

Abstract
The northern tree shrew (Tupaia belangeri) possesses high potential as an animal model of human diseases and biology, given its genetic similarity to primates. Although genetic information on the tree shrew has already been published, some of the entire coding sequences (CDSs) of tree shrew genes remained incomplete, and the reliability of these CDSs remained difficult to determine. To improve the determination of tree shrew CDSs, we performed sequencing of the whole-genome, mRNA, and total RNA and integrated the resulting data. Additionally, we established criteria for the selection of reliable CDSs and annotated these sequences by comparison to the human transcriptome, resulting in the identification of complete CDSs for 12,612 tree shrew genes and yielding a more accurate tree shrew genome database (TupaiaBase: http://tupaiabase.org). Transcriptome profiles in hepatitis B virus infected tree shrew livers were analyzed for validation. Gene ontology analysis showed enriched transcriptional regulation at 1 day post-infection, namely in the “type I interferon signaling pathway”. Moreover, a negative regulator of type I interferon, SOCS3, was induced. This work, which provides a tree shrew CDS database based on genomic DNA and RNA sequencing, is expected to serve as a powerful tool for further development of the tree shrew model.
Subject terms: Genomic analysis, Genome
Introduction
The northern tree shrew (Tupaia belangeri), which belongs to the family Tupaiidae, has a body weight ranging between 100–150 g, and is similar in appearance to squirrels1. The natural habitat of Tupaia spp. consists of the tropical rainforest in South East Asia where the animals feed on fruits, insects, and small vertebrates1. One of the appealing features of the tree shrew is its genetic closeness to human2. Thus, tree shrews are widely used as experimental animals in various research fields3–6. Recently, generation of a transgenic tree shrew has been reported7. The importance of tree shrew as an alternative animal model is growing.
Tree shrews have been employed in viral infection studies8–10, especially for hepatitis B virus (HBV)11,12 and hepatitis C virus (HCV)13,14, viruses for which the only other natural non-human infection model is chimpanzee. HBV, a member of the family Hepadnaviridae, causes acute and chronic hepatitis, and is a major worldwide public health concern15. Chronic HBV infection is strongly associated with an increased risk of cirrhosis, which in turn can lead to hepatocellular carcinoma15,16. Worldwide, 2 billion people have been reported to be infected with HBV, and more than 350 million have been reported to be chronically infected15,17. Urgent measures are required, but the development of drugs and vaccines has been hampered by the lack of an efficient animal model for infection by this virus. Since HBV causes human-like symptomology (including hepatitis and persistent infection) in tree shrews11,12,18, tree shrew could be a powerful animal model for HBV infection.
Genomic information is essential for various studies such as gene expression analysis and immunological analysis. Recently, an analysis of the tree shrew genome was reported2,19, and associated genomic information has been published in Ensembl (http://asia.ensembl.org) and the Tree shrew Database (TreeshrewDB: http://www.treeshrewdb.org). However, some of the entire coding sequences (CDSs) of tree shrew genes still remained incomplete. Additionally, even when CDSs have been identified, it can be difficult to determine how reliable those CDSs are. In the present study, we performed whole-genome sequencing, mRNA sequencing, and total RNA sequencing, and used the resulting data to select reliable complete tree shrew CDSs by utilizing defined criteria. Based on the obtained data, we have developed an enhanced tree shrew genome database (TupaiaBase: http://tupaiabase.org). In addition, we have validated this database using HBV-infected tree shrew liver specimens at an early stage of infection.
Results
Genome sequencing
In our initial work, we determined the whole-genome sequence of the tree shrew using next-generation sequencing. Starting from a tree shrew DNA sample, we constructed a series of libraries with different insert sizes (170 bp–20 Kb) (Table 1). Altogether, 14 libraries were generated, yielding a total of approximately 335 Gb of sequencing data. Assembly employed a subset of the data representing 240 Gb of high-quality sequence. The final N50 contig and total contig sizes were 33 Kb and 2.7 Gb, respectively (Table 2). The N50 scaffold and total scaffold sizes were 1.1 Mb and 2.7 Gb, respectively. As a next step, these sequencing data were annotated by homology-based gene prediction and de novo gene prediction, and a comprehensive gene set was constructed using GLEAN20. Based on GLEAN gene models, the tree shrew genome was predicted to contain a total of 19,320 protein-coding genes (Table 3).
Table 1.
Statistics of whole-genome sequencing data.
| Pair-end libraries | Insert size | Read length (bp) | Total Data (Gb) | Sequence depth (fold)* | Physical depth (fold)* |
|---|---|---|---|---|---|
| Illumina Reads | 170 bp | 100 | 60.96 | 19.82 | 16.84 |
| 500 bp | 100 | 72.67 | 23.63 | 59.06 | |
| 800 bp | 100 | 51.25 | 16.66 | 66.65 | |
| 2 Kb | 57 | 56.64 | 18.41 | 322.49 | |
| 5 Kb | 49 | 35.94 | 11.68 | 596.03 | |
| 10 Kb | 49 | 41.24 | 13.41 | 1368 | |
| 20 Kb | 49 | 16.38 | 5.33 | 1087 | |
| Total | 335.08 | 108.93 | 3516.07 |
*The genome size is assumed to be 3.08 Gb.
Table 2.
Statistics of the assembled sequence length of whole-genome sequencing.
| Contig | Scaffold | |||
|---|---|---|---|---|
| Size (bp) | Number | Size (bp) | Number | |
| N50 | 33,380 | 24,001 | 1,149,110 | 679 |
| Longest | 267,380 | — | 13,631,494 | — |
| Total Size | 2,709,670,168 | — | 2,746,321,810 | — |
| Total number (>=100 bp) | — | 585,492 | — | 447,618 |
| Total number (>=2 kb) | — | 121,063 | — | 7,608 |
Table 3.
General statistics of predicted protein-coding genes of whole-genome sequencing.
| Gene set | Number | Average gene length (bp) | Average CDS length (bp) | Average exon per gene | Average exon length (bp) | Average intron length (bp) |
|---|---|---|---|---|---|---|
| GLEAN | 19,320 | 24,193 | 1,419 | 7.68 | 184.85 | 3,411 |
Identification of CDSs by genome and RNA sequence analysis
To enhance the quality of gene annotation, HBV-infected tree shrews were used for RNA sequence (seq) analysis. Since an immune response should be induced under normal conditions, the use of HBV-infected tree shrew liver and spleen samples for RNA sequencing was expected to permit the identification of genes associated with an immune response to viral infection. In addition, different set of genes are expected to be expressed in each phase of infection. Therefore, RNA sequencing was performed on RNA samples obtained from HBV-infected tree shrew tissue samples at the acute phase of infection (at 1, 3, and 21 days post-infection (dpi)) and at the chronic phase of infection (at 8 months post-infection). In mRNA-seq analysis, over 200 million reads per group were sequenced, with over 90% of the bases of all samples exceeding Q30 (Table 4). Across all samples, 77 to 84% of the reads were mapped to the tree shrew genome. In total RNA-seq analysis, 88 to 126 million reads per sample were sequenced, with approximately 95% of the bases of all samples exceeding Q30. The percentages of total mapping reads of all samples were 86 to 91% (Table 5).
Table 4.
mRNA-seq overview.
| Sample | Total reads | % of >= Q30 bases | Trimmed reads | Total mapped reads | % of total mapped reads | Unmapped reads | % of unmapped reads |
|---|---|---|---|---|---|---|---|
| Uninfected tree shrew liver (#1) | 51.46 | 91.12 | 49.70 | 40.02 | 80.53 | 9.68 | 19.47 |
| Uninfected tree shrew liver (#2) | 52.91 | 90.82 | 50.98 | 45.73 | 82.96 | 9.39 | 17.04 |
| Uninfected tree shrew liver (#3) | 57.20 | 90.99 | 55.12 | 40.86 | 80.15 | 10.12 | 19.85 |
| Uninfected tree shrew liver (#4) | 53.87 | 90.86 | 51.85 | 42.14 | 81.26 | 9.72 | 18.74 |
| HBV-C 1 dpi tree shrew liver (#1) | 94.47 | 90.76 | 91.16 | 76.55 | 83.97 | 14.61 | 16.03 |
| HBV-C 1 dpi tree shrew liver (#2) | 111.28 | 90.31 | 107.00 | 87.40 | 81.68 | 19.60 | 18.32 |
| HBV-C 3 dpi tree shrew liver (#1) | 93.80 | 90.60 | 90.49 | 72.73 | 80.37 | 17.76 | 19.63 |
| HBV-C 3 dpi tree shrew liver (#2) | 129.30 | 90.44 | 124.43 | 99.89 | 80.27 | 24.54 | 19.73 |
| HBV-A 21 dpi tree shrew liver (#1) | 200.98 | 90.15 | 191.47 | 149.06 | 77.85 | 42.41 | 22.15 |
Read values represent millions of reads.
Table 5.
Total RNA-seq overview.
| Sample | Total reads | % of >= Q30 bases | Trimmed reads | Total mapped reads | % of total mapped reads | Unmapped reads | % of unmapped reads |
|---|---|---|---|---|---|---|---|
| Uninfected tree shrew liver (#1) | 117.38 | 94.87 | 115.39 | 100.37 | 86.99 | 15.02 | 13.01 |
| Uninfected tree shrew liver (#2) | 125.70 | 95.04 | 123.59 | 108.03 | 87.40 | 15.57 | 12.60 |
| Uninfected tree shrew liver (#3) | 92.43 | 94.74 | 90.28 | 82.01 | 90.84 | 8.27 | 9.16 |
| Uninfected tree shrew liver (#4) | 99.86 | 95.39 | 98.30 | 87.11 | 88.62 | 11.19 | 11.38 |
| HBV-C 1 dpi tree shrew liver (#1) | 106.45 | 95.33 | 104.75 | 92.07 | 87.90 | 12.68 | 12.10 |
| HBV-C 1 dpi tree shrew liver (#2) | 97.06 | 95.31 | 95.45 | 84.01 | 88.01 | 11.44 | 11.99 |
| HBV-C 3 dpi tree shrew liver (#1) | 99.51 | 95.51 | 97.87 | 85.30 | 87.15 | 12.57 | 12.85 |
| HBV-C 3 dpi tree shrew liver (#2) | 100.26 | 95.61 | 98.72 | 85.53 | 86.64 | 13.18 | 13.36 |
| HBV-A 21 dpi tree shrew liver (#1) | 94.93 | 95.57 | 93.48 | 80.42 | 86.02 | 13.07 | 13.98 |
| Uninfected tree shrew spleen (mix; #1, #2, and #4) | 91.34 | 95.43 | 89.69 | 79.00 | 88.09 | 10.69 | 11.91 |
| HBV-C 1 and 3 dpi tree shrew spleen (mix; 1 dpi [#1, #2], 3 dpi [#2]) | 88.74 | 95.55 | 87.09 | 78.24 | 89.83 | 8.85 | 10.17 |
| HBV-A 8 mpi tree shrew spleen (mix; #1, #2, and #3) | 93.64 | 95.56 | 91.72 | 82.19 | 89.62 | 9.52 | 10.38 |
Read values represent millions of reads.
A total of 74,425 transcripts and 37,817 genes were detected in the mRNA-seq analysis (Fig. 1). In the total RNA-seq analysis, a total of 105,158 transcripts and 50,824 genes were detected. The combination of these data yielded a total of 117,687 transcripts and 53,953 genes. We assessed the transcriptome assemblies using Benchmarking Universal Single-Copy Orthologs (BUSCO) software (Table 6). The number of total BUSCOs searched was 6,192. The number of complete BUSCOs in the combined transcriptome was 5,518 (89.1%), a value that exceeded the number (5,319; 85.9%) obtained using the previously available database results. Given the genetic similarity to human, these transcripts were annotated based on the Uniprot human protein database21. We then selected transcripts from each gene to identify the CDSs. To select only high-quality transcript data and determine the CDS of each gene, we set the following criteria for selection. (i) Start codon and stop codon existed in the transcript sequence. (ii) The lengths of the presumed gene product and the corresponding human orthologous gene product differed by less than 10%. (iii) The length of the overlap between the presumed tree shrew gene sequence and the corresponding human orthologous gene sequence (as aligned by BLASTX) constituted a distance of more than 50% of the presumed gene sequence. If there were multiple transcripts that met the above criteria for a given gene, the transcript that was most-strongly expressed was selected. Based on the criteria, we identified CDSs for a total of 12,612 genes.
Figure 1.
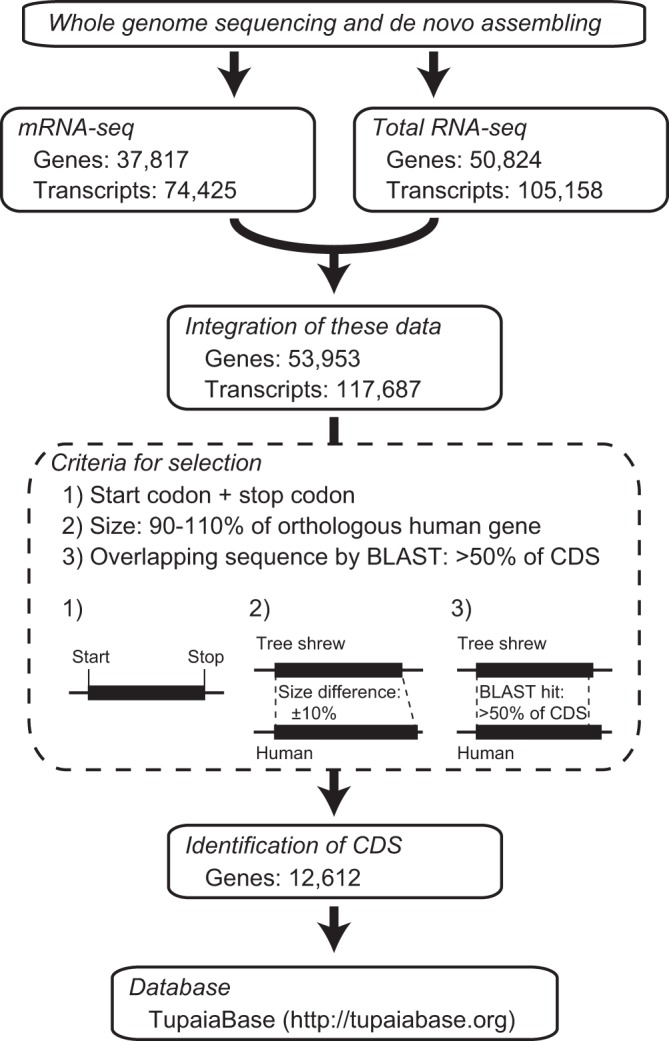
Schematic diagram of CDS identification of tree shrew genes.
Table 6.
Evaluation of assemblies by BUSCO.
| TupaiaBase (Sanada et al. 2019) | TreeshrewDB (Fan et al. 2014) | |
|---|---|---|
| Number of predicted genes | 53,935 | 119,898 |
| Number of predicted transcripts | 117,687 | 192,459 |
| BUSCO analysis | ||
| Complete BUSCOs (%) | 5,518 (89.1%) | 5,319 (85.9%) |
| Fragmented BUSCOs (%) | 398 (6.4%) | 562 (8.1%) |
| Missing BUSCOs (%) | 276 (4.5%) | 311 (5.0%) |
| Total BUSCOs (%) | 6,192 (100%) | 6,192 (100%) |
Database construction
Using whole-genome sequencing, mRNA sequencing and total RNA sequencing data, we constructed a tree shrew genome database (TupaiaBase: http://tupaiabase.org). The JBrowse22 genome browser was used for the visualization of genome annotations.
Gene sequence verification of database
To analyze the accuracy of the gene sequences predicted on the basis of the combined genome and RNA sequencing, we cloned a subset of genes, sequenced the resulting clones, and compared these sequences with the predicted versions. A total of 64 of these genes were successfully cloned and sequenced. Among the CDSs predicted solely on the basis of the genome sequence, 30 of 64 (46.9%) sequences were identical to the actual cloned sequence (Fig. 2a,b, and Supplementary Table S1); among the CDSs predicted by a combination of genome sequencing and RNA sequencing, 51 of 64 (79.7%) gene sequences were identical to actual cloned sequences. For example, the CDSs of the CD8 alpha (CD8A) and interleukin-7 (IL7) -encoding genes were not identified by genome sequencing alone, but these CDSs were detected by the combination of genome and RNA sequencing data. These predicted mRNA sequences did indeed match those of the actual cloned cDNA sequences (Fig. 2c,d). In a previous report, Yu et al. also determined the tree shrew IL7 mRNA sequences23, and the CDS predicted by our method matched the canonical form of tree shrew IL7 mRNA transcript (accession number: JQ182399). These results showed that gene sequence predictions based on the combination of genome and RNA sequencing were more accurate than those based on genome sequencing alone.
Figure 2.

Analysis of the accuracy of the gene sequences predicted on the basis of the combined genome and RNA sequencing. (a) Identities of nucleotide sequences of each gene when comparing between the cloned sequence and the sequence predicted from whole-genome sequencing or between the cloned sequence and the sequence predicted from the combined sequence data from whole-genome sequencing and RNA sequencing (b) Percentage of sequence-completely-matched genes between the cloned sequence and sequence predicted from whole-genome sequencing or combined sequence data. (c,d) Comparison of predicted and actual gene sequences for CD8A (c) and IL7 (d). Upper sequence: predicted sequence based on genome sequencing. Middle sequence: predicted sequence based on genome and RNA sequencing. Lower sequence: Cloned sequence.
Analysis of genes expressed in liver
To validate the 12,612 protein-coding genes identified in our analysis, we analyzed the expression of genes in liver. First, we determined how many genes were identified among genes expressed in liver. Among 426 genes categorized (in the human protein atlas: https://www.proteinatlas.org) as having “elevated expression in liver”, 366 genes were annotated in 1,766 transcripts from 117,687 transcripts not subjected to selection, and 274 genes (64.3%) with 290 transcripts from 12,612 selected transcripts were identified by our CDS selection criteria.
Next, to evaluate the accuracy of the CDSs selected by our criteria, we compared the expression levels of 1,766 tree shrew transcripts with those of homologous human genes. Expression of functional transcripts in tree shrew was expected to correlate with the expression of the homologous human genes in liver. The genes expressed in human liver were analyzed using the livers of chimeric mice harboring humanized livers. The expression levels of tree shrew transcripts (290 in total) that met the criteria correlated well with the expression levels of transcripts of the homologous human genes (274 genes) (R = 0.5597) (Fig. 3a). On the other hand, the expression levels of tree shrew transcripts (1,476 transcripts) that failed to meet the criteria showed a poor correlation with the expression levels of transcripts of homologous human genes (R = 0.2923) (Fig. 3b). These data suggested that the identified CDSs selected by our criteria were more reliable than were excluded CDSs.
Figure 3.
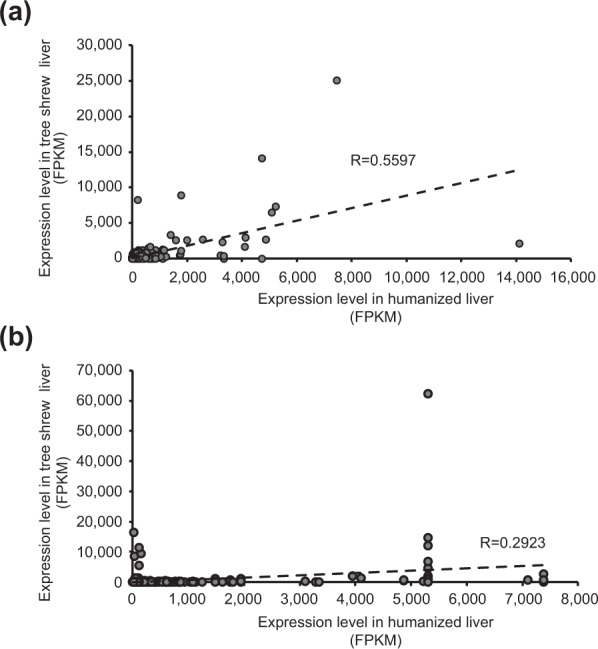
Expression level of liver-specific genes in tree shrew liver and in humanized liver in mouse. Correlation between gene expression level in humanized liver and homologous gene in tree shrew liver assessed for selected transcripts (a) or transcripts that failed to meet our criteria (b). Broken lines indicate regression curves.
Expression analysis of HBV-infected tree shrew
It is difficult to identify accurately when HBV patients become initially infected; therefore, host response at the initial stage of HBV infection remains poorly documented. Hence, to have more insight in this regard, we analyzed the transcriptome profile of the identified genes in the early stage of HBV infection in tree shrew. We infected tree shrews by intravenous injection with HBV genotype C and sacrificed animals at 1 or 3 dpi (Fig. 4a). At 1 dpi, HBV viral loads were 1.0 × 102 to 2.2 × 102 copies/ml in serum samples, and 5.7 × 100 to 1.1 × 101 copies/μg liver DNA (Fig. 4b,c). At 3 dpi, HBV viral loads were 4.2 × 101 to 1.8 × 102 copies/ml in serum samples, and 1.4 × 100 to 3.2 × 100 copies/μg liver DNA. No viral DNA was detected from the livers of two tree shrews at 1 dpi, and from one tree shrew at 3 dpi. HBV infections caused nominal but non-significant elevation of serum ALT levels in some tree shrews (Fig. 4d). Interestingly, abnormal architecture of liver-cell cords was observed at 3 dpi, and lymphocytic infiltration also was observed by histochemical analysis (Fig. 4e).
Figure 4.

HBV infection in tree shrew. (a) Experimental schedule of HBV infection in tree shrew. (b–d) Viral DNA titers in sera (b) and liver (c), and serum ALT level (d), at 1 dpi or 3 dpi in HBV-infected tree shrew, or in uninfected tree shrew. Heavy bars indicate means of each group. (e) Histological analysis (hematoxylin-eosin staining; representative images) of liver from uninfected and HBV-infected (at 1 and 3 dpi) tree shrews. Bar, 100 μm.
Transcriptome analysis showed that the number of differentially expressed genes (DEGs) at 1 and 3 dpi were 35 and 28, respectively. To characterize the DEGs, we performed GO term analysis of the DEGs at each time point (Fig. 5). The GO term characteristics of DEGs at 1 and 3 dpi were distinct. At 1 dpi, the GO terms of DEGs were primarily immune response-related (e.g., “Type I interferon signaling pathway” and “Regulation of inflammatory response”) (Fig. 5a). At 3 dpi, the GO terms of DEGs were mainly related to “Cholesterol biosynthetic process” and “Glycogen biosynthetic process” (Fig. 5b). These results implied that the primary immune response of tree shrew to HBV infection was attenuated rapidly (within 3 days).
Figure 5.
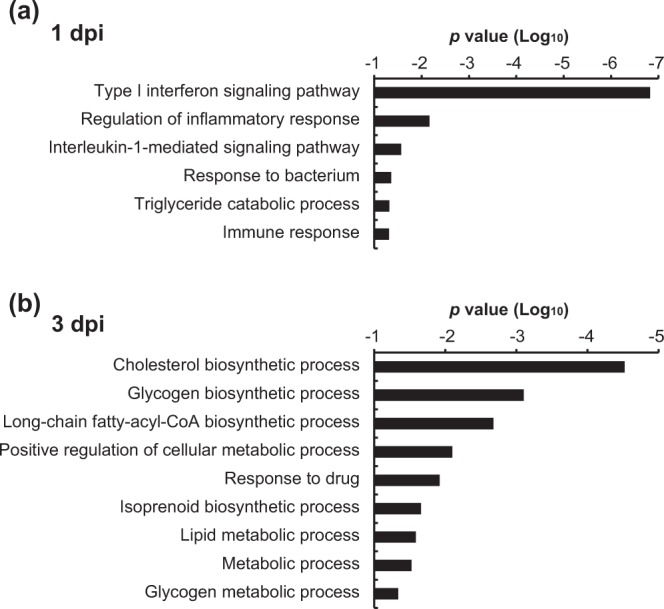
GO term analysis of differentially expressed genes in HBV-infected tree shrew at 1 dpi (a) and 3 dpi (b).
Analysis of genes related to type I interferon signaling
Since GO term analysis revealed expression changes at 1 dpi in genes related to the type I interferon signaling pathway, we analyzed the expression level of DEGs categorized as part of the “type I interferon signaling pathway”. A total of 6 DEGs related to the type I interferon signaling pathway were observed at 1 dpi (Fig. 6a). The gene designations (using the names of the orthologous human genes) were as follows: interferon-induced protein with tetratricopeptide repeats 3 (IFIT3), interferon regulatory factor 7 (IRF7), ubiquitin-like protein interferon-stimulated gene 15 (ISG15), early growth response protein 1 (EGR1), interferon alpha-inducible protein 27 (IFI27), and HLA class I histocompatibility antigen, A-69 alpha chain (HLA-A). Notably, following viral infection, the levels of these transcripts (with the exception of EGR1) decreased at 1 dpi, with the levels of IFIT3 and ISG15 remaining significantly depleted at 3 dpi.
Figure 6.

Expression analysis of genes related to type I interferon signaling. (a) Expression levels (in uninfected tree shrew and in HBV-infected tree shrew at 1 and 3 dpi) of genes whose GO included the term “type I interferon signaling pathway” and exhibited the strongest differential expression genes at 1 dpi. (b) Expression levels (in uninfected tree shrew and in HBV-infected tree shrew at 1 and 3 dpi) of genes known to be central to the type I interferon signaling pathway. Horizontal bars indicate mean values in each group. Asterisks indicate significant differences (p < 0.05).
Our previous study showed that HBV infection suppressed or did not induce the expression of the interferon beta-encoding gene (IFNB) in tree shrew at 4 and 31 weeks post-infection24. To clarify whether early-stage HBV infection induced the expression of genes that are key factors in the type I interferon signaling pathway, we analyzed the mRNA expression levels of IFNB and the genes encoding interleukin-6 (IL6), and tumor necrosis factor alpha (TNFA). Notably, expression of these genes did not differ significantly when comparing between uninfected and HBV-infected (at 1 and 3 dpi) tree shrews (Fig. 6b). A recent study showed that the HBx protein suppresses type I interferon signaling by upregulating expression of the suppressor of cytokine signaling 3-encoding gene (SOCS3)25. Indeed, in HBV-infected tree shrew, expression of the SOCS3 transcript was significantly upregulated at 1 dpi (Fig. 6b). These data suggested that the initial phase of HBV infection in tree shrew induces the expression of the SOCS3 gene, resulting in suppression of the type I interferon response.
Discussion
Recently, tree shrew has been widely used for various biological studies, including investigations of viral infection11,12,14, depression3, the visual system4, and so on. However, research tools (e.g., antibody, PCR system) are limited for the establishment of tree shrew as an experimental animal model. Although these tools are based on genomic information, some of the entire CDSs of genes still had not been completed. Even in cases for which the CDS had been determined, it remained difficult to determine how reliable that CDS was. In the present study, we combined whole-genome analysis with RNA sequencing data, and selected 12,612 genes for which the CDS seemed to be accurate. We then published these tree shrew genomic data as part of a publicly available tree shrew database.
Based on whole-genome analysis alone, it is difficult to predict the entire CDSs of a genome. Comparing the predicted sequences based on whole-genome analysis with actual cDNA sequences revealed that approximately half of the cloned sequences did not match the predicted sequences. This observation reflects, in part, how difficult it is to determine the sequences of long genes and those of genes that contain short exons. To improve the accuracy of predicted CDSs, we combined whole-genome analysis and RNA sequencing data. RNA sequencing reads were mapped to the tree shrew genome, enabling identification of the CDSs of individual genes. About 80% of the cDNA sequences predicted on the basis of the combination of whole-genome sequencing and RNA sequencing were identical to actual cloned gene sequences; the remaining 20% of the predicted cDNA sequences were not recovered as actual cDNA sequences, presumably reflecting the low level of expression of these genes under the conditions of our experiment. Since gene expression levels may differ in various organs and under other conditions, experiments to sequence the RNAs collected from other organs and under other conditions will be needed.
In the present study, we focused on tree shrew genes sharing high homology with orthologous human genes, and determined the CDSs of 12,612 genes. To validate the protein-coding genes identified in our analysis, we analyzed the expression of transcripts annotated as liver-specific genes. Expression of selected transcripts (i.e., those that met the stated criteria) in tree shrew liver showed better correlation with that of homologous human genes in humanized liver (R = 0.5597) than with that of excluded transcripts (i.e., those that did not meet these criteria) (R = 0.2923). These data suggested that CDSs selected by our criteria are more reliable than unselected CDSs. Of course, our selection is far from being perfect, given that we focused on the tree shrew genes sharing high sequence similarity with the corresponding human genes. We are currently performing an optimization of the criteria for transcript selection, with the expectation that this analysis will permit identification of tree shrew-specific genes.
In the present study, we constructed a tree shrew genome database (TupaiaBase: http://tupaiabase.org). The first feature of our database is the quality of data. Complete BUSCOs of this study is 89.1%; thus, our database is thought to be based on high-quality data. The second feature of our database is that the database uses JBrowse for the visualization of genome annotations. JBrowse has been used extensively in other databases, including Mouse Genome Informatics (MGI; http://www.informatics.jax.org), Rat Genome Database (RGD; https://rgd.mcw.edu), and FlyBase (https://flybase.org), and is known to be fast and easy to use. The third feature of our database is the availability of the CDSs. Cloned sequences and selected genes in this study are displayed, permitting the user to readily identify reliable CDSs. Though a tree shrew database (TreeshrewDB) is already available, these features of our tree shrew genome database (TupaiaBase) are expected to help facilitate advances in research in this organism. The utility of our database should extend the potential of tree shrew as an animal model in various research fields.
In practice, it is difficult to identify when HBV patients become initially infected, and thus the collection of samples from humans at the initial stage of HBV infection has not been possible. This shortcoming has precluded the determination of the host response in the early stages of HBV infection in humans. Tree shrew infected with HBV exhibits hepatitis that resembles human cases, and therefore is expected to serve as a useful tool for clarifying the host response to HBV infection. Indeed, by using the tree shrew model, we were able to perform a preliminary analysis of the dynamics of transcript accumulation in the initial phase of HBV infection. Kouwaki et al. also have shown that early-stage HBV infection induces hepatic interferon gamma expression in tree shrew26. These results indicate that the tree shrew HBV infection model will be of great value in clarifying the early in vivo response to HBV infection in animals with intact immune systems.
The present study revealed that the expression of tree shrew genes related to the type I interferon signaling pathway is downregulated at 1 dpi. Our study also showed that genes encoding key factors (e.g., IFNB) known to be involved in the type I interferon response were not induced in the earliest stage of HBV infection of tree shrew. However, upregulation of SOCS3 at 1 dpi was observed in our study, an observation consistent with a recent report that SOCS3 is induced by HBx protein in human cell culture, thereby suppressing the type I interferon response25. Although the upregulation of SOCS3 expression has been detected in the liver of chronically HBV-infected patients25,27, the role and kinetics of SOCS3 expression during the initial phase of HBV infection in vivo remains unknown. Our study suggests that upregulation of SOCS3 may facilitate the initial propagation of HBV in vivo. Further studies using the tree shrew model are expected to elucidate the role of SOCS3 in HBV infection and to reveal other key factors involved in the initial phases of HBV infection.
In conclusion, we have constructed a database that identifies tree shrew CDSs based on a combination of genome and RNA sequencing. To improve this database, we are planning to perform long-read sequencing to enhance recovery of the complete genome with the correct order; we will continue to incorporate the new sequencing data as part of this database. Since tree shrew is increasingly being used in various research fields, this database is expected to serve as a powerful tool for further development of the tree shrew model and thus for enhancement of associated research.
Methods
Ethics statement
This study was carried out in strict accordance with the Guidelines for Animal Experimentation of the Japanese Association for Laboratory Animal Science and the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All protocols were approved by the Committee on the Ethics of Animal Experiments of the Tokyo Metropolitan Institute of Medical Science.
Animals
Northern tree shrews (T. belangeri) were purchased from the Kunming Institute of Zoology, Chinese Academy of Sciences. The animals were bred at Kagoshima University and the Tsukuba Primate Research Center for further experimental use.
Chimeric mice with humanized livers were purchased from PhoenixBio (Hiroshima, Japan).
Genome sequencing
A male four-year-old tree shrew was used for whole-genome sequencing. The whole-genome sequencing of tree shrew was performed by BGI (Shenzhen, China). Using a tree shrew DNA sample, libraries of different insert sizes (170 bp, 500 bp, 800 bp, 2 Kb, 5 Kb, 10 Kb, and 20 Kb) were constructed by standard Illumina library preparation protocols. The tree shrew genome was assembled from massive reads using software in the SOAPdenovo (Short Oligonucleotide Assembly Program; version 1.05)28 package by the following steps. First, sequencing errors on raw reads were corrected based on K-mer frequencies using KmerFreq and Corrector. Then, contigs and scaffolds were built using the SOAPdenovo assembler. Finally, gaps were closed using GapCloser. To predict genes in the tree shrew genome, homology-based gene prediction and de novo gene prediction were performed. For the homology-based gene prediction, human (Homo sapiens), gorilla (Gorilla gorilla), rhesus macaque (Macaca mulatta), lemur (Microcebus murinus), and gibbon (Nomascus leucogenys) proteins were mapped onto the tree shrew genome to define the putative genes. For de novo gene prediction, two de novo prediction programs (GENSCAN29 and Augustus30 (version 2.5.5)) were used. Using GLEAN20 (version 1.0.1), these gene sets were combined to yield a comprehensive gene set.
HBV inoculum
The HBV inoculum was the serum of a chimeric mouse with humanized liver that had been infected with HBV genotype A2 and C (C_JPNAT; accession number: AB246345.1).
HBV infection of animals
One-year-old tree shrews (n = 11) were inoculated intravenously with 1.0 × 107 viral DNA copies of HBV. Animals were sacrificed at 1 dpi (n = 4), 3 dpi (n = 3), 21 dpi (n = 1), and 8 months post-infection (mpi) (n = 3). Animals at 1 and 3 dpi were used for RNA sequencing and transcriptome analysis. Animals at 21 dpi and 8 mpi were used only for RNA sequencing.
RNA extraction and sequencing
Total RNA was isolated from liver and spleen tissues from uninfected (n = 4) and HBV-infected tree shrews at 1 dpi (n = 4), 3 dpi (n = 3), 21 dpi (n = 1), and 8 mpi (n = 3). Total RNA was purified using the AGPC and RNeasy kit (Qiagen, Hilden, Germany). For mRNA sequencing, the mRNA in each total RNA sample was converted into a cDNA library using the TruSeq Standard mRNA prep kit (Illumina, San Diego, CA, United States). For total RNA sequencing, each RNA sample was converted into a cDNA library using the TruSeq Standard Total RNA prep kit (Illumina). These libraries then were sequenced using a HiSeq2000 sequencer (Ilumina). All reads were mapped to the tree shrew genome using TopHat (version 2.0.10)31 with an option that enabled the identification of micro-exons (–microexon-search). Transcripts were assembled and abundances were estimated using Cufflinks (version 2.1.1)32. The data from the mRNA sequencing and total RNA sequencing were merged using Cuffmerge, an application that is included as part of the Cufflinks package. Transcripts were annotated by comparison to human Uniprot protein data using BLAST+ (version 2.2.29+)33.
Total RNA from the livers of uninfected chimeric mice with humanized livers (n = 3) was isolated, purified, and used for mRNA sequencing as described above. Reads were mapped to the human reference genome (GRCh37 release 75) using TopHat (version 2.0.14). Transcripts were assembled and abundances were estimated using Cufflinks (version 2.2.1).
Comparison with publicly available data
Publicly available RNA sequencing data for the Northern tree shrew were obtained from NCBI SRA listed in TreeshrewDB. The accession numbers of the data obtained were as follows: SRX1009946, SRX1017387, SRX125163, SRX157960, SRX157961, SRX157962, SRX157963, SRX157964, SRX157965, SRX157966, SRX3341772, SRX3358315, SRX3358316, SRX3358317, SRX3358318, and SRX3358319. These data were analyzed by the same informatics process as described above.
The completeness of transcript sets of our data and publicly available data were assessed using BUSCO software34 (version 3.0.2).
Identification of gene CDSs
To select only high-quality transcript data and determine the CDS of each gene, we used the following criteria for selection. (i) Both a start codon and a stop codon were present in the transcript sequence. (ii) The lengths of the presumed gene product and the corresponding orthologous human gene product differed by less than 10%. (iii) The length of the overlap between the presumed tree shrew gene sequence and the corresponding orthologous human gene sequence (as aligned by BLASTX with an option that set the maximum expectation value to 10−5) constituted a length of more than 50% of the presumed gene sequence. If there were multiple transcripts that met the above criteria for a given gene, the transcript showing highest expression was selected.
Prediction of CDSs by genome sequencing
To predict the CDSs of genes by genome sequencing, the tree shrew genome sequencing data were searched for gene sequences by BLASTX with human gene sequences. Since exons of each gene were separated, sequences that showed overlap by BLASTX were connected and considered as a single predicted CDS in the genome sequence.
Cloning of genes
To perform the cloning of genes, we predicted both the 5′- and 3′ -end sequences of each gene CDS. Sequences at both ends were predicted from the genome sequence as described above. Gene cloning was performed by Takara Bio (Shiga, Japan). The target sequences were reverse transcribed and amplified by PCR. The resulting PCR fragments were cloned into the pCAGGS vector.
Analysis of gene expression
To analyze the mRNA expression level in tree shrew liver, FPKM (fragments per Kb of exon model per million mapped fragments) values were used for normalization. Differentially expressed genes were categorized by gene ontology (GO)35 and analyzed by enriched GO using DAVID software (version 6.8)36.
HBV-DNA quantification
Viral DNA was extracted using SMItest EX-R&D kits (Nippon Genetics, Tokyo, Japan) according to the manufacturer’s instructions. Quantification of HBV-DNA was performed using real-time detection PCR, as previously described37. The primers and probes for the S gene consisted of forward primer HB-166-S21 (nucleotides (nts) 166–186: 5′-CACATCAGGATTCCTAGGACC-3′), reverse primer HB-344-R20 (nts 344–325: 5′-AGGTTGGTGAGTGATTGGAG-3′), and TaqMan probe HB-242-S26FT (nts 242–267: 5′-CAGAGTCTACACTCGTGGTGGACTTC-3′).
Measurement of serum alanine aminotransferase (ALT) activity
Serum ALT activity in tree shrew was determined using the Transaminase CII-test Wako (Wako Pure Chemical Industries, Osaka, Japan) according to the manufacturer’s instructions.
Histological analysis
Tree shrew liver tissues were fixed with 10% phosphate-buffered formalin and embedded in paraffin. The samples then were sectioned and stained with hematoxylin and eosin using standard methodologies.
Statistical analyses
p values lower than 0.05 were considered significant. Cuffdiff, included in Cufflinks, was used to estimate the statistical significance of gene expression changes between sample groups.
Accession numbers
The raw data of the whole-genome sequencing and RNA sequencing have been deposited in the DNA Data Bank of Japan (DDBJ) with accession numbers DRR155071-DRR155099.
Supplementary information
Acknowledgements
The authors thank Dr. Yukiko Yamazaki for construction of database and Dr. Hayying Chi, Dr. Bouchra Kitab, Rika Matsuyama, Takumi Haraguchi, and Takuya Kato for their help in tree shrew care. This study was supported by grants from the Ministry of Health, Labour and Welfare of Japan (H24-HBV-general-014) and the Research Program on Hepatitis from the Japanese Agency for Medical Research and Development, AMED (JP16fk0310513, JP16fk0210108, and JP17fk0310111). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Contributions
T.S., K.T.-K., Y.Y., K.I., T.G., M.M. and M.K. designed the study. T.S., K.T.-K., N.Y., M.E.H.K., J.T., Y.S. and M.K. performed the experiments. T.S., K.T.-K., T.S.-I., M.E.H.K., D.Y., M.M. and M.K. analyzed the data. T.S., K.T.-K., M.E.H.K. and M.K. wrote the manuscript. All authors reviewed the manuscript.
Data Availability
All data that support the findings of this study are available from the corresponding authors upon reasonable request.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Kyoko Tsukiyama-Kohara, Email: kkohara@vet.kagoshima-u.ac.jp.
Michinori Kohara, Email: kohara-mc@igakuken.or.jp.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-48867-x.
References
- 1.Tsukiyama-Kohara K, Kohara M. Tupaia belangeri as an experimental animal model for viral infection. Exp. Anim. 2014;63:367–374. doi: 10.1538/expanim.63.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fan Y, et al. Genome of the Chinese tree shrew. Nat. Commun. 2013;4:1426. doi: 10.1038/ncomms2416. [DOI] [PubMed] [Google Scholar]
- 3.Hai-Ying C, et al. Establishment of an intermittent cold stress model using Tupaia belangeri and evaluation of compound C737 targeting neuron-restrictive silencer factor. Exp. Anim. 2016;65:285–292. doi: 10.1538/expanim.15-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee KS, Huang X, Fitzpatrick D. Topology of ON and OFF inputs in visual cortex enables an invariant columnar architecture. Nature. 2016;533:90–94. doi: 10.1038/nature17941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu L, et al. Loss of RIG-I leads to a functional replacement with MDA5 in the Chinese tree shrew. Proc. Natl. Acad. Sci. USA. 2016;113:10950–10955. doi: 10.1073/pnas.1604939113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yao YG. Creating animal models, why not use the Chinese tree shrew (Tupaia belangeri chinensis)? Zool. Res. 2017;38:118–126. doi: 10.24272/j.issn.2095-8137.2017.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li CH, et al. Long-term propagation of tree shrew spermatogonial stem cells in culture and successful generation of transgenic offspring. Cell Res. 2017;27:241–252. doi: 10.1038/cr.2016.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li L, et al. Herpes Simplex Virus 1 Infection of Tree Shrews Differs from That of Mice in the Severity of Acute Infection and Viral Transcription in the Peripheral Nervous System. J. Virol. 2016;90:790–804. doi: 10.1128/JVI.02258-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanada T, et al. Avian H5N1 influenza virus infection causes severe pneumonia in the Northern tree shrew (Tupaia belangeri) Virology. 2019;529:101–110. doi: 10.1016/j.virol.2019.01.015. [DOI] [PubMed] [Google Scholar]
- 10.Zhang, N. N. et al. Zika Virus Infection in Tupaia belangeri Causes Dermatological Manifestations and Confers Protection against Secondary Infection. J. Virol. 93, 10.1128/JVI.01982-18 (2019). [DOI] [PMC free article] [PubMed]
- 11.Walter E, Keist R, Niederost B, Pult I, Blum HE. Hepatitis B virus infection of tupaia hepatocytes in vitro and in vivo. Hepatology. 1996;24:1–5. doi: 10.1002/hep.510240101. [DOI] [PubMed] [Google Scholar]
- 12.Sanada T, et al. Property of hepatitis B virus replication in Tupaia belangeri hepatocytes. Biochem. Biophys. Res. Commun. 2016;469:229–235. doi: 10.1016/j.bbrc.2015.11.121. [DOI] [PubMed] [Google Scholar]
- 13.Xie ZC, et al. Transmission of hepatitis C virus infection to tree shrews. Virology. 1998;244:513–520. doi: 10.1006/viro.1998.9127. [DOI] [PubMed] [Google Scholar]
- 14.Amako Y, et al. Pathogenesis of hepatitis C virus infection in Tupaia belangeri. J. Virol. 2010;84:303–311. doi: 10.1128/JVI.01448-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seeger C, Mason WS. Molecular biology of hepatitis B virus infection. Virology. 2015;479-480:672–686. doi: 10.1016/j.virol.2015.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cougot D, Neuveut C, Buendia MA. HBV induced carcinogenesis. J. Clin. Virol. 2005;34(Suppl 1):S75–78. doi: 10.1016/S1386-6532(05)80014-9. [DOI] [PubMed] [Google Scholar]
- 17.Lavanchy D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J. Viral Hepat. 2004;11:97–107. doi: 10.1046/j.1365-2893.2003.00487.x. [DOI] [PubMed] [Google Scholar]
- 18.Yang C, et al. Chronic hepatitis B virus infection and occurrence of hepatocellular carcinoma in tree shrews (Tupaia belangeri chinensis) Virol J. 2015;12:26. doi: 10.1186/s12985-015-0256-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan Y, Yu D, Yao YG. Tree shrew database (TreeshrewDB): a genomic knowledge base for the Chinese tree shrew. Sci. Rep. 2014;4:7145. doi: 10.1038/srep07145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elsik CG, et al. Creating a honey bee consensus gene set. Genome Biol. 2007;8:R13. doi: 10.1186/gb-2007-8-1-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Consortium TU. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45:D158–D169. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buels R, et al. JBrowse: a dynamic web platform for genome visualization and analysis. Genome Biol. 2016;17:66. doi: 10.1186/s13059-016-0924-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu D, et al. Diverse interleukin-7 mRNA transcripts in Chinese tree shrew (Tupaia belangeri chinensis) PLoS One. 2014;9:e99859. doi: 10.1371/journal.pone.0099859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kayesh MEH, et al. Interferon-beta response is impaired by hepatitis B virus infection in Tupaia belangeri. Virus Res. 2017;237:47–57. doi: 10.1016/j.virusres.2017.05.013. [DOI] [PubMed] [Google Scholar]
- 25.Tsunematsu S, et al. Hepatitis B virus X protein impairs alpha-interferon signaling via up-regulation of suppressor of cytokine signaling 3 and protein phosphatase 2A. J. Med. Virol. 2017;89:267–275. doi: 10.1002/jmv.24643. [DOI] [PubMed] [Google Scholar]
- 26.Kouwaki T, et al. Extracellular Vesicles Including Exosomes Regulate Innate Immune Responses to Hepatitis B Virus. Infection. Front. Immunol. 2016;7:335. doi: 10.3389/fimmu.2016.00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koeberlein B, et al. Hepatitis B virus overexpresses suppressor of cytokine signaling-3 (SOCS3) thereby contributing to severity of inflammation in the liver. Virus Res. 2010;148:51–59. doi: 10.1016/j.virusres.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 28.Li R, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20:265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- 30.Stanke M, Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19(Suppl 2):ii215–225. doi: 10.1093/bioinformatics/btg1080. [DOI] [PubMed] [Google Scholar]
- 31.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Camacho C, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 35.Consortium TGO. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017;45:D331–D338. doi: 10.1093/nar/gkw1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 37.Tanaka T, et al. Virological significance of low-level hepatitis B virus infection in patients with hepatitis C virus associated liver disease. J. Med. Virol. 2004;72:223–229. doi: 10.1002/jmv.10566. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data that support the findings of this study are available from the corresponding authors upon reasonable request.