

Abstract
4-Substitution on proline directly impacts protein main chain conformational preferences. The structural effects of N-acyl substitution and of 4-substitution were examined by NMR spectroscopy and X-ray crystallography on minimal molecules with a proline 4S-nitrobenzoate. The effects of N-acyl substitution on conformation were attenuated in the 4S-nitrobenzoate context, due to the minimal role of the n→π* interaction in stabilizing extended conformations. By X-ray crystallography, an extended conformation was observed for most molecules. The formyl derivative adopted a δ conformation that is observed at the i+2 position of β-turns. Computational analysis indicated that the structures observed crystallographically represent the inherent conformational preferences of 4S-substituted prolines with electron-withdrawing 4-position substituents. The divergent conformational preferences of 4R- and 4S-substituted prolines suggest their wider structure-specific application in molecular design. In particular, the proline endo ring pucker favored by 4S-substituted prolines uniquely promotes the δ conformation ((ϕ, ψ) ~ (–80°, 0°)) found in β-turns. In contrast to other acyl capping groups, the pivaloyl group strongly promoted trans amide bond and polyproline II helix conformation, with a close n→π* interaction in the crystalline state, despite the endo ring pucker, suggesting its special capabilities in promoting compact conformations in ϕ due to its strongly electron-donating character.
Graphical Abstract
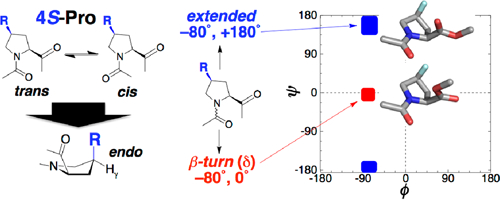
Conformational control: 4S-substituted prolines with electron-withdrawing 4-substitutents strongly promote the endo ring pucker and the δ conformation central to β-turns (ϕ, ψ = −80°, 0°), as determined by X-ray crystallography and calculations. This work suggests their broader application in protein design, medicinal chemistry, and catalysis.
Introduction
The proline exo and endo ring puckers have distinct effects on conformational preferences in the protein main chain (torsion angles ϕ, ψ, and ω).[1] The exo ring pucker (Cγ puckered away from the proline carbonyl; sometimes referred to as the “up” ring pucker) is associated with more compact conformations in ϕ (PPII, α-helix) (Figure 1).[1c, 2] In contrast, the endo ring pucker (Cγ puckered toward the proline carbonyl; “down” ring pucker) is associated with more extended conformations in ϕ. In addition, the endo ring pucker is observed to favor the δ (ϕ, ψ ~ −90°, 0°) conformation that is observed at the i+2 position of β-turns.
Figure 1.

Proline conformational equilibria. (a) Proline exo and endo ring puckers. Puckering of proline at Cγ results in two envelope conformations of the pyrrolidine ring, the exo and endo ring puckers. Nomenclature is based on puckering of the γ-carbon toward (endo) or away from (exo) the carbonyl of the same residue. (b) Proline trans and cis amide conformations. Proline cis–trans isomerization has an activation barrier of ~20 kcal mol–1 (timescale of seconds to minutes at room temperature), whereas proline ring pucker interconversion has an activation barrier of 2–5 kcal mol–1 (timescale of picoseconds at room temperature).[1c, 2]
The n→π* interaction between consecutive carbonyls in proteins stabilizes compact conformations, but only minimally stabilizes more extended conformations.[3] In the n→π* interaction of the protein main chain, the lone pair (n) of one carbonyl is positioned in a manner that allows orbital overlap with the π* antibonding orbital of the subsequent carbonyl, with resultant interresidue electron delocalization. This n→π* interaction is most readily identified by Oi…Ci+1 intercarbonyl distances that are substantially below the 3.22 Å sum of the van der Waals radii of carbon (1.70 Å) and oxygen (1.52 Å). The most favorable n→π* interactions also have an OiCi+1Oi+1 angle close to the Bürgi-Dunitz trajectory (O…C=O angle ~ 109°) that allows maximal orbital overlap with the π* orbital. In addition, in high-resolution structures, pyramidalization can be observed in the acceptor carbonyl, in a manner analogous to early stages of a nucleophilic attack of one carbonyl oxygen on the other carbonyl carbon.[4]
We previously demonstrated that the interaction strength of an n→π* interaction could be modulated via the identity of acyl capping groups that change the electronic properties of the donor carbonyl.[5] These experiments were conducted using the 4R-nitrobenzoate ester of hydroxyproline, which strongly promotes an exo ring pucker and thus promotes conformations with a favorable n→π* interaction.[6] The observed conformations of 4-substituted prolines are dependent on the stereochemistry and the balance of the electronic versus steric effects of the 4-substituent.[3a, 6b, 7] The conformational effects of the nitrobenzoate, similar to those of fluorine,[6a, 6b] are due to the highly electron-withdrawing nature of the nitrobenzoate group, which leads to its strong preference to be in a pseudo-axial position on the pyrrolidine ring. Effects were observed energetically, via van’t Hoff enthalpies of trans-cis isomerization equilibria (ω torsion angle)), as well as crystallographically and computationally, via direct effects on the ϕ and ψ torsion angles. Thus, electron-donating pivaloyl, iso-butyryl, and propionyl groups exhibited conformations (α-helix, polyproline II helix) with more compact values of ϕ (–45° to −60°) and shorter intercarbonyl distances (as close as 2.68 Å), compared to structures with the acetyl group. In contrast, less electron-donating acyl capping groups (fluoroacetyl, formyl, trifluoroacetyl) exhibited larger intercarbonyl distances (3.05–3.34 Å) and significantly more extended conformations, despite the presence of the 4R-nitrobenzoate. These data indicated that the strength of the n→π* interaction directly impacts the ϕ and ψ main chain torsion angles, and that the identity of acyl capping groups represents an additional approach to promote defined conformational preferences.
4R-Substituted prolines stabilize the polyproline II helix (PPII) conformation present at the Yaa position of collagen, and more generally stabilize the PPII helix via the n→π* interaction.[3a, 7g, 8] In contrast, 4S-substituted prolines disfavor α-helix and PPII conformations and adopt more extended conformations, including the Xaa position in collagen, where the n→π* interaction minimally contributes energetically.[7a, 7b, 7e, 7f, 9] However, a broader understanding of the conformational preferences of 4S-substituted prolines could lead to greater application of these amino acids beyond collagen-mimetic peptides or proteins with cis-proline amide bonds.[7d] In order to examine this structural landscape, the effects of the acyl capping group were investigated within the context of 4S-substituted prolines, which promote conformations that are not significantly stabilized by n→π* interactions.
Results and Discussion
Prolines that are 4S-substituted with electron-withdrawing groups promote the endo ring pucker.[7a, 10] This ring pucker preference is due to a gauche effect, in which the 4S-substituent is gauche to the adjacent Cβ–Cα or Cδ–N bonds, which results in the substituent being pseudo-axial on the pyrrolidine ring (Figure 2). This sterically disfavored conformation is preferred because it results in the 4S-substituent being anti-periplanar to two C–H bonds (C–Hβ and C–Hδ) (Figure 2b).[11] This conformation is stabilized due to hyperconjugation between the σC–H of the electron-rich C–H bonds and the σ* antibonding orbital of the C–X bond, which are syn-periplanar (eclipsing) to one another, maximizing orbital overlap and electron delocalization. Thus, more electron-withdrawing proline 4-substituents are better electron acceptors both due to more polarized C–X bonds and due to lower energies of the C–X σ*, resulting in greater stabilization due to hyperconjugation and a greater preference for the gauche conformation. Within 4S-substituted prolines, the stabilizing effects of hyperconjugation thus lead to a stronger preference for the endo ring pucker. Because ring pucker in prolines is associated with main chain conformation, a greater preference for an endo ring pucker thus leads to both (1) greater preference for extended or δ conformations and (2) a greater likelihood of cis proline amide bond.
Figure 2.

Side chain conformational preferences of 4S-hydroxyproline nitrobenzoate (hyp(4-NO2-Bz), hnb). (a) The nitrobenzoate preferentially adopts the sterically disfavored endo conformation, in which the nitrobenzoate is pseudo-axial on the pyrrolidine ring. (b) Hyperconjugative effects promote the endo ring pucker due to the presence of two C–H bonds (C–Hβ and C–Hδ) anti to the nitrobenzoate. This conformation results in electron delocalization, via favorable orbital overlap between σC–H of the electron-rich C–H bonds and the σ* antibonding orbital of the C–O bond. Localized depictions of key orbital lobes and limiting gauche conformations (CCCO = 60° or HCCO = 180°) are shown. Observed HCCO torsion angles are typically 150–170° in 4S-substituted prolines (vide infra and ref. [6c]).
The effects of 4-substitution on conformation have been extensively examined within collagen model peptides, simple acetylated amino acid methyl esters, and other short model peptides, typically using the ω torsion angle (Ktrans/cis) to quantify conformational effects in model compounds and Tm to quantify conformational effects in collagen model peptides.[3a, 6a, 6b, 7a, 7b, 7d, 7e, 10a, 12] In addition, 4-substituted prolines have been examined within a limited number of globular proteins.[7d, 9b, 13] In these cases, matching of the stereochemistry of the 4-substituent to the crystallographically observed ring pucker and main chain conformation typically leads to stabilization of the protein structure. In contrast, a mismatch of the proline stereochemistry and the observed conformation at proline leads to protein destabilization. In addition, 4-substituted prolines have been incorporated in pharmaceuticals and within asymmetric catalysts.[11b, 14]
Despite the increasing usage of 4-substituted prolines in medicinal chemistry, protein design, and catalysis, the broad effects of 4-substitution on main chain conformation (particularly ϕ and ψ) have not yet been fully appreciated outside of work in collagen model peptides and other examples in protein design. We sought to more generally investigate the conformational landscape of 4S-substituted prolines and to examine the role of capping groups on conformation within 4S-substituted prolines. Therefore, a series of molecules was synthesized based on 4S-hydroxyproline nitrobenzoate methyl ester (Scheme 1). Molecules were synthesized with pivaloyl, iso-butyryl, acetyl, trifluoroacetyl, formyl, and Boc N-acyl groups, which represent a range of steric and electronic properties, as well as with the free amine.
Scheme 1.

Synthesis of 4S-hydroxyproline nitrobenzoate methyl ester derivatives.
All molecules were analyzed by NMR spectroscopy, in order to quantify the effect of the acyl capping group in solution on the equilibrium between trans and cis amide bonds (Ktrans/cis) (Table 1). As expected and as has been observed previously in various contexts, most molecules with the 4S-nitrobenzoate diastereomer exhibited a significantly higher population of cis amide bond compared to molecules with the 4R-nitrobenzoate. In contrast to prior data with 4R-nitrobenzoates, here, a smaller dependence of Ktrans/cis on donor carbonyl electronic properties was also observed, with the differences in Ktrans/cis more readily explained by steric effects, whereby larger acyl groups sterically disfavor the cis amide conformation. The endo ring pucker exhibits weaker n→π* interactions, due to the more extended conformation associated with the endo ring pucker, consistent with the data herein. In contrast to other molecules, the pivaloyl derivative exhibited exclusively trans amide bond by NMR spectroscopy. The size of the pivaloyl group strongly disfavors a cis amide bond, which would introduce a large steric clash with the proline carbonyl (Figure 1b).[15]
Table 1.
Summary of the NMR data of the (2S,4S)-4-nitrobenzoyl-hydroxyproline methyl ester derivatives, with relative populations of trans and cis conformations determined via 1H NMR spectroscopy in CDCl3 at 298 K.
| compound | X(C=O)-hyp(Nbz)-OCH3X= | Ktrans/cisa | ΔGtrans/cis,bkcal mol−1 |
| 5 | C(CH3)3 (Piv) | > 20c | < −1.8 |
| 6 | CH(CH3)2 (i-But) | 2.5 | −0.54 |
| 7 | CH3 (Ac) | 1.9 | −0.38 |
| 8 | CF3 (Tfa) | 2.8 | −0.61 |
| 9 | H (For) | 0.7 | +0.21 |
| 3 | OC(CH3)3 (Boc) | 0.8d | +0.12 |
Ktrans/cis = ratio of the population of the species with trans amide bond to the population of the species with cis amide bond.
ΔGtrans/cis = – RT ln Ktrans/cis.
Peaks corresponding to the cis isomer were not observed via 1H NMR spectroscopy.
Analysis of the Hα and other chemical shifts is consistent with the cis isomer being the major species.
The nitrobenzoate group promotes crystal formation in compounds. Within 4R-substituted hydroxyproline nitrobenzoates, we previously observed that the electronic properties of the acyl capping group dramatically impacted the ϕ and ψ torsion angles and n→π* interaction intercarbonyl distances, consistent with stronger n→π* interactions with more electron-rich acyl donor carbonyls.[5b] Therefore, crystallization was attempted on all compounds, in order to identify the inherent conformational effects of 4S-substituted nitrobenzoates and to examine the ability of acyl donor groups to modulate the conformational preferences.
Single-crystal X-ray structures were solved for compounds 3–9 (Figure 3, Table 2).[16] In general, crystal assembly was mediated by aromatic slip-stacking of the nitrobenzoate groups.[17] In addition, in all structures, C–H/O interactions with multiple H…O distances significantly (0.2–0.4 Å) below the 2.72 Å sum of the van der Waals radii of H and O were observed at the intermolecular interfaces in the crystals (Figure S26).[18] Proline residues exhibit favorable C–H/O interactions in proteins, due in part to the presence of polarized C–H bonds at Hα and at both Hδ.[19] In 4-substituted prolines, Hγ is also significantly polarized, rendering these molecules particularly rich in C–H bonds for intermolecular assembly via C–H/O interactions.
Figure 3.

X-ray crystal structures of derivatives of 4S-hydroxyproline nitrobenzoate methyl ester. ϕ and ψ torsion angles are indicated in magenta. Intercarbonyl distances (d, Oi…Ci+1) are indicated in red for molecules with trans amide bonds. All molecules exhibited an endo ring pucker crystallographically.
Table 2.
Summary of conformational data from crystal structures of 4S- and 4R- hydroxyproline nitrobenzoate methyl esters and other 4-substituted proline derivatives.a
| 4S-substituted | 4R-substituted | |||||||
|---|---|---|---|---|---|---|---|---|
| X-(C=O)- | X–ray crystallography | X-(C=O)- | X–ray crystallography | |||||
| hyp(Nbz)-OMe | Hyp(Nbz)-OMeb | molecule 1 | molecule 2 | |||||
| X= | ϕ, ψ ° | ω ° | d, Å | X= | ϕ, ψ ° | d, Å | ϕ, ψ ° | d, Å |
| C(CH3)3 (5) (Piv) | −57, +157 | +176 | 2.759 | C(CH3)3 (Piv) | −56, +138 | 2.681 | – | – |
| CH(CH3)2 (6) (i-But) | −76, −165 | +5 | – | CH(CH3)2 (i-But) | −45, −44 | 2.687 | −47, −40 | 2.738 |
| CH3 (7) (Ac) | −85, −171 | +10 | – | CH3 (Ac) | −65, −34 | 2.991 | −69, −29 | 3.049 |
| CF3 (8) (Tfa) | −73, +174 | −179 | 3.095 | CF3 (Tfa) | −74, +167 | 3.099 | – | – |
| H (9) (For) | −84, +14 | −1 | – | H (For) | −76, +163 | 3.197 | −84, +173 | 3.336 |
| OC(CH3)3 (3) (Boc) | −71, −173 | −7 | – | |||||
| Fmoc-hyp(4-I-Ph)-OHc | −83, +177 | +9 | – | |||||
| Boc-hyp(4-I-Ph)-OHc | −76, +1 | +1 | – | |||||
| Boc-Hyp(4-I-Ph)-OMec | −52, −39 | 2.836 | ||||||
| Ac-hyp-OMed | −84, +18 | 0 | – | |||||
| Ac-Hyp-OMee | −51, +145 | 2.751 | −63, +156 | 2.898 | ||||
| Ac-Hyp-OHf | −58, −32 | 2.832 | ||||||
| Boc-Hyp-OMeg | −55, −;32 | 2.886 | ||||||
| Boc-hyp(Me)-OMed | −85, +18 | −1 | – | |||||
| Ac-Hyp(Me)-OMeh | −58, +148 | 2.842 | ||||||
| Ac-Flp-OMee | −55, +140 | 2.752 | −56, +141 | 2.778 | ||||
Torsion angles and intercarbonyl distances were measured from the crystallographic structures. d = distance from Oi to Ci+1. hyp = (2S,4S)-hydroxyproline. Hyp = (2S,4R)-hydroxyproline. Nbz = nitrobenzoate ester. 4-I-Ph = 4-iodophenyl ether.
Ref. [5b]. Most structures had two molecules in the unit cell; both structures are indicated in these cases.
Ref. [10b], CSD EMITEA and EMITIE.
Ref. [20b], CSD RISDAY and RISDEC.
Ref. [20c], CSD EXOBIE.
Ref. [20d], CSD INEMIX.
Ref. [12b], CSD SODJEB.
These structures were analyzed for their conformational features (Figure 3) and also were compared to the equivalent 4R-hydroxyproline nitrobenzoates (Figure 4, Table 2).[5b, 10b, 12b, 20] All proline 4S-nitrobenzoates exhibited an endo ring pucker, consistent with the strong stereoelectronic effects of the nitrobenzoate group. Indeed, the non-acylated molecules, which lack a donor carbonyl capable of inducing an n→π* interaction, also exhibited an endo ring pucker crystallographically, both as the free amine and as the ammonium. Of the other molecules, two had a trans amide bond, while four had a cis amide bond, consistent with the weak trans-cis amide rotameric preference of proline with an endo ring pucker.
Figure 4.

Comparison of the structures of the 4S- (blue text, white carbons, top) and 4R- (red text, grey carbons, bottom) diastereomers of 4-hydroxyproline nitrobenzoate methyl esters, as a function of acyl N-capping group. Diastereomers were subjected to superposition of the N, Cα, and Cδ atoms using Chimera.[30] Nbz = 4-nitrobenzoyl.
Among the derivatives with a cis amide bond, an extended conformation was observed with the iso-butyryl, acetyl, and Boc N-acyl groups. These data are consistent with the significant preference for an extended conformation in proline with an endo ring pucker, as well as the weaker n→π* interaction inherent to the endo ring pucker.[1a, 3c] Notably, the formyl derivative exhibited the δ conformation (~ −90°, 0°), which is observed at the i+2 position of β-turns.[21] Moreover, the conformations observed and the differences between the 4S- and the 4R- diastereomers were similar to those found in previously reported diastereomeric pairs of 4-substituted prolines (Table 2), suggesting that these results are general.[5b, 10b, 12b, 20] β-Turns are important protein conformations and are common epitopes for protein recognition.[22] These crystallographic data suggest the specific utility of 4S-substituted prolines to promote β-turn conformations, as well as more extended proline conformations, with potential applications in medicinal chemistry and protein design.[9b, 23]
Among molecules with a trans amide bond, the trifluoroacetyl derivative exhibited an extended conformation and a longer intercarbonyl distance, consistent with a weak n→π* interaction that is due to both an endo ring pucker and an electron-poor donor carbonyl. In contrast, the pivaloyl derivative exhibited a compact value of ϕ (–57°) and a close intercarbonyl distance (d = 2.76 Å). This conformation is unusually compact for a proline endo ring pucker, suggesting that the pivaloyl group promotes particularly favorable n→π* interactions. Indeed, in our prior work on 4R-hydroxyproline nitrobenzoates, we observed an unusually close n→π* interaction for the pivaloyl derivative. Collectively, these data indicate that the pivaloyl group is unique in its ability to induce conformations with compact values of ϕ, in addition to its known preference for trans amide bonds.
In order to further understand the effects of 4S-substitution and acyl capping group on proline conformation, computational investigations were conducted.[1b, 1c, 2, 5b, 24] In these calculations, fluorine was used in place of the hydroxyproline nitrobenzoate, due to the similar effects on conformation of both groups[6a, 6b] and the substantially greater computational simplicity of fluorine compared to a nitrobenzoate. While the nitrobenzoate group is substantially larger than fluorine, the aromatic ring is rotated away from the proline main chain in the crystal structures herein and observed previously[5b], and thus is not likely to significantly impact the proline conformational preferences. We similarly observed in crystal structures of 4R- and 4S-iodophenyl hydroxyprolines that the aromatic ring was rotated away from the peptide main chain.[20a] Thus, we expect that these computational results should be general for a significant number of 4-substituted prolines with electron-withdrawing 4-substituents. Indeed, in our analysis of the conformational effects of a wide range of 4-substitued prolines within peptides, we saw broad similarities in the NMR spectra of peptides with the same stereochemsitry and similar electronic properties.[6b]
Molecules were analyzed with 4S-fluoroproline methyl ester as a function of acyl group (pivaloyl, iso-butyryl, acetyl, trifluoroacetyl, formyl), amide bond conformation (trans versus cis), and quadrant of the Ramachandran plot. These calculations were all conducted using the endo ring pucker preferred by 4S-fluoroproline. These calculations confirmed that, for 4S-substituted prolines, the identity of the acyl donor only minimally impacts conformation for most acyl groups, consistent with the weak n→π* interaction associated with the endo ring pucker (Table S55). In contrast, the pivaloyl group computationally exhibited a preference for a more compact conformation of ϕ and closer intercarbonyl distances, as had been observed crystallographically. Natural bond orbital (NBO) analysis[25] confirmed these conclusions on n→π* interactions, with significantly greater orbital overlap between the donor carbonyl oxygen p-like orbital (np) and the acceptor carbonyl π* orbital for the pivaloyl derivative compared to the formyl derivative (Figure 5). Collectively, the data indicate that the pivaloyl group uniquely promotes compact values of ϕ due to particularly favorable n→π* interactions that result from the electron-rich nature of the pivaloyl carbonyl, consistent with the computational results on fluoroprolines and the crystallographic results observed for 4R- and 4S-hydroxyproline nitrobenzoates.
Figure 5.

Natural bond orbital (NBO) analysis[25] of geometry-optimized structures of Piv-flp-OMe and For-flp-OMe, with a trans amide bond, endo ring pucker, and PPII/β conformation. By this analysis, the conformation in the pivaloyl derivative is stabilized by 0.52 kcal mol–1 by the n→π* interaction, while in the formyl derivative the conformation is only stabilized by 0.12 kcal mol–1. These energy differences are reflected in the differing extents of orbital overlap and the divergent Oi…Ci+1 intercarbonyl distances (Piv: d = 2.92 Å; For: d = 3.17 Å). Additional computational analysis as a function of acyl capping group and conformation is included in the Supporting Information.
In order to further understand the effects of proline 4-substitution on conformation, computational investigations were conducted on acetyl 4R- and 4S-fluoroproline methyl ester, as a function of proline amide rotamer (trans versus cis), ring pucker (exo versus endo), and quadrant of the Ramachandran plot (αR/δ versus PPII/β).[1b, 1c, 2, 5b, 24] Each combination of diastereomer and conformation was subjected to geometry optimization, and the molecules analyzed for optimized conformation and relative energies. (Figure 6, Table 3). These data confirm the previously observed inherent strong preference of 4R-fluoroproline (Flp) for a trans amide bond and an exo ring pucker, as well as the strong preference of 4S-fluoroproline (flp) for an endo ring pucker and its weaker preference for a trans amide bond. In addition, these calculations are consistent with the previously observed preference of the exo ring pucker for more compact conformations, and of the endo ring pucker for more extended conformations, independent of fluoroproline stereochemistry. Moreover, these data are consistent with the ability of the fluoroprolines to adopt both PPII/β conformations and α-helical conformations, suggesting broad applications of 4-substituted prolines for conformational control in medicinal chemistry and protein design.
Figure 6.

Conformational landscape of 4S- and 4R-fluoroprolines as determined by computational investigations. Summary of computational results on (a) Ac-flp-OMe and (b) Ac-Flp-OMe. 4S-Fluoroproline (flp) (a, top) prefers an endo ring pucker, with both trans and cis amide bonds similar in energy. In contrast, 4R-fluoroproline (Flp) (b, bottom) prefers an exo ring pucker and a trans amide bond. Notably, for Ac-Flp-OMe, the exo ring is preferred in the cis amide conformation, while for Ac-flp-OMe the endo ring pucker typically observed for cis-proline is preferred, leading to substantially different preferred main chain conformations with either the trans or the cis amide conformation. Boxes indicate the lowest energy conformations for each diastereomer (conformations with energies within 0.6 kcal mol–1 of the lowest energy conformation identified via DFT calculations). Conformations with energies > 2.8 kcal mol–1 higher than the lowest energy conformation are not shown.
Table 3.
Summary of computational results on calculations on acetylated methyl esters of 4S-fluoroproline (flp) and 4R-fluoroproline (Flp).a
| O…C=O | Erel, | ||||||
|---|---|---|---|---|---|---|---|
| molecule | amide | pucker | quadrant | d, Å | ϕ, ° | ψ, ° | kcal mol−1 |
| Ac-flp-OMe | trans | endo | αP/δ | 3.19 | −75 | −3 | 0.4 |
| trans | endo | PPII/β | 3.10 | −72 | 173 | 0.0 | |
| cis | endo | αP/δ | −80 | 6 | 0.6 | ||
| cis | endo | PPII/β | −78 | −175 | 0.2 | ||
| trans | exo | αP/δ | 2.79 | −54 | −37 | 2.1 | |
| trans | exo | PPII/β | 2.78 | −57 | 144 | 1.9 | |
| cis | exo | αP/δ | −59 | −34 | 3.7 | ||
| cis | exo | PPII/β | −61 | 158 | 3.3 | ||
| Ac-Flp-OMe | trans | exo | αP/δ | 2.79 | −54 | −38 | 0.2 |
| trans | exo | PPII/β | 2.78 | −56 | 142 | 0.0 | |
| cis | exo | αP/δ | −58 | −35 | 1.6 | ||
| cis | exo | PPII/β | −60 | 157 | 1.4 | ||
| trans | endo | αP/δ | 2.89 | -60 | -34 | 2.4 | |
| trans | endo | PPII/β | 2.93 | −65 | 153 | 1.9 | |
| cis | endo | αP/δ | −72 | −21 | 3.5 | ||
| cis | endo | PPII/β | −75 | 167 | 2.9 |
Calculations were conducted at the DFT level of theory with the M06–2X method and the 6–311++G(3d,3p) basis set in implicit water. All structures are the result of geometry optimizations with the given combination of amide conformation (trans or cis), ring pucker (exo or endo), and quadrant of the Ramachandran plot (α-helix(αR)/δ conformation or PPII/extended(β) conformation). All energies are relative to the lowest energy conformation of the indicated diastereomer. All conformations within 0.6 kcal mol–1 of the lowest energy conformation are indicated in color.
Notably, the fluoroproline diastereomers adopt relatively similar conformations with an exo ring pucker, with a compact value of ϕ and similar intercarbonyl n→π* interaction distances observed in either the α-helical or PPII conformation. In contrast, the observed conformations were different with an endo ring pucker. Whereas 4R-fluoroproline adopts a somewhat more extended αR conformation with an endo ring pucker compared to that observed with an exo ring pucker, 4S-fluoroproline adopts distinct conformations with the endo ring pucker. 4R-Fluoroproline prefers α-helical and PPII conformations with an endo ring pucker. In contrast, 4S-fluoroproline was observed in particular to prefer a more extended or β-turn/δ conformation, with significantly more extended values of ϕ (~−80°), and with ψ either ~ 0° or +180°. These conformational preferences were observed in both the trans and cis amide bonds. Combined with the divergent relative energies of these conformations for the 4R- and 4S-fluoroprolines, these calculations and the analogous structures determined by X-ray crystallography provide strong experimental and computational support that proline diastereomers with electron-withdrawing 4-substituents exhibit distinct conformational landscapes.
4R-Substituted prolines prefer more compact conformations with both the trans and cis amide rotamers, with α-helical and polyproline helix conformation and compact values of ϕ stabilized. In contrast, 4S-substituted prolines exhibit more extended values of ϕ and ψ. The conformational preferences observed computationally are similar to crystallographic data herein and previously reported for diastereomers of 4R- and 4S-substituted prolines (Figure 4, Table 2). Notably, this work indicates that 4S-substituted prolines strongly promote the δ conformation central to β-turns (ϕ, ψ = −80°, 0°). This conformation (or its mirror image, which would be accessible via the commercially available D-hydroxyproline) is present in type I, I’, II, II’, and VIa1 β-turns, which collectively represent the vast majority of β-turns.[21] β-Turns are central to protein structure and function, and optimized β-turns are widely employed in molecular design.[26] β-Turns are also common recognition elements in proteins, including being a major conformation recognized by GPCRs, and thus are of significant interest in medicinal chemistry.[22] The work herein demonstrates that 4S-substituted prolines are unique in their ability to favor this conformation. Collectively, these results suggest the specific consideration of the distinct conformational preferences of 4-substituted prolines as a central component to their application.
Conclusion
The conformational preferences of proline with an electron-withdrawing 4S-substituent were examined by X-ray crystallography, NMR spectroscopy, and computational investigations. A series of derivatives of 4S-hydroxyproline nitrobenzoate methyl ester was synthesized with acyl N-capping groups that varied in their steric and electronic properties. All molecules exhibited an endo ring pucker crystallographically. For most acyl derivatives, only a modest preference for a trans versus cis amide bond was observed, with minimal evidence of a significant n→π* interaction with the endo ring pucker. In this context, steric effects were the major determinant of trans versus cis amide bond. More extended conformations were observed for 4S-substitution than were observed with 4R-substituion, with ϕ ~ −80° and ψ ~ 0° or 180° seen crystallographically for the 4S-hydroxyproline nitrobenzoates and computationally for 4S-fluoroprolines. The δ conformation adopted in β-turns in particular was observed both crystallographically and computationally as a preferred conformation of 4S-substituted prolines with an endo ring pucker. Given the importance of β-turns in protein structure and as recognition motifs in biology and medicinal chemistry, these results suggest the specific application of 4S-substituted prolines in these contexts. Direct comparison of the structures of the 4R- and 4S- diastereomers of a series of derivatives emphasizes the distinct conformational preferences of the 4-substituted prolines. The 4R-substituted prolines prefer an exo ring pucker and promote compact conformations that are stabilized by n→π* interactions. In contrast, the 4S-substituted prolines prefer an endo ring pucker and promote more extended conformations in ϕ and/or ψ. Notably, even in the 4S-hydroxyproline nitrobenzoate, the pivaloyl group uniquely induced a compact value of ϕ and a close n→π* interaction, with the molecule in a polyproline II helix conformation. These results suggest a special ability of the pivaloyl group to promote the n→π* interaction and polyproline II helix.
Experimental
Synthesis.
All compounds were synthesized using variations on previously described methods. Details are in the Supporting Information.
NMR spectroscopy.
Compounds 3–9 were analyzed by NMR spectroscopy in CDCl3, with 32,768 data points and a relaxation delay of 2.0 s. The populations of the trans and cis rotamers were quantified via the Hα resonances using baseline-corrected spectra. NOESY experiments were conducted on 7 and 9 to confirm the assignments of the trans and cis amide rotamers. Additional details, expansions of key spectral regions, and NOESY data are in the Supporting Information.
X-ray crystallography.
Compounds crystallized from solutions in ethyl acetate or from solutions of ethyl acetate in hexanes. Structures were determined by X-ray diffraction and have been deposited in the Cambridge Structural Database under CCDC 1914568–1914575.[16] Additional details are in the Supporting Information.
Computational chemistry.
Calculations were conducted with Gaussian09.[27] Natural bond orbital (NBO) analysis[25] was conducted using the implementation of NBO within Gaussian. Visualization was conducted with GaussView 5, using isovalues of 0.02 for molecular orbitals. Geometry optimization was conducted using the M06–2X DFT method.[28] Initial models were generated using proline derivatives with each combination of the following conformational pairs: trans or cis amide bonds; exo or endo ring pucker; and combinations of ϕ and ψ corresponding to α-helix or polyproline II helix. Models were initially optimized using the 6–311++G(d,p) basis set.[29] These initial models were subsequently optimized using the 6–311++G(2d,2p), then 6–311++G(3d,3p), basis sets, using implicit water solvation (IEFPCM continuum polarization model). These models were subjected to frequency analysis. If necessary, geometry optimization was continued until there were zero imaginary frequencies. All final models had zero imaginary frequencies.
Supplementary Material
Acknowledgements
We thank NSF (CHE-1412978) for funding. We thank the David A. Plastino Undergraduate Research Scholars Program for support for NAW. Instrumentation support was provided by NIH (GM110758) and NSF (CHE-1229234). Molecular graphics in Figure 4 were created using UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311.
Footnotes
Supporting Information Available
Experimental procedures, NMR data on 3–9, NOESY spectra for 7 and 9, X-ray crystallographic data, additional analysis of computational data, 1H and 13C NMR spectra for all new compounds, and coordinates for all geometry-optimized structures.
References
- [1].a Milner-White EJ, Bell LH, Maccallum PH, J. Mol. Biol 1992, 228, 725–734; [DOI] [PubMed] [Google Scholar]; b Vitagliano L, Berisio R, Mastrangelo A, Mazzarella L, Zagari A, Protein Sci. 2001, 10, 2627–2632; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kang YK, Choi HY, Biophys. Chem 2004, 111, 135–142; [DOI] [PubMed] [Google Scholar]; d Ho BK, Coutsias EA, Seok C, Dill KA, Protein Sci. 2005, 14, 1011–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Aliev AE, Bhandal S, Courtier-Murias D, J. Phys. Chem. A 2009, 113, 10858–10865. [DOI] [PubMed] [Google Scholar]
- [3].a Bretscher LE, Jenkins CL, Taylor KM, DeRider ML, Raines RT, J. Am. Chem. Soc 2001, 123, 777–778; [DOI] [PubMed] [Google Scholar]; b Bartlett GJ, Choudhary A, Raines RT, Woolfson DN, Nat. Chem. Biol 2010, 6, 615–620; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Newberry RW, Raines RT, Acc. Chem. Res 2017, 50, 1838–1846; [DOI] [PMC free article] [PubMed] [Google Scholar]; d Singh SK, Das A, Phys. Chem. Chem. Phys 2015, 17, 9596–9612; [DOI] [PubMed] [Google Scholar]; e Singh SK, Mishra KK, Sharma N, Das A, Angew. Chem., Int. Ed 2016, 55, 7801–7805. [DOI] [PubMed] [Google Scholar]
- [4].Choudhary A, Newberry RW, Raines RT, Org. Lett 2014, 16, 3421–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a Newberry RW, VanVeller B, Guzei IA, Raines RT, J. Am. Chem. Soc 2013, 135, 7843–7846; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wenzell NA, Ganguly HK, Bhatt MR, Yap GPA, Zondlo NJ, ChemBioChem 2019, 20, 963–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a Thomas KM, Naduthambi D, Tririya G, Zondlo NJ, Org. Lett 2005, 7, 2397–2400; [DOI] [PubMed] [Google Scholar]; b Pandey AK, Naduthambi D, Thomas KM, Zondlo NJ, J. Am. Chem. Soc 2013, 135, 4333–4363; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Pandey AK, Yap GPA, Zondlo NJ, J. Org. Chem 2014, 79, 4174–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a Hodges JA, Raines RT, J. Am. Chem. Soc 2003, 125, 9262–9263; [DOI] [PubMed] [Google Scholar]; b Horng JC, Raines RT, Protein Sci. 2006, 15, 74–83; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Shoulders MD, Hodges JA, Raines RT, J. Am. Chem. Soc 2006, 128, 8112–8113; [DOI] [PMC free article] [PubMed] [Google Scholar]; d Renner C, Alefelder S, Bae JH, Budisa N, Huber R, Moroder L, Angew. Chem., Int. Ed 2001, 40, 923–925; [PubMed] [Google Scholar]; e Umashankara M, Babu IR, Ganesh KN, Chem. Commun 2003, 2606–2607; [DOI] [PubMed] [Google Scholar]; f Kim W, Hardcastle KI, Conticello VP, Angew. Chem., Int. Ed 2006, 45, 8141–8145; [DOI] [PubMed] [Google Scholar]; g Sonntag LS, Schweizer S, Ochsenfeld C, Wennemers H, J. Am. Chem. Soc 2006, 128, 14697–14703. [DOI] [PubMed] [Google Scholar]
- [8].a Babu IR, Ganesh KN, J. Am. Chem. Soc 2001, 123, 2079–2080; [DOI] [PubMed] [Google Scholar]; b Persikov AV, Ramshaw JAM, Kirkpatrick A, Brodsky B, J. Am. Chem. Soc 2003, 125, 11500–11501. [DOI] [PubMed] [Google Scholar]
- [9].a Doi M, Nishi Y, Uchiyama S, Nishiuchi Y, Nakazawa T, Ohkubo T, Kobayashi Y, J. Am. Chem. Soc 2003, 125, 9922–9923; [DOI] [PubMed] [Google Scholar]; b Kim W, McMillan RA, Snyder JP, Conticello VP, J. Am. Chem. Soc 2005, 127, 18121–18132; [DOI] [PubMed] [Google Scholar]; c Lin Y-J, Horng J-C, Amino Acids 2014, 46, 2317–2324; [DOI] [PubMed] [Google Scholar]; d Tressler CM, Zondlo NJ, J. Org. Chem 2014, 79, 5880–5886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a Erdmann RS, Kumin M, Wennemers H, Chimia 2009, 63, 197–200; [Google Scholar]; b Shoulders MD, Kotch FW, Choudhary A, Guzei IA, Raines RT, J. Am. Chem. Soc 2010, 132, 10857–10865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a Thiehoff C, Rey YP, Gilmour R, Isr. J. Chem 2017, 57, 92–100; [Google Scholar]; b Aufiero M, Gilmour R, Acc. Chem. Res 2018, 51, 1701–1710. [DOI] [PubMed] [Google Scholar]
- [12].a Jenkins CL, McCloskey AI, Guzei IA, Eberhardt ES, Raines RT, Biopolymers 2005, 80, 1–8; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kotch FW, Guzei IA, Raines RT, J. Am. Chem. Soc 2008, 130, 2952–2953; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Taylor CM, Hardré R, Edwards PJB, Park JH, Org. Lett 2003, 5, 4413–4416; [DOI] [PubMed] [Google Scholar]; d Taylor CM, Hardre R, Edwards PJB, J. Org. Chem 2005, 70, 1306–1315; [DOI] [PubMed] [Google Scholar]; e Chiang YC, Lin YJ, Horng JC, Protein Sci. 2009, 18, 1967–1977; [DOI] [PMC free article] [PubMed] [Google Scholar]; f Kubyshkin V, Pridma, Budisa N, New J. Chem 2018, 42. [Google Scholar]
- [13].a Naduthambi D, Zondlo NJ, J. Am. Chem. Soc 2006, 128, 12430–12431; [DOI] [PubMed] [Google Scholar]; b Zheng T-Y, Lin Y-J, Horng J-C, Biochemistry 2010, 49, 4255–4263; [DOI] [PubMed] [Google Scholar]; c Holzberger B, Marx A, J. Am. Chem. Soc 2010, 132, 15708–15713; [DOI] [PubMed] [Google Scholar]; d Holzberger B, Obeid S, Welte W, Diederichs K, Marx A, Chem. Sci 2012, 3; [Google Scholar]; e Tang HC, Lin YJ, Horng JC, Proteins Struct. Funct. Bioinform 2014, 82, 67–76; [DOI] [PubMed] [Google Scholar]; f Crespo MD, Rubini M, PLoS One 2011, 6, e19425; [DOI] [PMC free article] [PubMed] [Google Scholar]; g Roderer D, Glockshuber R, Rubini M, ChemBioChem 2015, 16, 2162–2166. [DOI] [PubMed] [Google Scholar]
- [14].a Chandler CL, List B, J. Am. Chem. Soc 2008, 130, 6737–6739; [DOI] [PubMed] [Google Scholar]; b Myers EL, Palte MJ, Raines RT, J. Org. Chem 2019, 84, 1247–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a Liang G-B, Rito, Gellman SH, Biopolymers 1992, 32, 293–301; [DOI] [PubMed] [Google Scholar]; b Rai R, Aravinda S, Kanagarajadurai K, Raghothama S, Shamala N, Balaram P, J. Am. Chem. Soc 2006, 128, 7916–7928; [DOI] [PubMed] [Google Scholar]; c Reddy DN, Prabhakaran EN, Biopolymers 2014, 101, 66–77; [DOI] [PubMed] [Google Scholar]; d Meng HY, Thomas KM, Lee AE, Zondlo NJ, Biopolymers (Peptide Sci.) 2006, 84, 192–204. [DOI] [PubMed] [Google Scholar]
- [16].Groom CR, Bruno IJ, Lightfoot MP, Ward SC, Acta Cryst. 2016, B72, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a Hunter CA, Sanders JKM, J. Am. Chem. Soc 1990, 112, 5525–5534; [Google Scholar]; b Salonen LM, Ellermann M, Diederich F, Angew. Chem., Int. Ed 2011, 50, 4808–4842. [DOI] [PubMed] [Google Scholar]
- [18].a Desiraju GR, Acc. Chem. Res 1996, 29, 441–449; [DOI] [PubMed] [Google Scholar]; b Gu YL, Kar T, Scheiner, J. Am. Chem. Soc 1999, 121, 9411–9422; [Google Scholar]; c Jones CR, Baruah PK, Thompson AL, Scheiner S, Smith MD, J. Am. Chem. Soc 2012, 134, 12064–12071; [DOI] [PubMed] [Google Scholar]; d Adhikari U, Scheiner S, J. Phys. Chem. A 2013, 117, 10551–10562; [DOI] [PubMed] [Google Scholar]; e Steiner T, Angew. Chem., Int. Ed 2002, 41, 48–76. [Google Scholar]
- [19].a Horowitz S, Trievel RC, J. Biol. Chem 2012, 287, 41576–41582; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Senes A, Ubarretxena-Belandia I, Engelman DM, Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 9056–9061; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bella J, Berman HM, J. Mol. Biol 1996, 264, 734–742. [DOI] [PubMed] [Google Scholar]
- [20].a Forbes CR, Pandey AK, Ganguly HK, Yap GPA, Zondlo NJ, Org. Biomol. Chem 2016, 14, 2327–2346; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Panasik N, Eberhardt ES, Edison AS, Powell DR, Raines RT, Int. J. Peptide Protein Res. 1994, 44, 262–269; [DOI] [PubMed] [Google Scholar]; c Woinska M, Grabowsky S, Dominiak PM, Wozniak K, Jayatilaka D, Sci. Adv 2016, 2, e1600192; [DOI] [PMC free article] [PubMed] [Google Scholar]; d Clegg W, Deboves HJC, Elsegood MRJ, Acta. Cryst 2003, E59, o1987–01989. [Google Scholar]
- [21].Hutchinson EG, Thornton JM, Protein Sci. 1994, 3, 2207–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tyndall JDA, Pfeiffer B, Abbenante G, Fairlie DP, Chem. Rev 2005, 105, 793–826. [DOI] [PubMed] [Google Scholar]
- [23].Quancard J, Karoyan P, Lequin O, Wenger E, Aubry A, Lavielle S, Chassaing G, Tetrahedron Lett. 2004, 45, 623–625. [Google Scholar]
- [24].DeRider ML, Wilkens SJ, Waddell MJ, Bretscher LE, Weinhold F, Raines RT, Markley JL, J. Am. Chem. Soc 2002, 124, 2497–2505. [DOI] [PubMed] [Google Scholar]
- [25].Glendening CR, Landis CR, Weinhold F, WIREs Comput. Mol. Sci 2012, 2, 1–42. [Google Scholar]
- [26].a Haque TS, Little JC, Gellman SH, J. Am. Chem. Soc 1994, 116, 4105–4106; [Google Scholar]; b Struthers MD, Cheng RP, Imperiali B, Science 1996, 271, 342–345; [DOI] [PubMed] [Google Scholar]; c Stanger HE, Gellman SH, J. Am. Chem. Soc 1998, 120, 4236–4237; [Google Scholar]; d Tatko CD, Waters ML, J. Am. Chem. Soc 2002, 124, 9372–9373; [DOI] [PubMed] [Google Scholar]; e Schneider JP, Pochan DJ, Ozbas B, Rajagopal K, Pakstis L, Kretsinger J, J. Am. Chem. Soc 2002, 124, 15030–15037. [DOI] [PubMed] [Google Scholar]
- [27].Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery J, J. A., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ, Gaussian, Inc, Wallingford, CT, 2013. [Google Scholar]
- [28].Zhao Y, Truhlar DG, Theor. Chem. Act 2008, 120, 215–241. [Google Scholar]
- [29].Raghavachari K, Binkley JS, Seeger R, Pople JA, J. Chem. Phys 1980, 72, 650–654. [Google Scholar]
- [30].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, J. Comput. Chem 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.