

Abstract
Glutamate receptor interacting protein (GRIP) is a neuronal scaffolding protein that anchors GluA2-containing AMPA receptors to the cell membrane. GRIP plays a critical role in activity-dependent synaptic plasticity, including that which occurs after drug exposure. Given that cocaine administration alters glutamate receptor trafficking within the prefrontal cortex (PFC), a better understanding of the role of receptor trafficking proteins could lead to a more complete understanding of addictive phenotypes. AMPA receptor trafficking in general, and GRIP specifically, is known to play a role in cocaine seeking and conditioned reward in the nucleus accumbens, but its role in the PFC has not been characterized. The current study demonstrates that conditional deletion of GRIP1 in the medial prefrontal cortex increases the motivation for cocaine and potentiates cue-induced reinstatement of cocaine seeking in male and female mice. As no effects of PFC GRIP1 deletion were seen in reinstatement of food seeking, strategy set-shifting, or reversal learning the effects on cocaine seeking are not related to generalized alterations in cognitive function. While disrupting GRIP1 might be expected to lead to decreased AMPA transmission, our electrophysiological data indicate an increase in sEPSC amplitude in the prefrontal cortex and a corresponding decrease in paired pulse facilitation in the nucleus accumbens. Taken together this suggests a strengthening of the PFC to NAc input following prefrontal GRIP1 deletion that may mediate the enhanced drug seeking behavior.
Keywords: Glutamate receptor interacting protein, AMPA, cocaine, reinstatement, prefrontal cortex
1. Introduction
Disruptions within glutamatergic pathways may underlie the development of uncontrollable drug seeking and relapse. Much work has focused on the role of the glutamatergic pathway from the medial prefrontal cortex (mPFC) to the nucleus accumbens in cocaine seeking (McFarland et al., 2003; Moorman et al., 2015; Park et al., 2002; Stefanik et al., 2016; Stefanik et al., 2013). After extended drug exposure, deficits in accumbal glutamate reuptake lead to reduced plasticity associated with addiction (Kalivas, 2009). Alterations in glutamate signaling within the PFC also play a role in addiction-like behaviors. Abnormalities in prefrontal functioning lead to compulsive drug taking and impairments in the executive function needed for a number of self-regulatory behaviors (Goldstein and Volkow, 2011).
Cocaine self-administration leads to both a decrease in basal glutamate levels within the mPFC and a decrease in cocaine-evoked glutamate release (Ben-Shahar et al., 2012). This decrease in release is accompanied by an increase in glutamate receptor expression, specifically GluN2B expression in the mPFC during withdrawal. Increased GluN2B expression is seen with or without cue-induced drug seeking, suggesting that the elevated GluN2B levels are the result of cocaine experience and/or withdrawal, and not other prefrontal mediated processes such as motivation (Szumlinski et al., 2016). These alterations in glutamate signaling have functional consequences such as increasing glutamate levels within the infralimbic cortex leading to attenuation of incubation of cocaine craving (Shin et al., 2018). While it is clear that dysfunction in glutamate signaling in the PFC plays a role in drug-associated behaviors, the mechanisms underlying these alterations in glutamate signaling within the PFC remain largely unexplored.
Glutamate Receptor Interacting Protein (GRIP) is a scaffolding protein that regulates the trafficking of GluA2-containing α-amino-3-hydroxy-5-methyl-4-isoazolepropionic acid receptors (AMPARs) in and out of the cell membrane (Dong et al., 1997). As such, GRIP has been shown to be involved in activity-dependent synaptic plasticity throughout the brain (Summa et al., 2011; Takamiya et al., 2008; Xue et al., 2010). Although the effects of GRIP on receptor trafficking have been characterized, its role in behavior is less clear. There is some evidence of a role for GRIP in social behavior (Mejias et al., 2011). Additionally, there has been some research on the role of GRIP in addictive phenotypes. Exposure to drug-paired cues decreases GRIP expression in the nucleus accumbens core (Liang et al., 2017). Furthermore, GRIP deletion from the nucleus accumbens enhances cue-induced cocaine seeking (Briand et al., 2014) and impairs the ability of calpain to disrupt reconsolidation of cocaine conditioned reward (Liang et al., 2017). However, little is known on the behavioral effects of GRIP in the prefrontal cortex. In this experiment, we sought to elucidate the effects of prefrontal GRIP knockout on addiction-like behaviors. There are two forms of GRIP, GRIP1 and GRIP2, and both regulate activity dependent AMPAR internalization and recycling (Mao et al., 2010). Although GRIP1 and GRIP2 are homologous and highly conserved, GRIP1 can completely rescue function in GRIP2 KO mice, whereas GRIP2 can only partially restore GRIP1 function (Tan et al., 2015). Therefore, to examine the influence of GRIP function, we utilized a mouse with a floxed GRIP1 gene on a background of GRIP2 knockout. In the absence of cre recombinase, the GRIP1 performs all GRIP functions, thereby rendering this mouse indistinguishable from wildtype. However, the GRIP2 knockout background is needed in order to prevent GRIP2 from rescuing the function of GRIP1 following the inducible knockout. Based on the role of prefrontal glutamate signaling and addiction, we hypothesized that prefrontal GRIP1 knockout would lead to the development of an addictive phenotype in mice. We found that PFC GRIP1 knockout led to increased motivation for cocaine and increased cue-induced cocaine seeking, without altering motivation or seeking for sucrose.
2. Methods
2.1. Subjects
Mice homozygous for the Cre/lox-conditional allele of GRIP1 (flox/flox) and GRIP2 knockout (−/−) were bred on a C57bl/6J background. Male and female mice (2–6-months old, age matched across group) were housed individually following stereotaxic surgery and during experimental paradigms. All animals were housed in a temperature- and humidity-controlled animal care facility with a 12-h light/dark cycle (lights on at 0700 hours). All procedures were approved by the Temple University Animal Care and Use Committee. Cocaine was obtained from the National Institute on Drug Abuse Drug Supply Program (Bethesda, MD) and dissolved in sterile 0.9% saline.
2.2. Prefrontal Microinjections and Adeno-Associated Virus Constructs
The adeno-associated virus (AAV) expressing Cre recombinase (AAV2/9.CMV.PI.CRE, titer 2.84 × 1013 vgc/µl) and the AAV expressing green fluorescent protein (eGFP) (AAV2/9.CMV.eGFP, titer 3.74 × 1013 vgc/µl) were generated by the University of Pennsylvania Vector Core. GRIP1 flox/flox mice (6–8 weeks) were anesthetized with isoflurane and 0.4µl of the viral construct (Cre or GFP) was injected bilaterally into the prefrontal cortex through a 30-gauge needle at a rate of 0.1 µl/min. Stereotaxic coordinates for the prefrontal cortex are (from Bregma) anterior-posterior 2.4, lateral +/− 0.3, dorso-ventral −2.3. Following recovery, mice remained in the home cage for 6 weeks prior to behavioral testing. The procedures involving the AAV viruses have all been approved by the Temple University Institutional Biosafety committee. Knockout was confirmed via western blot, and animals removed from study if knockout was less than a 30% decrease from average GFP control levels (n=2).
2.3. Operant Food Training
Before catheterization, mice were trained to perform an operant response for sucrose pellets. The mice were placed in operant chambers (Med-Associates) and trained to spin a wheel manipulandum to receive a sucrose pellet, with one-quarter spin measured as a single active response. Mice performed 5 days of FR1 responding followed by 5 days of FR5 responding, and a single day of a progressive ratio schedule (5*EXP(0.2*P)-5, where P=previous ratio; eg. 1, 2, 4, 6, 9, 12, 15, 20, 25, 32, 40…). A compound cue stimulus consisting of a cue light above the active wheel, a 2900-Hz tone, and house light off was concurrent with each pellet administration, followed by an additional 8 s time-out when responding had no programmed consequences and the house light remained off. Mice were allowed to self-administer a maximum of 50 pellets per 60 min operant session. During the food training phase, mice were food restricted to ∼90% of their free-feeding weight. Mice returned to ad libitum food access 3 days following the start of the cocaine self-administration phase.
2.4. Jugular Catheterization Surgery
Prior to surgery, mice were anesthetized with 80 mg/kg ketamine and 12 mg/kg xylazine. An indwelling silastic catheter was placed into the right jugular vein and sutured in place. The catheter was then threaded subcutaneously over the shoulder blade and was routed to a mesh backmount platform (Strategic Applications, Inc) that secured the placement. Catheters were flushed daily with 0.1 ml of an antibiotic (Timentin, 0.93 mg/ml) dissolved in heparinized saline. The catheters were sealed with plastic obturators when not in use.
2.5. Cocaine Self-Administration
Mice were tested for cocaine self-administration behavior in 2-hour sessions in the same chamber used for sucrose pellet self-administration. During testing, responding on the wheel now delivered an intravenous cocaine injection (0.6 mg/kg/infusion), paired with the same compound cue, under the same schedule as the food training. Following 10 days of cocaine self-administration on an FR1 schedule, mice underwent one day of cocaine self-administration on a progressive ratio schedule (5*EXP(0.2*P)-5, where P=previous ratio; eg. 1, 2, 4, 6, 9, 12, 15, 20, 25, 32, 40…) in which the same compound cue was presented. Breakpoint criteria were defined as failure to acquire an infusion of cocaine within 1800 seconds of the last infusion. The following day, mice began extinction training, in which cocaine-seeking behavior was extinguished by replacing the cocaine with 0.9% saline. During this time the light and tone cues paired with cocaine delivery were not present. Daily 2-h extinction sessions continued until animals met the extinction criterion of less than 25% of their self-administration responding (average of last 3 days). Twenty-four hours following meeting the extinction criterion, animals underwent a cue-induced reinstatement session. During the cue-induced reinstatement session, the light and tone cues were presented non-contingently for 20 seconds every 2 minutes during the first 10 minutes of the session. After this time period, the cues were presented contingent with operant responding, just as was done during the cocaine self-administration phase. During the reinstatement session, animals received saline infusions following responses on the active wheel.
2.6. Operant Set Shifting Task
Mice were run in a cognitive flexibility task as described in Parikh et al., 2016. A standard mouse operant conditioning chamber (MED Associates), containing grid floor, houselight, two large cue lights, central port with fluid dipper, and retractable levers was used. Mice were first trained on an FR1 schedule to acquire the lever press response, which provided 10µl of .066% saccharin solution. Once mice had completed a minimum of 30 lever presses within a 30-minute session, they began a pretraining phase. During pretraining, one of the levers (left or right of the central port) was extended for 10 seconds. Pressing the lever resulted in presentation of the reward and retraction of the lever. After the lever press or omission, an ITI of 9 ± 3 seconds began. Once mice met criteria (30 rewards and ≤ 20% omissions for 3 consecutive days), they were moved on to the visual discrimination phase. In this phase, both levers were presented for 5 seconds and mice were required to press the one underneath the illuminated cue light in order to receive a reward. After 3 consecutive days of 80% correct responses, mice were moved on to the set shifting phase. During this phase, mice were assigned to a “right lever” or “left lever” condition in a counterbalanced manner. The lever condition denoted which lever (right or left) was the active lever, and mice were required to press that lever to receive a reward, regardless of the location of the illuminated cue light. Once mice again were responding at least 80% correct for three consecutive days, they were moved on to the final stage, reversal. During reversal, mice were assigned the opposite lever condition as their set shift assignment. Once they had responded at 80% correct for three consecutive days, they were removed from the task and water returned ad libitum.
2.7. Western Blot
GRIP1 levels in the prefrontal cortex were measured using a western blot, as described in Briand et al., 2014. Briefly, animals were decapitated, and the prefrontal cortex dissected using a brain block (Braintree Scientific). Protein quantification was performed using a Pierce BCA Protein Assay Kit (Thermo Scientific). Equal amounts of protein (30 µg) were loaded into each well of a Tris-glycine gel (Lonza) and transferred to nitrocellulose membranes (Immobilon). Membranes were blocked with Li-Cor blocking buffer and allowed to incubate in primary antibody solution (GRIP1, 1:2000 (BD Biosciences) and GAPDH, 1:5000 (Cell Signaling)) for 24 hours at 4°C. Membranes were then incubated with fl uorescent secondary antibodies (1:20,000; IR-dye 680 or IR-dye 800, Li-Cor) and imaged on an Odyssey fluorescent scanner (Li-Cor). Western blots were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and the percent knockout calculated as a fraction of the average of the GRIP1 levels in GFP-infused mice.
2.8. Electrophysiology
Slice Preparation.
Following prefrontal injection of AAV-Cre or GFP, naïve mice were cervically dislocated and decapitated. The brain was removed and coronal slices of prefrontal cortex and nucleus accumbens were cut with a Vibratome (VT1000S, Leica Microsystems) in an ice-cold artificial cerebrospinal fluid solution (ACSF), in which NaCl was replaced by an equiosmolar concentration of sucrose. ACSF consisted of 130 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 26 mM NaHCO3, 10 mM glucose, 1 mM MgCl2, and 2 mM CaCl2 (pH 7.2–7.4 when saturated with 95% O2/5% CO2). Slices were incubated in ACSF at 32–34 °C for 25 min and kept at 22–25 °C thereafter, until transfer to the recording chamber. The osmolarity of all extracellular solutions was 300–315 mOsm. Slices were viewed using infrared differential interference contrast optics under an upright microscope (Slice Scope Pro, Scientifica) with a 40 × water-immersion objective.
Recordings.
The recording chamber was continuously perfused (1–2 ml/min) with oxygenated ACSF heated to 32±1 °C using an automatic temperatu re controller (Warner Instruments). Picrotoxin (100 µM) was added to all solutions to block the GABAA receptor-mediated currents. Recording pipettes were pulled from borosilicate glass capillaries (World Precision Instruments) to a resistance of 4–7 MΩ when filled with the intracellular solution (whole-cell recordings) or to a resistance of 1–2 MΩ when filled with extracellular solution (field recordings). All recordings were conducted with a MultiClamp700B amplifier (Molecular Devices). Whole-cell recordings. Intracellular solution contained (in mM): 100 CsCH3O3S, 50 CsCl, 3 KCl, 0.2 BAPTA, 10 HEPES, 1 MgCl2, 2.5 phosphocreatine-2Na, 2 Mg-ATP, 0.25 GTP-Tris, 1 QX-314 (pH 7.2–7.3 with CsOH, osmolarity 280–290 mOsm). All sEPSC recordings were conducted in whole-cell voltage-clamp mode (Vh = −70 mV). Currents were low-pass filtered at 2 kHz and digitized at 20 kHz using a Digidata 1440A acquisition board and pClamp10 software (both from Molecular Devices). Access resistance (10–30 MΩ) was monitored throughout the recordings by injection of 10 mV hyperpolarizing pulses and data were discarded if access resistance changed by >25% over the course of data acquisition. sEPSCs were detected using an automated sliding-template-based algorithm in pClamp 10. This method compares the shape of the detected current to that of a template and has been shown to detect events with amplitude of at least 3 times the square deviation of the noise (Clements and Bekkers, 1997). All detected events were verified by visual confirmation of a fast rise time and slower exponential decay to baseline. Mean sEPSC amplitude was analyzed from an average sEPSCs trace computed from a minimum of 150 individual sEPSCs. Mean sEPSC frequencies were analyzed from 180-s long trace segments. Evoked responses were triggered by 100 µs constant-current pulses generated by an A310 Accupulser (World Precision Instruments) and delivered at 0.1 Hz via a bipolar tungsten stimulation electrode positioned within 100 µm of the recorded cell. The amplitude of the current pulses was controlled by a stimulus isolator (WPI Linear Stimulus Isolator A395) and was adjusted to elicit monosynaptic responses in the range of 100–300 pA (the required stimulus intensity ranged from 15 to 80 µA). For all measures, cells from at least 3 animals, within each group, were used. Recordings were taken from cells within the accumbens core. Field Recordings. A bipolar tungsten stimulating electrode was placed within 100–300 µm from the recording electrode and used to stimulate excitatory afferents at 0.1 Hz. The field recordings were performed within the core of the nucleus accumbens. The amplitude of current pulses was set at the intensity required to evoke a 70% maximal response. Stimulations were applied as paired pulses (interval 20–420 ms) at 0.06Hz. The initial slope of fEPSPs was used as a measure of synaptic response.
2.9. Statistical analysis.
All self-administration experiments were analyzed with two-way ANOVAs with viral injection and day as the independent variables and pellets/responses/infusions as the dependent variable. Sidak’s post hoc comparisons were made when main effects or interactions were detected (p < 0.05). The protein quantification, progressive ratio, extinction responding and days to criterion, cue-induced reinstatement responding and cognitive flexibility data were analyzed using unpaired t-tests with viral injection as the independent variable. The sEPSC data were also analyzed using an upaired t-test with viral injection as the independent variable. The paired pulse recordings were analyzed using a two-way ANOVA with viral injection and interpulse interval as the independent variables.
3. Results
3.1. Viral Mediated Deletion of GRIP1 in the Medial Prefrontal Cortex
Mice used in this experiment were GRIP1-floxed mice bred on a GRIP2-null background, as the elimination of both GRIP isoforms is necessary (Takamiya et al, 2008). Six weeks following the injection of AAV-Cre into the mPFC, we elicited a significant knockout in GRIP1 levels in this region compared to AAV-GFP injected controls [t(35) = 4.79, p < 0.0001; Fig. 1]. The available antibodies only allow us to quantify the extent of the knockout using western blot techniques; this does not allow us to differentiate between GRIP1 knockout in neurons versus GRIP1 knockout in glial cells. As AAV9 preferentially targets neurons, the lack of complete knockout may be due to glial expression of GRIP1. To insure the behavioral effects seen here were not due to anterograde AAV expression, we confirmed that AAV-Cre injection into the mPFC did not affect GRIP1 expression in the nucleus accumbens [t(29) = 0.337, p =.74; Fig. 1f].
Figure 1. Cre recombinase injection into the mPFC leads to a significant decrease in GRIP1 protein levels.
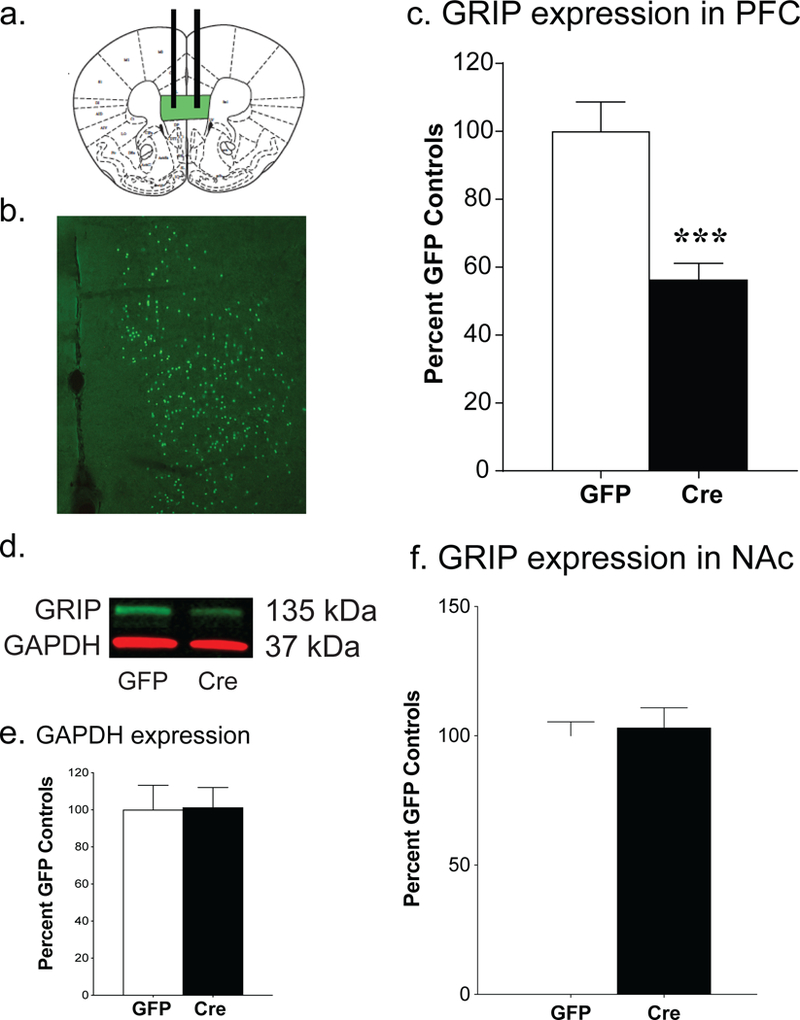
Green areas indicate the location of the bilateral injections of 0.4 µg of GFP or AAV-Cre recombinase into the medial prefrontal cortex (a). Coronal section of the mouse brain showing the viral expression of GFP within the mPFC (b). Quantification of western blot reveals a significant decrease in GRIP1 protein within the mPFC following AAV-Cre injection, as normalized to glyceraldehyde 3-phosphate dehydrogenase [GAPDH; t(35)=4.79, ***p<.0001, n=15–24; c]. Representative western blots showed GRIP1 knockout in the mPFC (d). There was no effect of AAV-Cre injection on either GAPDH expression in the PFC (e) or GRIP1 expression in the nucleus accumbens (NAc; f). Bars represent average ± SEM.
3.2. Prefrontal GRIP1 Knockout Does Not Affect Fixed Ratio Self-Administration of Sucrose or Cocaine
Six weeks after viral injections, GFP controls and Cre GRIP1 knockout mice underwent ten days of sucrose self-administration to acquire the operant response. An ANOVA for training day and viral injection revealed that there were no differences in number of pellets received [F(1, 83)=0.37, p=0.54; Fig. 2a], number of responses on the active response wheel [F(1, 83)=1.37, p=0.25; Fig. 2b], nor in percent of responses on the active response wheel between the two groups [F(1, 75)=2.27, p= 0.14; Fig. 2c]. After acquiring the operant response for food, mice received jugular catheterization surgery and began the cocaine self-administration phase. Again, ANOVA revealed that there were no differences between controls and prefrontal GRIP1 knockout mice in number of infusions received [F(1,59)=0.01, p=0.91; Fig. 3a], number of responses on the active response wheel [F(1,59)=1.11, p=0.30; Fig. 3b], nor in percent of active wheel responses [F(1,57)=0.21, p=0.65]. As no effect of sex was found for either food or cocaine self-administration (Table 1), sex is collapsed across groups.
Figure 2. GRIP1 knockout in the mPFC does not alter operant learning during food self-administration.
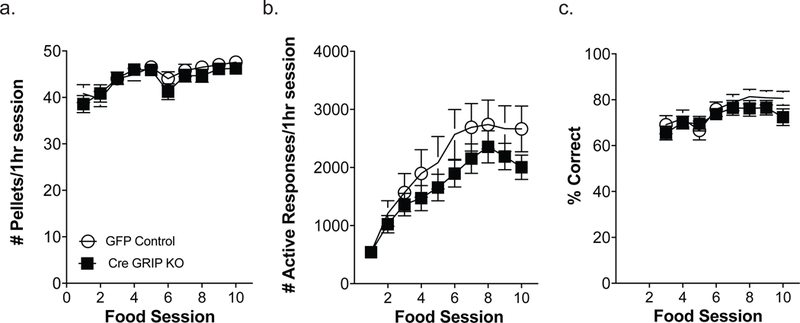
Over the 10 days of food self-administration, there were no significant differences between GFP control mice (n=39) and Cre GRIP1 KO mice (n=46) in the number of pellets consumed (a), number of responses on the active wheel (b), or percent of active responses during food training (c). Boxes represent average ± SEM.
Figure 3. GRIP1 knockout in the mPFC does not alter cocaine self-administration on a fixed ratio schedule of reinforcement.
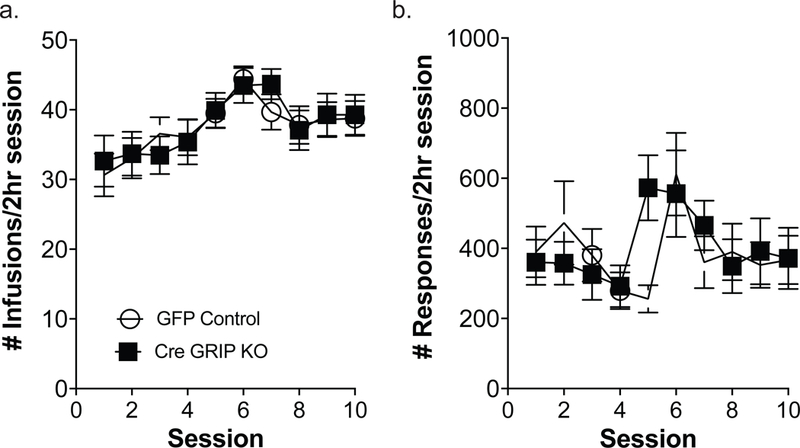
Over 10 days of cocaine self-administration, there were no significant differences between GFP control mice and Cre GRIP1 KO mice in the number of cocaine infusions or the number of responses on the active wheel (a, b; n=27–34). Boxes represent average ± SEM.
Table 1.
Raw data for behavioral tests separated by sex. No significant effects of sex were seen in any of the behavioral measured and therefore the data were collapsed across sex for all analyses. There was a significant interaction between sex and knockout on the number of food pellets earned on day 10 but the pairwise comparisons did not reveal any significant differences.
| Behavioral Variable | GFP Control Males | GFP Control Females | GRIP KD Males | GRIP KD Females | Effect of Sex | Interaction |
|---|---|---|---|---|---|---|
| Food Pellets D10 | 45±2 n=17 |
49±0.8 n=22 |
48±1 n=21 |
46 ±1 n=25 |
F(1,81) = 0.35, p = 0.56 | F(1,81) = 4.30, p = 0.04* |
| Food Responses D10 | 2911±559 n=17 |
2111±314 n=22 |
2173±603 n=21 |
2181±284 n=25 |
F(1,81) = 0.82, p = 0.37 | F(1,81) = 0.85, p = 0.36 |
| Food PR | 2191±419 n=11 |
1311±261 n=13 |
1384±264 n=12 |
2273±644 n=14 |
F(1,46) < 0.01, p = 0.99 | F(1,46) = 3.88, p = 0.06 |
| Food RI | 2462±1410 n=5 |
3182±1203 n=7 |
1274±222 n=7 |
3219±1100 n=7 |
F(1,22) = 1.62, p = 0.22 | F(1,22) = 0.34, p = 0.56 |
| Cocaine Infusions D10 | 42±3 n=15 |
40±2 n=19 |
36±5 n=10 |
37±4 n=17 |
F(1,58) < 0.01, p = 0.96 | F(1,58) = 0.14, p = 0.71 |
| Cocaine Responses D10 | 356±71 n=15 |
316±59 n=19 |
256±55 n=10 |
515±147 n=17 |
F(1,58) = 0.99, p = 0.32 | F(1,58) = 1.84, p = 0.18 |
| Cocaine PR | 183±64 n=10 |
144±34 n=13 |
435±190 n=6 |
369±161 n=11 |
F(1,37) = 0.19, p = 0.67 | F(1,37) = 0.01, p = 0.91 |
| Cocaine RI | 307±56 n=10 |
387±98 n=10 |
668±344 n=5 |
779±301 n=9 |
F(1,30) = 0.21, p = 0.65 | F(1,30) < 0.01, p = 0.94 |
p<.05.
3.3. Prefrontal GRIP1 Knockout Enhances Responding for Cocaine but not Sucrose on a Progressive Ratio Schedule
After 10 days of fixed ratio cocaine self-administration, GFP control and Cre GRIP1 knockout mice ran on a progressive ratio schedule of reinforcement to assess their willingness to work for cocaine. An unpaired t-test showed that Cre GRIP1 knockout mice exhibited a higher breakpoint compared to the GFP control mice [t(38)=2.18, p=0.04; Fig. 4a]. In contrast, GFP control and Cre GRIP1 knockout mice exhibit similar willingness to work for sucrose on a PR schedule [t(48)=0.33, p=0.75; Fig. 4b]. Sex is collapsed across groups as no sex differences were observed (Table 1).
Figure 4. Prefrontal GRIP1 knockout increases progressive ratio breakpoint and reinstatement responding for cocaine but not sucrose.
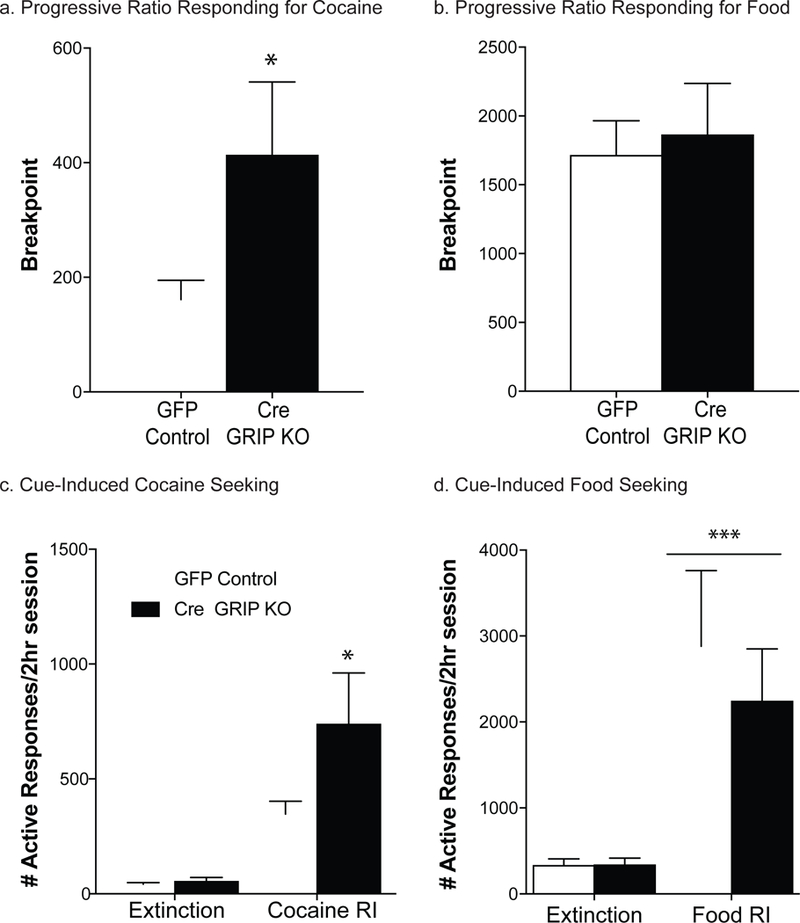
Following prefrontal GRIP1 knockout, mice exhibited an increased breakpoint on a progressive ratio schedule for cocaine [a; t(38) = 2.18, *p = 0.04; n=18–24] but not for food (b; n=24–26). Breakpoint is defined as the final ratio the mice achieved before timing out of the session, i.e. the number active responses required to move onto the next step. Further, PFC GRIP1 knockout mice exhibit greater cocaine seeking during cue-induced reinstatement [c; t(31) = 2.27, *p = 0.03; n=14–20] but not greater reinstatement of food seeking (d; n=13–14). Bars represent average ± SEM.
3.4. Prefrontal GRIP1 Knockout Enhances Responding for Cocaine but not Sucrose During Cue-Reinstatement
Following the cocaine self-administration phase, a subset of mice began extinction training. No differences were seen in the responding on the first day of extinction [GFP Control= 206.21±60.94; Cre GRIP1 KO= 293.67±91.18; t(40)=0.83, p=0.41] or in the days to reach the extinction criterion [GFP Control=6.63±0.78; Cre GRIP1 KO=6.07±0.95; t(31)=0.46, p=0.65]. However, during the cue-induced reinstatement session, mPFC GRIP1 knockout mice exhibit significantly greater responding compared to GFP control mice [t(31) = 2.27, p = 0.03; Fig. 4c], indicating higher cue-derived cocaine seeking. In a separate cohort of mice, we examined extinction and cue-induced reinstatement of food seeking. Similar to what was seen with extinction of cocaine seeking, we did not see any effect of mPFC GRIP1 knockout on extinction of food seeking (GFP Control= 483.69±66.44; Cre GRIP1 KO= 481.79±113.15; t(25)=0.01, p=0.99]. Additionally, we did not see a viral mediated increase in cue-induced food seeking following mPFC GRIP1 knockout [t(24) = 0.61, p = 0.30; Fig. 4d]. There were no sex differences in these behavioral measures (Table 1), so sex is collapsed across groups.
3.5. Prefrontal GRIP1 Knockout Does Not Lead to Deficits in Cognitive Flexibility
To determine whether the increased cocaine seeking during reinstatement was due to deficits in cognitive flexibility, we ran a separate cohort of mice on a cognitive flexibility task (Parikh et al., 2016). There were no differences between GFP controls and Cre GRIP1 KO on the number of trials to reach visual discrimination criteria [t(81)=1.74, p=0.09]. We did not detect any effect of mPFC GRIP1 knockout on the trials to criterion [t(84)=1.47, p=.15; Fig. 5a] or errors to criterion [t(84)=0.24, p=0.81; Fig. 5b] on the set-shift phase of the task. We also did not see any differences between the groups during the reversal phase of the task in either the trials to criterion [t(84)=0.70, p=.49; Fig. 5c] or the errors to criterion [t(84)=0.25, p=.81; Fig. 5d]. As no sex differences were observed (Table 1), sex is collapsed across groups for analysis.
Figure 5. Prefrontal GRIP1 knockout does not impact cognitive flexibility.
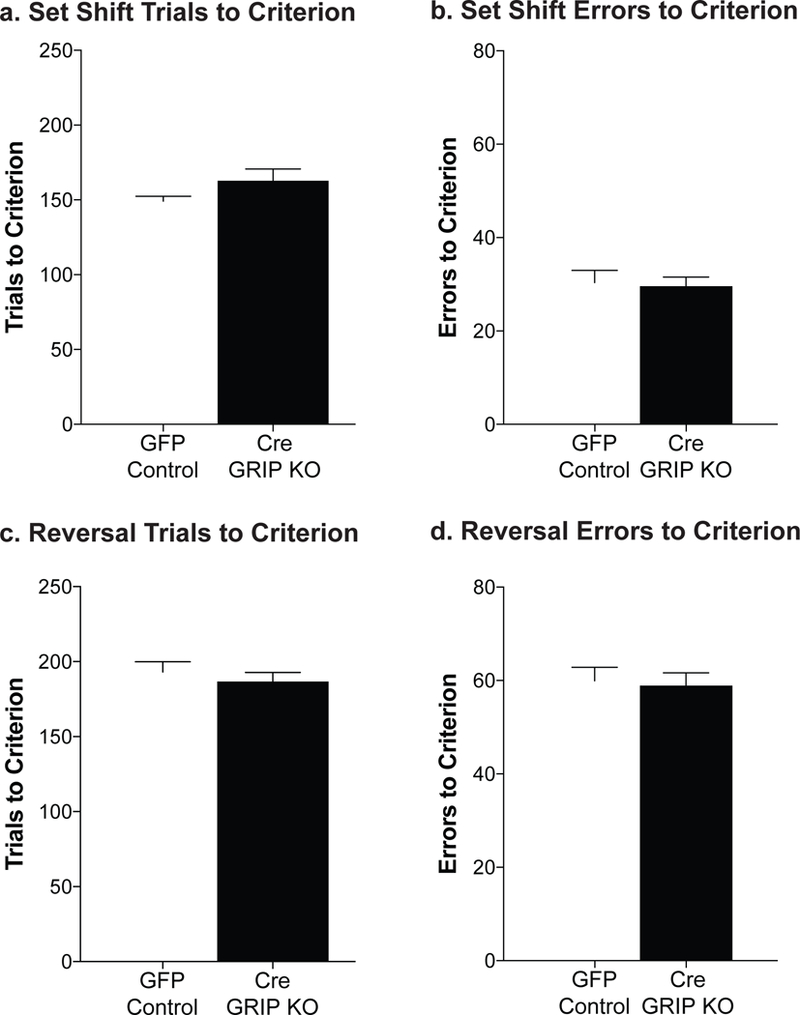
No differences in were seen following prefrontal GRIP1 knockout in set-shifting performance, either trials to criterion or errors to criterion (a, b; n=40–45). Further, no differences were seen between the groups in trials to criterion or errors to criterion in the reversal learning task (c,d; n=40–45). Bars represent average ± SEM.
3.6. Prefrontal GRIP1 Knockout alters glutamate transmission in the PFC and the NAc.
Six to eight weeks after viral-mediated knockout of GRIP1 in the prefrontal cortex, we examined spontaneous excitatory transmission in drug-naïve mice. Following mPFC GRIP1 knockout we see an increase in the amplitude of spontaneous excitatory postsynaptic currents (sEPSCs) compared to GFP-injected controls [t(15)=3.24, p<0.01; Fig. 6a]. No differences were seen between the groups in sEPSC frequency [t(15) = 0.91, p = 0.38; Fig. 6b]. To determine whether these physiological effects within the PFC altered downstream transmission in the nucleus accumbens, we examined paired-pulse ratio (PPR) in both cocaine-experienced and naïve mice. Medial PFC GRIP1 knockout led to a decrease in PPR in the nucleus accumbens regardless of drug history [F(1,33)=4.35, p=0.04; Fig. 6c]. No sex differences were observed.
Figure 6. Disrupting GRIP1 in the PFC augments glutamate transmission in the PFC and nucleus accumbens.
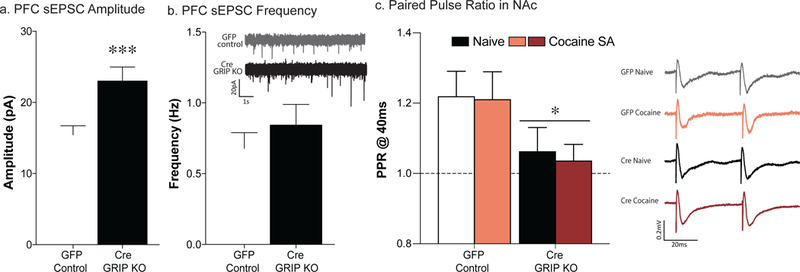
Quantification of sEPSC amplitude reveals an increase in prefrontal GRIP1 knockout mice compared to GFP controls [a; t(15)=3.24, **p<0.01; n=15–20]. No differences were seen between the groups in sEPSC frequency (b; n=15–19). Prefrontal GRIP1 knockout led to a decrease in the paired-pulse ratio (40ms IPI) in both naïve and cocaine-experienced mice [c; F(1,33)=4.35, p=0.04; n=12–13]. Bars represent average ± SEM.
4. Discussion
Overall, we find that the scaffolding protein, GRIP1, plays a critical role within the prefrontal cortex in mediating cocaine seeking. Our data demonstrate that knockout of prefrontal GRIP1 increases motivation for cocaine and cocaine seeking during cue-induced reinstatement, while not affecting sucrose seeking or consumption. Furthermore, these alterations are not simply the result of alterations in cognitive function. Prefrontal GRIP1 knockout does not alter set-shifting or reversal learning. Electrophysiological recordings demonstrate that GRIP1 knockout leads to increased sEPSC amplitude within the PFC and downstream alterations in presynaptic transmission in the NAc.
4.1. GRIP1 knockout in the prefrontal cortex increases motivation for cocaine and potentiates cocaine seeking in both males and females.
The glutamatergic projection from the PFC to the NAc is critically involved in the reinstatement of drug seeking in part due to its role in consolidation of cue-driven reward memory (Berke and Hyman, 2000; Kalivas et al., 2005). Inactivation of the medial PFC disrupts reinstatement of drug seeking (Martin-Garcia et al., 2014; McLaughlin and See, 2003; Palombo et al., 2017; Rocha and Kalivas, 2010; Zavala et al., 2003). However, the mechanisms underlying this involvement are less well understood. The current study demonstrates that glutamate trafficking within the PFC, specifically mediated by GRIP1 function, plays a role in cocaine seeking.
Our findings build upon previous work showing cocaine-induced alterations in glutamate signaling in the prefrontal cortex. Prefrontal glutamatergic transmission, and especially the pathway between the PFC and nucleus accumbens, is involved in reinstatement of cocaine-seeking (McFarland et al., 2003). Within the PFC itself, glutamate release plays a role in protracted withdrawal from cocaine and the development of incubation of craving (Shin et al., 2018). An increase in prefrontal glutamate is also seen during the first day of withdrawal after chronic cocaine treatment (Williams and Steketee, 2004). Within 24 hours of cocaine self-administration, prefrontal glutamate levels are decreased (Ben-Shahar et al., 2012). However, while basal glutamatergic activity within the PFC is decreased, burst firing in response to cocaine is increased in drug-experienced rats (Sun and Rebec, 2006). This increase, not seen in drug-naïve rats, suggests a mechanism by which cocaine usurps prefrontal circuits during addiction (Sun and Rebec, 2006).
In the current study we found that GRIP1 knockout led to an increase in sEPSC amplitude, suggesting our manipulation has increased excitatory transmission in the PFC. Cocaine is known to increase excitability in the PFC (Nasif et al., 2005), so these changes may underlie the potentiated cocaine seeking seen here. GRIP1 is specifically involved in anchoring GluA2-containing AMPARs, therefore, we hypothesize that this increased activity is due to a relative increase in the amount of GluA2-lacking AMPA receptors at the membrane. Cocaine experience can increase in the contribution of GluA2-lacking AMPARs in the PFC (Nic Dhonnchadha et al., 2013; Pena-Bravo et al., 2017), and these molecular changes are hypothesized to underlie cocaine withdrawal symptoms (Nasif et al., 2005). As we also see a downstream decrease in paired pulse ratio in the accumbens, GRIP1 KO in the PFC may be priming the brain into an addictive-like state.
4.2. Prefrontal GRIP1 does not play a role in natural reward taking or seeking.
We did not find any effect of prefrontal GRIP1 knockout on natural reward taking or seeking. This is consistent with past work showing that glutamatergic afferents from the PFC are active during drug, but not food, reinstatement (McFarland et al., 2003). Moreover, experimental manipulation of AMPA receptors has consistently failed to affect food seeking (Anderson et al., 2008; Briand et al., 2014; Famous et al., 2008). Therefore, we must conclude that glutamatergic plasticity within the PFC is more sensitive to drug use than to natural reward. Given that specific neuronal ensembles within the vmPFC are responsible for encoding food reward and extinction of food seeking, we may have seen different results if we had manipulated GRIP1 expression after the formation of these memories (Warren et al., 2016). However, it is also possible that we would have found different results had we examined a high fat food reward that can lead to more compulsive food seeking (Decarie-Spain et al., 2018; Ghitza et al., 2006; Johnson and Kenny, 2010).
The differing effects between drug seeking and natural reward are also seen in the extracellular accumbal glutamate levels of rats trained to self-administer cocaine (McFarland et al., 2003). Extracellular glutamate levels are increased after cocaine self-administration but not food self-administration. Additionally, food self-administration has been shown to create a reversible potentiation of glutamatergic signaling within the VTA, whereas cocaine self-administration leads to VTA potentiation stable for at least 3 weeks (Chen et al., 2008). Thus, our data is congruent with the existing literature on the effects of glutamate during drug versus natural reward.
4.3. Knocking down GRIP1 in the PFC does not alter cognitive function.
The PFC is involved in executive behavior, including whether a rodent should engage in or suppress an action based on context (Moorman and Aston-Jones, 2015). Prefrontal lesions impair set-shifting in mice as well as rats (Bissonette et al., 2008). Additionally, lesions of the mPFC disrupt the formation of an attentional set (Bissonette et al., 2008), impair both sustained attention and response inhibition (Broersen and Uylings, 1999), and impair performance on a delay discounting task in both rodents (Déziel and Tasker, 2017) and humans (Bechara et al., 2000). More specifically, glutamate within the PFC is critical for maintenance of these cognitive functions. Disrupting either AMPAR or NMDAR function in the prefrontal cortex leads to deficits in behavioral flexibility in male rats retrieving a food reward from a T-maze (Stefani et al., 2003). Similarly, AMPA antagonists injected into the mPFC of male rats causes deficits in extradimensional shifting when digging for a food reward in pots containing two different odors and digging mediums (Jett et al., 2017).
Given the role of the PFC in cognitive function (Miller, 2000), it is perhaps surprising that the current study did not find any effects of prefrontal GRIP1 knockout on strategy set-shifting, reversal learning or the ability of mice to learn an operant task. However, prefrontal GRIP1 knockout did not lead to an overall disruption in glutamate signaling in the PFC. To the contrary, we saw increased AMPA transmission. Therefore, our findings are consistent with previous work highlighting the role of the PFC in cognition. In rats, microinfusion of an AMPAR positive allosteric modulator into the prelimbic cortex has been shown to enhance cognition on an odor-reward association task (Yefimenko et al., 2013). Likewise, injection of an AMPAR antagonist into the mPFC of rats impairs discrimination learning and set-shifting due to general learning deficits (Stefani and Moghaddam, 2006). Consistent with our findings, an increase in prefrontal AMPA transmission would not be predicted to inhibit cognitive learning on an operant task.
4.4. Altering AMPA trafficking in the PFC has downstream effects on accumbal physiology.
Disrupting GRIP1 function leads to a decrease in the anchoring of GluA2-containing AMPARs to the synapse (Mejias et al., 2011). Therefore, one might expect a decrease in prefrontal glutamate transmission following site-specific GRIP1 deletion. In contrast, we found that prefrontal GRIP1 knockout led to an increase in sEPSC amplitude, measured in layer 5 of the PFC. While GluA2-containing AMPARs are the primary subtype, GluA2-lacking AMPARs are also present in the PFC. In fact, cocaine exposure can lead to an increase in the contribution of GluA2-lacking AMPARs in the prefrontal cortex (Pena-Bravo et al., 2017). GluA2-lacking AMPARs are calcium permeable and therefore exhibit a higher conductance than the GluA2-containing AMPARs. By knocking out GRIP1 in the PFC and disrupting the insertion of GluA2-containing AMPARs into the membrane, it’s possible that GluA2-lacking AMPA were preferentially inserted in the synapse, leading to an enhanced sEPSC amplitude. As the sEPSC recordings are influenced by not only AMPA-mediated currents but also spontaneous action potential firing, it is possible that the effects we see on sEPSC amplitude are mediated by other glutamate receptor subtypes (i.e. NMDARs). However, as GRIP1 has not been demonstrated to be involved in NMDAR trafficking but plays an established role in AMPAR trafficking, the effects we see are likely due to differences in AMPAR signaling.
As cocaine increases excitability in the prefrontal cortex (Nasif et al., 2005), the increased sEPSC amplitude following GRIP1 knockout could contribute to the increased cocaine seeking. The current study also found that prefrontal GRIP1 knockout led to a decrease in paired pulse ratio within the nucleus accumbens of both naïve and cocaine-experienced mice. The decrease in paired-pulse ratio suggests that prefrontal GRIP1 knockout leads to an increase in glutamate release probability in the nucleus accumbens (Fioravante and Regehr, 2011; Regehr, 2012). This increase in release probability could lead to an increase in cue-evoked glutamate release in the nucleus accumbens, perhaps driving the increases in reinstatement behavior.
Conclusion
In the current study, we have shown that conditional deletion of GRIP1 in the mPFC leads to a specific increase in cocaine seeking and motivation for cocaine in both male and female mice. Disrupting GRIP1 in the mPFC does not alter intake or seeking of natural rewards nor does it affect cognitive flexibility. Furthermore, GRIP1 knockout leads to an increase in AMPA transmission in the mPFC as well as alterations in glutamate transmission downstream in the nucleus accumbens. These results suggest that pharmacotherapies aimed at augmenting the interaction between GRIP1 and GluA2 could be effective in treating cocaine use disorder.
Supplementary Material
Highlights.
Disruption of GRIP in the prefrontal cortex potentiates motivation for cocaine in male and female mice.
Disruption of GRIP in the prefrontal cortex potentiates cue-induced cocaine seeking in male and female mice.
Disruption of GRIP in the prefrontal cortex does not alter motivation for food or cue-induced cocaine seeking
Prefrontal GRIP expression is not necessary for strategy set shifting or reversal learning.
Selective deletion of GRIP in the PFC leads to an increase in sEPSC amplitude in the PFC and a decrease in paired pulse ratio in the nucleus accumbens
Acknowledgements
We thank Julia Kirkland, Anne Fosnocht, and Jeffrey Lenz for providing assistance in running the behavioral experiments in this study.
Funding
This work was supported by National Institute on Drug Abuse (NIDA) Grant R00 DA033372 (L.A.B.), T32 DA007273 (M.M.W.), a Brain & Behavior Research Foundation NARSAD award (L.A.B.) and a Deutsche Forschungsgemeinschaft (DFG) grant DE 2828/1–1 (A.U.D).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure
The authors declare no conflict of interest.
References
- Anderson SM, Famous KR, Sadri-Vakili G, Kumaresan V, Schmidt HD, Bass CE, Terwilliger EF, Cha JH, Pierce RC, 2008. CaMKII: a biochemical bridge linking accumbens dopamine and glutamate systems in cocaine seeking. Nat Neurosci 11, 344–353. [DOI] [PubMed] [Google Scholar]
- Bechara A, Tranel D, Damasio H, 2000. Characterization of the decision-making deficit of patients with ventromedial prefrontal cortex lesions. Brain 123 ( Pt 11), 2189–2202. [DOI] [PubMed] [Google Scholar]
- Ben-Shahar OM, Szumlinski KK, Lominac KD, Cohen A, Gordon E, Ploense KL, DeMartini J, Bernstein N, Rudy NM, Nabhan AN, Sacramento A, Pagano K, Carosso GA, Woodward N, 2012. Extended access to cocaine self-administration results in reduced glutamate function within the medial prefrontal cortex. Addiction biology 17, 746–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berke JD, Hyman SE, 2000. Addiction, dopamine, and the molecular mechanisms of memory. Neuron 25, 515–532. [DOI] [PubMed] [Google Scholar]
- Bissonette GB, Martins GJ, Franz TM, Harper ES, Schoenbaum G, Powell EM, 2008. Double dissociation of the effects of medial and orbital prefrontal cortical lesions on attentional and affective shifts in mice. J Neurosci 28, 11124–11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briand LA, Kimmey BA, Ortinski PI, Huganir RL, Pierce RC, 2014. Disruption of glutamate receptor-interacting protein in nucleus accumbens enhances vulnerability to cocaine relapse. Neuropsychopharmacology 39, 759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broersen LM, Uylings HB, 1999. Visual attention task performance in Wistar and Lister hooded rats: response inhibition deficits after medial prefrontal cortex lesions. Neuroscience 94, 47–57. [DOI] [PubMed] [Google Scholar]
- Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM, Chou JK, Bonci A, 2008. Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron 59, 288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Bekkers JM, 1997. Detection of spontaneous synaptic events with an optimally scaled template. Biophysical journal 73, 220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decarie-Spain L, Sharma S, Hryhorczuk C, Issa-Garcia V, Barker PA, Arbour N, Alquier T, Fulton S, 2018. Nucleus accumbens inflammation mediates anxiodepressive behavior and compulsive sucrose seeking elicited by saturated dietary fat. Mol Metab 10, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Déziel RA, Tasker RA, 2017. Effects of endothelin-induced prefrontal cortical lesions on delay discounting in the rat. Behav Neurosci 131, 11–19. [DOI] [PubMed] [Google Scholar]
- Dong H, O’Brien RJ, Fung ET, Lanahan AA, Worley PF, Huganir RL, 1997. GRIP: a synaptic PDZ domain-containing protein that interacts with AMPA receptors. Nature 386, 279–284. [DOI] [PubMed] [Google Scholar]
- Famous KR, Kumaresan V, Sadri-Vakili G, Schmidt HD, Mierke DF, Cha JH, Pierce RC, 2008. Phosphorylation-dependent trafficking of GluR2-containing AMPA receptors in the nucleus accumbens plays a critical role in the reinstatement of cocaine seeking. J Neurosci 28, 11061–11070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioravante D, Regehr WG, 2011. Short-term forms of presynaptic plasticity. Curr Opin Neurobiol 21, 269–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghitza UE, Gray SM, Epstein DH, Rice KC, Shaham Y, 2006. The anxiogenic drug yohimbine reinstates palatable food seeking in a rat relapse model: a role of CRF1 receptors. Neuropsychopharmacology 31, 2188–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein RZ, Volkow ND, 2011. Dysfunction of the prefrontal cortex in addiction: neuroimaging findings and clinical implications. Nat Rev Neurosci 12, 652–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jett JD, Bulin SE, Hatherall LC, McCartney CM, Morilak DA, 2017. Deficits in cognitive flexibility induced by chronic unpredictable stress are associated with impaired glutamate neurotransmission in the rat medial prefrontal cortex. Neuroscience 346, 284–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PM, Kenny PJ, 2010. Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats. Nat Neurosci 13, 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, 2009. The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci 10, 561–572. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Volkow N, Seamans J, 2005. Unmanageable motivation in addiction: a pathology in prefrontal-accumbens glutamate transmission. Neuron 45, 647–650. [DOI] [PubMed] [Google Scholar]
- Liang J, Li JL, Han Y, Luo YX, Xue YX, Zhang Y, Zhang LB, Chen ML, Lu L, Shi J, 2017. Calpain-GRIP Signaling in Nucleus Accumbens Core Mediates the Reconsolidation of Drug Reward Memory. J Neurosci 37, 8938–8951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao L, Takamiya K, Thomas G, Lin DT, Huganir RL, 2010. GRIP1 and 2 regulate activity-dependent AMPA receptor recycling via exocyst complex interactions. Proc Natl Acad Sci U S A 107, 19038–19043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Garcia E, Courtin J, Renault P, Fiancette JF, Wurtz H, Simonnet A, Levet F, Herry C, Deroche-Gamonet V, 2014. Frequency of cocaine self-administration influences drug seeking in the rat: optogenetic evidence for a role of the prelimbic cortex. Neuropsychopharmacology 39, 2317–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Lapish CC, Kalivas PW, 2003. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 23, 3531–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin J, See RE, 2003. Selective inactivation of the dorsomedial prefrontal cortex and the basolateral amygdala attenuates conditioned-cued reinstatement of extinguished cocaine-seeking behavior in rats. Psychopharmacology (Berl) 168, 57–65. [DOI] [PubMed] [Google Scholar]
- Mejias R, Adamczyk A, Anggono V, Niranjan T, Thomas GM, Sharma K, Skinner C, Schwartz CE, Stevenson RE, Fallin MD, Kaufmann W, Pletnikov M, Valle D, Huganir RL, Wang T, 2011. Gain-of-function glutamate receptor interacting protein 1 variants alter GluA2 recycling and surface distribution in patients with autism. Proceedings of the National Academy of Sciences of the United States of America 108, 4920–4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EK, 2000. The prefrontal cortex and cognitive control. Nat Rev Neurosci 1, 59–65. [DOI] [PubMed] [Google Scholar]
- Moorman DE, Aston-Jones G, 2015. Prefrontal neurons encode context-based response execution and inhibition in reward seeking and extinction. Proc Natl Acad Sci U S A 112, 9472–9477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman DE, James MH, McGlinchey EM, Aston-Jones G, 2015. Differential roles of medial prefrontal subregions in the regulation of drug seeking. Brain Res 1628, 130–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasif FJ, Sidiropoulou K, Hu XT, White FJ, 2005. Repeated cocaine administration increases membrane excitability of pyramidal neurons in the rat medial prefrontal cortex. J Pharmacol Exp Ther 312, 1305–1313. [DOI] [PubMed] [Google Scholar]
- Nic Dhonnchadha B, Lin A, Leite-Morris KA, Kaplan GB, Man HY, Kantak KM, 2013. Alterations in expression and phosphorylation of GluA1 receptors following cocaine-cue extinction learning. Behav Brain Res 238, 119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palombo P, Leao RM, Bianchi PC, de Oliveira PEC, Planeta CDS, Cruz FC, 2017. Inactivation of the Prelimbic Cortex Impairs the Context-Induced Reinstatement of Ethanol Seeking. Front Pharmacol 8, 725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Cole RD, Patel PJ, Poole RL, Gould TJ, 2016. Cognitive control deficits during mecamylamine-precipitated withdrawal in mice: Possible links to frontostriatal BDNF imbalance. Neurobiol Learn Mem 128, 110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park WK, Bari AA, Jey AR, Anderson SM, Spealman RD, Rowlett JK, Pierce RC, 2002. Cocaine administered into the medial prefrontal cortex reinstates cocaine-seeking behavior by increasing AMPA receptor-mediated glutamate transmission in the nucleus accumbens. J Neurosci 22, 2916–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena-Bravo JI, Reichel CM, Lavin A, 2017. Abstinence from Cocaine-Induced Conditioned Place Preference Produces Discrete Changes in Glutamatergic Synapses onto Deep Layer 5/6 Neurons from Prelimbic and Infralimbic Cortices. eNeuro 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, 2012. Short-term presynaptic plasticity. Cold Spring Harb Perspect Biol 4, a005702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha A, Kalivas PW, 2010. Role of the prefrontal cortex and nucleus accumbens in reinstating methamphetamine seeking. Eur J Neurosci 31, 903–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin CB, Templeton TJ, Chiu AS, Kim J, Gable ES, Vieira PA, Kippin TE, Szumlinski KK, 2018. Endogenous glutamate within the prelimbic and infralimbic cortices regulates the incubation of cocaine-seeking in rats. Neuropharmacology 128, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani MR, Groth K, Moghaddam B, 2003. Glutamate receptors in the rat medial prefrontal cortex regulate set-shifting ability. Behavioral neuroscience 117, 728–737. [DOI] [PubMed] [Google Scholar]
- Stefani MR, Moghaddam B, 2006. Distinct contributions of glutamate receptor subtypes to cognitive set-shifting abilities in the rat. Annals of the New York Academy of Sciences 1003, 464–467. [DOI] [PubMed] [Google Scholar]
- Stefanik MT, Kupchik YM, Kalivas PW, 2016. Optogenetic inhibition of cortical afferents in the nucleus accumbens simultaneously prevents cue-induced transient synaptic potentiation and cocaine-seeking behavior. Brain Struct Funct 221, 1681–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanik MT, Moussawi K, Kupchik YM, Smith KC, Miller RL, Huff ML, Deisseroth K, Kalivas PW, LaLumiere RT, 2013. Optogenetic inhibition of cocaine seeking in rats. Addiction biology 18, 50–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summa M, Di Prisco S, Grilli M, Marchi M, Pittaluga A, 2011. Hippocampal AMPA autoreceptors positively coupled to NMDA autoreceptors traffic in a constitutive manner and undergo adaptative changes following enriched environment training. Neuropharmacology 61, 1282–1290. [DOI] [PubMed] [Google Scholar]
- Sun W, Rebec GV, 2006. Repeated cocaine self-administration alters processing of cocaine-related information in rat prefrontal cortex. J Neurosci 26, 8004–8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szumlinski KK, Wroten MG, Miller BW, Sacramento AD, Cohen M, Ben-Shahar O, Kippin TE, 2016. Cocaine Self-Administration Elevates GluN2B within dmPFC Mediating Heightened Cue-Elicited Operant Responding. J Drug Abuse 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamiya K, Mao L, Huganir RL, Linden DJ, 2008. The glutamate receptor-interacting protein family of GluR2-binding proteins is required for long-term synaptic depression expression in cerebellar Purkinje cells. J Neurosci 28, 5752–5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HL, Queenan BN, Huganir RL, 2015. GRIP1 is required for homeostatic regulation of AMPAR trafficking. Proc Natl Acad Sci U S A 112, 10026–10031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren BL, Mendoza MP, Cruz FC, Leao RM, Caprioli D, Rubio FJ, Whitaker LR, McPherson KB, Bossert JM, Shaham Y, Hope BT, 2016. Distinct Fos-Expressing Neuronal Ensembles in the Ventromedial Prefrontal Cortex Mediate Food Reward and Extinction Memories. J Neurosci 36, 6691–6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JM, Steketee JD, 2004. Cocaine increases medial prefrontal cortical glutamate overflow in cocaine-sensitized rats: a time course study. Eur J Neurosci 20, 1639–1646. [DOI] [PubMed] [Google Scholar]
- Xue L, Zhang F, Chen X, Lin J, Shi J, 2010. PDZ protein mediated activity-dependent LTP/LTD developmental switch at rat retinocollicular synapses. Am J Physiol Cell Physiol 298, C1572–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yefimenko N, Portero-Tresserra M, Martí-Nicolovius M, Guillazo-Blanch G, Vale-Martínez A, 2013. The AMPA receptor modulator S18986 in the prelimbic cortex enhances acquisition and retention of an odor-reward association. Neurosci Lett 548, 105–109. [DOI] [PubMed] [Google Scholar]
- Zavala AR, Weber SM, Rice HJ, Alleweireldt AT, Neisewander JL, 2003. Role of the prelimbic subregion of the medial prefrontal cortex in acquisition, extinction, and reinstatement of cocaine-conditioned place preference. Brain Res 990, 157–164. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.