis than the more common KRas G12D allele (Haigis, 2017). In cultured cells, KRas G13D lacks the characteristic anti-apoptotic potential of KRas G12V (Guerrero et al., 2000, 2002). Furthermore, isogenic CRC cell lines expressing G12D or G13D have distinct phosphoproteomic profiles (Hammond et al., 2015). Despite their chemical similarity, G12D and G13D show different biochemical properties: G13D is more prone to the intrinsic exchange of guanosine diphosphate (GDP) for GTP and enhances the nucleotide exchange reaction catalyzed by Son of Sevenless (SOS), whereas G12D and G12V do not (Hunter et al., 2015; Smith et al., 2013). The molecular mechanism that differentiates G13D, particularly in KRas versus other isoform contexts, is key to understanding its etiology and druggability.
Ras bound to GTP is regulated by conformational states associated with the G-domain (Lu et al., 2016). We have previously shown that differences in structure and conformation underlie the transformative potential of different Q61 mutants of HRas, and that these differences are related to both local and global effects that the Q61 mutations have on the G-domain (Buhrman et al., 2007; Fetics et al., 2015). More recently, we have shown that the G12D mutant in the GTP-bound form of KRas also samples conformational states differently from wild-type KRas (Parker et al., 2018). Here, we present the structures, biochemical properties, and conformational states associated with the G13D mutant in the three Ras isoforms, focusing on KRas G13D for comparison with KRas G12D. Unlike D12, D13 stabilizes an open active site in the GTP-bound form in all three isoforms. Our collection of structures reveals mechanistic detail for the nucleotide exchange in the G13D mutant and points to structural features of KRas that correlate with the higher appearance of the G13D allele in KRAS than in NRAS or HRAS associated with human cancers. We then examine how these two mutants affect cellular and tissue homeostasis in the colon epithelium using genetically engineered mouse models. We find that KRas G13D shows an attenuated proliferative phenotype in vivo in comparison to KRas G12D, which is consistent with the expected outcome, given the structural and biochemical differences between the two oncogenic KRas proteins. With this work, we link isoform specificity and conformational states stabilized by D13 and D12 in KRas oncogenic mutants with molecular and phenotypic descriptions from in vivo analyses of KRas G13D and KRas G12D in mice.
RESULTS
The G-domain in the three Ras isoforms (HRas, KRas, and NRas) is divided into the effector lobe containing the switch regions and the allosteric lobe containing the isoform-specific residue differences (Johnson et al., 2017). In active Ras, switch I samples an open conformation, state 1, that is associated with nucleotide exchange and does not interact with effector proteins, and a closed conformation, state 2, associated with GTP hydrolysis and effector binding (Figure 1A) (Spoerner et al., 2010). Helix 3, loop 7, and helix 4 in the allosteric lobe work concertedly to regulate the stability and conformation of switch II associated with R and T states when switch I is in state 2 (Bandaru et al., 2017; Buhrman et al., 2010; Gorfe et al., 2008; Holzapfel et al., 2012; Johnson and Mattos, 2013). The balance of conformational states at equilibrium changes in the Ras isoforms and their oncogenic mutants, as can be detected by NMR (Parker et al., 2018; Spoerner et al., 2010), and this leads to distinct biochemical properties (Johnson et al., 2017). While HRas-GppNHp prefers state 2, the balance of conformational states in wild-type (WT) KRas-GppNHp is shifted toward state 1 (Parker et al., 2018) (Figure 1A). KRas G12D bound to GppNHp is in state 2 (Parker et al., 2018). Structures and conformational states associated with G13 mutants in the GTP-bound state have not been published for any of the Ras isoforms, although there is a crystal structure of KRas G13D bound to GDP (PDB: 4TQA) (Hunter et al., 2015).
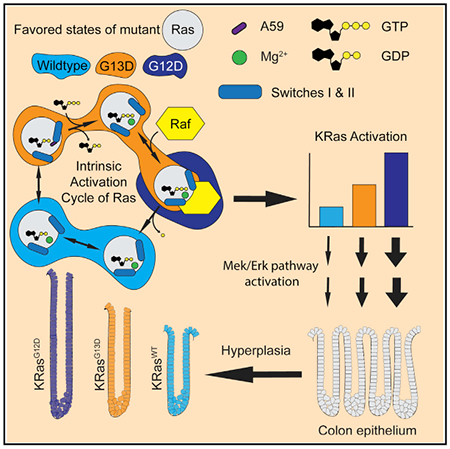
Figure 1. Ras G13D in the GTP-Bound State Favors an Open Active Site.
(A) The G-domain of Ras with effector (light gray) and allosteric lobes (light blue). Wild-type KRas-GppCH2p with switch I in state 1 (PDB: 5UK9, magenta). HRas-GppNHp in state 2 can have switch II in the disordered T-state (PDB: 2RGE, gray) or the R-state (PDB: 3K8Y, green).
(B) Ras G13D structures in state 1: H13GNP (orange), Ha13GNP (bright orange), Hb13GNP (light orange), Hc13GNP (yellow), Ka13GNP (cyan), Kb13GNP (light teal), and N13GNP (purple), with wild-type KRas as in (A).
(C) Ramachandran plot showing the ϕ,ψ dihedral angles for P loop residues 10–14 in wild type (gray), G12D (blue), and G13D (orange) for the Ras structures shown in Table S1. Generally allowed regions are contoured in blue and those allowed for glycine are in green (empirical data compiled by Lovell et al., 2003). Yellow-filled circles enclose ϕ,ψ angles for residues other than D13 and red circles enclose ϕ,ψ angles for D13 in the G13D structures.
(D) Ras G13D disrupts the interaction between switches I and II at the beginning of the β2 and β3 sheets. The β2 and β3 H-bonding interactions are shown for wild-type HRas as black dashed lines and for the G13D mutant as yellow dashed lines. Wild-type HRas is in green and the Ha13GNP model is in bright orange.
We solved the crystal structures of the G13D mutant G-domains of HRas, KRas, and NRas (residues 1–166) bound to the GTP analog GppNHp or to GDP (Table 1). Two crystal forms of HRas G13D bound to GppNHp were obtained, with one and three molecules in the asymmetric unit (AU), respectively. We thus have four models of HRas G13D bound to GppNHp (referred to as H13GNP, Ha13GNP, Hb13GNP, and Hc13GNP). The crystal structure of HRas G13D bound to GDP has three molecules in the AU (Ha13GDP, Hb13GDP, and Hc13GDP). KRas G13D bound to GppNHp has two molecules in the AU (Ka13GNP and Kb13GNP) and KRas G13D bound to GDP has one molecule in the AU (K13GDP). Crystals of NRas G13D bound to GppNHp also have one molecule in the AU (N13GNP). We did not obtain a structure of NRas G13D bound to GDP. The data collection and structure refinement statistics are shown in Table 1 for each of the six crystal structures, with their respective PDB identification numbers.
Table 1.
Data Collection and Structure Refinement Statistics
| H13GNP Form 1 | H13GNP Form 2 | N13GNP | K13GNP | H13GDP | K13GDP | |
|---|---|---|---|---|---|---|
| PDB | 6E6C | 6E6P | 6E6H | 6E6F | 6DZH | 6E6G |
| Molecules | H13GNP | Ha-c13GNP | N13GNP | Ka-b13GNP | Ha-c13GDP | K13GDP |
| WAVELENGTH (Å) | 1.54178 | 1.54178 | 1.54178 | 1.54178 | 1.54178 | 1.54178 |
| Resolution | 25.1–1.9 (1.96–1.9) |
40.1–1.93 (2–1.93) |
32.5–1.99 (2.07–1.99) |
36.1–3.40 (3.52–3.40) |
35.7–1.95 (2.02–1.95) |
34.0–1.93 (2–1.93) |
| Space group | C121 | P212121 | C121 | C121 | P212121 | P212121 |
| a, b, c (Å) | 54.6, 47.8, 57.0 | 61.6, 75.6, 94.7 | 59.6, 39.6, 65.0 | 114.5, 39.1, 89.3 | 60.6, 71.4, 94.9 | 36.1, 37.9, 100.2 |
| α, β, γ (°) | 90, 118.3, 90 | 90, 90, 90 | 90, 106.4, 90 | 90, 124.4, 90 | 90, 90, 90 | 90, 90, 90 |
| Total reflections | 33,830 | 217,382 | 42,385 | 22,660 | 175,532 | 63,590 |
| Unique reflections | 9,755 (817) | 32,621 (2,740) | 9,752 (854) | 4,526 (445) | 25,256 (1,138) | 10,737 (977) |
| Multiplicity | 3.5 | 6.7 | 4.3 | 5 | 7 | 5.9 |
| Completeness (%) | 95 | 96 | 97 | 97 | 82 | 99 |
| Mean I/sigma(I) | 14.8 (1.1) | 23.7 (1.6) | 13.9 (1.5) | 7.9 (1.6) | 17.5 (1.5) | 14.9 (1.5) |
| Wilson B-factor | 26.21 | 29.78 | 24.28 | 54.29 | 19.97 | 21.1 |
| R-merge | 0.088 | 0.085 | 0.098 | 0.14 | 0.092 | 0.101 |
| R-meas. | 0.10 (0.67) | 0.09 (0.69) | 0.11 (0.67) | 0.16 (0.54) | 0.104 (0.7) | 0.11 (0.46) |
| CC1/2 | 0.93 (0.67) | 0.96 (0.83) | 0.93 (0.55) | 0.93 (0.73) | 0.92 (0.69) | 0.96 (0.80) |
| CC* | 0.9799 (0.895) | 0.99 (0.95) | 0.98 (0.84) | 0.98 (0.92) | 0.98 (0.90) | 0.99 (0.94) |
| Reflections | ||||||
| For refinement | 9,751 (815) | 32,598 (2735) | 9,745 (852) | 4,524 (445) | 25,252 (1138) | 10,733 (976) |
| For R-free | 974 (82) | 1,998 (167) | 976 (85) | 453 (46) | 2,000 (90) | 1,068 (99) |
| R-work (%) | 16.8 (23.7) | 18.4 (25.1) | 17.9 (24.4) | 18.2 (21.0) | 16.3 (25.9) | 16.5 (22.4) |
| R-free (%) | 23.0 (34.1) | 23.7 (31.3) | 23.6 (31.1) | 25.8 (30.5) | 21.8 (30.0) | 22.5 (25.3) |
| Number of Atoms | ||||||
| Non-hydrogen | 1,403 | 4,131 | 1,332 | 2,287 | 4,072 | 1,481 |
| Macromolecules | 1,284 | 3,777 | 1,218 | 2,214 | 3,713 | 1,315 |
| Ligands | 34 | 132 | 33 | 65 | 112 | 29 |
| Protein residues | 164 | 483 | 154 | 304 | 472 | 166 |
| RMSD (bonds) | 0.006 | 0.007 | 0.007 | 0.011 | 0.007 | 0.006 |
| RMSD (°) | 0.92 | 0.92 | 0.90 | 1.39 | 0.98 | 0.90 |
| Clash score | 3.12 | 3.14 | 2.44 | 26.64 | 7.09 | 5.74 |
| Ramachandran (%) | ||||||
| Favored | 98 | 97 | 98 | 83 | 97 | 99 |
| Allowed | 1.9 | 2.9 | 1.9 | 14 | 2.6 | 1.2 |
| Outliers | 0 | 0.21 | 0 | 3.7 | 0.43 | 0 |
| Rotamer outliers (%) | 0 | 1.5 | 1.5 | 2.4 | 2.5 | 0.71 |
| B-factors | ||||||
| Average | 27.61 | 36.59 | 26.36 | 40.46 | 27.42 | 23.27 |
| Macromolecule | 27.41 | 36.36 | 26.04 | 40.47 | 27.20 | 22.63 |
| Ligands | 24.27 | 35.15 | 23.78 | 41.16 | 25.55 | 16.58 |
| Solvent | 31.95 | 41.29 | 32.23 | 31.97 | 31.52 | 30.84 |
CC, Pearson correlation coefficient; RMSD, root mean square deviation.
Values are dataset averages, and values in parentheses are from data in the highest-resolution shell.
Ras G13D Bound to GppNHp Favors an Open Active Site
The structures of Ras G13D bound to GppNHp showed greater variation in switches I and II, as well as in loop 8 (residues 120–128) and helix 3/loop 7 (residues 102–108), relative to wild-type, G12, and Q61 structures in the PDB (Berman et al., 2000) (Table S1; Figure S1). Of the seven GppNHp-bound protein models of Ras G13D in the three isoforms, only Hc13GNP showed a closed active site in state 2 (discussed below). The other six models show switch I in an open state 1 conformation, with Y32 flipped outward and T35 pulled away from the nucleotide and the Mg2+ ion (Figure 1B). In all but Hb13GNP, D13 is positioned over the nucleotide near its most favored rotamer (Figure S2). The D13 residues in our structures have different ϕ,ψ values (Figure 1C, red circle) than those found in structures with G13, where the ϕ,ψ values are in a region of the Ramachandran plot allowed for glycine but not for other residues (Lovell et al., 2003). This is not the case for the G12 mutants, as the ϕ,ψ torsions for G12 are already in a generally allowed region. The change in ϕ,ψ for residue D13 results in a small adjustment in ϕ,ψ values in neighboring P loop residues to varying extents (Figure 1C, residues 11 and 12). In the HRas G13D models, the adjustment is localized to D13, and G60 interacts with the γ-phosphate in all but the Ha13GNP model. In the two models of KRas G13D and in NRas G13D bound to GppNHp, adjustments in the P loop backbone torsion angles propagate from G13 to G10, and the carbonyl of G60 no longer interacts with the amide of G12 or with the nucleotide. This is associated with a highly disordered switch II. The two switch regions are linked by the β2-β3 interswitch β sheet, which normally begins with an H-bond between the carbonyl of I36 and the amide of A59 (Figure 1D, black-boxed residues and dashed lines). In all of the Ras G13D models (regardless of isoform), I36 is far from A59 and the β-ladder is “unzipped” by two H-bonds, starting with an H-bond between the carbonyl of D38 and the amide of D57 (Figure 1D, yellow-boxed residues and dashed lines).
KRas G13D Is Unique in Destabilizing the Nucleotide-Binding Pocket Beyond State 1
The KRas nucleotide-binding pocket is more open in our models of the GppNHp-bound KRas G13D mutant than in the G13D mutants of HRas and NRas isoforms due to key side chains being disordered. The Ka13GNP model is similar in overall conformation to that observed in the wild-type KRas-GppCH2p structure (PDB: 5UK9) in spite of the different crystal forms from which the two structures were solved (Figure 2A). However, in Ka13GNP, there is significant adjustment of ϕ,ψ torsions associated with P loop residues, the γ-phosphate of GppNHp is shifted toward switch I, and switch II is disordered from residue 59 to 70. The side chain of K16, a critical nucleotide-binding residue, is turned toward the carbonyl groups of P loop residues G10 and A11 and does not interact with the shifted β- and γ-phosphates of the nucleotide (Figure 2A, yellow versus black dashed lines).
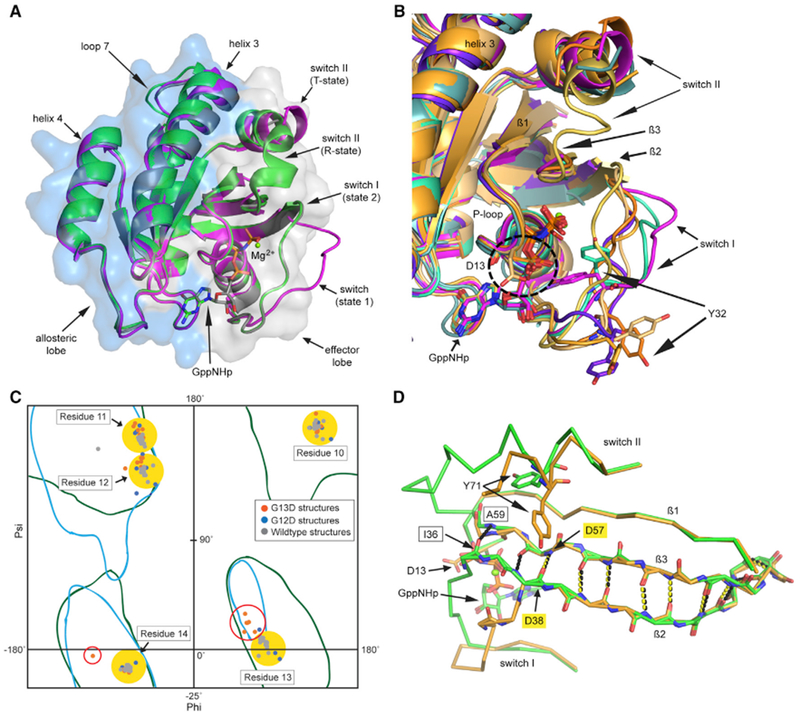
Figure 2. KRas G13D Structures in the GTP-Bound and GDP-Bound Forms.
(A) H95 in KRas G13D (Ka13GNP, brown), nestled between Q99 and Y96, affects residues in the nucleotide-binding pocket relative to wild-type KRas (PDB: 5UK9, magenta). H-bonds are shown as black dashed lines for wild-type KRas and as yellow dashed lines for Ka13GNP.
(B) View of the active site with electron density (blue wire mesh) for the Kb13GNP structure (yellow), with disordered F28 and K147.
(C) The K13GDP model (bright brown) shows D13 coordinated to a Na+ ion (purple) and linked to switch II and helix 3 residue H95 through water-mediated H-bonding interactions (yellow dashed lines).
(D) The N-terminal end of switch II in K13GDP (bright brown, yellow dashed lines) with A59 in the Mg2+ binding site, superimposed on switch II of wild-type HRas bound to SOS (PDB: 1BKD, green sticks, black dashed lines).
Electron density contoured at the 1 σ level is shown in (B) and (D) (blue wired mesh).
See also Figure S2.
The nucleotide-destabilizing features seen in Ka13GNP are exacerbated in Kb13GNP, where switch I is away from crystal contacts. In this model, switch I is disordered, with little or no electron density for residues 28 to 40. Both F28 and K147, which together normally help stabilize the guanine base, are disordered in the Kb13GNP model (Figure 2B). The γ-phosphate is shifted further from its binding pocket with a shift also in the β-phosphate and the remainder of the nucleotide. In this structure, the K16 side chain is exposed to solvent, disordered beyond the Cγ carbon, and the Mg2+ is absent. Switch II is disordered starting at residue A59, with electron density reappearing at S65. In spite of different crystallographic environments, the two molecules of KRas G13D bound to GppNHp are remarkably similar with respect to nucleotide shift and active site disorder, as well as in the role of isoform-specific residues. In particular, H95, which is a residue unique to KRas (Q95 in HRas and L95 in NRas), is found in a similar conformation in both the G13D mutant KRas structures and in wild-type KRas-GppCH2p, intercalated between Y96 and Q99 (Figure 2A). In contrast, Q95 in HRas and L95 in NRas are turned away from Q99 in all of the available wild-type and mutant structures. In KRas G13D, the presence of D13 and the resulting shifts in P loop ϕ,ψ angles (observed in all G13D structures to varying extents) appear to work in synergy with the Q99, H95, Y96 trio to affect the side chain of K16, the position of the nucleotide, and the F28 and K147 side chains. Thus, the position of H95 and the interactions that result contribute to the nucleotide-destabilizing features unique to KRas G13D in the context of an open switch I, facilitating the shifts observed for the guanine base and its interacting residue D119.
The Structure of Ras G13D Poised for GDP Release
The crystal structure of HRas G13D bound to GDP has three molecules in the AU and is in the same crystal form as our structure of HRas G13D bound to GppNHp (Figure S2). The features common to the three molecules modeled in this structure include the presence of the Mg2+ ion, as in the wild-type HRas-GDP structure (PDB: 4Q21), and adjustment of the P loop ϕ,ψ torsion angles due to the D13 side chain. Switch I is either disordered or in an open conformation, different from that seen in the wild-type HRas-GDP structure, where switch I is closer to the nucleotide (PDB: 4Q21). Switch II is also found in a more open conformation. The presence of D13 destabilizes residues T35, I36, and E37, leading to a loss of the first H-bond in the β2-β3 ladder between I36 and A59, as observed in the GppNHp bound Ras G13D structures.
The crystal structure of KRas G13D bound to GDP was obtained from a full crystallization screen and is very different from the previously published structure of this mutant bound to GDP (PDB: 4TQA) (Hunter et al., 2015), which is nearly identical to the wild-type KRas-GDP structure (PDB: 4OBE) and wild-type HRas-GDP structure (PDB: 4Q21). In the crystal structure represented by the K13GDP model (Figure 2C), the D13 side chain is over the nucleotide interacting with a Na+ ion, and the P loop ϕ,ψ adjustments propagate to G10. Switch I is in an open conformation, with water molecules bridging the backbone of Y32 to the phosphate groups of the nucleotide. Residues I36 and E37 are disordered, and the first H-bond in the β2-β3 ladder is between the carbonyl of D38 and amide of D57 as we observe in all of our G13D structures. This allows a unique and highly ordered conformation of switch II in K13GDP, with several of its residues interacting intimately with residues on helix 3 around the KRas isoform-specific residue H95. Residues Y96, H95, and Q99 have the same relation to each other as described for the models of KRas G13D bound to GppNHp, with a similar relation to the P loop G10 carbonyl group, confirming the effect of these residues with this high-resolution structure. The C-terminal end of switch II forms the α2 helix involved in an extensive network of H-bonding interactions (Figure 2C). The N-terminal end of switch II is in a unique conformation in K13GDP, where it wraps around the side chain of R68, with the backbone carbonyl groups of Q61, E62, and, potentially, E63 interacting with the R68 side chain and with the hydroxyl group of T58 bridged to R68 through a water molecule. These interactions position the side chain of Q61 to make an amino-aromatic interaction with Y71, linking the two ends of switch II. The side chain of Y64 is turned toward helix 3, with its OH group forming an H-bond with H95 and the aromatic ring stacking with the aliphatic portion of the E63 side chain (Figure 2C). The interaction between switch II and helix 3 is further stabilized by a salt bridge between D69 (switch II) and R102 (helix 3). Overall, the D13 side chain near the nucleotide, the KRas-specific residue H95 on helix 3, and the salt bridge at the end of switch II, linked by interactions with R68 at the center of this network, converge to stabilize a conformation where residues 58–61 form a tight β-turn that places A59 in the Mg2+-binding pocket, precluding the binding of Mg2+, which is essential for nucleotide binding by Ras (Figure 2D) (John et al., 1993). This is the location of A59 in the complex between HRas and the nucleotide exchange factor SOS with no Mg2+ or nucleotide in the active site (PDB: 1BKD) (Boriack-Sjodin et al., 1998). In this complex, residues 58–61 form a tight β-turn similar to the one we observe in K13GDP. The structure we have captured for KRas G13D bound to GDP suggests a mechanism for the expulsion of Mg2+ in KRas G13D, which, together with the high concentration of GTP in the cell, could explain the more prominent appearance of the G13D mutant in the KRas isoform.
Conformational States, GTP Hydrolysis, and Binding to Raf-RBD
We recently published 1H-NMR downfield chemical shifts of KRas and HRas proteins bound to GppNHp, where resonance peaks Z and X are probes of the conformational states associated with switch I and switch II, respectively (see Method Details) (Figures 3A and 3B) (Parker et al., 2018). In addition to determining that switch I in KRas shifts toward state 1 relative to HRas and that the interaction between switch II and the γ-phosphate remains for HRas but not for KRas in the state 1 conformation, we showed that the G12D mutant stabilizes a state 2 conformation in KRas, insensitive to increases in temperature (Parker et al., 2018), which is consistent with molecular dynamics (MD) simulations that found a particularly stable state 2 for this mutant (Sayyed-Ahmad et al., 2017). Here, we report the 1H-NMR spectra for KRas G13D and HRas G13D, shown with the respective wild-type spectra for comparison (Figures 3A and 3B). The most striking feature for KRas G13D is that the spectrum between 10 and 13 ppm is devoid of peaks at all temperatures collected, in contrast to wild-type KRas where peaks X and Z are observed, indicating that state 2 is populated in the wild-type protein (Figure 3A) (Parker et al., 2018). The situation for HRas G13D is very different. Peak Z is attenuated relative to the wild-type HRas protein, consistent with a more prominent state 1, but even at the higher temperatures, state 2 is detected in HRas G13D (Figure 3B). Peak X is prominent in the HRas G13D spectrum, consistent with an intact interaction between the γ-phosphate and G60 (switch II) in our crystal structures of HRas G13D.
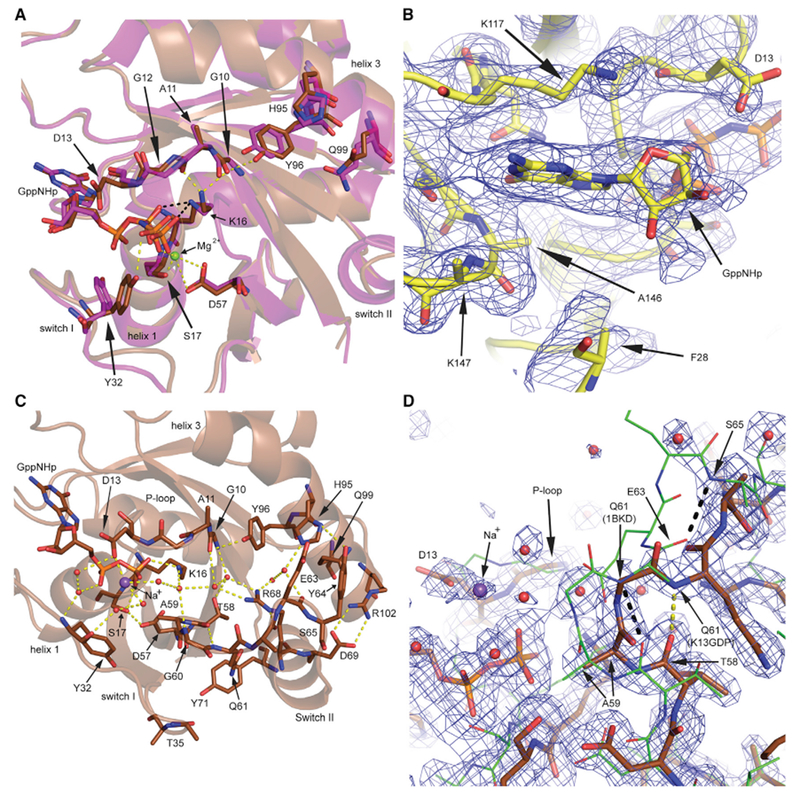
Figure 3. G13D Favors State 1 More Prominently Than Wild Type in Both KRas and HRas.
(A and B) 1H-NMR spectra for (A) wild-type KRas and KRas G13D and (B) wild-type HRas and HRas G13D bound to GppNHp, showing the temperature dependence for each peak. The spectra for the wild-type proteins (black) were previously discussed, along with peak contributions (Parker et al., 2018), also described in Method Details.
(C) Superposition of the Hc13GNP model (gold) with that of the Ras/Raf-RBD complex (PDB: 4G0N; HRas green, Raf-RBD magenta).
(D) D13 of Hc13GNP (gold) clashes with the position of Y32 in wild-type HRas in state 2 (PDB: 3K8Y, green). D13 helps coordinate a Na+ ion (purple) in the active site. Water molecules are shown in the black sphere for wild-type HRas and in red for Hc13GNP, with orange dashed lines representing H-bonding interactions in the mutant.
See also Figures S3, S4, S5, and S6.
For completeness, we report rate constants for GTP hydrolysis (Figure S3) and nucleotide exchange (Figure S4) for HRas G13D and KRas G13D measured side by side. The hydrolysis rate constants were obtained by measuring the release of 32Pi from γ-32P-GTP loaded Ras proteins (Johnson et al., 2017), thus avoiding the 2(3)-O-(N-methylanthraniloyl) (mant) fluorescent tag that may affect hydrolysis in unpredictable ways (Figure S3) (Mazhab-Jafari et al., 2010). We measured the rate constants for the intrinsic exchange of GDP for mant-GDP or mant-GppNHp for wild-type and mutant KRas and HRas proteins side by side (Figure S4), as had been done previously for KRas and its mutants (Hunter et al., 2015). These experiments come with the inherent problem that the mant-nucleotide may affect the rate constants as detailed in the supplemental information file (Figure S4). We also determined the dissociation constant (Kd) and thermodynamic parameters for the interaction between Raf-RBD (Ras-binding domain) and the three isoforms of Ras G13D using isothermal titration calorimetry (ITC) (Table 2), collected concurrently with our previously published values of 100 nM for wild-type KRas and wild-type HRas and 200 nM for wild-type NRas (Johnson et al., 2017). It appears that the presence of an increased population of state 1 in wild-type KRas does not impair Raf-RBD binding, which stabilizes the state 2 conformation, as previously shown for HRas (Spoerner et al., 2010) and KRas (Parker et al., 2018). The thermodynamic parameters for the interaction in the wild-type Ras proteins showed that the increase in Kd for wild-type NRas was primarily due to an entropic factor. The Kd for interaction with Raf-RBD is increased for Ras G13D in all three isoforms as expected, given a more open switch I (Table 2). The slightly greater Kd values for mutant KRas and NRas are due to entropic contributions, consistent with our observations from the crystal structures and NMR experiments for KRas G13D shown in Figure 3. In spite of the somewhat lower affinity, our ITC experiments indicate that Ras G13D interacts favorably with Raf-RBD (negative ΔG values in Table 2) and thus would be expected to activate Raf in cells. Relative affinities (not absolute affinities) of various KRas mutants for Raf-RBD have been reported (Hunter et al., 2015). The measurements were made using a competition assay with some inherent limitations (Figure S5) and therefore cannot be compared to the absolute Kd values in Table 2.
Table 2.
ITC Data for Complex Formation between RAS G13D and RAF-RBD
| HRAS G13D | KRAS G13D | NRAS G13D | |
|---|---|---|---|
| Ka (106 M−1) | 2.5 ± 0.08 | 2.07 ± 0.07 | 2.13 ± 0.06 |
| Kd (nM) | 409.2 ± 12.74 | 483.4 ± 16.34 | 468.9 ± 12.43 |
| N | 1.0 ± 0.05 | 1.0 ± 0.01 | 1.0 ± 0.01 |
| ΔS (J • mol/K) | 59.7 ± 3.72 | 56.7 ± 6.25 | 55.8 ± 1.67 |
| ΔH (kcal/mol) | −4.5 ± 0.28 | −4.6 ± 0.44 | −4.7 ± 0.11 |
| ΔG (kcal/mol) | −8.7 ± 0.02 | −8.6 ± 0.02 | −8.6 ± 0.02 |
| T • ΔS (kcal/mol) | 4.3 ± 0.27 | 4.0 ± 0.44 | 4.0 ± 0.12 |
Of the seven GppNHp-bound Ras G13D models presented here, the Hc13GNP model of HRas G13D is the only one in which a state 2 conformation of switch I is observed, required for both signaling through effector proteins and hydrolysis of GTP. Superposition of this structure with that of the HRas-GppNHp/Raf-RBD structure (PDB: 4G0N) (Fetics et al., 2015) shows that the conformation observed in the Hc13GNP molecule is expected to allow the binding of Raf-RBD, with E31 and D33 in position to interact with Raf-RBD residue K84, with only minor adjustments in switch I (Figure 3C). In the Hc13GNP model, the D13 side chain is positioned over the nucleotide in its most favored rotamer, with a Na+ ion directly interacting with it, precisely at the location where Y32 is found in the wild-type HRas GppNHp-bound structure (PDB: 3K8Y) (Figure 3D). There are several water molecules interacting with the Na+ ion and one of them H-bonds to the backbone carbonyl of Y32. Thus, it is possible that Na+ helps catalyze the hydrolysis of GTP in the G13D mutants by neutralizing the negative D13 charge to facilitate a conformational change of the negatively charged switch I to state 2 and by helping to stabilize the transition state of the GTP hydrolysis reaction (Du et al., 2004). This is consistent with the fact that the G13D mutation does not alter the intrinsic hydrolysis reaction as much as mutations at positions G12 and Q61 (Smith et al., 2013). A Na+ ion at this position was also reported to be persistent in Ras G13D simulations, with no exchange with solvent throughout the microsecond trajectory (Sayyed-Ahmad et al., 2017). Cation-dependent GTPases, which have aspartate, serine, or threonine instead of glycine at residue 13 in the P loop, use monovalent ions in a similar way, providing precedence for this kind of mechanism (Kuhle and Ficner, 2014). Furthermore, hydrolysis experiments in the presence of NaCl versus CaCl2 show a greater decrease in rate for KRas G13D relative to wild-type KRas in the absence of Na+ (Figure S6).
K-Ras G13D Differently Perturbs the Homeostasis of Murine Colonic Epithelium Compared to K-Ras G12D
Missense mutations coding for aspartate instead of glycine are the most frequent codon 12 and 13 alterations for KRas-driven colorectal cancers, with KRas G12D mutants considerably more common than KRas G13D (Haigis, 2017). Moreover, these two mutations are associated with significantly different clinical outcomes in CRC patients (De Roock et al., 2010; Haigis, 2017; Imamura et al., 2012; Messner et al., 2013). Here, we explored the possibility that the biochemical differences in activation mechanism for G12D (impairment of GTP hydrolysis) and G13D (enhancement of nucleotide exchange) offer an explanation for the differences in frequency and clinical phenotypes between these two alleles. To this end, we examined the biochemical and histological phenotypes of mice expressing KRas G12D or KRas G13D in colonic epithelium. Tissue-specific expression of KRas G13D was achieved by mating KRasLSL-G13D animals to those with the Fabpl-Cre driver, resulting in G13D expression in the epithelium of the colon and the distal small intestine of the mouse, as we have previously done for KRas G12D (Haigis et al., 2008). When we assessed the gross colonic morphology of colons expressing KRas G13D (Figure 4A), we observed crypt hyperplasia that was intermediate between wild-type and KRas G12D colons (Figures 4B and 4C). In agreement with our histologic analysis, we observed that colons expressing KRas G13D had an intermediate level of Ras-GTP, as assayed by pulldown with Raf-RBD (Figure 4D).
Figure 4. KRas G13D Has a Distinct Phenotype from that of Wild-Type KRas and KRas G12D in the Murine Colonic Epithelium.
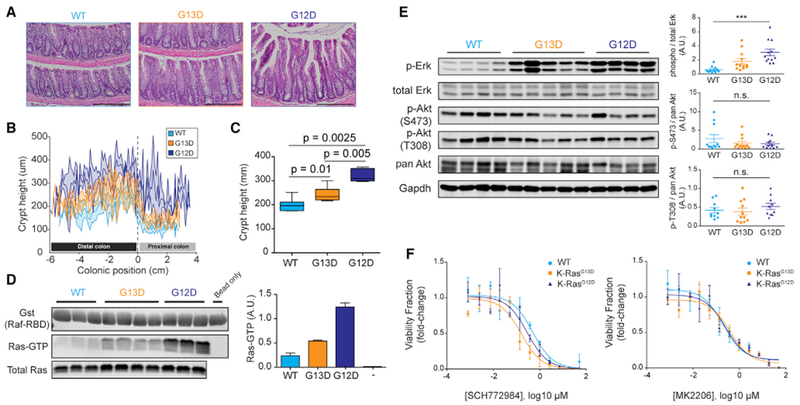
(A) H&E staining of formalin-fixed and paraffin-embedded colon Swiss rolls (5 μm) from 8- to 12-week-old mice. Scale bar, 200 μm.
(B) Crypt height measurements across the entire length of the colon from H&E stains in (A). Measurements were taken every 5 crypts, and the curves are composites of measurements from 7 wild type (Fabpl-Cre), 7 G13D, and 5 G12D.
(C) Quantification of data in (B).
(D) Affinity precipitation of Ras-GTP using Raf-RBD. Each pull-down used 500 μg colon lysate from 8- to 12-week-old mice, while total Ras blot was done with 50 μg lysate. Right, quantification of band intensities from 3 wild-type, 4 G13D, and 3 G12D pull-down experiments.
(E) Representative western blot of wild-type, G13D, and G12D colon lysate for phosphorylated-to-total protein from Erk1/2, pan-Akt, and its two phosphorylation sites T308 and S473. Right, quantitation of western blot bands from 11 wild type, 12 G13D, and 12 G12D biological replicates for Erk and pAkt S473, and 10 wild-type, 11 G13D, and 10 G12D biological replicates for pAkt T308; ***p < 0.0001, 1-way ANOVA (Kruskal-Wallis). Error bars of quantitated western blot data represent SEMs (±SEMs).
(F) Response of murine colonic organoids of various KRas alleles to 6 days of Akt (MK2206) and Erk (SCH772984) treatment. Data show luminescence averages from three biological replicates per genotype, and curves were fit with nonlinear regression. Error bars of luminescence data ± SEMs.
We also assessed KRas signaling by interrogation of Erk and Akt phosphorylation via quantitative western blotting. While Erk is a readout of Raf activation, we chose Akt phosphorylation as a means to assess the activation of PI3K (Haigis et al., 2008; Saxton and Sabatini, 2017). G13D, like G12D, activated Erk above wild-type levels. However, KRas G13D showed an intermediate level of pErk compared to G12D and wild type (Figure 4E). In contrast, we did not observe significant differences between G12D and G13D induction of pAkt at residues T308 and S473 (Figure 4E). These data demonstrate that quantitative differences in steady-state KRas activation translate into proportional differences in signaling and cell readouts in an in vivo setting.
Because of the differential Erk activation between G12D and G13D and the fact that we observed putative attenuation of p-Ser473 Akt in G12D colons (Figure 4E, top panel), we wanted to determine whether KRas mutant epithelial cells would respond differently to the inhibitors of PI3K and MAPK pathways. To this end, we treated wild-type, KRas G12D, and KRas G13D murine colonic organoids with SCH772984, an Erk inhibitor, and MK2206, an Akt inhibitor, and assessed their effect on cell viability after 6 days. We observed that while mutant KRas alleles did not alter sensitivity to the inhibition of Akt in this system, organoids expressing mutant KRas were more sensitive to Erk inhibition than wild type (Figure 4F). The difference in the half-maximal inhibitory concentration (IC50) between KRas mutants and wild type suggested allele-specific sensitivity and reflected the observations we made previously (Haigis et al., 2007; Poulin et al., 2019) that G12D-mediated hyperproliferation in the colon was mitogen-activated protein kinase (MAPK) dependent. In this vein, the sensitivity of KRas G13D organoids to Erk inhibition indicates that its intermediate activation of Erk (Figure 4E) is enough to cause addiction to MAPK activity.
DISCUSSION
Protein structures are dynamic in solution and sample a variety of conformational states populated according to their respective energies. Given the very high sequence similarity between the G-domains in the three isoforms of Ras, it is likely that they share a set of accessible conformations associated with biological function, which are populated more or less, depending on isoform-specific residues. Oncogenic mutants disfavor certain conformational states and stabilize others (Fetics et al., 2015), and their effects vary depending on the specific context of each isoform. This is clearly shown here for the G13D mutants. We have obtained GppNHp- and GDP-bound structures of Ras G13D, most of them in multiple crystallographic environments. This gives us a rare perspective for analysis, allowing the visualization of features common to the presence of the G13D side chain, regardless of isoform and crystal form. We also see certain features in the KRas G13D structures bound to GppNHp appear in distinct crystallographic environments that do not appear for HRas G13D and NRas G13D, and those features correlate well with our solution studies. Given the lower resolution of this structure (Table 1), we focus on general trends such as disorder of entire side chains in the active site or general proximity of residues, rather than specific distances between residues. The fact that we observe features in Ka13GNP and Kb13GNP common to all of our G13D structures gives us confidence in aspects that are unique to these models, analyzed at the appropriate resolution. In support of our crystal structures, we have shown that conformational states in KRas G13D (Figure 3) are accessed differently than in KRas G12D (Parker et al., 2018) in a way that is consistent with the biological effects that we observe in our in vivo experiments.
The common features in the structures of Ras G13D bound to GppNHp are that (1) switch I is predominantly in an open state 1 conformation; (2) the P loop adjusts to relieve strain, resulting in weaker interactions between G60 and the nucleotide; and (3) the first two H-bonds in the β2-β3 ladder that links the two switch regions are broken (Figure 1). These features together lead to a more open and disordered switch II, with Y71 turned toward the solvent, in contrast to its position in the core of the protein seen in most HRas-GppNHp structures. Our structures of Ras G13D bound to GDP share these general characteristics. The more open and disordered active sites due to the D13 side chain promote fast intrinsic nucleotide exchange as measured for HRas and KRas compared to their wild-type counterparts (Hunter et al., 2015; Smith et al., 2013) (Figure S4). KRas G13D is unique in promoting further destabilization of the nucleotide-binding pocket in both GppNHp- and GDP-bound structures, and we have linked this to a synergy between the D13 side chain and the isoform-specific residue H95, sandwiched between Y96 and Q99 on helix 3 (Figures 2A and 2C). This trio of residues is an essential feature in the binding pocket occupied by an inhibitor covalently bound to the KRas mutant M72C, uncovering a new mode of binding between switch II and helix 3, likely to be particular to KRas (Gentile et al., 2017). In KRas G13D bound to GppNHp, this cluster of residues perturbs interactions between the P loop and the nucleotide γ-phosphate, destabilizing the nucleotide-binding pocket and weakening interaction with the Mg2+ ion (Figure 2A). In KRas G13D bound to GDP, the cluster is part of an extensive network that promotes a β-turn conformation at the beginning of switch II, in which A59 is positioned to displace the Mg2+ ion in a fashion similar to that observed in the HRas-SOS complex (PDB: 1BKD) (Figures 2C and 2D).
Our structures of Ras G13D represent snapshots of conformational states accessed and populated differently in each isoform. Thus, we use our collection of models, as well as the previously published structure of KRas G13D bound to GDP (PDB: 4TQA) (Hunter et al., 2015), to piece together a structure-guided mechanism through which KRas G13D promotes self-activation via nucleotide exchange (Figure 5). In a “closed” GDP bound state (step 1, Figure 5), D13 is shifted away from the diphosphate moiety of GDP and the Mg2+ is bound in the active site, with an intact H-bonding ladder between β2-β3, similar to that observed for the wild-type KRas-GDP structure (PDB: 4OBE). In an important catalytic step, the D13 side chain promotes significant sampling of an “open” switch I conformation of the GDP bound state, represented by our HRas G13D model Hb13GDP, in which the Mg2+ ion is seen in the active site (Figure 5, step 2). Next, with D13 turned toward the diphosphate moiety (as in our model K13GDP), the C terminus of switch I, including I36 and E37, moves away from the active site, breaking the first β2-β3 backbone H-bond and relieving switch I and II interactions. Switch II, now disconnected from switch I, is stabilized by the R68/H95 network in KRas linking helix 3, switch II, and the side chain of D13, with the N terminus of switch II in a β-turn conformation that places the Cβ of A59 in the Mg2+ binding site (Figure 5, step 3). Thus, a major effect of the D13 side chain appears to be stabilization of KRas-GDP in a conformation poised for nucleotide exchange in a way not observed for wild type, unless in complex with SOS (Boriack-Sjodin et al., 1998). This major catalytic step ejects Mg2+ from the active site, thereby promoting the release of GDP, which is readily replaced by GTP, present at a concentration that is 10-fold higher in the cell (Traut, 1994). The model K13GDP representing step 3 appears to be poised for interaction with SOS, possibly aiding the significant increase in SOS-catalyzed nucleotide exchange in the G13D mutants of Ras (Smith et al., 2013). The fact that we captured this structure suggests that it may be favored in KRas relative to the other two isoforms. Presumably, SOS works in concert with the G13D mutation to maintain the high levels of GTP-bound Ras, although in KRas G13D the intrinsic stabilization of the conformation promoted by SOS may make its role less important than in other isoforms or mutants. The active GTP-bound state of KRas G13D (Figure 5, step 4) is represented by our model Ka13GNP, with the GTP analog and Mg2+ bound in the active site. While GTP binding may be less stable in KRas G13D than in the other two isoforms, the abundance of GTP likely ensures it to be the dominant state in this mutant. Binding of an effector protein such as Raf (Figure 5, step 5) promotes a state 2 conformation of switch I, as represented by Hc13GNP (Parker et al., 2018; Spoerner et al., 2010). Hydrolysis of GTP to GDP, possibly catalyzed by Na+, combined with the back exchange of nucleotide due to a destabilized active site (as in Kb13GNP), completes the cycle. We speculate that the destabilized GTP-binding pocket, as observed in KRas G13D bound to GppNHp, allows a small but significant fraction of the protein to be bound to GDP, which is consistent with the sensitivity of KRas G13D-driven cancers to epidermal growth factor receptor (EGFR) activation and inhibition (Bandaru et al., 2017; De Roocket al., 2010; Haigis, 2017; Imamura et al., 2012; Margonis et al., 2015; Messner et al., 2013).
Figure 5. Mechanism for Cycling between GDP- and GTP-Bound Forms of Ras G13D.
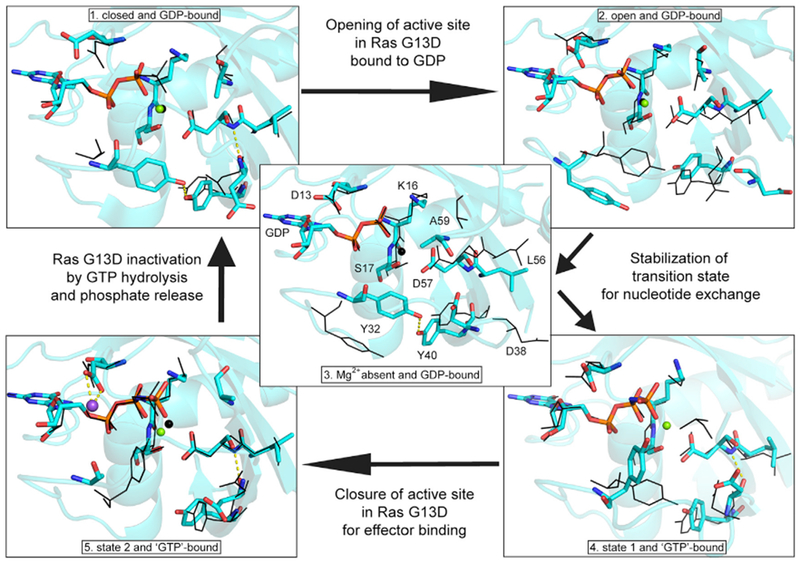
Models in cyan depict each step of the catalytic cycle, with the preceding step shown in black. Residues are labeled in the central panel (3), and these residues are shown in their respective conformations in each panel. (1) PDB: 4TQA, (2) Hb13GNP, (3) K13GDP, (4) Ka13GNP, and (5) Hc13GNP. Na+ is shown in purple and Mg2+ is shown in green. Note that A59 is disordered in (4) and therefore not present in the model shown in cyan.
Early attempts to distinguish the clinical outcome of KRas-specific alleles showed that G12 mutants were often found in metastatic tissue samples, whereas G13D mutations were not, suggesting that G13D is a weaker oncogenic allele of KRas (Finkelstein et al., 1993). These suspicions were confirmed later in vitro, where it was demonstrated that G13D was less prone to contact and anchorage independence, as well as more susceptible to apoptosis, compared to a KRas G12 mutant (Guerrero et al., 2000). Likewise, these in vitro data were recapitulated in xenograft experiments using nude mice (Guerrero et al., 2002). Finally, codon 13 mutants of KRas clearly favor the G13D mutation, whereas NRas and HRas have a much broader distribution of mutations at codon 13 (Forbes et al., 2017). The cause of these mutation-allele differences is less obscure in light of our structural and biochemical analyses. Interactions involving H95 in KRas, absent in the other two isoforms, in concert with D13, promote key conformations associated with the destabilization of the active site in favor of nucleotide exchange. In all, our structures show that the KRas G13D mutation promotes a Ras-activated state in a distinctly unique manner such that, in combination with unknown cellular factors associated with CRC, this mutant in KRas has a selective advantage over G13D in other isoforms.
In mouse experiments, we observe that KRas G12D and KRas G13D activating alleles are not functionally equivalent; G12D is a more aggressive driver of hyperproliferation in the colon crypt compared to G13D (Figure 4). This correlates with the ability of these mutants to recognize and bind Raf-RBD. Furthermore, we find that G12D and G13D mutant murine colonic organoids are similarly more sensitive to the blockade of Erk than wild type (Figure 4F), suggesting the presence of a signaling threshold for MAPK activity that drives the hyperproliferative phenotype observed in vivo, similar to what we have seen for KRas A147T (Haigis et al., 2007; Poulin et al., 2019). It is reasonable to suggest that signaling differences in the mouse colonic epithelium between KRas G13D and KRas G12D are due to differences in direct effector binding (Table 2; Figure 3). However, we showed that the binding reaction between Raf-RBD and Ras G13D is favorable and spontaneous (Table 2) and propose that although the 4-fold decrease in affinity relative to wild type may play some role toward the observed phenotype, this effect is overwhelmed in the KRas G13D mutant by destabilization of the active site in both the GDP- and GTP-bound states. The different activated states of KRas G12D and KRas G13D and their downstream effect on Erk1/2 activation in vivo likely represent the true ratio of GTP-/GDP-bound states of these proteins in the colon. Given that the two mutants are equally insensitive to GAPs and have similar hydrolysis rates (Hunter et al., 2015), the lower activated state of KRas G13D compared to KRa G12D likely reflects the upper limit of activation due to the back exchange of GTP for GDP.
Overall, the structural and biochemical data presented here for KRas G13D are consistent with the intermediate phenotype observed in the mouse colon and in cells for this mutant, suggesting a molecular mechanism for the prominence of the G13D allele in the KRas isoforms with unique clinical outcomes. Our findings are consistent with the previously suggested idea that in the context of the entire population of KRas molecules in a cell, the overall signaling activity is graded rather than binary, as it is for each individual KRas molecule (Haigis, 2017). Different mutants affect GTP hydrolysis, nucleotide exchange, and affinity to binding partners in unique ways, and this translates to distinct levels of activity in cells, as we observe for KRas G13D versus KRas G12D in mice. Although both mutants are drivers of cancer progression, differences in their biochemical properties modulated by mutation-specific chemical characteristics and conformational states provide opportunities for mutation-specific design of rational inhibitors. The concept has already been tested for KRas G12D with an inhibitor that has moderate specificity for this mutant (Kauke et al., 2017). In the case of KRas G13D, an open active site with unique features at the interface between switch I and switch II may be selectively targeted. Furthermore, destabilization of the nucleotide-binding pocket may result in lower affinity for nucleotides, providing a unique window of opportunity for compounds to compete with GTP/GDP at this site. This kind of mechanism-based approach is needed for the treatment of cancer in a mutation-specific manner, based on personalized medicine.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Carla Mattos (c.mattos@northeastern.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Biochemical assays involving recombinant protein used the Top10 Escherichia coli strain for amplification and propagation of mutant plasmids of human truncated Ras and Raf genes. Expression and purification of recombinant protein was done with the BL21 strain of E. coli.
All mouse work using the KRasLSL-G13D/+, KRasLSL-G12D/+, Fabp1-Cre lines was reviewed and approved by the Institutional Animal Care and Use Committee at Beth Israel Deaconess Medical Center. Animals were housed in a specific-pathogen free facility with 12-hour light-dark cycles, and food and water available ad libitum. Animals of both genders were used for tissue harvest. For organoid establishment, whole distal colons were harvested from Fabp1-Cre; KRasLSL-G12D/+ and Fabp1-Cre; KRasLSL-G13D/+ mice, and crypts were dissociated by incubation in 8 mM EDTA, resuspended in Matrigel (Corning #356235) and plated in 24-well plates (Pastula and Quante, 2014). KRas alleles were confirmed by Sanger sequencing. Organoid cultures were maintained in Matrigel domes submerged in Advanced D-MEM/F-12 culture medium, L-WRN-conditioned medium, 1% (v/v) B-27 supplement, 1% (v/v) GlutaMax, 1 mM HEPES, 10 mM nicotinamide, 0.5% (v/v) N-2 supplement, 500 μM N-Acetylcysteine, 50 nM [Leu15]-Gastrin I Human, 500 nM A83-01, 10 μM SB202190, 50 ng/mL recombinant human EGF, 1 μM PGE2, and 100 μg/mL primocin. To passage and for plating for drug treatments, Matrigel was digested with 10% (v/v) dispase and organoids dissociated to a single cell suspension with two rounds of incubation in TrypLE. L-WRN cells were propagated and used to make conditioned medium as previously described (Miyoshi and Stappenbeck, 2013).
METHOD DETAILS
Protein purification hydrolysis and binding assays
All biochemical and crystallization experiments were performed using the G-domain of human H, K, and NRas (1-166; EC 3.6.5.2). The RBD domain of Raf1 (or CRaf) kinase (51-131; EC 2.7.11.1) was used for the ITC experiments. Mutagenesis, protein expression and purification of wild-type Ras and Ras G13D in the three isoforms, as well as of Raf-RBD, was performed using standard and previously described protocols (Johnson et al., 2015, 2017; Kearney et al., 2014). Mutagenesis of the G-domain DNA sequences of H, N, and KRas inserted into the pET21a(+) plasmid vector was done using primers optimized for QuikChange™ (Agilent) parameters modified according to a two-stage mutagenesis protocol (Wang and Malcolm, 2001). Primer sequences for generating the G13D mutants in the three Ras isoforms are shown in Table S2.
Once purified, Ras proteins were stored as either bound to GDP or guanylyl-5′-imidodiphosphate (GppNHp), buffer exchanged into stabilization buffer (20 mM HEPES, 50 mM NaCl, 20 mM MgCl2, 1-2mM DTT at pH 7.5), and flash frozen in 25-100 μL aliquots at a concentration of 7-18 mg/mL. Raf-RBD was concentrated to 8-11 mg/mL, flash frozen in 55μL aliquots, and stored at −80°C.
Ras G13D crystallization, data collection and structure refinement
All protein crystals were grown using the vapor-diffusion hanging drop method (Unge, 1999) in VDX™ plates on siliconized glass slides from Hampton Research. All HRas G13D crystals were grown in 2:2 μL drops of protein:reservoir solution, while KRas G13D and NRas G13D crystals were grown in 1:1 μL drops. Data collection for all crystals on a home source MicroMax007HF with Cu2+ anode and tungsten filament, and a R-AxisIV2+ detector from Rigaku. Indexing, integration and scaling data processing steps were done using the HKL3000 package (Otwinowski and Minor, 1997; Minor et al., 2006) and molecular replacement and structure refinement was done using PHENIX software package (Adams et al., 2010) and COOT (Emsley et al., 2010). Data collection and refinement statistics are found in Table 1. Extraction ϕ,ψ values shown in Figure 1C was done using the VADAR web-based program (Willard et al., 2003).
HRas G13D GppNHp crystals with C121 symmetry were grown in the presence of 128 mM Ca(OAc)2, 20.8% PEG 3350 and 20% stabilization buffer at 18°C. They contained 1 molecule in the asymmetric unit and the structure was solved to 1.9 Å resolution. Crystals of HRas G13D GppNHP with P212121 symmetry were grown in 152 mM Ca(OAc)2, 22.8% PEG 3350, and 9.5% stabilization buffer at 18°C. They resulted in a structure solved with three molecules in the asymmetric unit and to a resolution of 1.93Å. Crystals of HRas G13D bound to GDP grew in 188.2 mM Ca(OAc)2, 18.8% PEG 3350, and 5.9% stabilization buffer at 18°C with P212121 symmetry and three molecules in the asymmetric unity (they were isomorphous with the P212121 HRas G13D GppNHp crystals). The structure was solved to 1.95Å resolution. Wild-type HRas bound to GppNHp (PDB ID 3K8Y) or GDP (PDB ID 2RGE) with switches I and II removed, were used in molecular replacement as initial phasing models for HRas G13D crystallized with symmetry of the C121 space group (Adams et al., 2010), and once solved, this solution was used to phase the other HRas, KRas and NRas GppNHp bound crystal structures. Likewise, the structure from HRas G13D crystals with P212121 symmetry bound to GppNHp, once solved, was used as a phasing model in molecular replacement for P212121 HRas G13D bound to GDP.
KRas G13D GppNHp crystals were grown at pH 5.6 in 5 mM Na(OAc), 91 mM sodium citrate tribasic, 183 mM NH4(OAc), 27.2% PEG 4000, and 4.3% stabilization buffer at 18°C. These crystals showed C121 symmetry, had two molecules in the asymmetric unit, and resulted in a structure solved to 3.4 Å resolution. The GDP bound crystals of KRAS G13D grew with P212121 symmetry at 18°C, at pH 5.6, in the presence of 27.5% PEG 3350, 125 mM Na(OAc), 122 mM sodium citrate, and 4.0% stabilization buffer. These crystals have one molecule in the asymmetric unit and the structure was solved to 1.9Å resolution.
NRas G13D bound to GppNHp crystals were grown at pH 7.5 in 9% 2-propanol, 72 mM HEPES, 20% PEG 4000, and 15% stabilization buffer, at 18°C. These crystals have C121 symmetry with one molecule in the asymmetric unity, and the structure was solved to 2.0 Å resolution. In general, the G-domain of wild-type NRas and its G13D mutant were less stable compared to their KRas and HRas counterparts. We were unable to crystalize NRas G13D bound to GDP.
Hydrolysis assays to determine rate constants
Hydrolysis of GTP was measured by production of radioactive inorganic phosphate from γ32P-GTP (Perkin Elmer) loaded Ras proteins, as previously published (Johnson et al., 2017). Intrinsic GTP hydrolysis experiments on KRas G13D and HRas G13D were performed in at least triplicate. Nucleotide exchange was done by incubating 5μM Ras bound to GDP with 50nM γ32P-GTP in the presence of 1mM EDTA for 5 min at 37°C. Hydrolysis γ32P-GTP was then started by addition of 4-fold excess hydrolysis buffer (20 mM Tris pH 8.0, 100 mM NaCl, 5 mM MgCl2, 2 mM DTT), pre-warmed to 37°C, to the nucleotide exchanged Ras sample. The GTP hydrolysis progress was measured by collecting samples at time points: 0, 3, 6, 9, 12, 15, 18, 25, 30, 35, 40, 45, 50, 60, 90, 120, 150, 180, 210, 240, 270, 300, 360, 420, 480, 540, 600, 660, 720 minutes. The reaction was quenched at each specific time point by adding 20 μL reaction to 200 μL stop solution containing 5mM silicotungstate and 1mM sulfuric acid. Each quenched reaction was stored on ice until the experiment was completed. Scintillant (1:1 isobutanol/toluene, 0.5% w/v 2,5-diphenyloxazole, 0.01% bis-(2-methylstyryl)benzene) was added to each reaction along with 40 μL of extraction solution (5% ammonium molybdate and 2M sulfuric acid). Samples were vortexed for 10 sand centrifuged at room temperature for 30 sat 14,000 rpm to separate the organic from the aqueous layers. For scintillation counting, 150 μL of the organic phase was added to scintillation vials corresponding to its reaction time. Counts were measured using a HIDEX 300SL. The rate constants for each reaction were determined by fitting the hydrolysis data to a first-order exponential curve using DynaFit (Kuzmic, 1996, 2009).
Initial hydrolysis rates with and without Na+
Measurement of KRas wild-type and KRas G13D in the presence of 100mM NaCl or 100mM CaCl2 were done in quadruplicate and were performed as the single turnover intrinsic hydrolysis reactions described in the main text, but followed for a much shorter total time to capture the initial rates given by the linear portion of the curves. Thus, what we report are initial reaction rates, not rate constants as these would require much more lengthy experiments. Protein for these experiments were first buffer exchanged into20mM Tris pH 7.6, 20mM MgCl2, and 1nM GDP. Ras protein was then loaded with γ32P-GTP, and hydrolysis was carried out using a hydrolysis buffer with 20mM Tris pH 8.0, 5mM MgCl2, and either 100mM NaCl or 100mM CaCl2. Reactions were measured up to 18 minutes with data points taken at 3-minute intervals.
Nucleotide exchange experiments
Determination of the intrinsic nucleotide exchange kinetics of HRas and KRas mutants were done using 2′-(or-3′)-O-(N-Methylanthraniloyl) guanosine 5′-diphosphate (mant-GDP) and the non-hydrolysable GTP analog 2′-(or-3′)-O-(N-methylanthraniloyl)-β:γ-imidoguanosine 5′-Triphosphate (mant-GppNHp) using a 96-well plate reader. All experiments were done in at least triplicate. For each experiment, the reaction mixture and the appropriate HRas or KRas protein pre-loaded with GDP in stabilization buffer (40mM HEPES pH 7.4, 150mM NaCl, 10mM MgCl2 and 1mM DTT) were allowed to sit on ice and in the dark for 30 minutes prior to mixing and measurement of nucleotide exchange. After 30 minutes, samples were pre-heated to 37°C over 10 minutes in a BioTek Synergy H1 microplate reader before addition of 6μL of either KRas or HRas protein at 1mg/mL to 139.5μL of reaction mixture. The final volume of 145.5μL had the following makeup: 26mM Tris pH 7.5, 7.6mM MgCl2, 2mM DTT, containing 10mM of either mant-GDP or mant-GppNHp, and 2.2μM of HRas or KRas. A control reaction without Ras protein was also included by adding 6μL of stabilization buffer to a reaction mixture of either mant-GDP or mant-GppNHp. Exchange was monitored every 70 s for 2 hours by the change in solution fluorescence at 440nm. To calculate the rate constant of each exchange reaction, readings were first normalized by subtracting the fluorescence change from the control wells from the Ras containing wells.
ITC assays
Thermodynamic properties of the GppNHp-bound Ras G13D and Raf-RBD binding interactions were done as previously published (Johnson et al., 2017). Binding interactions were measured at 25°C using a low volume isothermal titration calorimeter (Nano ITC, TA Instruments). All ITC binding experiments were carried out in triplicate. As shown in Figure 3 the exchange efficiency is nearly 100% for KRas and HRas G13D, as measured by the absence of the D peak at 13.4 ppm in the H1 NMR spectrum (see NMR methods below). Ras G13D mutants were extensively dialyzed into filtered and degassed buffer containing 20 mM HEPES (pH 7.5), 150mM NaCl, 10mM MgCl2, 0.5% (w/v) n-octyl-β-D-glucopyranoside, and 0.5 mM Tris-(2-carboxyethyl)phosphine. Protein concentrations post-dialysis were calculated using an average of the absorbance at 280nm (A280), and confirmed via standard Bradford assay for accuracy. Proteins were placed in the cell at between 35-50 μM and titrated with Raf-RBD at approximately 10-fold higher concentration to achieve saturation and maintain a 1:1 stoichiometry (N = 1). A total of 20 injections at 2 μL per injection were added to the cell volume of 171 μL for each experiment. Concentrated Raf-RBD injected into ITC buffer was subtracted from each titration experiment prior to data analysis for normalization.
1H NMR of KRas G13D and HRas G13D
1H NMR on Ras G13D samples was performed as recently described (Parker et al., 2018). Samples used for NMR analyses were dialyzed into 40 mM HEPES pH 7.4, 10 mM MgCl2, 150 mM NaCl, and 1 mM DTT and concentrated to approximately 2 mM. 1D 1H NMR spectra were acquired at 700 MHz with a Bruker AVANCE II NMR spectrometer equipped with a 5 mm triple resonance inverse probe at 37°C. For optimal detection of downfield exchangeable proton resonances, the 3-9-19 WATERGATE (Sklenar et al., 1993) pulse sequence (p3919fpgp) with gradients and additional flipback pulse was used with the center of the maximal excitation region at 13.9 ppm. The calculated delay for binomial water suppression was 39 μs at 700 MHz. Routinely, 4K scans were accumulated.
The resonance peaks were assigned as previously described (Parker et al., 2018). Briefly, we assigned a resonance peak at 13.2 ppm to the N1 on the guanine base of the nucleotide. This resonance (peak T) is a sensor of whether Ras is bound to GppNHp or GDP, where it is shifted slightly downfield to 13.4 ppm (peak D) upon GDP binding. A resonance at 10.9 ppm (peak X) is associated with the proton on one of the γ-phosphate oxygen atoms, with a high pKa when bound to Ras (Knihtila et al., 2015), and senses the conformation of switch II, possibly the interaction with G60. A third resonance at 10.3 ppm (peak Z) is likely due to the amino protons of K16 and senses the conformation of switch I (Parker et al., 2018). Peaks D, B and A are associated with the GDP-bound form of Ras and appear in the WT KRas spectrum (Figure 3) due to incomplete nucleotide exchange. In state 2 the K16 and the γ-phosphate are protected by closed conformations of switch I and switch II respectively, giving rise to peaks X and Z. The flat spectrum between 11 and 10 ppm for KRas G13D in Figure 3 indicates a very open active site in state 1 for this mutant in KRas. For HRas G13D there is a decrease in peak Z indicating a shift toward state 1, but state 2 is significantly sampled. Peak X is stable at all temperatures, consistent with a more closed switch II conformation. Overall, the KRas G13D spectrum is consistent with a highly exposed γ-phosphate and K16 in the active site, resulting in fast proton exchange with solvent and disappearance of peaks X and Z. This is as expected based on the Ka13GNP and Kb13GNP models, where the γ-phosphate is slightly displaced from its usual position, the side chain of K16 is disordered and the interaction between the γ-phosphate and switch II is not present. The extent of nucleotide exchange was determined by deconvolution of the spectra, using a mathematical model of the Gaussian linewidths and adjusting parameters until a good fit to the experimental spectrum was achieved.
CRISPR for KRAS G13D mouse
Using the online design tool (http://zlab.bio/guide-design-resources), a sgRNA was identified to target the G12D site in the heterozygous KRAS G12D C57BL/6 mouse. A 155 base ultramer was synthesized and used for homologous directed repair (HDR). The designed ultramer replaced the KRAS G12D mutation with a wild-type sequence at amino acid 12 and inserted a G13D mutation. A silent mutation was also added in the ultramer at amino acid 14, GTG to GTC, which along with the G13D mutation created a unique AatII restriction site used for screening and genotyping purposes. The ultramer and SgRNA for sequences can be found in Table S2.
B6C3F1/J female donors, B6D2F1/J recipient females and B6D2F1/J vasectomized males were purchased from Jackson Laboratories. KRas-LSL-G12D sperm donors were obtained from the Mouse Models of Human Cancers Consortium (MMHCC). Donor mice were superovulated with injection of 5 IU PMSG, followed by injection of 5 IU HCG 48 hours later. Eggs were removed and used for in vitro fertilization with sperm from the donor animals. Pronuclei of the resulting zygotes were injected with 50 ng/μL Cas9 mRNA, 25 ng/μL sgRNA and 100 ng/uL template. Surviving eggs were cultured overnight in Cooks Vitro Cleave to the two-cell stage. 13-17 two-cells were implanted into each pseudopregnant female. All animal experiments were performed in accordance with institutional IACUC protocols.
Lox-stop-lox inducible KRasG13D/+
Primers used for genotyping mouse strains are listed in Table S2. KRasLSL-G13D animals were backcrossed to wild-type C57BL/6J mice for at least five generations and the genetic background of these mice were confirmed to be the same as those of the existing mice in our colony by SNP genotyping (Jackson Laboratory). Eight to twelve-week old mice were sacrificed and their entire colons removed, flushed with PBS, and opened longitudinally. After removing a longitudinal strip for protein lysate, the rest of the colon was Swiss rolled from distal to proximal ends and formalin-fixed.
For crypt measurements, 5 μm paraffin sections were cut and stained by hematoxylin and eosin according to standard protocols, and measurements were made using Olympus slide scanner software (Olympus VS-ASW version 2.7). Full description of crypt measurement protocol was previously published (Lyons et al., 2018) and described here in brief. An arbitrary line was drawn from crypt base to apex every five crypts along the entire length of the colon. Off-axis crypts were denoted with short lines (< 10 μm) and their measurements removed from subsequent analysis, while a long (> 1000 μm) line marked the proximal-distal junction. To generate scatterplots, all measurements were distributed evenly across the distal and proximal regions, using region lengths that were measured by polylines. Averages were taken of all measurements in the distal and proximal regions separately.
Ras-GTP pulldown was performed as previously described (Taylor et al., 2001). Briefly, BL21 bacterial pellets were re-suspended in 10 mL 1X MLB buffer (Millipore 20-168) and sonicated 8 times 20 s on, 20 s off. The suspension was clarified by centrifugation at 13, 200 rpm in 1.2 mL eppendorf tubes for 5 minutes, and the supernatant was then rocked for 2-hours at 4°C with 150 μL of Glutathione Sepharose 4B beads (GE Healthcare #17075601) pre-washed with MLB buffer. Raf-GST beads were washed in 1X MLB buffer and stored as a 1:1 slurry with buffer at 4°C before use. Snap frozen mouse colons were lysed in 1X MLB buffer containing protease inhibitor cocktail (Roche #11697498001) and phosphatase inhibitor cocktails 2 and 3 (Sigma #P5726 & #P0044) with dissection scissors and homogenized by aspiration 3-5 times through a 20G needle. 10 μL Raf-GST beads were used to pulldown Ras-GTP from 500 μg protein per sample for 2 hours, rocking at 4°C. 15 μL of 6X loading buffer was added to the beads after washing and the entire volume loaded in a 12.5% polyacrylamide gel for western blotting.
Western blotting was done with 40 μg protein per lane on 12.5% polyacrylamide gels and transferred overnight at 4°C and 37V to PVDF membranes. Membranes were blocked for 1 hour and then incubated overnight at 4°C with primary antibodies diluted 1:1000 (unless noted otherwise) in Odyssey PBS blocking buffer (LiCOR 927-40000). Primary antibodies used were as follow, Ras10 (Millipore 05-516), phospho-Erk (Cell Signaling #4377), total Erk (Cell Signaling #4696), phospho-Akt S473 (Cell Signaling #4060), phospho-Akt S308 (Cell Signaling #2965), pan Akt (1:2000, Cell Signaling #2920). The following day, secondary antibody was diluted 1:10,000 in Odyssey blocking buffer and incubated with membranes for 1 hour at room temperature prior to visualization on an Odyssey LiCOR CLx machine. Secondary antibodies used were Alexa Fluor 680 goat anti-mouse IgG (Invitrogen #A21058) and Alexa Fluor Plus 800 goat anti-rabbit IgG (Invitrogen #A32735).
Small molecule inhibitor treatments in murine colonic organoids
For small molecule inhibitor studies, organoids were dissociated to single cells and 500 viable cells were plated per well of an ultra-low attachment 384-well plate (Corning #4588) in 20 μL media with 10% Matrigel by volume. Drug treatment media was made with advanced DMEM/F-12 (Thermo Fisher #12634028) that was supplemented with 1mM HEPES, 1X Glutamax (Thermo Fisher #35050061), 1mM N-acetylcysteine, B-27 (Thermo Fisher #17504044), N-2 (Thermo Fisher #17502048) and Primocin (Invivogen #ant-pm-1). After 24 hours, compounds were added to each well over 12-point dose curves along with DMSO controls using a D300e digital drug printer (Tecan LifeSciences). Cells were cultured for 6 days in the presence of compounds before assessing viability by adding 20 μL of CellTiter-Glo 3D (Promega) to each well, incubating for 1 hour at room temperature on a shaker, and measuring luminescence using an EnVision plate reader. Each condition was performed in triplicate, and each dose point was normalized to DMSO controls to estimate relative viability.
QUANTIFICATION AND STATISTICAL ANALYSIS
Rate constants for single turnover hydrolysis and nucleotide exchange, and their averages, were calculated using DynaFit4 (Kuzmic, 1996, 2009). Experiments examining the effect of different cations on KRas catalyzed hydrolysis of GTP were quantitated by first determining the slope of each reaction curve (fmol of Pi per minute) in Microsoft Excel. These data were then compiled and averaged in GraphPad.
ITC data analysis was conducted via manufacturer software (NanoAnalyze, TA Instruments) to give the stoichiometry (N), binding affinity (Ka), enthalpy change (ΔH°) for the Ras G13D and Raf-RBD interaction. Thermodynamic relationships were then used to calculate the total free energy (ΔG°) and entropy (ΔS°) changes upon binding, using the equations ΔG° = −RTInKa and ΔG° = ΔH°-TΔS°, respectively.
All averaging and statistical analysis of mouse crypt measurements, western blotting, and nucleotide exchange were done using GraphPad Prism 5.0. Statistical analyses of crypt height measurements and western blot quantitation were done using the Mann-Whitney and 1-way ANOVA (Kruskal-Wallis) tests, respectively. To determine cell viability following small molecule inhibitor treatment, a three-parameter log logistic function was fit to the data (viability versus dose) with GraphPad Prism. The number of replicates and statistical details for each experiment, where appropriate, are found in the respective figure legends. Error bars in all figures reflect standard error of the mean (±SEM) as calculated by GraphPad.
DATA AND CODE AVAILABILITY
The accession numbers for the Ras G13D structures reported in this paper are H13GNP, PDB: 6E6C; Ha-c13GNP, PDB: 6E6P; N13GNP, PDB: 6E6H; Ka-b13GNP, PDB: 6E6F; Ha-c13GDP, PDB: 6DZH; and K13GDP, PDB: 6E6G. KRasLSL-G13D/+ mice will be deposited to Jackson Laboratories for distribution.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-p-Akt T308 | Cell Signaling | Cat# 2965; RRID# AB_2255933 |
| Anti-pan-Akt | Cell Signaling | Cat# 2920; RRID# AB_1147620 |
| Anti-p-Akt S473 | Cell signaling | Cat# 4060; RRID# AB_2315049 |
| Anti-p-Erk1/2 T202/Y204 | Cell signaling | Cat# 4377; RRID# AB_331775 |
| Anti-Erk1/2 | Cell signaling | Cat# 4696; RRID# AB_390780 |
| Anti-GAPDH | Cell signaling | Cat# 5174; RRID# AB_10622025 |
| Ras10 | Millipore | Cat# 05-516; RRID# AB_2121151 |
| Alexa Fluor 680 goat anti-mouse IgG | Invitrogen | Cat# A21058; RRID# AB_2535724 |
| Alexa Fluor Plus 800 goat anti-rabbit IgG | Invitrogen | Cat# A32735; RRID# AB_2633284 |
| Bacterial and Virus Strains | ||
| Escherichia coli (E. Coli) BL21(DE3) competent cells | New England Biolabs | Cat#C2527I |
| E. Coli One Shot® Top10 competent cells | Invitrogen | Cat#C404010 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| KRAS(1-166) G13D | This paper | N/A |
| KRAS(1-166) wildtype | This paper | N/A |
| HRAS(1-166) G13D | This paper | N/A |
| HRAS(1-166) wildtype | This paper | N/A |
| NRAS(1-166) G13D | This paper | N/A |
| RAF1-RBD (51-131) | This paper | N/A |
| GTP [γ-32P] | Perkin Elmer | BLU004Z250UC |
| 50% Polyethylene glycol 3350 | Hampton Research | HR2-527 |
| PEG/Ion #28 / PEG/Ion HT C4 185mL | Hampton Research | HR2-922-28 |
| Reagent 9 Crystal screen | Hampton Research | HR2-110 |
| Reagent 41 Crystal screen | Hampton Research | HR2-110 |
| Hematoxylin | Sigma | MHS16 |
| Eosin | Thermo Fisher | 6766008 |
| Permount | Fisher Scientific | SP15-100 |
| Mg2+ Lysis/Wash buffer (MLB) | Millipore | 20-168 |
| Glutathione Sepharose 4B beads | GE Healthcare | 17075601 |
| cOmplete™ protease inhibitor cocktail | Roche | 11697498001 |
| Phosphatase inhibitor cocktails 2 | Sigma | P5726 |
| Phosphatase inhibitor cocktails 2 | Sigma | P0044 |
| Benzamidine | Sigma | 434760 |
| Leupeptin | Fisher scientific | L2884 |
| Antipain | Fisher scientific | 10791 |
| Odyssey® phosphate buffered saline blocking buffer | LICOR | 927-40000 |
| MK2206 (Akt inhibitor) | ChemieTek | CT-MK2206 |
| SCH772984 (Erk inhibitor) | ChemieTek | CT-SCH772 |
| GIBCO Advanced DMEM/F-12 media | Thermo Fisher | 12634028 |
| GIBCO HEPES (1M) | Thermo Fisher | 15630080 |
| GIBCO GlutaMAX™ Supplement | Thermo Fisher | 35050061 |
| GIBCO B-27™ Supplement (50X) | Thermo Fisher | 17504044 |
| GIBCO N-2™ Supplement (100X) | Thermo Fisher | 17502048 |
| N-Acetyl-L-cysteine | Sigma | A9165 |
| Nicotinamide | Sigma | N3376 |
| Human [Leu15]-Gastrin I | Sigma | G9145 |
| A83-01 | Sigma | SML0788 |
| SB 202190 | Sigma | S7067 |
| Recombinant human EGF | Thermo Fisher | PHG0311 |
| PGE2 | Sigma | P5640 |
| Primocin | InvivoGen | ant-pm-1 |
| Matrigel® Matrix | Corning | 356235 |
| GIBCO FBS, Lot 1883434 | Thermo Fisher | 16000-044 |
| Corning™ DMEM with L-Glutamine, 4.5g/L glucose and sodium pyruvate | Fisher Scientific | MT10013CV |
| Antibiotic Antimycotic Solution (100 ×), Stabilized | Sigma | A5955 |
| TrypLE Express Enzyme (1X), no phenol red | Thermo Fisher | 12604021 |
| Corning™ Dispase | Fisher Scientific | 354235 |
| Critical Commercial Assays | ||
| Crystal Screen | Hampton Research | HR2-110 |
| Bicinchoninic acid assay (BCA) | Pierce | 23225 |
| CellTiter-Glo® 3D Cell Viability Assay | Promega | G9683 |
| Deposited Data | ||
| HRAS G13D GppNHp—C121 | This paper | PDB: 6E6C |
| HRAS G13D GppNHp— P212121 | This paper | PDB: 6E6P |
| HRAS G13D GDP | This paper | PDB: 6DZH |
| KRAS G13D GppNHp | This paper | PDB: 6E6F |
| KRAS G13D GDP | This paper | PDB: 6E6G |
| NRA SG13D GppNHp | This paper | PDB: 6E6H |
| Experimental Models: Cell Lines | ||
| Fabp1-Cre colon organoids | This paper | N/A |
| Fabp1-Cre; KRasLSL-G12D/+ colon organoids | This paper | N/A |
| Fabp1-Cre; KRasLSL-G13D/+ colon organoids | This paper | N/A |
| L-WRN | ATCC | CRL-3276 |
| Experimental Models: Organisms/Strains | ||
| Mouse: KRasLSL-G13D/+: N/A | Laboratory of Matthew Steensma |
N/A |
| Mouse: Fabp1-Cre: FVB/N-Tg(Fabp1-Cre)1Jig/Nci | NCI Mouse Repository | 01XD8 |
| Mouse: KRasLSL-G12D: B6.129-Krastm4Tyj/Nci | NCI Mouse Repository | 01XJ6 |
| Mouse: B6C3F1/J: female donors | Jackson Laboratories | 100010 |
| Oligonucleotides | ||
| Primers for mutagenesis can be found in Table S2 | This paper | N/A |
| Primers for genotyping can be found in Table S2 | This paper | N/A |
| Primers and oligonucleotides for CRISPR can be found in Table S2 | This paper | N/A |
| Recombinant DNA | ||
| Human: HRAS(1-166) cDNA | This paper—three stop codons are present after | N/A |
| Human: KRAS(1-166) cDNA | Genewiz—Full-length KRAS4B with 3 codons after residue 166 | https://www.genewiz.com/en |
| Human: NRAS(1-166) cDNA | Genewiz—Full-length NRAS with 3 codons after residue 166 | https://www.genewiz.com/en |
| Human: Raf1 GST-RBD 1-149 | Addgene | Plasmid #: 13338 |
| Human: Raf1-RBD 51-131 | Mattos lab | N/A |
| Software and Algorithms | ||
| PHENIX | Adams et al., 2010 | https://www.phenix-online.org/ |
| Coot | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| HKL-3000R | Minor et al., 2006 | https://www.hkl-xray.com/hkl-3000 |
| TopSpin™ ver 3.5 | Bruker Corporation | https://www.bruker.com |
| PRISM ver 5-7 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| DynaFit4 | Kuzmic, 2009 | http://www.biokin.com/index.html |
| MikroWin 300 SL | HIDEX 300SL | http://hidex.com |
| Other | ||
| Corning® 384-well Black/Clear Bottom Ultra-Low Attachment Microplate | Corning | 4588 |
Highlights.
Ras G13D proteins have open active sites with disconnected switches I and II
KRas G13D shows unique destabilization of the nucleotide-binding pocket
KRas G13D has attenuated oncogenic phenotype relative to KRas G12D
KRas G13D and KRas G12D are more sensitive to Erk than to Akt inhibition
ACKNOWLEDGMENTS
The structural biology and biochemistry aspects of this research were supported by NSF grants MCB-1244203 and MCB-1517295 to C.M., which supported C.W.J.’s PhD work at Northeastern University. The mouse work was supported by NCI grants (R01CA195744 and R01CA232372) to K.M.H. Y.-J.L. was supported by a pre-doctoral fellowship from the Landry Family Cancer Consortium. C.W.J. is currently in the Haigis lab at the Beth Israel Deaconess Medical Center supported by the postdoctoral fellowship 130428-PF-17-066-01-TBG from the American Cancer Society. The MicroMax007HF used to collect the X-ray data at Northeastern University was purchased in part with funds from the NSF MRI-1228897 grant to C.M. Our thanks to Radha Kalekar in A.J.A.’s group for help with the organoid inhibitor experiments and to Senthil Muthuswamy for use of the BioTek Synergy H1 Hybrid Multi-Mode microplate reader to perform the nucleotide exchange assays.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.026.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandaru P, Shah NH, Bhattacharyya M, Barton JP, Kondo Y, Cofsky JC, Gee CL, Chakraborty AK, Kortemme T, Ranganathan R, and Kuriyan J (2017). Deconstruction of the Ras switching cycle through saturation mutagenesis. eLife 6, e27810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, and Bourne PE (2000). The Protein Data Bank. Nucleic Acids Res 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boriack-Sjodin PA, Margarit SM, Bar-Sagi D, and Kuriyan J (1998). The structural basis of the activation of Ras by Sos. Nature 394, 337–343. [DOI] [PubMed] [Google Scholar]
- Buhrman G, Wink G, and Mattos C (2007). Transformation efficiency of RasQ61 mutants linked to structural features of the switch regions in the presence of Raf. Structure 15, 1618–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhrman G, Holzapfel G, Fetics S, and Mattos C (2010). Allosteric modulation of Ras positions Q61 for a direct role in catalysis. Proc. Natl. Acad. Sci. USA 107, 4931–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhrman G, Kumar VS, Cirit M, Haugh JM, and Mattos C (2011). Allosteric modulation of Ras-GTP is linked to signal transduction through RAF kinase. J. Biol. Chem 286, 3323–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Roock W, Jonker DJ, Di Nicolantonio F, Sartore-Bianchi A, Tu D, Siena S, Lamba S, Arena S, Frattini M, Piessevaux H, et al. (2010). Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 304,1812–1820. [DOI] [PubMed] [Google Scholar]
- Du X, Black GE, Lecchi P, Abramson FP, and Sprang SR (2004). Kinetic isotope effects in Ras-catalyzed GTP hydrolysis: evidence for a loose transition state. Proc. Natl. Acad. Sci. USA 101, 8858–8863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta. Crystallogr. D. Biol. Crystallogr 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetics SK, Guterres H, Kearney BM, Buhrman G, Ma B, Nussinov R, and Mattos C (2015). Allosteric effects of the oncogenic RasQ61L mutant on Raf-RBD. Structure 23, 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein SD, Sayegh R, Christensen S, and Swalsky PA (1993). Genotypic classification of colorectal adenocarcinoma. Biologic behavior correlates with K-ras-2 mutation type. Cancer 71, 3827–3838. [DOI] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, et al. (2017). COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res 45 (D1), D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile DR, Rathinaswamy MK, Jenkins ML, Moss SM, Siempelkamp BD, Renslo AR, Burke JE, and Shokat KM (2017). Ras Binder Induces a Modified Switch-II Pocket in GTP and GDP States. Cell Chem. Biol 24,1455–1466.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorfe AA, Grant BJ, and McCammon JA (2008). Mapping the nucleotide and isoform-dependent structural and dynamical features of Ras proteins. Structure 16, 885–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero S, Casanova I, Farré L, Mazo A, Capellà G, and Mangues R (2000). K-ras codon 12 mutation induces higher level of resistance to apoptosis and predisposition to anchorage-independent growth than codon 13 mutation or proto-oncogene overexpression. Cancer Res 60, 6750–6756. [PubMed] [Google Scholar]
- Guerrero S, Figueras A, Casanova I, Farré L, Lloveras B, Capellà G, Trias M, and Mangues R. (2002). Codon 12 and codon 13 mutations at the K-ras gene induce different soft tissue sarcoma types in nude mice. FASEB J. 16, 1642–1644. [DOI] [PubMed] [Google Scholar]
- Haigis KM (2017). KRAS Alleles: The Devil Is in the Detail. Trends Cancer 3, 686–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis KM, Wistuba II, and Kurie JM (2007). Lung premalignancy induced by mutant B-Raf, what is thy fate? To senesce or not to senesce, that is the question. Genes Dev 21, 361–366. [DOI] [PubMed] [Google Scholar]
- Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, Niwa-Kawakita M, Sweet-Cordero A, Sebolt-Leopold J, Shannon KM, et al. (2008). Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat. Genet 40, 600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond DE, Mageean CJ, Rusilowicz EV, Wickenden JA, Clague MJ, and Prior IA (2015). Differential reprogramming of isogenic colorectal cancer cells by distinct activating KRAS mutations. J. Proteome Res 14, 1535–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzapfel G, Buhrman G, and Mattos C (2012). Shift in the equilibrium between on and off states of the allosteric switch in Ras-GppNHp affected by small molecules and bulk solvent composition. Biochemistry 51, 6114–6126 [DOI] [PubMed] [Google Scholar]
- Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, and Westover KD (2015). Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res 13, 1325–1335. [DOI] [PubMed] [Google Scholar]
- Imamura Y, Morikawa T, Liao X, Lochhead P, Kuchiba A, Yamauchi M, Qian ZR, Nishihara R, Meyerhardt JA, Haigis KM, et al. (2012). Specific mutations in KRAS codons 12 and 13, and patient prognosis in 1075 BRAF wild-type colorectal cancers. Clin. Cancer Res 18, 4753–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John J, Rensland H, Schlichting I, Vetter I, Borasio GD, Goody RS, and Wittinghofer A (1993). Kinetic and structural analysis of the Mg(2+)-binding site of the guanine nucleotide-binding protein p21H-ras. J. Biol. Chem 268, 923–929. [PubMed] [Google Scholar]
- Johnson CW, and Mattos C (2013). The Allosteric Switch and Conformational States in Ras GTPase Affected by Small Molecules In Inhibitors of the Ras Superfamily G-proteins, Part A and Fuyuhiko T, eds. (Academic Press; ), pp. 41–67. [DOI] [PubMed] [Google Scholar]
- Johnson CW, Buhrman G, Ting PY, Colicelli J, and Mattos C (2015). Expression, purification, crystallization and X-ray data collection for RAS and its mutants. Data Brief 6, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson CW, Reid D, Parker JA, Salter S, Knihtila R, Kuzmic P, and Mattos C (2017). The small GTPases K-Ras, N-Ras, and H-Ras have distinct biochemical properties determined by allosteric effects. J. Biol. Chem 292, 12981–12993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauke MJ, Traxlmayr MW, Parker JA, Kiefer JD, Knihtila R, McGee J, Verdine G, Mattos C, and Wittrup KD (2017). An engineered protein antagonist of K-Ras/B-Raf interaction. Sci. Rep 7, 5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney BM, Johnson CW, Roberts DM, Swartz P, and Mattos C (2014). DRoP: a water analysis program identifies Ras-GTP-specific pathway of communication between membrane-interacting regions and the active site. J. Mol. Biol 426, 611–629. [DOI] [PubMed] [Google Scholar]
- Knihtila R, Holzapfel G, Weiss K, Meilleur F, and Mattos C (2015). Neutron Crystal Structure of RAS GTPase Puts in Question the Protonation State of the GTP γ-Phosphate. J. Biol. Chem 290, 31025–31036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhle B, and Ficner R (2014). A monovalent cation acts as structural and catalytic cofactor in translational GTPases. EMBO J 33, 2547–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmic P (1996). Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal. Biochem 237, 260–273. [DOI] [PubMed] [Google Scholar]
- Kuzmic P (2009). DynaFit-a software package for enzymology. Methods Enzymol 467, 247–280. [DOI] [PubMed] [Google Scholar]
- Lovell SC, Davis IW, Arendall WB 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, and Richardson DC (2003). Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins 50, 437–450. [DOI] [PubMed] [Google Scholar]
- Lu S, Jang H, Muratcioglu S, Gursoy A, Keskin O, Nussinov R, and Zhang J (2016). Ras Conformational Ensembles, Allostery, and Signaling. Chem. Rev 116, 6607–6665. [DOI] [PubMed] [Google Scholar]
- Lyons J, Brubaker DK, Ghazi PC, Baldwin KR, Edwards A, Boukhali M, Strasser SD, Suarez-Lopez L, Lin YJ, Yajnik V, et al. (2018). Integrated in vivo multiomics analysis identifies p21-activated kinase signaling as a driver of colitis. Sci. Signal 11, eaan3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margonis GA, Kim Y, Spolverato G, Ejaz A, Gupta R, Cosgrove D, Anders R, Karagkounis G, Choti MA, and Pawlik TM (2015). Association Between Specific Mutations in KRAS Codon 12 and Colorectal Liver Metastasis. JAMA Surg 150, 722–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazhab-Jafari MT, Marshall CB, Smith M, Gasmi-Seabrook GM, Stambolic V, Rottapel R, Neel BG, and Ikura M (2010). Real-time NMR study of three small GTPases reveals that fluorescent 2’(3’)-O-(N-methylanthraniloyl)-tagged nucleotides alter hydrolysis and exchange kinetics. J. Biol. Chem 285, 5132–5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messner I, Cadeddu G, Huckenbeck W, Knowles HJ, Gabbert HE, Baldus SE, and Schaefer KL (2013). KRAS p.G13D mutations are associated with sensitivity to anti-EGFR antibody treatment in colorectal cancer cell lines. J. Cancer Res. Clin. Oncol 139, 201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, and Chruszcz M (2006). HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta. Crystallogr. D. Biol. Crystallogr 62, 859–866. [DOI] [PubMed] [Google Scholar]
- Miyoshi H, and Stappenbeck TS (2013). In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc 8, 2471–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Parker JA, Volmar AY, Pavlopoulos S, and Mattos C (2018). K-Ras Populates Conformational States Differently from Its Isoform H-Ras and Oncogenic Mutant K-Ras G12D. Structure 26, 810–820.e4. [DOI] [PubMed] [Google Scholar]
- Pastula A, and Quante M (2014). Isolation and 3-dimensional Culture of Primary Murine Intestinal Epithelial Cells. Bio-protocol 4 10.21769/BioProtoc.1125. [DOI] [Google Scholar]
- Poulin EJ, Bera AK, Lu J, Lin YJ, Strasser SD, Paulo JA, Huang TQ, Morales C, Yan W, Cook J, et al. (2019). Tissue-Specific Oncogenic Activity of KRASA146T. Cancer Discov 9, 738–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior IA, Lewis PD, and Mattos C (2012). A comprehensive survey of Ras mutations in cancer. Cancer Res 72, 2457–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, and Sabatini DM (2017). mTOR Signaling in Growth, Metabolism, and Disease. Cell 169, 361–371. [DOI] [PubMed] [Google Scholar]
- Sayyed-Ahmad A, Prakash P, and Gorfe AA (2017). Distinct dynamics and interaction patterns in H- and K-Ras oncogenic P-loop mutants. Proteins 85, 1618–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffzek K, Ahmadian MR, Kabsch W, Wiesmüller L, Lautwein A, Schmitz F, and Wittinghofer A (1997). The Ras-Ras GAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 277, 333–338. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A (2018). Cancer statistics, 2018. CA Cancer J. Clin 68, 7–30. [DOI] [PubMed] [Google Scholar]
- Sklenar V, Piotto M, Leppik R, and Saudek V (1993). Gradient-tailored water suppression for H-1-N-15 Hsqc experiments optimized to retain full sensitivity. J. Magn. Reson. A 102, 241–245. [Google Scholar]
- Smith MJ, Neel BG, and Ikura M (2013). NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proc. Natl. Acad. Sci. USA 110, 4574–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spoerner M, Hozsa C, Poetzl JA, Reiss K, Ganser P, Geyer M, and Kalbitzer HR (2010). Conformational states of human rat sarcoma (Ras) protein complexed with its natural ligand GTP and their role for effector interaction and GTP hydrolysis. J. Biol. Chem 285, 39768–39778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SJ, Resnick RJ, and Shalloway D (2001). Nonradioactive determination of Ras-GTP levels using activated ras interaction assay. Methods Enzymol 333, 333–342. [DOI] [PubMed] [Google Scholar]
- Traut TW (1994). Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem 140, 1–22. [DOI] [PubMed] [Google Scholar]
- Unge T (1999). Crystallization methods In Protein Crystallization: Techniques, Strategies, and Tips, Bergfors TM, ed. (International University Line; ), pp. 7–18. [Google Scholar]
- Wang W, and Malcolm BA (2001). Two-Stage Polymerase Chain Reaction Protocol Allowing Introduction of Multiple Mutations, Deletions, and Insertions, UsignQuickChange (™) Site-Directed Mutagenesis In In Vitro Mutagenesis Protocols, Braman J, ed. (Humana Press; ), pp. 37–43. [DOI] [PubMed] [Google Scholar]
- Wang Y, Velho S, Vakiani E, Peng S, Bass AJ, Chu GC, Gierut J, Bugni JM, Der CJ, Philips M, et al. (2013). Mutant N-RAS protects colorectal cancer cells from stress-induced apoptosis and contributes to cancer development and progression. Cancer Discov 3, 294–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willard L, Ranjan A, Zhang H, Monzavi H, Boyko RF, Sykes BD, and Wishart DS (2003). VADAR: a web server for quantitative evaluation of protein structure quality. Nucleic Acids Res 31, 3316–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the Ras G13D structures reported in this paper are H13GNP, PDB: 6E6C; Ha-c13GNP, PDB: 6E6P; N13GNP, PDB: 6E6H; Ka-b13GNP, PDB: 6E6F; Ha-c13GDP, PDB: 6DZH; and K13GDP, PDB: 6E6G. KRasLSL-G13D/+ mice will be deposited to Jackson Laboratories for distribution.