

Abstract
Purpose of Review
Functional decline of hematopoiesis that occurs in the elderly, or in patients who receive therapies that trigger cellular senescence effects, results in a progressive reduction in the immune response and an increased incidence of myeloid malignancy. Intracellular signals in hematopoietic stem cells and progenitors (HSC/P) mediate systemic, microenvironment, and cell-intrinsic effector aging signals that induce their decline. This review intends to summarize and critically review our advances in the understanding of the intracellular signaling pathways responsible for HSC decline during aging and opportunities for intervention.
Recent Findings
For a long time, aging of HSC has been thought to be an irreversible process imprinted in stem cells due to the cell intrinsic nature of aging. However, recent murine models and human correlative studies provide evidence that aging is associated with molecular signaling pathways, including oxidative stress, metabolic dysfunction, loss of polarity and an altered epigenome. These signaling pathways provide potential targets for prevention or reversal of age-related changes.
Summary
Here we review our current understanding of the signalling pathways that are differentially activated or repressed during HSC/P aging, focusing on the oxidative, metabolic, biochemical and structural consequences downstream, and cell-intrinsic, systemic, and environmental influences.
Keywords: Hematopoietic Stem Cells and Progenitors, Aging, metabolism, mitochondria, reactive oxygen species, polarity, epigenomics
Introduction
Organismal aging is an inevitable physiological process that leads to the dysfunction of various tissues resulting in increased morbidity and mortality. Adult somatic stem cells play a central role in the homeostasis of tissues where high cellular turnover must be maintained for functionality, such as the epidermis, gut epithelium and blood. These stem cells, along with germinal stem cells, are susceptible to a time-dependent functional decline [1]. Hematopoietic stem cells (HSC) represent a rare and highly quiescent population of cells that reside in the adult bone marrow (BM) cavity and are responsible for the lifelong production of blood. HSC divide very infrequently, an estimated four to five traceable divisions in the cells lifespan before losing repopulation abilities, and are defined by two functional properties: maintenance of self-renewal and multilineage differentiating potential [2*, 3, 4]. In the hematopoietic system, aging reduces stem cell function by hindering mobilization, homing, engraftment and lineage commitment, manifesting as immunosenescence and an increased propensity for myeloid malignancies [5–9]. Such phenotypes displayed in aged HSC are the result of a combination of cell intrinsic, systemic and microenvironmental influences (Figure 1). The prospect of either preventing stem cell aging or restoring normal stem cell function and tissue homeostasis is of considerable interest for future clinical intervention. This review intends to discuss critically some of the most novel approaches that may potentially ameliorate or even revert the aging-associated hallmarks of HSC. Before we review our current understanding of the mechanisms involved in HSC damage during aging, we will discuss the consequences of aging in HSC.
Figure 1. Phenotypic and functional changes associated with aging of hematopoietic stem cells (HSC).
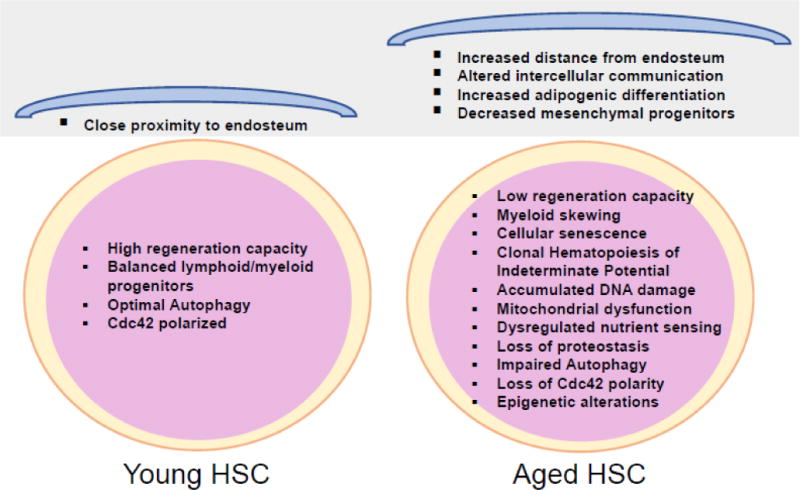
HSC and BM microenvironmental niche undergo age-related changes due to oxidative stress, genomic instability and epigenetic shift during the aging process. In young HSC compartment, Cdc42 is polarized with tubulin and the lymphoid and myeloid differentiation as well as basal autophagy are homeostatically maintained, enabling a balanced production of blood progeny. Aged HSC however exhibit myeloid skewing, loss of Cdc42 polarity, impaired autophagy and replicative senescence. Moreover, aging of the hematopoietic microenvironment including adipogenesis and loss of mesenchymal progenitors results enhanced HSC mobilization and decreased HSC homing and engraftment. In addition, young HSC localize in close proximity to the BM endosteum, while aged HSC reside further away from the endosteal stem cell niche.
Consequences of HSC/P aging
1. HSC depletion, senescence and myeloid lineage commitment bias
The hallmarks of aging bone marrow are increased frequency of immunophenotypically identifiable HSC with loss of hematopoietic activity in normal and especially stress conditions, and a significant bias to myeloid differentiation [10], which all depend on genetic traits [11, 12]. These changes are believed to happen secondary to an intrinsic effect of cellular senescence. Cellular senescence represents an irreversible state of growth arrest characterized by the accumulation of cell cycle regulators, including p53, p21, p16Ink4a and p19Arf, while the loss of these cell cycle checkpoints are the most frequent cytogenetic events in cancers [13]. Replicative senescence results from the combination of DNA damage response and telomere shortening, which leads to the activation of p53, p19Arf, p16Ink4a and BCL2 family members and is associated with stem cell exhaustion in older animals [14–16].
Mounting evidence indicates that increased expression of p16Ink4a in aged HSC is linked to compromised self-renewal and repopulating activity as well as myeloid skewing through differentiation (Figure 2) [17–19*]. Furthermore, increased senescence and loss of HSC function in mice deficient in the polycomb repression complex protein Bmi-1 has been shown to be associated with p16Ink4a and p19Arf overexpression [20], and rejuvenation of cell-cycle activity and engraftment capacity has been observed in p16Ink4a deficient aged HSC [17]. Nevertheless, this rejuvenation was associated with an increased incidence of cancer [13]. In contrast, other investigators have failed to detect a significant role for p16Ink4a in aged HSC and proposed that it is an inconsequential intrinsic regulator of steady-state HSC aging [21]. Furthermore, p16Ink4a-positive senescent cells accumulate in various tissues and organs over time, and have a role in aging or age-associated diseases, likely through the depletion of HSC/P and the adverse actions of the senescence-associated secretory phenotype (SASP), including inflammatory cytokines, matrix metalloproteinases and growth factors [19*, 22*]. Selective elimination of senescent cells in INK-ATTAC transgenic mice, which express FK-506-binding-protein-caspase 8 fusion protein (FKBP-Casp8) under control of the Ink4a promoter, extended the lifespan of the mice, with improved function of multiple tissues, as well as delayed tumorigenesis [19]. Interestingly, depleting senescent cells using the senolytic drug ABT263 (a specific inhibitor of BCL-2 and BCL-xL) significantly improved the function of aged HSC in serial transplant experiments (Table 1) [22*]. Conversely, detection of p16Ink4a/SAβG-positive macrophages in old mice suggest that the antiaging effects following eradication of p16Ink4a positive senescent cells also depends on the clearance of non-senescent p16Ink4a/SAβG-positive macrophages [23, 24].
Figure 2. Cell intrinsic and extrinsic mechanisms regulating hematopoietic stem cells aging.
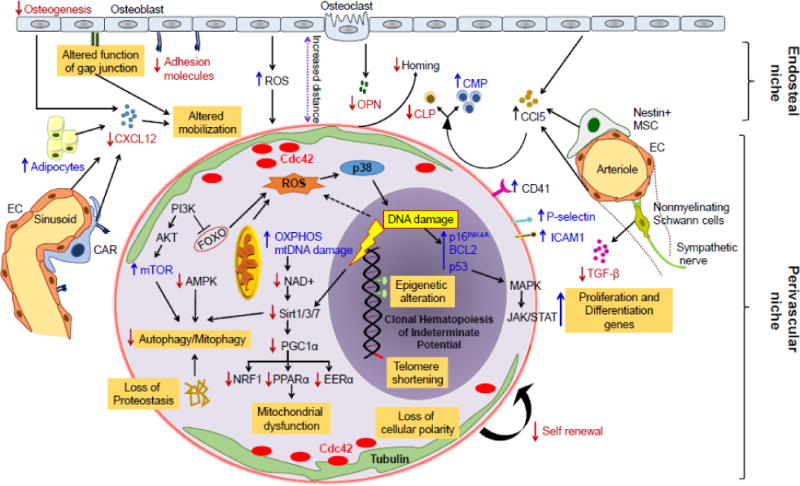
Aging negatively affects HSC function through both intrinsic and hematopoietic microenvironment mediated mechanisms. Intrinsic effects include increased ROS levels leading to DNA damage and replicative senescence, epigenomic alterations with H4K16ac, H4K20me3 and H3K4me3 and telomere attrition. Increased ROS in aged HSC consequently activates p38-MAPK, followed by induction of cell cycle regulators p53, BCL2 and p16Ink4a, resulting in an age-associated decline in HSC function and increased senescence. Moreover, aging associated amplification of nutrient sensing PI3K/Akt/mTOR pathway and the inhibition of downstream FOXO (1, 3 and 4) transcription factors, as well as metabolic alterations comprising decrement in NAD+ or Sirtuins (Sirt1/3/7) and consequent alleviation of transcription factors Nrf1, ERRα, and PPARα are involved in loss of proteostasis, autophagy/mitophagy and mitochondrial biogenesis in aged HSC. Low calorie intake however activate AMPK pathway, and promotes HSC quiescence and self-renewal. Increase in Cdc42 activity and subsequently loss of Cdc42 and tubulin polarization in aged HSC associated with defective HSC self-renewal activity. Aging associated changes in hematopoietic microenvironment including high oxidative stress, decreased adhesion to bone marrow stromal cells, increased adipogenic differentiation and decreased mesenchymal progenitors also linked with reduced HSC self-renewal and differentiation, homing as well as HSC mobilization. Various niche cells including osteoblasts, endothelial cells (EC), Nestin+ mesenchymal stem cells (MSC), CXCL12 abundant retricular (CAR) cells and nonmyelinating Schwann cells have been implicated for their roles in HSC aging. In addition, aging of HSC increased CD41 expression, which modulates HSC long-term repopulation and survival as well as myeloid skewing, while increased levels of surface adhesion molecules P-selectin (CD62P) and intercellular adhesion molecule 1 (ICAM1/CD54) have been associated with enhanced HSC mobility and attenuated engraftment. Red down arrows represent repressive and blue up arrows represents activating signals. Reactive oxygen species (ROS), osteopontin (OPN), common myeloid progenitor (CMP), common lymphoid progenitor (CLP), mitochondrial DNA (mtDNA), oxidative phosphorylation (OXPHOS), Forkhead O (FOXO), nuclear respiratory factor 1 (Nrf1), estrogen-related receptor alpha (ERRα), Peroxisome proliferator-activated receptor alpha (PPARα), CXC-chemokine ligand 12 (CXCL12), CC-chemokine ligand 5 (CCL5).
Table 1.
Interventions that contribute to HSC rejuvenation or prevent HSC aging
| Rejuvenation Approach | Mechanism | Rejuvenation Phenotype | References |
|---|---|---|---|
| Pharmacological interventions | |||
|
| |||
| Nicotinamide riboside | NAD+ | Elevates NAD+ levels through SIRT1 activation, Enhances mitochondrial function and proteostasis in aged HSC | [84] |
| Metformin | AMPK activator | Downregulates mTOR, Improves HSC numbers and competitive repopulation ability | [94] |
| Rapamycin | mTOR inhibitor | Reduces the number of HSC to normal (young) levels, Restores HSC self-renewal upon transplantation, Reduces the frequency of myeloid-biased HSC | [156] |
| Casin | Cdc42; H4K16 acetylation | Inhibits Cdc42 activity, Enhances competitive repopulation ability, Reduces the frequency of myeloid-biased HSC | [116] |
| Senolytic drug | Inhibitor of BCL- 2 and BCL-xL | Depletes senescent HSC from the bone marrow, Attenuates myeloid skewing, Improves reconstitution potential of HSC following serial transplantation | [19, 22] |
| NAC (N-acetyl L- cysteine) | ROS scavenger | Improves the replicative potential of HSC | [69, 65] |
|
Nutrient sensing | |||
| Caloric restriction | IGF-1, mTOR, AMPK, FOXO, Sirtuins | Reduces the frequency of myeloid-biased HSC, Restores youthful HSC self-renewal ability and regenerative capacity upon serial transplantation | [157, 75] |
| Resveratrol | Caloric mimetic | Increases mitochondrial biogenesis of HSC through activation of SIRT1 and PGC1α, Increases the total number of functional HSC | [93] |
|
Genetic modulator | |||
| Satb1 overexpression | Epigenetic modification | Overexpression enriches lymphoid progeny of aged HSC | [132] |
| Sirt3 overexpression | ROS, mitochondrial function | Restores long-term competitive repopulation ability during serial transplantation, Decreases ROS production | [158] |
| Sirt7 overexpression | Mitochondrial function | Overexpression reduces mitochondrial protein folding stress, Rescues myeloid-biased differentiation, Improves reconstitution potential of HSC | [102] |
Several studies highlight the pro-aging function of deregulated p53 activity in human and murine HSC (Figure 2) [25, 14]. p19Arf regulates p53 stability through inactivation of the ubiquitin ligase Mdm2. Constitutively active p53 in mice overexpressing truncated isoform of p53 (DNp53) [26], or the short isoform of p53 (p44) [27], accumulate senescent cells and exhibit accelerated aging and reduced HSC engraftment capability [28]. In contrast, mice with increased but normally regulated levels of Arf and p53, together with reduced Mdm2 activity, are resistant to tumorigenesis, live longer, and have reduced levels of age-associated replicative stress [29]. Similarly, super Ink4/Arf/p53 transgenic mice where p53 activity is enhanced but normal regulation is retained, and p53(S18A) transgenic mice, where serine 18 is mutated to a non-phosphorylatable alanine, have reduced levels of age-associated replicative stress, further demonstrating the anti-aging effect of p53 [29, 30]. Congruently, depletion of p53 target protein PUMA or forced expression of BCL-2 in the hematopoietic compartment of mice ameliorate aging phenotype and improves HSC repopulating potential [31, 32]. Thus, the potential role of p53 in HSC aging is still controversial and the modulation of p53 activity can have either pro- or anti-aging effects depending on context. Recently, Kirschner et al. using single-cell transcriptomics identified cellular heterogeneity in HSC aging, and demonstrated that around 25% of p53-activated old HSC co-expressed cell-cycle proliferative JAK/STAT signaling, resulting in prolonged cell proliferation, myeloid skewing and stem cell exhaustion [33*]. Additional studies show an increased number of osteoprogenitors exhibiting p53 phosphorylation in old mice which are associated with a decline in bone formation with age [34]. Collectively, these findings suggest that pharmacological and genetic inhibition of cell cycle regulators and senescence potentially ameliorate the aging phenotype, but extensive studies are required to delineate the adverse effects associated with these phenotypes, including cancer.
2. Clonal hematopoiesis of indeterminate potential and myelodysplastic syndrome
Hematopoiesis has been generally viewed as a polyclonal event, with the perception that most HSC are equipotent when contributing to blood production throughout life. Nonetheless, recent paradigm shifting research on HSC clonality and the heterogeneous contributions of HSC clones to hematopoiesis has challenged this view and current reports clearly demonstrate that HSC are heterogeneous with respect to function and activity [35, 36]. Higher levels of clonality have been observed in hematopoietic aging where only a few clones of HSC actively contribute to the production of peripheral blood cells [37–39]. Several groups have identified somatic mutations in the blood cells of healthy older adults using high-throughput targeted sequencing methodologies on large data sets [40–42**]. Certain rare somatic mutations can be detected in younger individuals, but their frequency increases with age, reaching approximately 10-20% of the clonal contribution among persons aged over 70 years [40**, 41**]. A clonal hematopoiesis of indeterminate potential (CHIP) has been described which is a risk factor for hematopoietic neoplasia, in which somatic mutations are found in the cells of the blood or bone marrow, but no other criteria for neoplasia are met (Figure 2) [43**]. It is believed to be a potential precursor of myeloid diseases. The annual risk of transformation of CHIP into a hematologic neoplasm is low (~0.5% to 1%) but due its cumulative effect, the hematologic neoplasm prevalence associated with CHIP increases with age. Such clonal hematopoiesis in these older adults is largely associated with mutations in the DNA methyltransferase enzyme DNMT3a, the DNA demethylase TET2, and the polycomb group protein transcriptional repressor ASXL1, which implies a causal relation for these genes in age-associated clonality. Interestingly, DNMT3a, TET2 and ASXL1 all encode proteins that epigenetically regulate transcription, suggest the possibility of epigenetic clonality with aging, where clones of certain epigenetic profiles are preferentially selected for and maintained by the aging microenvironment. Among the most frequently mutated genes in acute myeloid leukemia (AML) are DNMT3a, TET2, and ASXL1, rendering these genes of high importance. Leukemias are an extreme case of clonal hematopoiesis, however we cannot ignore the connection between somatic mutations during clonal hematopoiesis and hematopoietic malignancy.
Intracellular signals involved in HSC aging
1. DNA damage accumulation
DNA damage accumulates during the lifespan of normal stem cells. Aged HSC exhibit increased DNA damage as assessed by single-cell analysis of DNA breakage and levels of the DNA damage response protein, γ-H2AX (Figure 2) [44, 45]. The accumulation of γ-H2AX is correlated with the mislocalization of a γ-H2AX phosphatase, PP4c, in aged HSC, and is believed to contribute to a delayed and/or ineffective dephosphorylation of γ-H2AX and represents a major contributor to its accumulation [46]. A second factor responsible for increased DNA damage is cell cycle entry. Quiescent HSC efficiently repair their DNA damage once they enter the cell cycle [44]. We know that high levels of replication stress occurs as quiescent HSC populations re-enter the cell cycle and various studies have demonstrated that DNA damage induces attrition of HSC [46, 47**]. This age-dependent accumulation of DNA damage appears to be exclusive to stem cell populations as this phenomenon is not seen in more committed progenitors [48]. Two likely hypotheses have been proposed: a) proliferating progenitors are better equipped for DNA damage repair, therefore their DNA accumulates less damage during subsequent cell divisions; and/or b) DNA damaged progenitors are efficiently cleared by cells with phagocytic activity [48]. It is also possible that both mechanisms occur concurrently.
With each replication, telomeres are shortened due to the inability of DNA-polymerase to work on the single-stranded 3′-ends. Telomere shortening is a replication “timer”. Despite telomerase-dependent and independent mechanisms of telomere elongation, critical telomere shortening (12.8 repeats of repetitive clusters) occurs. The mechanism of telomere shortening is the loss of telomere-capping proteins during serial under-replication, resulting in telomere dysfunction with consequent chromosome fusion and genome instability [49]. Interestingly, nearly negligible telomere shortening has been observed when investigating telomere lengths in aged HSC [50]. This might reflect the fact that HSC, as opposed to most somatic cells, express significant levels of telomerase which ameliorate the telomere shortening issue [51]. However, excessive HSC cycling induced by serial myeloablative therapies or transplantation that model the HSC aging phenotype can lead to extensive telomere shortening that is ultimately associated with functional exhaustion (Figure 2) [52]. The overexpression of telomerase in serially transplanted HSC, resulting in an appropriate maintenance of telomere length, did not protect the HSC from exhaustion [53] which argues against telomere erosion as a primary mechanism of HSC aging, and suggests that the therapeutic interventions aimed strictly to maintain telomere length would not be an effective therapeutic avenue in aging HSC.
Compounding with the increase in DNA damage, reports suggest there is also a decrease in the DNA repair machinery/capacity of aged HSC [54–56]. DNA damage activates a cascade of cellular events known as the DNA damage response (DDR). Depending on the type of damage, a series of signaling pathways can result in transient cell-cycle arrest, apoptosis, senescence or differentiation [57, 58]. Differential gene expression analyses of young and aged murine HSC identified a number of DNA repair associated genes that were downregulated with age. These genes include Xab2 (XPA-binding protein 2, involved in the NER pathway), Rad52 (a ssDNA-binding protein involved in homologous recombination), Xrcc1 (involved in coordination of single-strand break repair and base excision repair pathways) and Blm (a req helicase involved in DSB repair whose deficiency is associated with the bone marrow failure in Bloom syndrome) [54]. Hypomorphic mutations of the murine ligase IV (Lig4y288c), a protein crucial for functional repair of double strand breaks through the non-homologous end-joining pathway (NHEJ), cause a progressive age-dependent defect in hematopoiesis during aging [55]. Analysis of human CD34+ cells have shown that aging is associated with downregulation of the NHEJ Ku70 protein. Related work using genetic mouse models for the deletion of the Ku80 and Ku70 proteins has yielded very similar findings with severe reconstitution and repopulation abilities [56, 59, 60]. Combined, these data confirm that defects in DNA double-strand break repair hinder HSC reconstitution ability. However, the precise role that the double-strand breaks play during hematopoietic reconstitution, and why repair of these breaks is so vital for stem cell survival have not been elucidated.
Studies using genetic mouse models deficient in genes involved in the nucleotide excision repair (NER) pathway, such as Xpd, Ercc11 or with hypomorphic mutations in the MRN complex component Rad50, have shown a loss in HSC reconstitution capacity, decreased self-renewal, increased apoptosis and hematopoietic exhaustion [61, 62]. Moreover, loss of p53 function in the context of the hypomorphic Rad50 mutation can create a partial rescue of stem cell survival, indicating that cell cycle and apoptotic signaling from the unrepaired double-strand break through p53 results stem cell death. Although the additional deletion of p53 protects these mice from severe anemia or HSC exhaustion, these animals were susceptible to malignant transformation and developed lymphomas due to the loss of genomic stability [61]. Additionally, mice with deletion of the DNA damage signal transducer ataxia-telangiectasia (ATM) show increased infrared (IR)-sensitivity and decreased T-cell numbers. HSC from these mice have increased reactive oxygen species (ROS) levels and present with a decrease in the number and function of HSC upon aging that results in progressive BM failure [63–65]. The likely paradigm here is that reduced expression and function of the DNA damage response genes disrupts DNA repair performance, resulting in genomic instability that manifests as a HSC functional-decline with age. However, it has also been reported that DNA damage response efficiency is not altered upon aging HSC with respect to cell-cycle checkpoint activation, apoptosis and the accumulation of DNA mutations in response to total body irradiation [66]. It could be possible that the DNA damaging effects from irradiation or other insults effect the aging process differently.
2. ROS/Oxidative stress
ROS produced predominantly during mitochondrial oxidative phosphorylation (OXPHOS) contributes significantly to stem cell function and fate [67]. HSC reside in a low oxygen bone marrow niche and maintain low ROS levels and long-term self-renewal activity. However, the frequency of HSC with low ROS, and long-term competitive repopulation ability declines with age [68, 69]. Oxidative stress in aged HSC leads to the activation of the DNA damage response pathways mediated by p53, the DNA damage sensing serine/threonine protein kinase, ATM, and forkhead box O (FOXO) 3a; they in turn activate the HSC cell cycle inhibitors p16Ink4a/p19Arf, and promote stem cell exhaustion and senescence (Figure 2) [16]. Conversely, antioxidant N-acetyl-L-cysteine (NAC) treatment significantly improves the replicative potential of mouse HSC in serial transplantation experiments (Table 1) [69, 65]. Similarly, inhibition of the oxidative stress regulating pathways in Atm−/− mice leads to premature HSC exhaustion due to the loss of quiescence and HSC self-renewal capacity [65]. Another study challenged the role of NAC in the recovery of aging phenotype, and highlighted the mitochondria targeted antioxidant 10-(6′-plastoquinonyl) decyltriphenylphosphonium (SkQ1) as a promising intervention to recover aging associated changes [70]. Interestingly, ROS also plays an adaptive role in stem cell function. Conditional deletion of AKT1/2 or the phosphatase and tensin homolog (PTEN) resulted in decreased intracellular ROS and attenuation of long-term engraftment after bone marrow transplantation [71, 72], suggesting dose and context dependent biphasic activity of ROS. These discrepancies warrant further research to clarify the potential therapeutic value of ROS modulating agents in stem cell regulatory actions and aging.
Increased ROS activates p38 mitogen-activated protein kinase (MAPK) signaling in aged HSC, which in turn induces HSC exhaustion and lineage skewing by upregulating p16 and/or Arf [65]. Inhibition of p38-MAPK however ameliorates age-associated defects, and rescues ROShigh HSC colony formation in aged mice [69]. Furthermore, p38 is critical for human mesenchymal stem cell (MSC) senescence and aging, and its inhibition abrogates aging phenotypes [73, 74]. Recently, Jung et al. highlighted the thioredoxin-interacting protein (TXNIP)-p38 axis as a regulatory mechanism in HSC aging, and showed that inhibition of p38 activity by cell-penetrating peptide (CPP)-conjugated peptide derived from the TXNIP-p38 interaction rejuvenated aged HSC [74].
3. Altered mitochondrial function, proteostasis and metabolism
Cumulating evidence suggests that mitochondria are critical for HSC fate determination and highlights the predominant link between dysregulated nutrient sensing, gradual mitochondrial dysfunction and aging [75–77]. Mitochondria regulate stem cell aging by modulating the metabolic profile of the cell. Young stem cells have relatively high numbers of metabolically inactive mitochondria and rely on glycolytic metabolism as the major source of ATP [78–80]. However, functional mitochondria are required for adult stem cells proper maintenance [81]. HSC aging is accompanied by a decline in mitochondrial function and accumulation of mitochondrial DNA (mtDNA) mutations as a consequence of oxidative stress [82, 78]. Mice carrying proofreading deficient mtDNA polymerase gamma (POLG) exhibit premature aging due to the accumulation of mtDNA mutations [83]. On the other hand, these mice are unable to recapitulate the physiological aging process and are insensitive to the effects of ROS on HSC function [78]. The discrepancy between the physiological aging and those observed in POLG mutant mice suggests that mtDNA mutations may not be a primary driver of stem cell aging, and reinforces the need for additional research to determine the mechanistic link between oxidative stress and mtDNA mutations in HSC aging.
Aging associated phenotypes were further linked with reductions in nicotinamide adenine dinucleotide (NAD+), which contribute in progressive mitochondrial dysfunction leading to accumulation of misfolded protein stress that trigger mitochondrial unfolded protein response (UPRmt) and stem cell exhaustion [84**, 75]. Imbalance between nuclear and mitochondria encoded respiratory chain subunits caused by a decrement in NAD+ disrupts OXPHOS in aged mice [84**, 85]. NAD+ supplementation or pharmacological interventions bolstering cellular NAD+ levels however restored the mitochondrial function by modulating mitochondrial proteostasis and functionally rejuvenate aged HSC (Table 1) [84**]. NAD+ depletion and defective mitochondrial and endoplasmic reticulum protein folding have also been noted in many age-related neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease [86, 87].
In addition to mtDNA mutation, secondary alterations in the mitochondrial function associated with metabolic alteration also supports aging. Nutrient sensing and energy homeostasis are the metabolic drivers of mitochondrial function and longevity. Nutrient sensors including PI3K/Akt/mTOR/FOXO/AMPK pathway modulate the balance between stem cell quiescence, self-renewal and proliferation during aging. Furthermore, activation of PI3K/AKT in aged HSC leads to the inhibition of the FOXO transcription factors, which crosstalk with AMPK and maintains the equilibrium between oxidative phosphorylation and glycolysis [85, 88]. A decrease nutrient uptake capacity in aged HSC indicates its role in the regulation of stem cell aging and longevity. Interestingly, p53 activation following replicative stress or downregulation of sirtuin 7 (SIRT7) in HSC from old individuals attenuates the expression of PGC1α, which consequently results in nuclear respiratory factor 1 (Nrf1), estrogen-related receptor alpha (ERRα), and PPARα- dependent inhibition of mitochondrial biogenesis, loss of quiescence and myeloid biased differentiation (Figure 2) [75].
Caloric restriction also maintains the stem cell function and protects against aging by reducing the mTOR pathway, while conditional deletion of the mTOR negative regulator, tuberous sclerosis 1 (Tsc1) accelerates senescence, resulting in loss of HSC quiescence and repopulation ability [72]. Increased mTORC1 signaling has been linked with many age-related pathologies, including Hutchinson-Gilford progeria syndrome, which is caused by dominant splice site mutation in the LMNA gene and resembles premature aging [89]. Inhibition of the activity of the metabolic sensor mTOR in hypomorphic mTOR(Δ/Δ) mutant mice, in mice heterozygous for both mTOR and mLST8, or in mice with deleted ribosomal S6 protein kinase 1 (S6K1), results in extended longevity and a reduction in aging biomarkers [90–92]. In addition, resveratrol, a caloric mimetic, or rapamycin, an mTOR inhibitor, increases mitochondrial biogenesis through activation of SIRT1 and PGC1α and increased reconstitution potential with balanced hematopoietic precursors (Table 1) [93, 75, 85]. Interestingly, inhibition of mTOR activity by AMPK activator metformin promotes longevity (Table 1) [94].
It is recognized that NAD dependent protein deacetylases, Sirtuins regulate HSC fate through the regulation of oxidative stress, mitochondrial biogenesis and metabolism. SIRT1 and SIRT6 are dispensable for HSC self-renewal and regulate HSC homeostasis through activation of FOXO3 or repression of Wnt target genes, respectively [95–97**]. Indeed, SIRT3 and SIRT7 regulate HSC homeostasis through their function on mitochondrial biogenesis and unfolded protein response stress [98, 99]. Age-dependent decline in nuclear NAD+ availability and consequent functional decline in mitochondrial OXPHOS by the SIRT1/HIF-1α/c-Myc/TFAM pathway have been associated with mitochondrial dysfunction and increased ROS production in HSC [100], while loss of SIRT6 results genomic instability and premature aging in mice [101]. Similarly, decreased SIRT7 expression in aged HSC leads to disproportionate NRF1-dependent mitochondrial biogenesis, increased mitochondrial stress, compromised regenerative capacity and myeloid skewing. Upregulation of SIRT3 and SIRT7 however relieves mitochondrial protein folding stress, reduces oxidative stress by enhancing the antioxidant activity of superoxide dismutase 2 (SOD2) and improves the regenerative capacity of aged HSC (Table 1) [102**].
Several studies have consistently revealed FOXO transcription factors as important determinants in aging and longevity via shuttling from cytoplasm to nucleus [103, 104]. They influence aging or age related disorders by increasing the key detoxification enzymes, SOD2 and catalase [105, 103]. Hematopoietic-specific deletion of FOXO1, FOXO3a, and FOXO4 correlates with a marked increase in ROS accumulation, mitochondrial fragmentation and mitochondrial protein misfolding stress, resulting in loss of HSC quiescence and compromised long-term repopulation ability [106–109].
Together, these studies suggest that HSC aging is potentially reversible by modulation of mitochondrial metabolism and unfolded protein stress, which in turn attenuate the intracellular ROS accumulation.
4. Impaired Autophagy
Autophagy is an essential proteostasis and stress response mechanism essential for the maintenance of cellular health. In quiescent HSC, basal autophagy maintains glycolysis, HSC stemness and regenerative potential by removing metabolically active mitochondria and controlling OXPHOS driven oxidative stress [110**–112]. Cumulative evidence suggests that impaired autophagy and autophagy-related genes including Atg1/5/6/7/8/12 in aged HSC results in proteostasis imbalance and mitochondrial dysfunction [110–112]. Deletion of Atg 5, Atg7 or Atg12 in the hematopoietic compartment display myeloid skewing, accumulation of mitochondria and ROS, increased proliferation and DNA damage as well as impaired HSC self-renewal activity and regenerative potential [110–112]. Notably, a recent study observed that although aged HSC remain competent for autophagy induction, approximately 30% of aged HSC exhibit high autophagy levels, low OXPHOS metabolism and retains long-term regeneration potential similar to young HSC [110**].
Further studies highlighted the link between caloric restriction, autophagy and stem cells aging. Caloric restriction act as an efficient inducer of autophagy and ameliorate age-associated phenotype through inhibition of insulin/insulin-like growth (IGF) factor, mTOR complex 1 and activation of AMPK (Figure 2) [113, 75]. Indeed, the pro-longevity effects elicited by caloric restriction also involves SIRT1 and FOXO3, which maintains transcription of many autophagy related genes to protect HSC during starvation or aging-associated metabolic stress [114, 75]. Recently, Ito et al. demonstrated that FOXO3 is a critical regulator of mitophagy (mitochondrial clearance by autophagy), a key process required for HSC self-renewal [115].
5. Effect of Polarity on HSC division and aging
Aged HSC show reduced polarity with respect to established polarity markers numb, tubulin and Cdc42 (Figure 2) [116, 117]. Polarity is associated with specialized and compartmentalized functions in HSC, such as migration or division while the loss of polarity has been correlated with reduced self-renewal and altered differentiation of HSC [116, 17]. These data hint toward a crucial role for polarity in stem cell maintenance. Mechanisms of cellular polarity establishment and maintenance are well characterized in neuronal stem cells and epithelial cells, but largely unknown in HSC. There is an accumulating body of evidence focusing on the mechanisms that control cellular asymmetry and division among eukaryotic single-celled microorganisms linking polarity directly with the aging process [118–121]. Polarity may have evolved as a mechanism to avoid clonal senescence by establishing an aging mother cell that accumulates oxidatively damaged or aggregated proteins while a rejuvenated daughter cell inherits limited amounts of toxic and deteriorated material, preserving the fitness of this daughter cell.
Unfortunately, very few polarity proteins have been functionally interrogated in HSC, and of those that have, many are dispensable for HSC function [122, 123]. Perhaps the most direct evidence connecting cellular polarity to aging HSC comes from work by Florian et al. in which they show that Cdc42 (a small Rho-GTPase) is polarized with tubulin in young HSC. Aged HSC exhibit an increase in Cdc42 activity and consequently lose Cdc42 polarization [116]. The function of these apolar HSC correlates with the observed age-associated decline in HSC function. Pharmacological intervention using a Cdc42 inhibitor, casin can rejuvenate aged HSC to be functionally equivalent to young HSC (Table 1) [116]. Further investigation into Cdc42 polarity revealed that a shift from canonical to non-canonical Wnt signaling leads to apolar HSC which are responsible for the age related deterioration in the hematopoietic system. Florian et al show that the levels of Wnt5a are significantly elevated in aged long-term HSC, while expression of canonical Wnt ligands remains unaltered during aging. Remarkably, reduction of the non-canonical Wnt5a signaling in old HSC successfully rejuvenated chronologically aged HSC and restored polar localization of Cdc42 [124**].
6. Nuclear Architecture and Epigenetic Alterations
In accordance with age-induced myeloid skewing, comparative transcriptional analysis of young and aged HSC indicates that myeloid differentiation genes, such as Runx1, Hoxb6 and Osmr are upregulated while lymphopoiesis-specific genes are transcriptionally downregulated in aged HSC [8]. Global gene expression changes such as these raise the possibility that aging may be a consequence of altered epigenetic transcriptional regulation. Indeed, HSC aging is associated with an altered epigenetic landscape (Figure 2) [125, 116, 126]. Age-associated epigenetic marks include increased H4K16 acetylation, H4K20 trimethylation or H3K4 trimethylation, as well as decreased H3K9 methylation or H3K27 trimethylation [54, 127, 128]. Specifically, epigenetic profiling of young and old HSC has revealed that aged HSC displayed broader H3K4 trimethylation peaks across HSC identity and self-renewal genes. Aged HSC also exhibit hypermethylation at transcription factor binding sites that pertain to differentiation-promoting genes, and reduced DNA methylation at genes related to HSC maintenance. Collectively, these epigenetic changes reinforce HSC self-renewal while occluding differentiation-promoting genes, which parallel phenotypic HSC aging behavior [126].
These age-associated epigenetic marks include both active and repressive changes without a clear directional genome-wide bias toward either activation or repression upon aging, and thus are termed “epigenetic drift”. Comparative gene expression analyses demonstrated deregulation of genes involved in chromatin organization and epigenetic maintenance, such as PolyComb Repression Complex 2 (PRC2) genes, validating the age-associated epigenetic shift theory [54]. Interestingly, areas of high H3K4 methylation in translocation prone regions of human HSC enable chromosomal breakage and increased translocation frequency [129*]. Additional studies are warranted to investigate to what extent epigenetic drift can impact aging related malignancies caused from such translocations.
A key emerging contributor to genome function, in addition to the rapidly evolving field of epigenetics, is the architectural organization of the cell nucleus. The cell nucleus is a spatially and functionally compartmentalized organelle containing numerous proteinaceous nuclear bodies as well as non-randomly positioned chromatin domains. The most-striking class of nuclear protein origin diseases are the laminopathies, characterized by nuclear morphologic defects and chromatin disorganization. This diverse class of diseases is caused by mutations in the structural lamin A and C proteins encoded by the LMNA gene. Interestingly, mutations in LMNA commonly manifest as the premature-aging disease, Hutchinson-Gilford Progeria Syndrome [130]. Interestingly, unpublished reports indicate that the LMNA gene is epigenetically silenced in aged HSC resulting in its downregulated expression. Continued investigations toward nuclear envelope structural proteins may provide further insights into the architectural characteristics and chromatin organization of the nucleus within aging HSC.
Subnuclear distribution of heterochromatin by H3K9 methylation has been shown to regulate HSC differentiation [131*]. Although no clear role for HSC nuclear architecture has yet been defined for aging stem cells, speculations can be made that it could regulate the aging process similar to how it controls HSC differentiation. In fact, Florian et al. observed that young HSC display nuclear polarization of AcH4K16 and this polarization is lost upon aged HSC. Inhibition of aberrant Cdc42 activity in aged HSC can restore the level and spatial distribution of AcH4K16 [116]. Rejuvenating agents such as Satb1 activators and casin, which can reprogram the epigenetic landscape and the overall nuclear architecture of aged HSC, provide promising translational approaches for attenuating hematopoietic aging (Table 1) [116, 132].
Extrinsic factors regulating HSC aging
Although most data indicate that HSC aging is a cell intrinsic phenomenon, HSC aging may also depend on, or be triggered by, extrinsic stimuli from systemic factors and/or supportive cells in their immediate proximity. Two sources of stimuli are of upmost interest.
1. HSC niche
It is likely that HSC aging encompasses both intrinsic changes within the stem cell pool itself and within the microenvironment in which they reside. Aging of niche cells or age-dependent alterations in locally secreted factors including stem cell factor (SCF), thrombopoietin (TPO) and the activators of Notch signaling cause maladaptive changes in stem cell function. Young and aged HSC have distinct niche selectivity and, contrary to young stem cells, aged HSC localize away from the endosteal stem cell niche (Figure 1, 2) [117]. Niche composition is also altered upon aging due to decreased bone formation, angiogenesis, endosteal thinning, trabecular degradation and changes in extracellular matrix (ECM) components [133, 134]. Aging seems to reduce the number of metabolically active MSC and induces their differentiation in adipocytes, which promotes myelopoiesis over lymphopoiesis and impair HSC function (Figure 2) [135]. In addition, aging associated decline in SCF coupled with decline of CD31hiEmcnhi endothelial cells, osteoprogenitors, PDGFRβ+ or NG2+ perivascular cells and arterioles formation [136**]. Activation of endothelial Notch signaling however induces arteriole formation, leads to the expansion of HSC niches and restores SCF levels [136].
The chemokine receptor CXCR4 and its ligand stromal-derived factor (SDF)-1 are indispensable for HSC quiescence, BM retention and protection against oxidative stress [16, 137]. Aging associated increased adipocytes in the bone marrow microenvironment decreased plasma IGF-1 and SDF-1 level, which negatively regulate hematopoiesis and delay engraftment [138]. Conversely, in a heterochronic transplantation setting, aged HSC environment associated with skewing of aged HSC towards a myeloid lineage, through increased secretion of RANTES/CCL5 [139]. Reduced HSC homing was also noted upon transplantation in old recipients compared to young ones [6]. Bone marrow macrophage-osteoblast niche regulate short term repopulating and myeloid progenitor cells egress through p62 mediated inhibition of IKK/NFκB/CCL4 pathway [140]. Recently, it was observed that decline in osteopontin (OPN) produced by pre-osteoblasts, osteoblasts and osteocytes in aged stroma promotes aging associated phenotypes on HSC including decreased regenerative capacity, impaired homing and loss of polarity [141**]. Exposure of OPN fragments activated by thrombin however completely attenuate the aging of old HSC [141**]. Furthermore, age related alteration in adhesion molecules modulates interaction of HSC with their niche. Bone marrow endothelial cells promote HSC regeneration by upregulating adhesion proteins. However, decreased self-renewal ability of aged HSC could be associated with impaired adhesive properties (Figure 2) [16]. Interestingly, G-CSF mediated mobilization of HSC increased with age.
Transforming growth factor-β (TGF-β), virtually released by a variety of BM niche cells, but majorly by non myelinating Schwann cells act as a significant contributor of age related functional decline of HSC (Figure 2) [142, 126, 143]. TGF-β signaling in HSC reduced with age, and transcriptome profiling of young and old HSC represent downregulation of TGF-β-regulated genes including Egr1; a regulator of HSC homeostasis, collagen and metalloproteinase genes; implicated in HSC-niche interactions and Nr4a1, Cepba, Jun, and Junb; negative regulator of myeloid differentiation in aged HSC [126]. However, the relationship of TGF-β1 with HSC aging is complex, and using hematopoietic specific deletion of Tif1γ, which controls TGF-β1 receptor (Tgfbr1) turnover, Quere et al. identified two distinct subsets of HSC: 1) more sensitive Tgfbr1hi HSC exhibiting an age-associated HSC/P expansion and myeloid bias, and; 2) Tgfbr1lo HSC with normal stem cell counts and balanced lymphopoiesis/myelopoiesis [142].
Aging also associated with increased ROS generation and decreased hypoxic condition of osteoblastic niche; however the underling mechanisms remain to be elucidated. During stressed hematopoietic regeneration gap junction protein, Connexin 43 (Cx43) preserved HSC function by transfer of ROS from HSC to stromal cells [144]. Interestingly, loss of Cx43 in osteoblasts and osteogenic progenitors disrupted trans-stromal migration and homing of HSC/Ps in myeloablated animals [145]. These findings define the role of environmental factors in the establishment of the aging-associated changes in HSC, while further investigations are necessary to dissect the mechanism of crosstalk between HSC and stem cell niche in aging, which could lead to a therapeutic basis for promoting stem cells rejuvenation.
2. Systemic factors
In addition to signals produced by HSC niche, circulating factors also profoundly affect the aging of hematopoietic and tissue stem cells, as well as the hematopoietic microenvironment. Inflamm-aging, which is a sterile chronic systemic inflammation, develops with age and acts as a major factor in development of cancer, metabolic diseases, and other age-related pathologies [146]. Production of chemokines like RANTES, MIP-1α, IL-8 and MCP-1 is increased in the elderly, as a consequence of inflammation [147]. Concurrently, chronic elevation of inflammatory mediators in aged tissue is associated with myeloid skewing and/or adipogenesis in the bone marrow, and contributes to the aging phenotype. Activation of TLR4 signaling, P-selectin, NFκB and RANTES-mTOR pathway in elderly also contributes to the production of pro-inflammatory cytokines and affects longevity by playing a role in inflamm-aging [148]. Downstream, the deficiency of the adaptor protein Lnk, which functions to dampen extrinsic cytokine signals, abrogates the phenotypes associated with HSC aging, and aged Lnk−/− HSC harbored superior reconstitution potential compared with aged WT HSC, with maintenance of a robust lymphoid differentiation potential [149, 150]. NFkB is the major stimulator of SASP, and the SASP factors such as TNF-α, MMP13, CXCL12, IL-6 and IL-1α are possibly involved in reduced functionality of HSC during aged hematopoiesis [151]. Indeed, accumulation of p16Ink4a positive senescent cells in aging tissues affect neighboring cells in the local environment by secreting SASP-associated inflammatory cytokines, growth regulators, proteases and other signaling molecules [85, 24, 23].
Several studies demonstrated that the gut microbiota act as a critical extrinsic regulator of hematopoiesis, and depletion of intestinal flora in mice treated with broad spectrum antibiotics show reduced number of stem and progenitor cells in the bone marrow and concomitant anemia, thrombocytosis, and leukopenia with pronounced pan-lymphopenia [152*, 153]. Interestingly, fecal microbiota transplantation significantly improves the hematopoietic changes associated with antibiotic treatment [152*]. Effects of antibiotics on stem cells depletion were further replicated in Stat1-deficient mice, suggesting that microbiota sustain steady-state hematopoiesis through activation of Stat1 signaling, leading to activation of pro-inflammatory interferon signaling [152*]. Further evidences highlighted that the changes in the gut microbiota in elderly people associated with aging, while diet driven microbiota alterations improve their health [154]. Recently, Han et al. demonstrated that bacterial genes influence host aging by modulating mTOR, JNK, and insulin signaling, as well as caloric restriction [155**]. Additionally, gut inhabitants promote longevity through increased secretion of the polysaccharide colanic acid, which regulates mitochondrial dynamics and UPRmt in the host [155**]. These findings further open new avenues to access the communication between gut microbiota, mitochondrial dynamics and aging of HSC, as well as the clinical significance of microbiota transplantation on chemo/immunotherapy and aging.
The findings described above argue that the aging environment and potentially the factors produced within it can also impact on the manifestation of the HSC aging state. However, as the transplantation of aged HSC into a young environment reconstructs an aged hematopoietic system, cell intrinsic alterations in aged HSC are sufficient, at least for the maintenance of the physiological HSC aging state.
Conclusions and future perspectives
Because physiological HSC aging is caused by the combinatorial action of numerous intrinsic and extrinsic alterations, current targeted rejuvenation attempts have failed to completely rejuvenate aging HSC function. Epigenetics, proteomics and biochemical analyses are hampered by the fact that HSC are rare cells and continue to be a major experimental obstacle to advance in our understanding of the causes and effects of aging in HSC. Therefore, as techniques are developed and adapted to allow for studies on these rare populations and subpopulations (e.g. quiescent, dormant, lymphoid-biased, platelet-biased), we anticipate that the detailed knowledge surrounding the molecular events that coincide with and drive HSC aging will continue to expand and potential regulators to be identified. However, the identification of regulators will face the challenge of investigating their interplay and physiological relevance. Therefore, to achieve full-fledged understanding of HSC aging, more systems biology information will undoubtedly be critical to thoroughly understand HSC aging and to eventually design appropriate rejuvenation approaches.
Acknowledgments
The authors want to thank Ms. Margaret O’Leary for editing this manuscript.
The authors also want to thank the Cincinnati Children’s Hospital Medical Center and Hoxworth Blood Center for their continued support. This study has been partly supported by the National Institutes of Health F31HL1324801 (M.J.A.), R01GM110628 (J.A.C), Leukemia and Lymphoma Society of North America (J.A.C.) and Williams Lawrence & Blanche Hughes Foundation (J.A.C.)
Footnotes
Compliance with Ethical Standards
Conflict of Interest
Abhishek K. Singh, Mark J. Althoff, and Jose A. Cancelas declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Cheng J, Turkel N, Hemati N, Fuller MT, Hunt AJ, Yamashita YM. Centrosome misorientation reduces stem cell division during aging. Nature. 2008;456(7222):599–604. doi: 10.1038/nature07386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2•.Bernitz JM, Kim HS, MacArthur B, Sieburg H, Moore K. Hematopoietic stem cells count and remember self-renewal divisions. Cell. 2016;167(5):1296–309 e10. doi: 10.1016/j.cell.2016.10.022. This article provides evidence that demonstrates age-related phenotypic changes within the HSC compartment are divisional history dependent. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135(6):1118–29. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 4.Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441(7097):1068–74. doi: 10.1038/nature04956. [DOI] [PubMed] [Google Scholar]
- 5.Bryder D, Rossi DJ, Weissman IL. Hematopoietic stem cells: the paradigmatic tissue-specific stem cell. Am J Pathol. 2006;169(2):338–46. doi: 10.2353/ajpath.2006.060312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liang Y, Van Zant G, Szilvassy SJ. Effects of aging on the homing and engraftment of murine hematopoietic stem and progenitor cells. Blood. 2005;106(4):1479–87. doi: 10.1182/blood-2004-11-4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morrison SJ, Wandycz AM, Akashi K, Globerson A, Weissman IL. The aging of hematopoietic stem cells. Nature medicine. 1996;2(9):1011–6. doi: 10.1038/nm0996-1011. [DOI] [PubMed] [Google Scholar]
- 8.Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ, et al. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A. 2005;102(26):9194–9. doi: 10.1073/pnas.0503280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xing Z, Ryan MA, Daria D, Nattamai KJ, Van Zant G, Wang L, et al. Increased hematopoietic stem cell mobilization in aged mice. Blood. 2006;108(7):2190–7. doi: 10.1182/blood-2005-12-010272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geiger H, Rudolph KL. Aging in the lympho-hematopoietic stem cell compartment. Trends Immunol. 2009;30(7):360–5. doi: 10.1016/j.it.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 11.de Haan G, Nijhof W, Van Zant G. Mouse strain-dependent changes in frequency and proliferation of hematopoietic stem cells during aging: correlation between lifespan and cycling activity. Blood. 1997;89(5):1543–50. [PubMed] [Google Scholar]
- 12.de Haan G, Van Zant G. Dynamic changes in mouse hematopoietic stem cell numbers during aging. Blood. 1999;93(10):3294–301. [PubMed] [Google Scholar]
- 13.Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127(2):265–75. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 14.Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging: the critical roles of p53. Oncogene. 2013;32(43):5129–43. doi: 10.1038/onc.2012.640. [DOI] [PubMed] [Google Scholar]
- 15.Geiger H, de Haan G, Florian MC. The aging haematopoietic stem cell compartment. Nature reviews Immunology. 2013;13(5):376–89. doi: 10.1038/nri3433. [DOI] [PubMed] [Google Scholar]
- 16.Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nature medicine. 2014;20(8):833–46. doi: 10.1038/nm.3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, et al. Stem-cell aging modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443(7110):421–6. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- 18.Jeck WR, Siebold AP, Sharpless NE. Review: a meta-analysis of GWAS and age-associated diseases. Aging cell. 2012;11(5):727–31. doi: 10.1111/j.1474-9726.2012.00871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19•.Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530(7589):184–9. doi: 10.1038/nature16932. This paper provides evidence of p16Ink4a positive cells accumulation with aging and it removal as an important therapeutic approach to extend longevity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423(6937):302–5. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- 21.Attema JL, Pronk CJ, Norddahl GL, Nygren JM, Bryder D. Hematopoietic stem cell aging is uncoupled from p16 INK4A-mediated senescence. Oncogene. 2009;28(22):2238–43. doi: 10.1038/onc.2009.94. [DOI] [PubMed] [Google Scholar]
- 22•.Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nature medicine. 2016;22(1):78–83. doi: 10.1038/nm.4010. This article shows that selective clearance of senescent cells rejuvenate aged stem cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, et al. p16(Ink4a) and senescence-associated beta-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging (Albany NY) 2017;9(8):1867–84. doi: 10.18632/aging.101268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, et al. Aging of mice is associated with p16(Ink4a)- and beta-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging (Albany NY) 2016;8(7):1294–315. doi: 10.18632/aging.100991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carrasco-Garcia E, Moreno M, Moreno-Cugnon L, Matheu A. Increased Arf/p53 activity in stem cells, aging and cancer. Aging cell. 2017;16(2):219–25. doi: 10.1111/acel.12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, et al. p53 mutant mice that display early aging-associated phenotypes. Nature. 2002;415(6867):45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- 27.Maier B, Gluba W, Bernier B, Turner T, Mohammad K, Guise T, et al. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004;18(3):306–19. doi: 10.1101/gad.1162404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dumble M, Moore L, Chambers SM, Geiger H, Van Zant G, Goodell MA, et al. The impact of altered p53 dosage on hematopoietic stem cell dynamics during aging. Blood. 2007;109(4):1736–42. doi: 10.1182/blood-2006-03-010413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matheu A, Maraver A, Klatt P, Flores I, Garcia-Cao I, Borras C, et al. Delayed aging through damage protection by the Arf/p53 pathway. Nature. 2007;448(7151):375–9. doi: 10.1038/nature05949. [DOI] [PubMed] [Google Scholar]
- 30.Armata HL, Garlick DS, Sluss HK. The ataxia telangiectasia-mutated target site Ser18 is required for p53-mediated tumor suppression. Cancer Res. 2007;67(24):11696–703. doi: 10.1158/0008-5472.CAN-07-1610. [DOI] [PubMed] [Google Scholar]
- 31.Liu D, Ou L, Clemenson GD, Jr, Chao C, Lutske ME, Zambetti GP, et al. Puma is required for p53-induced depletion of adult stem cells. Nat Cell Biol. 2010;12(10):993–8. doi: 10.1038/ncb2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Domen J, Cheshier SH, Weissman IL. The role of apoptosis in the regulation of hematopoietic stem cells: Overexpression of Bcl-2 increases both their number and repopulation potential. The Journal of experimental medicine. 2000;191(2):253–64. doi: 10.1084/jem.191.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33••.Kirschner K, Chandra T, Kiselev V, Flores-Santa Cruz D, Macaulay IC, Park HJ, et al. Proliferation drives aging-related functional decline in a subpopulation of the hematopoietic stem cell compartment. Cell Rep. 2017;19(8):1503–11. doi: 10.1016/j.celrep.2017.04.074. This article defines cellular heterogeneity in HSC aging, and highlighted the significance of JAK/STAT signaling in stem cell exhaustion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim HN, Chang J, Shao L, Han L, Iyer S, Manolagas SC, et al. DNA damage and senescence in osteoprogenitors expressing Osx1 may cause their decrease with age. Aging cell. 2017;16(4):693–703. doi: 10.1111/acel.12597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beerman I, Bhattacharya D, Zandi S, Sigvardsson M, Weissman IL, Bryder D, et al. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci U S A. 2010;107(12):5465–70. doi: 10.1073/pnas.1000834107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dykstra B, Olthof S, Schreuder J, Ritsema M, de Haan G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. The Journal of experimental medicine. 2011;208(13):2691–703. doi: 10.1084/jem.20111490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Busch K, Klapproth K, Barile M, Flossdorf M, Holland-Letz T, Schlenner SM, et al. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature. 2015;518(7540):542–6. doi: 10.1038/nature14242. [DOI] [PubMed] [Google Scholar]
- 38.Sun J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho YJ, et al. Clonal dynamics of native haematopoiesis. Nature. 2014;514(7522):322–7. doi: 10.1038/nature13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Verovskaya E, Broekhuis MJ, Zwart E, Ritsema M, van Os R, de Haan G, et al. Heterogeneity of young and aged murine hematopoietic stem cells revealed by quantitative clonal analysis using cellular barcoding. Blood. 2013;122(4):523–32. doi: 10.1182/blood-2013-01-481135. [DOI] [PubMed] [Google Scholar]
- 40••.Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477–87. doi: 10.1056/NEJMoa1409405. This article preformed whole exome sequencing on a large cohort and reveiled detectable clonal expansions most frequently involved somatic mutations in three genes (DNMT3A, ASXL1, and TET2) that have previously been implicated in hematologic cancers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41••.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–98. doi: 10.1056/NEJMoa1408617. This article highlights that detectable somatic mutations, partcularly in DNMT3A, TET2, and ASXL1, were rare in persons younger than 40 years of age but rose appreciably in frequency with age and was associated with an increase in the risk of hematologic cancer, an increase in all-cause mortality, with increases in the risks of incident coronary heart disease and ischemic stroke. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42••.McKerrell T, Park N, Moreno T, Grove CS, Ponstingl H, Stephens J, et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 2015;10(8):1239–45. doi: 10.1016/j.celrep.2015.02.005. This paper indicates that spliceosome gene mutations drive clonal expansion under selection pressures particular to the aging hemopoietic system and explains the high incidence of clonal disorders associated with these mutations in advanced old age. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43••.Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126(1):9–16. doi: 10.1182/blood-2015-03-631747. This article highlights the acquisition of somatic mutations that drive clonal expansion in the absence of cytopenias and dysplastic hematopoiesis can be considered clonal hematopoiesis of indeterminate potential (CHIP) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beerman I, Seita J, Inlay MA, Weissman IL, Rossi DJ. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell. 2014;15(1):37–50. doi: 10.1016/j.stem.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rossi DJ, Seita J, Czechowicz A, Bhattacharya D, Bryder D, Weissman IL. Hematopoietic stem cell quiescence attenuates DNA damage response and permits DNA damage accumulation during aging. Cell Cycle. 2007;6(19):2371–6. doi: 10.4161/cc.6.19.4759. [DOI] [PubMed] [Google Scholar]
- 46.Flach J, Bakker ST, Mohrin M, Conroy PC, Pietras EM, Reynaud D, et al. Replication stress is a potent driver of functional decline in aging haematopoietic stem cells. Nature. 2014;512(7513):198–202. doi: 10.1038/nature13619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47••.Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature. 2015;520(7548):549–52. doi: 10.1038/nature14131. This article establishes a novel link between physiological stress and DNA damage in normal HSC while providing a mechanistic explanation for the accumulation of DNA damage in HSC during aging and the accelerated failure of the haematopoietic system in Fanconi anaemia patients. [DOI] [PubMed] [Google Scholar]
- 48.Seita J, Rossi DJ, Weissman IL. Differential DNA damage response in stem and progenitor cells. Cell stem cell. 2010;7(2):145–7. doi: 10.1016/j.stem.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 49.Capper R, Britt-Compton B, Tankimanova M, Rowson J, Letsolo B, Man S, et al. The nature of telomere fusion and a definition of the critical telomere length in human cells. Genes Dev. 2007;21(19):2495–508. doi: 10.1101/gad.439107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wahlestedt M, Norddahl GL, Sten G, Ugale A, Frisk MA, Mattsson R, et al. An epigenetic component of hematopoietic stem cell aging amenable to reprogramming into a young state. Blood. 2013;121(21):4257–64. doi: 10.1182/blood-2012-11-469080. [DOI] [PubMed] [Google Scholar]
- 51.Morrison SJ, Prowse KR, Ho P, Weissman IL. Telomerase activity in hematopoietic cells is associated with self-renewal potential. Immunity. 1996;5(3):207–16. doi: 10.1016/s1074-7613(00)80316-7. [DOI] [PubMed] [Google Scholar]
- 52.Allsopp RC, Cheshier S, Weissman IL. Telomere shortening accompanies increased cell cycle activity during serial transplantation of hematopoietic stem cells. The Journal of experimental medicine. 2001;193(8):917–24. doi: 10.1084/jem.193.8.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Allsopp RC, Morin GB, Horner JW, DePinho R, Harley CB, Weissman IL. Effect of TERT over-expression on the long-term transplantation capacity of hematopoietic stem cells. Nature medicine. 2003;9(4):369–71. doi: 10.1038/nm0403-369. [DOI] [PubMed] [Google Scholar]
- 54.Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007;5(8):e201. doi: 10.1371/journal.pbio.0050201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nijnik A, Woodbine L, Marchetti C, Dawson S, Lambe T, Liu C, et al. DNA repair is limiting for haematopoietic stem cells during aging. Nature. 2007;447(7145):686–90. doi: 10.1038/nature05875. [DOI] [PubMed] [Google Scholar]
- 56.Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447(7145):725–9. doi: 10.1038/nature05862. [DOI] [PubMed] [Google Scholar]
- 57.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kenyon J, Gerson SL. The role of DNA damage repair in aging of adult stem cells. Nucleic Acids Res. 2007;35(22):7557–65. doi: 10.1093/nar/gkm1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qing Y, Wang Z, Bunting KD, Gerson SL. Bcl2 overexpression rescues the hematopoietic stem cell defects in Ku70-deficient mice by restoration of quiescence. Blood. 2014;123(7):1002–11. doi: 10.1182/blood-2013-08-521716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prall WC, Czibere A, Jager M, Spentzos D, Libermann TA, Gattermann N, et al. Age-related transcription levels of KU70, MGST1 and BIK in CD34+ hematopoietic stem and progenitor cells. Mech Aging Dev. 2007;128(9):503–10. doi: 10.1016/j.mad.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 61.Bender CF, Sikes ML, Sullivan R, Huye LE, Le Beau MM, Roth DB, et al. Cancer predisposition and hematopoietic failure in Rad50(S/S) mice. Genes Dev. 2002;16(17):2237–51. doi: 10.1101/gad.1007902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Prasher JM, Lalai AS, Heijmans-Antonissen C, Ploemacher RE, Hoeijmakers JH, Touw IP, et al. Reduced hematopoietic reserves in DNA interstrand crosslink repair-deficient Ercc1-/- mice. EMBO J. 2005;24(4):861–71. doi: 10.1038/sj.emboj.7600542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, et al. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86(1):159–71. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 64.Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431(7011):997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- 65.Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nature medicine. 2006;12(4):446–51. doi: 10.1038/nm1388. [DOI] [PubMed] [Google Scholar]
- 66.Moehrle BM, Nattamai K, Brown A, Florian MC, Ryan M, Vogel M, et al. Stem cell-specific mechanisms ensure genomic fidelity within HSCs and upon aging of HSCs. Cell Rep. 2015;13(11):2412–24. doi: 10.1016/j.celrep.2015.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takubo K, Nagamatsu G, Kobayashi CI, Nakamura-Ishizu A, Kobayashi H, Ikeda E, et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell stem cell. 2013;12(1):49–61. doi: 10.1016/j.stem.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hamilton ML, Van Remmen H, Drake JA, Yang H, Guo ZM, Kewitt K, et al. Does oxidative damage to DNA increase with age? Proc Natl Acad Sci U S A. 2001;98(18):10469–74. doi: 10.1073/pnas.171202698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110(8):3056–63. doi: 10.1182/blood-2007-05-087759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kolosova NG, Stefanova NA, Muraleva NA, Skulachev VP. The mitochondria-targeted antioxidant SkQ1 but not N-acetylcysteine reverses aging-related biomarkers in rats. Aging (Albany NY) 2012;4(10):686–94. doi: 10.18632/aging.100493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Juntilla MM, Patil VD, Calamito M, Joshi RP, Birnbaum MJ, Koretzky GA. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood. 2010;115(20):4030–8. doi: 10.1182/blood-2009-09-241000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen C, Liu Y, Liu R, Ikenoue T, Guan KL, Liu Y, et al. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. The Journal of experimental medicine. 2008;205(10):2397–408. doi: 10.1084/jem.20081297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Borodkina A, Shatrova A, Abushik P, Nikolsky N, Burova E. Interaction between ROS dependent DNA damage, mitochondria and p38 MAPK underlies senescence of human adult stem cells. Aging (Albany NY) 2014;6(6):481–95. doi: 10.18632/aging.100673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jung H, Kim DO, Byun JE, Kim WS, Kim MJ, Song HY, et al. Thioredoxin-interacting protein regulates haematopoietic stem cell aging and rejuvenation by inhibiting p38 kinase activity. Nat Commun. 2016;7:13674. doi: 10.1038/ncomms13674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lopez-Otin C, Galluzzi L, Freije JMP, Madeo F, Kroemer G. Metabolic control of longevity. Cell. 2016;166(4):802–21. doi: 10.1016/j.cell.2016.07.031. [DOI] [PubMed] [Google Scholar]
- 76.Wang Y, Hekimi S. Mitochondrial dysfunction and longevity in animals: Untangling the knot. Science. 2015;350(6265):1204–7. doi: 10.1126/science.aac4357. [DOI] [PubMed] [Google Scholar]
- 77.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Norddahl GL, Pronk CJ, Wahlestedt M, Sten G, Nygren JM, Ugale A, et al. Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell stem cell. 2011;8(5):499–510. doi: 10.1016/j.stem.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 79.Liang R, Ghaffari S. Mitochondria and FOXO3 in stem cell homeostasis, a window into hematopoietic stem cell fate determination. J Bioenerg Biomembr. 2017;49(4):343–6. doi: 10.1007/s10863-017-9719-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kohli L, Passegue E. Surviving change: the metabolic journey of hematopoietic stem cells. Trends Cell Biol. 2014;24(8):479–87. doi: 10.1016/j.tcb.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Luchsinger LL, de Almeida MJ, Corrigan DJ, Mumau M, Snoeck HW. Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature. 2016;529(7587):528–31. doi: 10.1038/nature16500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sun N, Youle RJ, Finkel T. The mitochondrial basis of aging. Mol Cell. 2016;61(5):654–66. doi: 10.1016/j.molcel.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, et al. Premature aging in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429(6990):417–23. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 84••.Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016;352(6292):1436–43. doi: 10.1126/science.aaf2693. This article demonstrates that NAD+ supplemantation delays senescence and rejuvanate aged HSC through induction of mitochondrial unfolded protein response and synthesis of prohibitin proteins. [DOI] [PubMed] [Google Scholar]
- 85.Oh J, Lee YD, Wagers AJ. Stem cell aging: mechanisms, regulators and therapeutic opportunities. Nature medicine. 2014;20(8):870–80. doi: 10.1038/nm.3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fang EF, Lautrup S, Hou Y, Demarest TG, Croteau DL, Mattson MP, et al. NAD+ in aging: Molecular mechanisms and translational implications. Trends in molecular medicine. 2017;23(10):899–916. doi: 10.1016/j.molmed.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bernales S, Soto MM, McCullagh E. Unfolded protein stress in the endoplasmic reticulum and mitochondria: a role in neurodegeneration. Front Aging Neurosci. 2012;4:5. doi: 10.3389/fnagi.2012.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shyh-Chang N, Daley GQ, Cantley LC. Stem cell metabolism in tissue development and aging. Development. 2013;140(12):2535–47. doi: 10.1242/dev.091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schreiber KH, Kennedy BK. When lamins go bad: nuclear structure and disease. Cell. 2013;152(6):1365–75. doi: 10.1016/j.cell.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326(5949):140–4. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wu JJ, Liu J, Chen EB, Wang JJ, Cao L, Narayan N, et al. Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of mTOR expression. Cell Rep. 2013;4(5):913–20. doi: 10.1016/j.celrep.2013.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012;335(6076):1638–43. doi: 10.1126/science.1215135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of aging and age-related disease. Nature. 2013;493(7432):338–45. doi: 10.1038/nature11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32(1):2–11. doi: 10.1128/MCB.06159-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rimmele P, Bigarella CL, Liang R, Izac B, Dieguez-Gonzalez R, Barbet G, et al. Aging-like phenotype and defective lineage specification in SIRT1-deleted hematopoietic stem and progenitor cells. Stem Cell Reports. 2014;3(1):44–59. doi: 10.1016/j.stemcr.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.O’Callaghan C, Vassilopoulos A. Sirtuins at the crossroads of stemness, aging, and cancer. Aging cell. 2017;16(6):1208–18. doi: 10.1111/acel.12685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97••.Wang H, Diao D, Shi Z, Zhu X, Gao Y, Gao S, et al. SIRT6 controls hematopoietic stem cell homeostasis through epigenetic regulation of Wnt signaling. Cell stem cell. 2016;18(4):495–507. doi: 10.1016/j.stem.2016.03.005. This study for the first time shows the link between SIRT6 and Wnt signaling in the regulation of HSC homeostasis and self-renewal. [DOI] [PubMed] [Google Scholar]
- 98.Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010;17(1):41–52. doi: 10.1016/j.ccr.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wrighton KH. Stem cells: SIRT7, the UPR and HSC aging. Nat Rev Mol Cell Biol. 2015;16(5):266–7. doi: 10.1038/nrm3981. [DOI] [PubMed] [Google Scholar]
- 100.Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155(7):1624–38. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124(2):315–29. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 102••.Mohrin M, Shin J, Liu Y, Brown K, Luo H, Xi Y, et al. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science. 2015;347(6228):1374–7. doi: 10.1126/science.aaa2361. This study identified SIRT7-NRF1 as a regulator of mitochondrial unfolded protein response and define its significance in rejuvanation of aged HSC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tothova Z, Gilliland DG. FoxO transcription factors and stem cell homeostasis: insights from the hematopoietic system. Cell stem cell. 2007;1(2):140–52. doi: 10.1016/j.stem.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 104.Mohrin M, Chen D. The mitochondrial metabolic checkpoint and aging of hematopoietic stem cells. Curr Opin Hematol. 2016;23(4):318–24. doi: 10.1097/MOH.0000000000000244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ito K, Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol. 2014;15(4):243–56. doi: 10.1038/nrm3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bigarella CL, Li J, Rimmele P, Liang R, Sobol RW, Ghaffari S. FOXO3 transcription factor is essential for protecting hematopoietic stem and progenitor cells from oxidative DNA damage. J Biol Chem. 2017;292(7):3005–15. doi: 10.1074/jbc.M116.769455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128(2):325–39. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 108.Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell stem cell. 2007;1(1):101–12. doi: 10.1016/j.stem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 109.Rimmele P, Liang R, Bigarella CL, Kocabas F, Xie J, Serasinghe MN, et al. Mitochondrial metabolism in hematopoietic stem cells requires functional FOXO3. EMBO Rep. 2015;16(9):1164–76. doi: 10.15252/embr.201439704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110••.Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature. 2017;543(7644):205–10. doi: 10.1038/nature21388. This study demonstrate that autophagy is necessary to preserve the regenerative capacity of old HSC by clearing active, healthy mitochondria and convincingly shows that about 30% of old HSC exhibit high autophagy and retain stemness like yong one. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. The Journal of experimental medicine. 2011;208(3):455–67. doi: 10.1084/jem.20101145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mortensen M, Watson AS, Simon AK. Lack of autophagy in the hematopoietic system leads to loss of hematopoietic stem cell function and dysregulated myeloid proliferation. Autophagy. 2011;7(9):1069–70. doi: 10.4161/auto.7.9.15886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Madeo F, Tavernarakis N, Kroemer G. Can autophagy promote longevity? Nat Cell Biol. 2010;12(9):842–6. doi: 10.1038/ncb0910-842. [DOI] [PubMed] [Google Scholar]
- 114.Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, et al. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature. 2013;494(7437):323–7. doi: 10.1038/nature11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ito K, Turcotte R, Cui J, Zimmerman SE, Pinho S, Mizoguchi T, et al. Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science. 2016;354(6316):1156–60. doi: 10.1126/science.aaf5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Florian MC, Dorr K, Niebel A, Daria D, Schrezenmeier H, Rojewski M, et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell stem cell. 2012;10(5):520–30. doi: 10.1016/j.stem.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kohler A, Schmithorst V, Filippi MD, Ryan MA, Daria D, Gunzer M, et al. Altered cellular dynamics and endosteal location of aged early hematopoietic progenitor cells revealed by time-lapse intravital imaging in long bones. Blood. 2009;114(2):290–8. doi: 10.1182/blood-2008-12-195644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Aguilaniu H, Gustafsson L, Rigoulet M, Nystrom T. Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science. 2003;299(5613):1751–3. doi: 10.1126/science.1080418. [DOI] [PubMed] [Google Scholar]
- 119.Erjavec N, Larsson L, Grantham J, Nystrom T. Accelerated aging and failure to segregate damaged proteins in Sir2 mutants can be suppressed by overproducing the protein aggregation-remodeling factor Hsp104p. Genes Dev. 2007;21(19):2410–21. doi: 10.1101/gad.439307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu B, Larsson L, Caballero A, Hao X, Oling D, Grantham J, et al. The polarisome is required for segregation and retrograde transport of protein aggregates. Cell. 2010;140(2):257–67. doi: 10.1016/j.cell.2009.12.031. [DOI] [PubMed] [Google Scholar]
- 121.Shcheprova Z, Baldi S, Frei SB, Gonnet G, Barral Y. A mechanism for asymmetric segregation of age during yeast budding. Nature. 2008;454(7205):728–34. doi: 10.1038/nature07212. [DOI] [PubMed] [Google Scholar]
- 122.Sengupta A, Duran A, Ishikawa E, Florian MC, Dunn SK, Ficker AM, et al. Atypical protein kinase C (aPKCzeta and aPKClambda) is dispensable for mammalian hematopoietic stem cell activity and blood formation. Proc Natl Acad Sci U S A. 2011;108(24):9957–62. doi: 10.1073/pnas.1103132108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wilson A, Ardiet DL, Saner C, Vilain N, Beermann F, Aguet M, et al. Normal hemopoiesis and lymphopoiesis in the combined absence of numb and numblike. J Immunol. 2007;178(11):6746–51. doi: 10.4049/jimmunol.178.11.6746. [DOI] [PubMed] [Google Scholar]
- 124••.Florian MC, Nattamai KJ, Dorr K, Marka G, Uberle B, Vas V, et al. A canonical to non-canonical Wnt signalling switch in haematopoietic stem-cell aging. Nature. 2013;503(7476):392–6. doi: 10.1038/nature12631. This paper highlights that Wnt5a treatment of young HSC induces aging-associated stem-cell apolarity, reduction of regenerative capacity and an aging-like myeloid-lymphoid differentiation skewing via activation of the small Rho GTPase Cdc42. They also identify that Wnt5a haploinsufficiency attenuates HSC aging, and stem-cell-intrinsic reduction of Wnt5a expression results in functionally rejuvenated aged HSC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Beerman I, Bock C, Garrison BS, Smith ZD, Gu H, Meissner A, et al. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell. 2013;12(4):413–25. doi: 10.1016/j.stem.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sun D, Luo M, Jeong M, Rodriguez B, Xia Z, Hannah R, et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell stem cell. 2014;14(5):673–88. doi: 10.1016/j.stem.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Fraga MF, Esteller M. Epigenetics and aging: the targets and the marks. Trends Genet. 2007;23(8):413–8. doi: 10.1016/j.tig.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 128.Han S, Brunet A. Histone methylation makes its mark on longevity. Trends Cell Biol. 2012;22(1):42–9. doi: 10.1016/j.tcb.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129•.Burman B, Zhang ZZ, Pegoraro G, Lieb JD, Misteli T. Histone modifications predispose genome regions to breakage and translocation. Genes Dev. 2015;29(13):1393–402. doi: 10.1101/gad.262170.115. This article identifies specific histone modifications as facilitators of chromosome breakage and translocations using a combination of bioinformatics, biochemical analysis, and cell-based assays. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Burke B, Stewart CL. Functional architecture of the cell’s nucleus in development, aging, and disease. Curr Top Dev Biol. 2014;109:1–52. doi: 10.1016/B978-0-12-397920-9.00006-8. [DOI] [PubMed] [Google Scholar]
- 131•.Ugarte F, Sousae R, Cinquin B, Martin EW, Krietsch J, Sanchez G, et al. Progressive chromatin condensation and H3K9 methylation regulate the differentiation of embryonic and hematopoietic stem cells. Stem Cell Reports. 2015;5(5):728–40. doi: 10.1016/j.stemcr.2015.09.009. This article identifies global chromatin rearrangements during stem cell differentiation and that heterochromatin formation by H3K9methylation regulates HSC differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Satoh Y, Yokota T, Sudo T, Kondo M, Lai A, Kincade PW, et al. The Satb1 protein directs hematopoietic stem cell differentiation toward lymphoid lineages. Immunity. 2013;38(6):1105–15. doi: 10.1016/j.immuni.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Vas V, Senger K, Dorr K, Niebel A, Geiger H. Aging of the microenvironment influences clonality in hematopoiesis. PLoS One. 2012;7(8):e42080. doi: 10.1371/journal.pone.0042080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Vas V, Wandhoff C, Dorr K, Niebel A, Geiger H. Contribution of an aged microenvironment to aging-associated myeloproliferative disease. PLoS One. 2012;7(2):e31523. doi: 10.1371/journal.pone.0031523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Siclari VA, Zhu J, Akiyama K, Liu F, Zhang X, Chandra A, et al. Mesenchymal progenitors residing close to the bone surface are functionally distinct from those in the central bone marrow. Bone. 2013;53(2):575–86. doi: 10.1016/j.bone.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136••.Kusumbe AP, Ramasamy SK, Itkin T, Mae MA, Langen UH, Betsholtz C, et al. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature. 2016;532(7599):380–4. doi: 10.1038/nature17638. This article demonstrated the role of vascular niches and niche forming vessels in HSC aging, and highlighted the significance of endothelial cells Notch signalling in niche expansion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang Y, Depond M, He L, Foudi A, Kwarteng EO, Lauret E, et al. CXCR4/CXCL12 axis counteracts hematopoietic stem cell exhaustion through selective protection against oxidative stress. Sci Rep. 2016;6:37827. doi: 10.1038/srep37827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tuljapurkar SR, McGuire TR, Brusnahan SK, Jackson JD, Garvin KL, Kessinger MA, et al. Changes in human bone marrow fat content associated with changes in hematopoietic stem cell numbers and cytokine levels with aging. J Anat. 2011;219(5):574–81. doi: 10.1111/j.1469-7580.2011.01423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ergen AV, Boles NC, Goodell MA. Rantes/Ccl5 influences hematopoietic stem cell subtypes and causes myeloid skewing. Blood. 2012;119(11):2500–9. doi: 10.1182/blood-2011-11-391730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chang KH, Sengupta A, Nayak RC, Duran A, Lee SJ, Pratt RG, et al. p62 is required for stem cell/progenitor retention through inhibition of IKK/NF-kappaB/Ccl4 signaling at the bone marrow macrophage-osteoblast niche. Cell Rep. 2014;9(6):2084–97. doi: 10.1016/j.celrep.2014.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141••.Guidi N, Sacma M, Standker L, Soller K, Marka G, Eiwen K, et al. Osteopontin attenuates aging-associated phenotypes of hematopoietic stem cells. EMBO J. 2017;36(7):840–53. doi: 10.15252/embj.201694969. This paper convincingly shows that reduced expression of osteopoetin upon aging limits HSC regeneation, and identified thrombin-cleaved osteopoetin as a novel rejuvenation approach. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Quere R, Saint-Paul L, Carmignac V, Martin RZ, Chretien ML, Largeot A, et al. Tif1gamma regulates the TGF-beta1 receptor and promotes physiological aging of hematopoietic stem cells. Proc Natl Acad Sci U S A. 2014;111(29):10592–7. doi: 10.1073/pnas.1405546111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Blank U, Karlsson S. TGF-beta signaling in the control of hematopoietic stem cells. Blood. 2015;125(23):3542–50. doi: 10.1182/blood-2014-12-618090. [DOI] [PubMed] [Google Scholar]
- 144.Taniguchi Ishikawa E, Gonzalez-Nieto D, Ghiaur G, Dunn SK, Ficker AM, Murali B, et al. Connexin-43 prevents hematopoietic stem cell senescence through transfer of reactive oxygen species to bone marrow stromal cells. Proc Natl Acad Sci U S A. 2012;109(23):9071–6. doi: 10.1073/pnas.1120358109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gonzalez-Nieto D, Li L, Kohler A, Ghiaur G, Ishikawa E, Sengupta A, et al. Connexin-43 in the osteogenic BM niche regulates its cellular composition and the bidirectional traffic of hematopoietic stem cells and progenitors. Blood. 2012;119(22):5144–54. doi: 10.1182/blood-2011-07-368506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Howcroft TK, Campisi J, Louis GB, Smith MT, Wise B, Wyss-Coray T, et al. The role of inflammation in age-related disease. Aging (Albany NY) 2013;5(1):84–93. doi: 10.18632/aging.100531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.De Martinis M, Franceschi C, Monti D, Ginaldi L. Inflamm-aging and lifelong antigenic load as major determinants of aging rate and longevity. FEBS Lett. 2005;579(10):2035–9. doi: 10.1016/j.febslet.2005.02.055. [DOI] [PubMed] [Google Scholar]
- 148.Kovtonyuk LV, Fritsch K, Feng X, Manz MG, Takizawa H. Inflamm-Aging of hematopoiesis, hematopoietic stem cells, and the bone marrow microenvironment. Front Immunol. 2016;7:502. doi: 10.3389/fimmu.2016.00502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bersenev A, Rozenova K, Balcerek J, Jiang J, Wu C, Tong W. Lnk deficiency partially mitigates hematopoietic stem cell aging. Aging cell. 2012;11(6):949–59. doi: 10.1111/j.1474-9726.2012.00862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Norddahl GL, Wahlestedt M, Gisler S, Sigvardsson M, Bryder D. Reduced repression of cytokine signaling ameliorates age-induced decline in hematopoietic stem cell function. Aging cell. 2012;11(6):1128–31. doi: 10.1111/j.1474-9726.2012.00863.x. [DOI] [PubMed] [Google Scholar]
- 151.Pietras EM. Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood. 2017;130(15):1693–8. doi: 10.1182/blood-2017-06-780882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152•.Josefsdottir KS, Baldridge MT, Kadmon CS, King KY. Antibiotics impair murine hematopoiesis by depleting the intestinal microbiota. Blood. 2017;129(6):729–39. doi: 10.1182/blood-2016-03-708594. This paper demonstrated that microbiota sustain steady-state hematopoiesis through activation of Stat1 signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Khosravi A, Yanez A, Price JG, Chow A, Merad M, Goodridge HS, et al. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe. 2014;15(3):374–81. doi: 10.1016/j.chom.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Heintz C, Mair W. You are what you host: microbiome modulation of the aging process. Cell. 2014;156(3):408–11. doi: 10.1016/j.cell.2014.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155••.Han B, Sivaramakrishnan P, Lin CJ, Neve IAA, He J, Tay LWR, et al. Microbial genetic composition tunes host longevity. Cell. 2017;169(7):1249–62 e13. doi: 10.1016/j.cell.2017.05.036. This study highlighted the significance of bacterial genes on host aging and identified mTOR, JNK, Insulin signaling, and caloric restriction as a novel molecular targets. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kennedy BK, Lamming DW. The mechanistic target of rapamycin: The grand conducTOR of metabolism and aging. Cell Metab. 2016;23(6):990–1003. doi: 10.1016/j.cmet.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cheng CW, Adams GB, Perin L, Wei M, Zhou X, Lam BS, et al. Prolonged fasting reduces IGF-1/PKA to promote hematopoietic-stem-cell-based regeneration and reverse immunosuppression. Cell stem cell. 2014;14(6):810–23. doi: 10.1016/j.stem.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Brown K, Xie S, Qiu X, Mohrin M, Shin J, Liu Y, et al. SIRT3 reverses aging-associated degeneration. Cell Rep. 2013;3(2):319–27. doi: 10.1016/j.celrep.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]