

Abstract
Background
In giant cell arteritis (GCA), vessel-wall infiltrating CD4 T-cells and macrophages form tissue-destructive granulomatous infiltrates and the artery responds with a maladaptive response-to-injury; leading to intramural neoangiogenesis, intimal hyperplasia and luminal occlusion. Lesion-residing T cells receive local signals, which represent potential therapeutic targets.
Objectives
We have examined how CD28 signaling affects vasculitis induction and maintenance and which pathogenic processes rely on CD28-mediated T-cell activation.
Methods
Vasculitis was induced by transferring PBMC from GCA patients into immunodeficient NSG mice engrafted with human arteries. Human artery-NSG chimeras were treated with anti-CD28 domain antibody (dAb) or control Ab. Treatment effects and immunosuppressive mechanisms were examined in vivo and in vitro applying tissue transcriptome analysis, immunohistochemistry, flow cytometry and immuno-metabolic analysis.
Results
Blocking CD28-dependent signaling markedly reduced tissue-infiltrating T-cells and effectively suppressed vasculitis. Mechanistic studies implicated CD28 in activating AKT signaling, T-cell proliferation and differentiation of IFN-γ and IL-21-producing effector T-cells. Blocking CD28 was immunosuppressive by disrupting T-cell metabolic fitness; specifically, the ability to utilize glucose. Expression of the glucose transporter Glut1 and of glycolytic enzymes as well as mitochondrial oxygen consumption were all highly sensitive to CD28 blockade. Also, induction and maintenance of CD4+CD103+ tissue-resident memory T-cells (TRM), needed to replenish the vasculitic infiltrates, depended on CD28 signaling. CD28 blockade effectively suppressed vasculitis-associated remodeling of the vessel wall.
Conclusions
CD28 stimulation provides a metabolic signal required for pathogenic effector functions in medium and large vessel vasculitis. Disease-associated glycolytic activity in wall-residing T-cell populations can be therapeutically targeted by blocking CD28 signaling.
Keywords: giant cell arteritis, vasculitis, CD28, T-cells, glycolysis, immunometabolism
Condensed Abstract
In giant cell arteritis, the PD-1/PD-L1 immunoinhibitory checkpoint is defective. Whether CD28-dependent co-stimulation is disease-relevant and unopposed is unknown. We engrafted human arteries into immunodeficient mice and induced vasculitis by transferring GCA immune cells. Treating such humanized mice with an anti-CD28 domain antibody was highly effective in suppressing vasculitis. Metabolic fitness of lesional T-cells, including their glycolytic machinery, required CD28 signaling. Also, tissue-resident memory T-cells that repopulate the inflammatory lesions, were highly sensitive to CD28 blockade. Anti-CD28 treatment inhibited intimal hyperplasia and intramural neoangiogenesis. Thus, CD28 signaling is critical in GCA and emerges as a promising therapeutic target.
Introduction
The wall of the aorta and its major branches is an immuno-privileged tissue site, typically protected from inflammatory attack (1,2). Breakdown of this immunoprivilege, as seen in vasculitis, leads to life-threatening complications, such as aortic aneurysm, aortic dissection and vaso-occlusive disease of the aortic branches. Giant cell arteritis (GCA) is the most frequent cause of aortitis and large/medium vessel vasculitis. GCA’s vascular complications include blindness, stroke and aortic arch syndrome. Extravascular complications, characterized by an intense acute phase response, have been attributed to excess innate cytokines (3), some of which can be therapeutically targeted (4). In the vessel wall, GCA presents with granulomatous infiltrates invading the media and intima, driving tissue damage and wall remodeling (5,6). Vascular GCA is relatively refractory to immunosuppressive therapy (7,8). More effective means to remove the vasculitic infiltrates are needed to prevent tissue ischemia and aortic wall destruction.
GCA’s immunopathology centers on CD4 T-cells and macrophages (9–11). GCA patients have a defect in a tissue-protective immune checkpoint, fail to express PD-L1 and permit unopposed activation of PD-1+ T-cells (12). Also, GCA patients lack anti-inflammatory T regulatory cells, evading proper suppression of CD4 T-cell immunity (13). Endothelial cells break the vascular immunoprivilege through aberrant Jagged1 expression that enables tissue entry of NOTCH1+ T-cells (14). Once in the adventitia and media, T-cells receive local stimulatory signals, especially from wall-endogenous dendritic cells (15), take up tissue residence and differentiate into CD103+ tissue-resident memory T-cells (TRM) (16). T-cells need assistance from MMP-9-producing monocytes for tissue invasion (11).
Lesional T-cells produce multiple effector cytokines (IFN-γ, IL-17, IL-21, IL-9) (8,12,16) indicative of generalized hyperresponsiveness. Accordingly, effective therapy may require inhibiting multiple T-cell effector functions. T-cells recognize antigen through the T-cell receptor (TCR) and require a “second signal”, often derived from CD28 interacting with CD80 and CD86 on antigen-presenting cells (17–19). Co-stimulation integrates TCR- and CD28-derived signaling, usually at the SLP76/VAV1/ITK signalosome level. CD28 stimulation increases glycolytic flux by PI3K recruitment and enhanced AKT/mTORC signaling, allowing antigen-stimulated T-cells to cope with increased energetic and biosynthetic needs (18,20). CD28 stimulation promotes optimal and sustained T-cell proliferation (21–24), survival and cytokine production. AKT/mTORC activation enhances glucose transporter 1 (Glut1) expression (20,25), preparing T-cells for increased glucose import and the biomass generation required for clonal expansion (26). CD28 ligation allows T-cells to uptake and utilization glucose for cellular proliferation and effector functions. T-cells without a costimulatory signal fail to increase fuel utilization (27–29).
Considering that co-inhibitory checkpoints are deficient in GCA, we have explored whether CD28-dependent co-stimulation is still intact and whether CD28-mediated signals are pathogenic; as seen in atherosclerotic disease (30). We have exploited a purely antagonistic antibody against CD28 (anti-CD28dAb) and have tested its efficacy in a human artery-SCID mouse chimeric model in which vasculitis is induced in human arteries in immune-reconstituted NSG mice. CD4 T-cells in vasculitic lesions were high expressers of Glut1 and glycolytic enzymes and were explicitly sensitive to CD28 blockade. Blocking CD28 suppressed T-cell proliferation, cytokine production, and most importantly, the induction of TRM, required to repopulate the vasculitic lesions. Interfering with CD28 signaling prevented vessel wall neoangiogenesis and intimal hyperplasia, identifying T-cell co-stimulation as a disease-driving checkpoint in medium and large vessel vasculitis.
Methods and Materials
Patient and tissue samples
Peripheral blood was collected from patients with a tissue diagnosis of GCA and active disease. Age and sex-matched healthy individuals (HC; >60 years of age) were recruited through the Stanford Blood Bank Research Program. A diagnosis of cancer, autoimmune disease and chronic viral infection were exclusion criteria. The patients’ demographic and clinical characteristics are summarized in Online Table 1. Non-inflamed temporal, subclavian and axillary arteries were collected from diagnostic biopsies and from organ donors (early postmortem harvest).
Human Artery-Severe Combined Immunodeficiency Mouse Chimeras
Human artery-mouse chimeras were generated as published using NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (11,12,16).
Statistics
Data are expressed as mean±SEM with individual values shown. GraphPad Prism 5.0 was used for all statistical analyses. Differences were assessed by Student’s t test or paired Wilcoxon signed-rank test as appropriate. Two-tailed p <0.05 was considered statistically significant. To adjust for multiple testing and control the false discovery rate (at a level of 0.05), the Benjamini-Hochberg procedure (BH step-up procedure) was applied.
Methods and materials are available in the online supplementary data.
Results
Blocking CD28-dependent signaling suppresses vasculitis
To examine whether CD28-dependent signals have pathogenic relevance in vasculitis, we treated human artery-NSG chimeras with a purely antagonistic anti-CD28 dAb or control Ab (Figure 1A). Anti-CD28 dAb treatment was profoundly immunosuppressive. Specifically, the density of wall-embedded T-cells fell as visualized by immunohistochemical staining of human CD3+ T-cells (Figure 1B–1C). We quantified the density of lesional T-cells through three methods; CD3+ T-cell enumeration in tissue sections (Figure 1D), TCR transcript quantification in tissue extracts (Figure 1E) and flow cytometry of T-cells isolated out of the artery grafts (Figure 1F–1G). All three methods revealed a reduction of vessel-wall infiltrating T-cells by 50-70% after inhibiting CD28 signalling.
Figure 1. Blocking CD28-dependent signaling suppresses vasculitis.

Vasculitis was induced in human arteries engrafted into NSG mice that were immuno-reconstituted with PBMCs from GCA patients. Chimeric mice were treated anti-CD28 dAb or control Ab (5mg/kg, 3x/week). Explanted arteries were processed for histology or tissue transcriptome analysis. (A) Treatment protocol. (B) H&E-stained arterial cross sections (original magnification: 200×). (C-D) Density of wall-infiltrating T-cells measured by immunolabeling of CD3+ T-cells. Representative images (C, original magnification: 200×) and enumeration of tissue-residing CD3+ T-cells in 8 paired arteries (paired Wilcoxon test). (E) Tissue-infiltrating T-cells quantified through TCR transcripts. Data from 8 paired arteries (paired Wilcoxon test). (F-G) Flow cytometry of wall-infiltrating T-cells in digested arteries. Representative dot blots (gated on live cells) and data from 5 arteries (paired t test). (H-I) Tissue transcriptome analysis in arteries by RT-PCR (paired Wilcoxon test). All data are mean ± SEM. Comparisons of T-bet, BCL-6, IFN-γ and IL-21 are statistically significant at the 0.05 level using Hochberg’s step-down adjustment for multiple comparisons. **p<0.01, ns: not significant. HPF: high-power field. BCL-6: B-cell lymphoma 6 protein; IFN: Interferon; IL: Interleukin; RT-PCR: Reverse transcription polymerase chain reaction; TCR: T-cell receptor; T-bet: T-box transcription factor.
We questioned whether disease-relevant T-cell effector cytokines were sensitive to CD28 blockade. Tissue transcriptome analysis yielded treatment-induced reduction of IFN-γ and IL-21 transcripts, but similar amounts of IL-17A mRNA in anti-CD28 and control-treated arteries (Figure 1H). Matching lineage-determining transcription factors displayed a similar pattern (Figure 1I). T-bet and BCL-6 (expressed in Th1 and Tfh cells, respectively) were high in control-treated tissues and suppressed after antibody injection. RORC, the marker transcription factor for Th17 cells, appeared unaffected by treatment. These data identified CD28-dependent signals as critical factors in determining the function of lesional T-cells.
CD28 signaling controls AKT-mTORC pathway activation, T-cell expansion and T-cell differentiation
In an effort to understand how T-cell biology in vasculitis is shaped by triggering CD28, we probed several functional domains of T-cell activation and function in vitro. CD28 surface expression was similar in healthy and patient-derived T-cells (Online Figure 1). First, we tested whether anti-CD28 dAbs interfered with AKT and mTOR pathway activation in CD4 T-cells. During 30 min of stimulation, patient-derived CD4 T-cells accumulated significantly higher amounts of phosphorylated AKT (p-AKT) and phosphorylated S6 (p-S6) than controls (Figure 2A–2B, Online Figure 2), indicative of more robust signal transmission in the AKT/mTOR pathway. Both, AKT and mTOR signaling, were CD28 dependent. In the presence of 1 ug/ml anti-CD28 dAb, p-AKT and p-S6 concentrations were significantly reduced.
Figure 2.
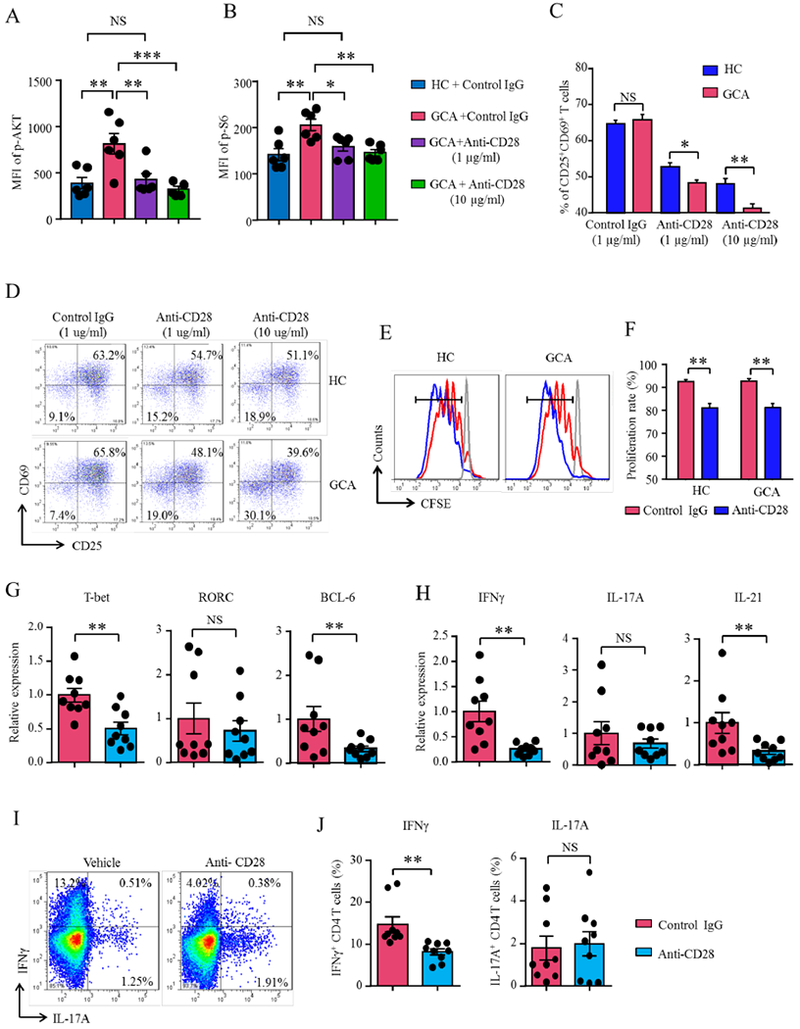
CD28 signaling controls AKT-mTORC pathway activation, T-cell expansion and T-cell differentiation. (A-B) GCA and control (HC) PBMCs were stimulated for 30 min in the presence of anti-CD28 dAb or control antibody. AKT and mTORC1 pathway signaling determined by phospho-flow for p-AKT and p-S6 on gated CD4 T-cells. Mean fluorescence intensity (MFIs) from 6 experiments (t test). (C-J) Patient-derived and control CD4 T-cells were stimulated in the presence of anti-CD28 or control Ab for 72 h. (C-F) Flow cytometric measurement of CD4 T-cell proliferation through CD4+CD25+ and CD4+CD69+ T-cell frequencies (C-D) or by CFSE dilution (E-F). (G-H) GCA CD4+ T-cells assessed through the induction of lineage-determining transcription factors (T-bet, RORγt, BCL-6) and T-cell effector cytokines (IFN-γ, IL-17A, IL-21). Gene expression of transcription factors and effector cytokines quantified by RT-PCR. (I-J) Intracellular cytokine stains. Representative dot blots and frequencies of IFN-γ- and IL-17A-producing CD4+ T-cells. Mean ± SEM are indicated. Paired Wilcoxon test. Comparisons of T-bet, BCL-6, IFN-γ and IL-21 are statistically significant at the 0.05 level using Hochberg’s step-down adjustment. *p<0.05, **p<0.01; ns: not significant. p-AKT: phosphorylated Protein Kinase B; BCL-6: B-cell lymphoma 6 protein; IFN: Interferon; IL: Interleukin; mTORC: Mammalian target of rapamycin; RORγ: Retinoic acid-related orphan receptor gamma, p-S6: phosphorylated Ribosomal protein S6; T-bet: T-box transcription factor.
Disruption of AKT-mTORC1 signaling affected multiple functional domains (31). Activation and proliferation of healthy and GCA CD4 T-cells were assessed in the presence of anti-CD28 dAb and control Ab. 60-70% of T-cells in the control cultures upregulated the activation markers CD25 and CD69. Increasing doses of anti-CD28 dAb reduced the frequencies of CD4+CD25+CD69+ T-cells to 40-50%. CD28 blockade was more effective in patient-derived T-cells, suggesting higher dependence on CD28 co-stimulation (Figure 2C–2D). CFSE staining confirmed suppression of T-cell proliferation that after antibody treatment (Figure 2E–2F); placing CD28-derived signals at the top of the T-cell activation cascade.
In T-cell differentiation assays, we monitored induction of lineage-determining transcription factors (T-bet, RORC, BCL-6), induction of IFN-γ, IL-17A and IL-21 transcripts and accumulation of intracellular cytokine stores containing IFN-γ or IL-17 (Figure 2G–2H). T-bet and BCL-6 induction required CD28 signaling (Figure 2G), but RORC was CD28-independent. In parallel, IFN-γ and IL-21 mRNA production declined with anti-CD28 dAb treatment, whereas IL-17A was un-affected (Figure 2H). Flow cytometry for intracellular cytokines demonstrated dependence of IFN-γ production and independence of IL-17A production from triggering of the CD28 receptor (Figure 2I–2J). These data were confirmed in T-cells from healthy individuals (Online Figure 3).
These findings supported a direct role of the CD28 co-stimulation pathway in AKT and mTORC activation and implicated this single receptor in controlling T-cell clonal expansion and lineage differentiation.
CD4 T-cells rely on CD28 co-stimulation to mobilize the glycolytic machinery
To understand how CD28 triggering interferes with T-cell function, we explored whether T-cells deprived of CD28 signalling were metabolically fit. CD4 T-cells collected from GCA patients were stimulated in the presence of anti-CD28 dAb or control Ab and metabolic fitness was assessed by the induction of the transcription factor HIF1α, the glucose transporter Glut1 and the glycolytic enzymes, HK2, PFK1, GAPDH and LDH. Cellular proliferation and differentiation imposes high energetic requirements which are fulfilled by glucose import and breakdown (32). The enzymes were chosen because of their strategic positioning in glycolysis and in the pyruvate-to-lactate transition (Figure 3A). Anti-CD28 dAb significantly lowered HIF1α and Glut1 transcripts (Figure 3B–3C). PFK transcripts were unaffected by blocking CD28, but all other glycolytic enzymes tested were downregulated by the CD28 antagonistic antibody (Figure 3D–3G). We confirmed the inhibitory effect on Glut1 and GAPDH by flow cytometry experiments (Figure 3H, 3K). We applied Seahorse analysis to quantify CD28’s impact on glycolytic activity (measured as extracellular acidification rate, ECAR) and mitochondrial activity (measured as oxygen consumption rate, OCR), (Figure 3L–3M). Lactate production, a correlate of glucose breakdown, was double as high in the control samples as in anti-CD28 dAb-treated cells. Similarly, blocking CD28 signaling diminished mitochondrial oxygen consumption, both under basal conditions as well as after uncoupling.
Figure 3. CD28-dependent signaling regulates glycolytic and mitochondrial activity in CD4 T-cells.

(A) Scheme of the glycolytic pathway. (B-G) GCA CD4+ T-cells were stimulated in the presence of anti-CD28 dAb or control Ab for 72h. Induction of transcripts for Glut1, HIF-1α and four glycolytic enzymes measured by RT-PCR (paired Wilcoxon test). (H-K) Glut1 and GAPDH protein expression measured by flow cytometry. Representative histograms and mean fluorescence intensity (MFIs) from 5 experiments (t test). (L-M) CD4 T-cells were stimulated for 72 h in the presence of anti-CD28 dAb or control Ab and analyzed by Seahorse Bioscience XF96 analyzer. Glycolytic activity was quantified through extracellular acidification rates (ECAR, L) and mitochondrial function was assessed by oxygen consumption rates (OCR, M). Mean±SEM compared by t test. *p<0.05, **p<0.01; ns: not significant. 1,3BPG: 1,3-bisphosphoglyceric acid; F1,6BP: Fructose1,6-bisphosphate; FCCP: Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; Glut1: Glucose transporter 1; G6P: glucose-6-phosphate; HIF-1α: Hypoxia-inducible factor 1-alpha; HK2: hexokinase-2; LDH: Lactate dehydrogenase; PFK1: Phosphofructokinase-1; TCA: Tricarboxylic acid.
These data identified the CD28-AKT-mTOR-HIF-1α pathway and the downstream bioenergetic machinery as a target of co-stimulatory signaling, embedding bioenergetic adaptation into the T-cell activation program.
CD28 signaling controls glycolytic activity of vasculitic T-cells
To investigate the metabolic status of T-cells in the vasculitic lesions, we induced vascular inflammation in the chimeras, collected the artery grafts, the spleens and the circulating blood and employed multi-color cytometry to determine expression of the glucose transporter Glut1 and the key glycolytic enzyme GAPDH (Figure 4A–4C). In vessel wall-infiltrating T-cells, amounts of Glut1 were 2.2-fold higher than in splenic and circulating T-cells. The difference between wall-embedded and extra-arterial T-cells was even higher for GAPDH expression (3.5-fold higher in wall-residing T-cells).
Figure 4. CD28 signaling regulates glucose utilization in tissue-residing CD4 T-cells.
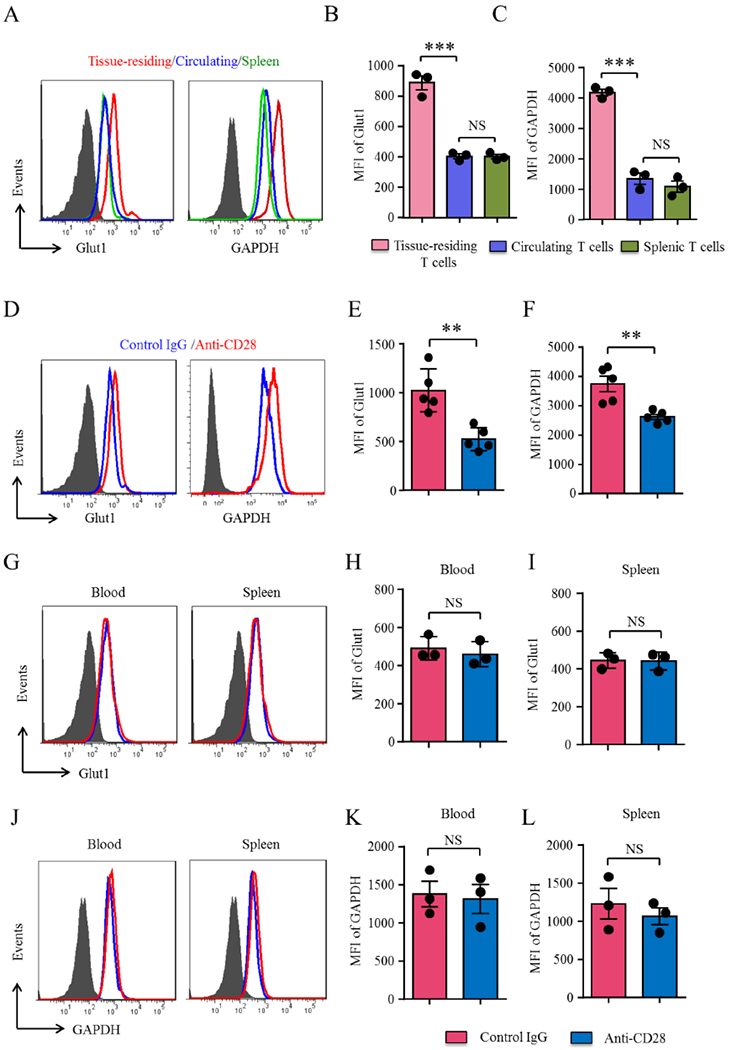
(A-C) Vasculitis was induced as in Figure 1. Spleens and arteries were harvested and digested. Protein expression of the glucose transporter Glut1 and the glycolytic enzyme GAPDH was analyzed by flow cytometry in splenic, peripheral blood and artery-infiltrating CD4+ T-cells. Representative histograms and mean fluorescence intensities (MFIs) from 3 experiments (t test). (D-F) Human artery-NSG chimeras were treated with anti-CD28 dAb or control Ab as in Figure 1. Cells were isolated from the explanted arteries, spleens and blood and Glut1 and GAPDH expression on CD4 T-cells was determined by multi-color flow cytometry. Representative histograms and MFIs from 5 experiments in E and F and 3 experiments in G to L (t test). All FACS plots were gated on CD45+CD4+ cells. Data are mean±SEM. **p<0.01, ***p<0.001. ns: not significant. GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; Glut1: Glucose transporter 1.
To explore whether CD28 signaling was relevant in adapting to higher energy demands in tissue-infiltrating T-cells, we treated with anti-CD28 dAb or control Ab and analyzed the explanted inflamed arteries, the blood and the spleens. Anti-CD28 dAb treatment affected both glucose uptake and glucose utilization in the vasculitic lesions (Figure 4D–4F). Blocking CD28 resulted in a significant reduction of Glut1 (47.1 % decline) and GAPDH expression (28.7% decline) in wall-infiltrating T-cells. This effect was selective for lesional T-cells; neither spleen-residing nor circulating T-cells were Glut1hi or GAPDHhi (Figure 4G–4L). Thus, glucose utilization and energy production in vasculitic T-cells depends on CD28 signaling.
CD28 provides survival signals for tissue-resident memory T cells
T-cells in the vasculitic lesions need to be replenished, either achieved by influx of fresh cells or by in-situ proliferation of tissue-residing T-cells. TRM cells are a specialized population characterized by tissue residence and fast and intense cytokine production (33). They express the CD103 marker and serve as a source of in-situ T-cells replenishment (16,34,35). To separate T-cells that enter the vasculitic lesions from outside from those residing in the tissue lesion, we have developed an experimental system in which vasculitis is induced in the chimera, inflamed arteries is then explanted and transengrafted into an “empty” mouse (Figure 5A). Treatment of the recipient mouse with anti-CD28 dAb or control Ab allowed assessment of signals relevant in T-cell survival and proliferation. Control-treated grafts contained 10,000 human T-cells/100 mg tissue and numbers declined to 4,060 T-cells/100 mg tissue after blocking CD28 (Figure 5B). A small population of human T-cells leave the inflammatory lesions, recirculate and re-populate the spleen (Online Figure 4). In anti-CD28 dAb treated chimeras recirculating T-cells were explicitly infrequent. Flow cytometric measurement of Ki67+ T-cell frequencies showed proliferation rates at 35-40% in control grafts and 19% in anti-CD28 dAb-treated arteries (Figure 5C–5D).
Figure 5. Tissue-resident memory T cells depend on CD28 signaling.
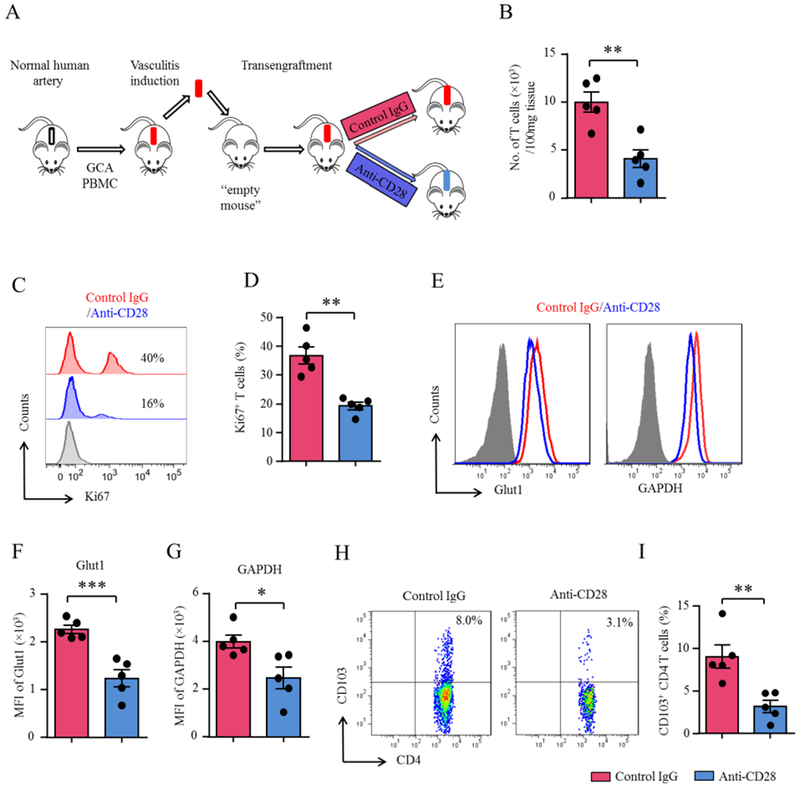
(A) Scheme of the mouse experiments. (B-I) Vasculitis was induced in human arteries as in Figure 1. To analyze cells contained within the vasculitic lesions, inflamed arteries were trans-engrafted into an “empty” NSG mouse, which were treated with anti-CD28 dAb or control Ab (5mg/kg, 4x/week). Single cell suspensions were prepared from explanted arteries. (B) Enumeration of tissue-residing CD4+ T-cells (t test). (C-D) Quantification of proliferating T-cells in the vasculitic lesions by Ki-67 flow cytometry. Representative histograms and percentages of Ki-67+ CD4+ T-cells (t test). (E-G) FACS analysis of Glut1 and GAPDH expression on tissue-residing CD4+ T-cells. Representative histograms and fluorescence intensity (MFIs) from 5 experiments (t test). (H-I) CD103+ tissue-resident memory CD4 T-cells analyzed by multi-color flow cytometry. Representative FACS plots and results from 5 experiments (t test). All FACS plots were gated on CD45+CD4+ cells. All data are mean ± SEM. *p<0.05, **p<0.01, ***p<0.001. GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; Glut1: Glucose transporter 1.
To investigate whether glycolytic activity in lesional T-cells was sensitive to CD28 inhibition, we determined Glut1 and GAPDH expression in wall-embedded T-cells (Figure 5E–5G). Interrupting CD28 signaling reduced expression of the glucose transport and the glycolytic enzyme by 30-50%.
To confirm that CD28 co-stimulation was needed to secure TRM cell survival, we quantified CD4+CD103+ T-cells amongst in wall-residing populations (Figure 5H–5I). In anti-CD28 dAb-treated mice, CD103+ TRM cell populations was reduced to one third compared to control grafts. Support for the notion that CD4+CD103+ T-cells expansion is CD28-dependent came from in vitro data (Online Figure 5). CD28-blocking antibodies strongly suppressed CD4+CD103+ TRM expansion.
These experiments implicated CD28 co-stimulation in securing the survival and the repopulation of T-cells populating the vasculitic lesions.
Intramural neoangiogenesis and intimal hyperplasia depend on CD28 signaling
Feared clinical consequences of GCA are related to vessel wall remodeling leading to luminal stenosis/occlusion and tissue ischemia (36,37). To examine whether CD28 co-stimulation participated in wall remodeling, we quantified intramural neoangiogenesis and intimal hyperplasia in explanted human arteries from anti-CD28 dAb and control Ab treated chimeras. Newly formed microvessels were identified by dual-color immunostaining of the endothelial marker vWF and a-SMA (Figure 6A–6C). Intimal layer thickness was measured as the lumen-to-lamina elastica interna distance (Figure 6B–6D). Arteries treated with control Ab contained abundant microvascular lumina and the intimal layer was expanded to 600 um. Blocking CD28 was highly effective in preventing neoangiogenesis, reducing the density of microvessels by 66% (Figure 6C). In parallel, CD28 blockade prevented intimal hyperplasia, with a 4.6-fold difference in intimal thickness amongst the anti-CD28 and control treated grafts (Figure 6D).
Figure 6. Vascular remodeling requires CD28-dependent T-cell co-stimulation.
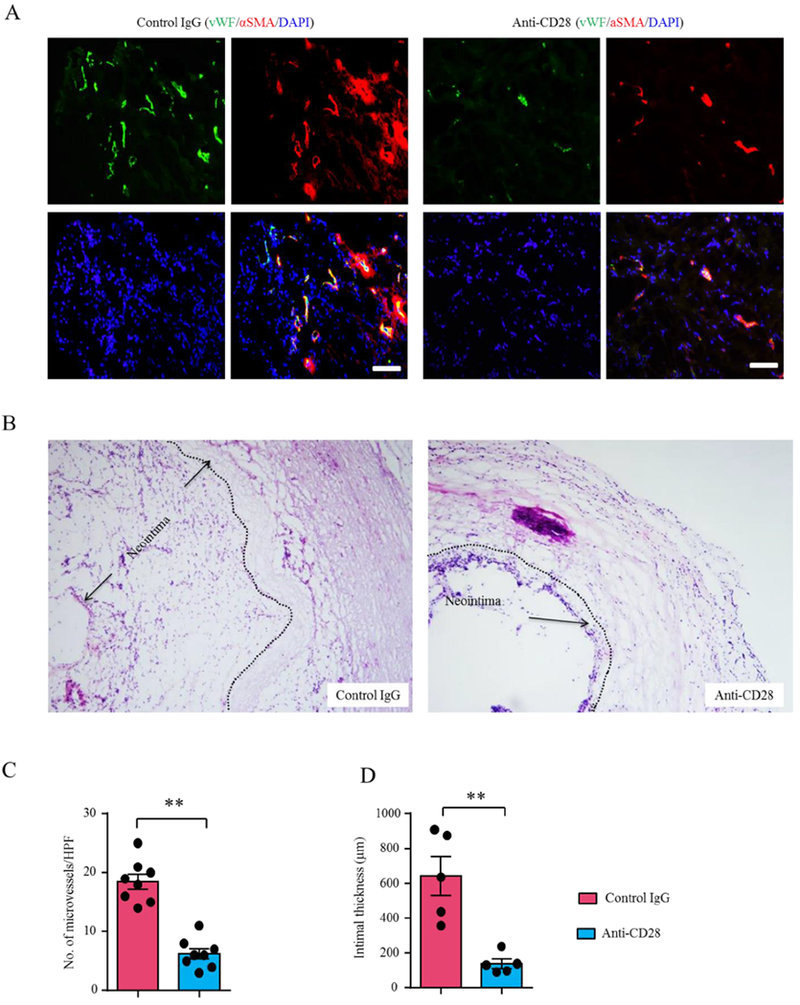
Vasculitis was induced in human arteries as in Fig 1. Mice carrying inflamed human arteries were assigned to the anti-CD28 dAb (5mg/kg) or control arm and treated for 2 weeks. Arteries were explanted for tissue analysis. (A) Intramural microangiogenesis determined by vWF (green) and αSMA (red) co-staining. Representative images. Scale bar: 100μm. (B) Intimal layer thickness measured in H&E-stained arterial cross-sections (lumen-to-lamina elastica interna distance). (Original magnification: 100×). (C) Enumeration of adventitial microvessels. Each dot represents one arterial graft (paired Wilcoxon test). (D) Intimal thickness measured in 5 experiments (paired Wilcoxon test). Mean ± SEM is indicated. *p<0.05, ***p<0.001. HPF: high-power field. αSMA: alpha smooth muscle actin; vWF: von Willebrand Factor.
Together these data provided a mechanistic link between T-cell co-stimulation and tissue-reparative processes, placing CD28-mediated signals at the top of the pathogenic cascade.
Discussion
Co-stimulatory and co-inhibitory signals shape protective and pathogenic T-cell responses, identifying molecules that amplify or suppress T-cell activation as superb therapeutic targets to modulate adaptive immunity (38). T-cells are key drivers in the immunopathogenesis of vasculitis, where they populate the vascular lesions (39). In GCA, CD4 T-cells and macrophages represent the key elements of the granulomatous infiltrates (40). Notably, GCA patients have a defect in the PD-1/PD-L1 co-inhibitory pathway, enabling unopposed T-cell activation inside and outside of the vasculitic lesions (12). In the absence of co-inhibition, costimulation, which we show here to promote multiple pathogenic domains of vasculitis, becomes even more important. Signaling through the co-stimulatory receptor CD28 proved necessary to induce and maintain glucose uptake, glycolytic breakdown and mitochondrial oxygen consumption in lesional T-cells (20,27).
By controlling glucose and oxygen utilization, CD28 functioned as a metabolic checkpoint determining life, death and functionality of T-cells (Central Illustration). Metabolic control translated into three pathogenic domains: proliferative expansion of CD4 T-cells, cytokine production of CD4 T-cells and survival of a specialized CD4 T-cell subset, CD4+CD103+ T-cells that act as tissue-resident memory cells and determine the lesion’s longevity. Lesional T-cells are the main driver of tissue inflammation and undergo proliferation and activation in the absence of circulating T-cells. Once differentiated into CD103+ TRM, tissue-residing T-cells survive in the absence of local antigen presentation (41). Such long-lived TRM cells mediate sustained inflammation and strong immune responses (33,35). Reducing TRM in the vasculitic lesions may lead to better control of disease activity and may change the course of GCA. Ultimately, pathogenic T-cells controlled the ischemia-inducing remodeling of the vessel wall, including intimal hyperplasia and microangiogenesis.
Central Illustration: CD28 signaling controls metabolic fitness of pathogenic T-cells in medium and large vasculitis.

CD28 ligation activates AKT-mTOR signaling, enhancing glucose utilization in T-cells. Glycolytic activity promotes vasculitis-inducing effector T-cells and enables differentiation of tissue-resident memory T-cells, which replenish the tissue infiltrates. Downstream effects include chronic-smoldering inflammation, neoangiogenesis, and lumen-compromising intimal hyperplasia.
Experiments in this project benefitted from two enabling resources: strictly antagonistic antibodies specific for CD28 and an animal model for human large vessel vasculitis. By treating chimeric mice carrying inflamed human arteries with anti-CD28 antibodies we collected data with immediate relevance for medium and large vessel vasculitis, specifically giant cell arteritis that affects the aorta and its branch vessels. Human T-cells entrapped in the inflamed arteries were Glut-1 high-expressors, identifying them as glucose-dependent. By regulating Glut1 expression, CD28 signaling ultimately controlled the longevity and functional competence of such T-cells. Notably, Glut1 expression distinguished circulating, spleen-residing and inflamed tissue-residing T-cells, possibly reflecting antigen recognition in the artery. Lesional T-cells produced an array of T-cell effector cytokines, including IFN-γ and IL-21, compatible with a lack of PD-1-mediated co-inhibition. IL-17 and the lineage-determining transcription factor RORC were independent of CD28 signaling. Given the steroid responsiveness of TH17 cells in the vasculitic lesions (8), combination therapy of corticosteroids plus CD28 blockade may be the most beneficial for GCA patients.
As previously shown, the vessel wall remodeling process, such as growth of new microvessels and intimal hyperplasia, is downstream of T-cell activation (12). CD28 blockade successfully prevented this disease complication, which is critical for the development of clinical complications of GCA. Whether neoangiogenesis and outgrowth of myofibroblasts results from direct T-cell induced signals or involves T-cell activated macrophages remains to be determined.
Study limitations
The study used a humanized mouse model, in which patient-derived PBMC cause vasculitis in a non-autologous artery to explore the role of CD28 signaling in medium and large vessel vasculitis. While the model enables mechanistic and therapeutic studies in vivo, we cannot assess possible systemic side effects of anti-CD28 therapy.
Conclusions
The study has consequences for the pathogenic understanding of medium and large vessel vasculitis and opens new therapeutic opportunities. The data reemphasize that the vascular component of GCA is ultimately an abnormality in adaptive immunity and needs to be targeted by modulating T-cell function. Vasculitic lesions require growth and expansion of T-cells in the vessel wall and tissue-residence of memory T-cell populations that promote and sustain the granulomatous infiltrates. Suppressing innate cytokines is unlikely to be sufficient to inhibit metabolically hyperactive T-cells in the vasculitic lesions. Mild therapeutic effects have been described for the CD28 signaling antagonist CTLA4Ig in GCA (42) and previous studies have reported defects in co-inhibitory pathways. Data reported here demonstrate the intactness and necessity of co-stimulation, linking pathogenic T-cell immunity to a disbalance of signals that control adaptive immune responses. Therapeutics blocking pro-inflammatory cytokines are unlikely to overcome hyperactive T-cells but limiting the metabolic fitness of lesional T-cells has the potential to disrupt critical pathogenic processes. Tissue-entrapped T-cells appear particularly dependent on glucose utilization, rendering them susceptible to metabolic interference. Abrogating CD28 signaling emerges as a novel means to target the metabolic fitness of disease-inducing T-cells and provides novel therapeutic opportunities for disease-modifying strategies in medium and large vessel vasculitis.
Supplementary Material
CLINICAL PERSPECTIVES.
Competency in Medical Knowledge
Signaling through the CD28 receptor controls glucose utilization and regulates multiple functions of T-cells. Blocking CD28 inhibits the activation, expansion and differentiation of tissue-resident memory T-cells.
Translational Outlook
Therapeutic targeting of CD28 suppresses vasculitis in medium-sized and large vessels.
Acknowledgments
Funding: This work was supported by the National Institutes of Health (R01 AR042527, R01 HL117913, R01 AI108906 and P01 HL129941 to CMW and R01 AI108891, R01 AG045779, U19 AI057266, R01 AI129191, and I01 BX001669 to JJG). HZ received fellowship support from the Govenar Discovery Fund.
Disclosures: This work was supported by a sponsored research agreement with Bristol-Myers Squibb. Dr. Nadler is employed by Bristol-Myers Squibb. Drs. Zhang, Watanabe, Berry, Goronzy and Weyand do not have relationships relevant to the contents of this paper to disclose.
Abbreviations:
- GCA
giant cell arteritis
- PBMC
peripheral blood monocyte
- TRM
tissue-resident memory T cells
- TCR
T-cell receptor
- Glut1
glucose transporter 1
- dAb
domain antibody
- anti-CD28
anti-CD28 antagonist dAb
- MFI
mean fluorescence intensity
- RT-PCR
Reverse transcription polymerase chain reaction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Tweet: Blocking CD28 suppresses metabolic fitness and inflammatory behavior of T cells. Is it a therapeutic alternative in large vessel vasculitis?
References
- 1.Dal Canto AJ, Swanson PE, O’Guin AK, Speck SH, Virgin HW. IFN-gamma action in the media of the great elastic arteries, a novel immunoprivileged site. J Clin Invest 2001;107:R15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tellides G, Pober JS. Inflammatory and immune responses in the arterial media. Circ Res 2015;116:312–22. [DOI] [PubMed] [Google Scholar]
- 3.Roche NE, Fulbright JW, Wagner AD, Hunder GG, Goronzy JJ, Weyand CM. Correlation of interleukin-6 production and disease activity in polymyalgia rheumatica and giant cell arteritis. Arthritis Rheum 1993;36:1286–94. [DOI] [PubMed] [Google Scholar]
- 4.Unizony S, Arias-Urdaneta L, Miloslavsky E et al. Tocilizumab for the treatment of large-vessel vasculitis (giant cell arteritis, Takayasu arteritis) and polymyalgia rheumatica. Arthritis Care Res. 2012;64:1720–9. [DOI] [PubMed] [Google Scholar]
- 5.Piggott K, Biousse V, Newman NJ, Goronzy JJ, Weyand CM. Vascular damage in giant cell arteritis. Autoimmunity 2009;42:596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sneller MC. Granuloma formation, implications for the pathogenesis of vasculitis. Cleve Clin J Med 2002;69 Suppl 2:SII40–3. [DOI] [PubMed] [Google Scholar]
- 7.Maleszewski JJ, Younge BR, Fritzlen JT et al. Clinical and pathological evolution of giant cell arteritis: a prospective study of follow-up temporal artery biopsies in 40 treated patients. Mod Pathol 2017;30:788–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng J, Younge BR, Olshen RA, Goronzy JJ, Weyand CM. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation 2010;121:906–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salvarani C, Cantini F, Boiardi L, Hunder GG. Polymyalgia rheumatica and giant-cell arteritis. N Engl J Med 2002;347:261–71. [DOI] [PubMed] [Google Scholar]
- 10.Salvarani C, Cantini F, Hunder GG. Polymyalgia rheumatica and giant-cell arteritis. Lancet 2008;372:234–45. [DOI] [PubMed] [Google Scholar]
- 11.Watanabe R, Maeda T, Zhang H et al. Matrix Metalloprotease-9 (MMP-9)-Producing Monocytes Enable T Cells to Invade the Vessel Wall and Cause Vasculitis. Circ Res 2018;123:700–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang H, Watanabe R, Berry GJ et al. Immunoinhibitory checkpoint deficiency in medium and large vessel vasculitis. Proc Natl Acad Sci U S A 2017;114:E970–E979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wen Z, Shimojima Y, Shirai T et al. NADPH oxidase deficiency underlies dysfunction of aged CD8+ Tregs. J Clin Invest 2016;126:1953–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wen Z, Shen Y, Berry G et al. The microvascular niche instructs T cells in large vessel vasculitis via the VEGF-Jagged1-Notch pathway. Sci Transl Med 2017;9: eaal3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pryshchep O, Ma-Krupa W, Younge BR, Goronzy JJ, Weyand CM. Vessel-specific Toll-like receptor profiles in human medium and large arteries. Circulation 2008;118:1276–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H, Watanabe R, Berry GJ, Tian L, Goronzy JJ, Weyand CM. Inhibition of JAK-STAT Signaling Suppresses Pathogenic Immune Responses in Medium and Large Vessel Vasculitis. Circulation 2018;137:1934–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 2013;13:227–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol 2001;1:220–8. [DOI] [PubMed] [Google Scholar]
- 19.Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1994;1:793–801. [DOI] [PubMed] [Google Scholar]
- 20.Frauwirth KA, Riley JL, Harris MH et al. The CD28 signaling pathway regulates glucose metabolism. Immunity 2002;16:769–77. [DOI] [PubMed] [Google Scholar]
- 21.Lucas PJ, Negishi I, Nakayama K, Fields LE, Loh DY. Naive CD28-deficient T cells can initiate but not sustain an in vitro antigen-specific immune response. J Immunol 1995;154:5757–68. [PubMed] [Google Scholar]
- 22.Shahinian A, Pfeffer K, Lee KP et al. Differential T cell costimulatory requirements in CD28-deficient mice. Science 1993;261:609–12. [DOI] [PubMed] [Google Scholar]
- 23.Green JM, Noel PJ, Sperling AI et al. Absence of B7-dependent responses in CD28-deficient mice. Immunity 1994;1:501–8. [DOI] [PubMed] [Google Scholar]
- 24.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol 1996;14:233–58. [DOI] [PubMed] [Google Scholar]
- 25.Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell 2007;18:1437–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerriets VA, Rathmell JC. Metabolic pathways in T cell fate and function. Trends Immunol 2012;33:168–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol 2005;5:844–52. [DOI] [PubMed] [Google Scholar]
- 28.Frauwirth KA, Thompson CB. Regulation of T lymphocyte metabolism. J Immunol 2004;172:4661–5. [DOI] [PubMed] [Google Scholar]
- 29.Jones RG, Thompson CB. Revving the engine: signal transduction fuels T cell activation. Immunity 2007;27:173–8. [DOI] [PubMed] [Google Scholar]
- 30.Lichtman AH. T cell costimulatory and coinhibitory pathways in vascular inflammatory diseases. Front Physiol 2012;3:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2002;2:489–501. [DOI] [PubMed] [Google Scholar]
- 32.Maciver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL, Rathmell JC. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J Leukoc Biol 2008;84:949–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol 2009;10:524–30. [DOI] [PubMed] [Google Scholar]
- 34.Amsen D, van Gisbergen K, Hombrink P, van Lier RAW. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat Immunol 2018;19:538–546. [DOI] [PubMed] [Google Scholar]
- 35.Park CO, Kupper TS. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med 2015;21:688–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaiser M, Weyand CM, Bjornsson J, Goronzy JJ. Platelet-derived growth factor, intimal hyperplasia, and ischemic complications in giant cell arteritis. Arthritis Rheum 1998;41:623–33. [DOI] [PubMed] [Google Scholar]
- 37.Makkuni D, Bharadwaj A, Wolfe K, Payne S, Hutchings A, Dasgupta B. Is intimal hyperplasia a marker of neuro-ophthalmic complications of giant cell arteritis? Rheumatology (Oxford) 2008;47:488–90. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Q, Vignali DA. Co-stimulatory and Co-inhibitory Pathways in Autoimmunity. Immunity 2016;44:1034–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weyand CM, Goronzy JJ. Medium- and large-vessel vasculitis. N Engl J Med 2003;349:160–9. [DOI] [PubMed] [Google Scholar]
- 40.Hilhorst M, Shirai T, Berry G, Goronzy JJ, Weyand CM. T cell-macrophage interactions and granuloma formation in vasculitis. Front Immunol 2014;5:432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mackay LK, Stock AT, Ma JZ et al. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci U S A 2012;109:7037–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Langford CA, Cuthbertson D, Ytterberg SR et al. A Randomized, Double-Blind Trial of Abatacept (CTLA-4Ig) for the Treatment of Giant Cell Arteritis. Arthritis Rheumatol 2017;69:837–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.