

Summary
High-grade serous ovarian cancers (HGSOCs) arise from exfoliation of transformed cells from the fallopian tube, indicating that survival in suspension, and potentially escape from anoikis, is required for dissemination. We report here the results of a multi-omic study to identify drivers of anoikis escape, including transcriptomic analysis, global non-targeted metabolomics, and a genome-wide CRISPR/Cas9 knockout (GeCKO) screen of HGSOC cells cultured in adherent and suspension settings. Our combined approach identified known pathways, including NOTCH signaling, as well as novel regulators of anoikis escape. Newly identified genes include effectors of fatty acid metabolism, ACADVL and ECHDC2, and an autophagy regulator, ULK1. Knockdown of these genes significantly inhibited suspension growth of HGSOC cells, and the metabolic profile confirmed the role of fatty acid metabolism in survival in suspension. Integration of our datasets identified an anoikis-escape gene signature that predicts overall survival in many carcinomas.
Subject Areas: Biological Sciences, Cell Biology, Cancer Systems Biology, Cancer
Graphical Abstract
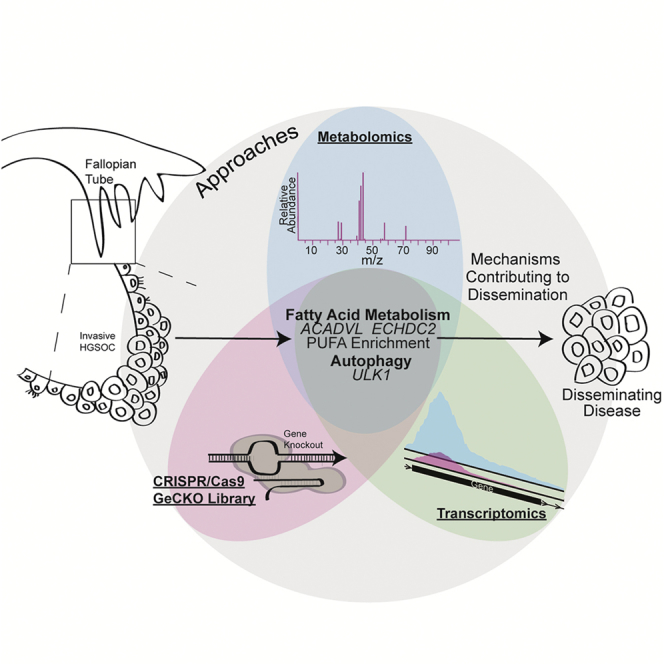
Highlights
-
•
Combined multi-omic screen identifies drivers of ovarian cancer dissemination
-
•
Fatty acid metabolism and autophagy identified as drivers of anoikis resistance
-
•
Suspension culture of ovarian cancer cells induces metabolic reprogramming
-
•
Anoikis resistance gene signature predicts survival in multiple carcinoma types
Biological Sciences; Cell Biology; Cancer Systems Biology; Cancer
Introduction
Epithelial ovarian cancer is the deadliest gynecologic malignancy, annually accounting for more than 220,000 deaths worldwide (Jayson et al., 2014). High-grade serous ovarian cancer (HGSOC) comprises the majority of ovarian cancer cases. Although a majority of patients with HGSOC initially respond to first-line therapy, in most patients the cancer will recur within 3 years (Jayson et al., 2014). Achieving optimal surgical resection of the tumors conveys the best prognosis but is challenging given that ovarian cancer, as well as fallopian and primary peritoneal carcinoma, tend to spread to the peritoneal cavity (Berek and Hacker, 2015). As tumor cells spread to the abdominal cavity they lead to the production of ascites, a collection of intraperitoneal fluid containing immune cells, tumor cells, and cytokines, among other cellular and acellular factors (Kim et al., 2016). Tumor cells in ascites are known to be highly invasive, to the extent that a majority of patient-derived xenograft ovarian cancer models are generated from injection of ascites fluid alone, indicating the presence of tumor-initiating cells (Kim et al., 2016, Lengyel et al., 2014). Tumor cells within ascites are thought to be the subpopulation of cells that lead to recurrent or metastatic disease (Kim et al., 2016).
Investigations over the past 10 years have revealed that HGSOC may, in fact, originate from transformed secretory fallopian tube epithelium (FTE) located on the fimbriated end of the fallopian tube. Precursor lesions include serous tubal intraepithelial carcinoma (STIC), which is focal and displays a cytologic appearance similar to HGSOC. HGSOC precursor lesions often exhibit a “p53 signature,” which includes normal secretory FTE cells that either lack p53 entirely or overexpress mutant p53 (Lee et al., 2007, Piek et al., 2001, Van Der Steen et al., 2017). Transformed cells within STIC lesions exfoliate from the fallopian tube extracellular matrix (ECM), escape detachment-induced cell death (anoikis), survive in an anchorage-independent manner, and disseminate to the ovary and peritoneum. For most cancers, such as breast and endometrial, anoikis escape followed by intravasation into the circulatory or lymphatic systems are critical steps in the metastatic process. However, ovarian, fallopian, and primary peritoneal carcinomas, or high-grade serous carcinoma of the pelvis, are unique in that cancer cells have direct access to the peritoneal cavity. HGSOC preferentially colonizes the fat-rich omentum, suggesting the metabolic environment contributes to dissemination. Anoikis escape is one of the only properties required by transformed FTE to disseminate. In this report, we systematically interrogate the relationship between survival in suspension and anoikis escape and almost every gene in the genome.
Given the limited understanding of the mechanisms promoting ovarian cancer dissemination, we performed a functional CRISPR/Cas9 genome-wide knockout screen to identify critical genes and pathways essential to escape anoikis or promote anchorage-independent survival. In parallel, we evaluated the metabolome and transcriptome (RNA-seq) of HGSOC cells cultured in forced suspension to study anoikis escape. Cross-referencing of the CRISPR/Cas9 knockout screen and global metabolomics revealed genes involved in fatty acid (FA) metabolism and enrichment of poly-unsaturated FAs. Specifically, ACADVL or ECHDC2 promoted poly-unsaturated FA (PUFA) accumulation in suspension cells, and knock down of these genes reduced the viability of cells grown in forced suspension culture. Analysis of the CRISPR/Cas9 knockout screen and RNA-seq data resulted in 108 genes associated with survival in suspension and therefore anoikis escape. Some of these play well-established roles in anchorage-independent survival; for example, NOTCH3 is a known regulator of anoikis escape (Brown et al., 2015). We more closely evaluated 13 ovarian cancer-related genes, including Unc-51 like autophagy activating kinase 1 (ULK1), which is a serine/threonine kinase that regulates autophagy (reviewed in Mizushima, 2010). We further examined the expression of the top 108 genes in the context of other cancer types by utilizing gene signature analysis. We cross-referenced the anoikis escape candidate gene list across a variety of carcinoma types, including ovarian, bladder, lung, prostate, breast, colorectal, and brain. We observed that the anoikis escape gene signature predicted overall survival in almost all cancers tested. The data also reveal several novel effectors of anoikis escape, which could be further investigated for therapeutic targeting or prognostic purposes.
Results
CRISPR/Cas9 Screen to Identify Effectors of Anoikis-Escape
During the progression of HGSOC tumor cells must acquire the ability to escape anchorage-independent cell death or anoikis. To identify genes that functionally contribute to anoikis escape we employed a genome-wide CRISPR/Cas9 screening approach in the HGSOC cell line, PEO1. PEO1 cells were selected as the primary cell line because of the HGSOC-like mutational profile (TP53 and BRCA2-mutated), transduction capacity, and moderate growth in suspension. To model HGSOC anoikis escape, cells were forced to grow in suspension by coating tissue culture dishes with poly-2-hydroxyethyl methacrylate (poly-HEMA), which prevents cell attachment and mimics an anchorage-independent microenvironment (Wheeler et al., 2018). PEO1 cells surviving culture on poly-HEMA-coated plates had a low proliferative rate (Figure S1A).
The Genome-Scale CRISPR Knock-Out libraries (GeCKO Libraries A and B, 119,000 individual gRNAs, 6 to 8 gRNA per gene) were lentivirally transduced into PEO1. Multiplicity of infection (MOI) and an effective puromycin concentration were optimized (Figures S1B and S1C). Transduced adherent cells were puromycin selected and allowed to recover. PEO1-GeCKO cells were then plated in adherent or forced suspension settings and harvested after 5 and 10 days (D5 and D10) from both culture conditions in triplicate and genomic DNA was extracted (Figure 1A). To allow for gRNA enrichment or depletion, cells were grown in suspension for 10 days. All samples were sequenced at a depth of at least 25 million reads (Figure S1D).
Figure 1.

Whole Genome CRISPR/Cas9 Screen to Identify Novel Drivers of Ovarian Cancer Dissemination
(A) Workflow of CRISPR/Cas9 screen. PEO1 cells (HGSOC cells, TP53/BRCA2-mutated) transduced with the Genome-Scale CRISPR Knock-Out (GeCKO) libraries.
(B) Schematic of expected results, which are to identify gRNA enriched (red) or depleted (yellow) in PEO1 cells grown in suspension.
(C) Scatterplot (dot = 1 single gRNA) of Fold Change (Suspension/Adherent, x axis) and average gRNA count (y axis). Red dots = Adjusted p value < 0.05.
(D) Scatterplot of gRNAs from Adherent Day 10 (x axis) and Suspension Day 10 (y axis). Red dots = gRNA with adjusted p < 0.05.
(E) The number of significant (adj. p < 0.05) gRNA with the same directionality associated with a gene.
(F) Significant (adj. p < 0.05) ACADVL or ECHDC2-specific gRNA in PEO1 cells and their respective change in abundance between adherent day 10 (A10) and suspension day 10 (S10).
(G) PEO1 cells were transduced with shControl (shCtrl) or shACADVL (#1 and #2) or shECHDC2 (#1 and #2). RNA was extracted from cells and used for RT-qPCR against ACADVL and ECHDC2. Internal control = 18S.
(H) Same as G, but cells were plated on tissue culture treated plastic (adherent) on day 0. Double-stranded DNA content was used as a surrogate for cell number and was measured on day 1 and day 7. Y axis represents the change in cell number from Day 1 to Day 7.
(I) Same as H, cells were plated in low adherent tissue culture plates (suspension), and double-stranded DNA content was measured on day 1 and day 7. Y axis represents the change in cell number from Day 1 to Day 7. Statistical test = ANOVA.
∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Error bars = S.E.M. See also Figure S1.
To identify genes critical for anoikis escape, we determined relative depletion or enrichment for each gRNA in HGSOC cells cultured in suspension conditions (Figure 1B). We focused primarily on day 10 to allow maximum degree of gRNA enrichment or depletion. The data presented represents the comparison of duplicates of Adherent Day 10 (A10) versus Suspension Day 10 (S10), based on principal component analyses and hierarchical clustering (Figures S1E–S1H). Greater than 95% of all genes had at least one gRNA detected (Figure S1I), demonstrating sufficient genome-wide coverage. In the A10 versus S10 comparisons, 15,636 gRNAs representing 11,571 genes were differentially abundant as determined by DESEq2 with Benjamin and Hoschberg multi-testing correction (Figures 1C and 1D). Considering gRNA directionality (enriched or depleted) among the 11,571 genes, there were a total of 2,206 genes with at least two significant targeting gRNAs with similar directionality (Figure 1E). Seventeen genes had four significant gRNA, 219 genes had three significant gRNA, and 1,970 genes had two significant gRNAs (Figure 1E). Functional analysis of the 2,206 genes with gene set enrichment analysis KEGG pathways revealed enrichments in MAPK signaling, focal adhesion, ErbB signaling pathways, and adherence junctions (Table 1). Gene ontology for biological processes related to the 2,206 genes showed significant enrichment in Response to External Stimulus, Regulation of Cell Proliferation, and Regulation of Intracellular Signal Transduction (Table 1). Gene ontology for the molecular function related to the 2,206 genes showed enrichment in Receptor Binding, Signal Transducer Activity, and Signaling Receptor Activity (Table 1). Enrichment analyses suggest a contribution of receptor tyrosine kinase signaling and external stimuli driving intracellular signaling in anoikis escape.
Table 1.
Pathway Analysis of Significant Genes Identified with CRISPR/Cas9 Screen
| Gene Set Name | # Genes in Gene Set (K) | # Genes in Overlap (k) | k/K | p Value | FDR Q-Value |
|---|---|---|---|---|---|
| KEGG | |||||
| Pathways in cancer | 328 | 54 | 16.5% | 4.47 × 10−17 | 8.32 × 10−15 |
| Focal adhesion | 201 | 35 | 17.4% | 2.67 × 10−12 | 2.48 × 10−10 |
| Mapk signaling pathway | 267 | 40 | 15.0% | 1.08 × 10−11 | 6.71 × 10−10 |
| Erbb signaling pathway | 87 | 20 | 23.0% | 8.10 × 10−10 | 3.63 × 10−8 |
| Calcium signaling pathway | 178 | 29 | 16.3% | 9.77 × 10−10 | 3.63 × 10−8 |
| Endometrial cancer | 52 | 15 | 28.9% | 3.62 × 10−9 | 1.12 × 10−7 |
| Chemokine signaling pathway | 190 | 29 | 15.3% | 4.59 × 10−9 | 1.22 × 10−7 |
| Adherens junction | 75 | 17 | 22.7% | 1.86 × 10−8 | 3.85 × 10−7 |
| B cell receptor signaling pathway | 75 | 17 | 22.7% | 1.86 × 10−8 | 3.85 × 10−7 |
| Insulin signaling pathway | 137 | 23 | 16.8% | 2.87 × 10−8 | 5.33 × 10−7 |
| GO Biological Processes | |||||
| Phosphate-containing compound metabolic process | 1,977 | 245 | 12.4% | 2.47 × 10−50 | 1.10 × 10−46 |
| Response to external stimulus | 1,821 | 229 | 12.6% | 4.74 × 10−48 | 1.05 × 10−44 |
| Regulation of transport | 1,804 | 227 | 12.6% | 1.15 × 10−47 | 1.70 × 10−44 |
| Positive regulation of gene expression | 1,733 | 217 | 12.5% | 3.45 × 10−45 | 3.83 × 10−42 |
| Protein localization | 1,805 | 222 | 12.3% | 5.69 × 10−45 | 5.05 × 10−42 |
| Positive regulation of response to stimulus | 1,929 | 231 | 12.0% | 7.09 × 10−45 | 5.24 × 10−42 |
| Positive regulation of biosynthetic process | 1,805 | 220 | 12.2% | 6.30 × 10−44 | 3.99 × 10−41 |
| Response to endogenous stimulus | 1,450 | 192 | 13.2% | 1.98 × 10−43 | 1.10 × 10−40 |
| Regulation of cell proliferation | 1,496 | 193 | 12.9% | 5.22 × 10−42 | 2.57 × 10−39 |
| Regulation of intracellular signal transduction | 1,656 | 204 | 12.3% | 2.20 × 10−41 | 9.74 × 10−39 |
| GO Molecular Function | |||||
| Ribonucleotide binding | 1,860 | 225 | 12.1% | 2.10 × 10−44 | 1.89 × 10−41 |
| Adenyl nucleotide binding | 1,514 | 197 | 13.0% | 1.96 × 10−43 | 8.81 × 10−41 |
| Receptor binding | 1,476 | 185 | 12.5% | 1.54 × 10−38 | 4.64 × 10−36 |
| Enzyme binding | 1,737 | 193 | 11.1% | 5.23 × 10−33 | 1.18 × 10−30 |
| Receptor activity | 1,649 | 185 | 11.2% | 3.52 × 10−32 | 6.35 × 10−30 |
| Kinase activity | 842 | 118 | 14.0% | 3.97 × 10−29 | 5.97 × 10−27 |
| Signal transducer activity | 1,731 | 183 | 10.6% | 1.21 × 10−28 | 1.56 × 10−26 |
| Molecular function regulator | 1,353 | 156 | 11.5% | 1.72 × 10−28 | 1.94 × 10−26 |
| Transferase activity transferring phosphorus-containing groups | 992 | 125 | 12.6% | 1.93 × 10−26 | 1.93 × 10−24 |
| Protein kinase activity | 640 | 91 | 14.2% | 3.63 × 10−23 | 3.27 × 10−21 |
Related to Figure 1.
Sixteen of the 17 genes with four significant (adj. p value < 0.05) gRNAs shared directionality and were depleted in S10 compared with A10, suggesting that the targeted genes are required for survival in the context of anchorage independence. Examining the top 17 genes using functional analysis failed to identify any enriched significant pathways; however, two of the 17 genes are associated with fatty acid metabolism. Four gRNAs specific for acyl-CoA dehydrogenase, very long chain (ACADVL) and Enoyl-CoA Hydratase Domain Containing 2 (ECHDC2) were depleted in S10 compared with A10 (Figure 1F). ACADVL promotes β-oxidation of long-chain fatty acids (FAs) (Kurtz et al., 1998), and ECHDC2 is involved in FA biosynthesis (Bahnson et al., 2002). In The Cancer Genome Atlas of ovarian cancer, ECHDC2 and ACADVL have a strong positive correlation (Spearman r = 0.25, p = 8.1 × 10−6, q = 1.655 × 10−4) suggesting that both genes are upregulated within the same tumor. The primary site of HGSOC dissemination is the fat-rich omentum, which has high levels of FA, including linoleic acid. These findings and previous reports suggest FA metabolism potentiates HGSOC dissemination (Liu et al., 2015, Miranda et al., 2016).
We further confirmed ACADVL and ECHDC2 are important in regulating anoikis escape through small hairpin knockdown (shRNA). We transduced PEO1 and OVCAR4 cell lines with a control plasmid (shCtrl) or two independent shRNA for either ACADVL or ECHDC2. Both shRNAs for ACADVL and ECHDC2 knocked down the mRNA expression of the target gene (Figures 1G and S1J). In adherent and suspension cell culture conditions over 7 days, we measured DNA double strand content as a surrogate for cell number of shCtrl, shACADVL, or shECHDC2 cells. In the adherent setting, PEO1 ACADVL knockdown cells slightly increased cell number, whereas in OVCAR4 ACADVL knockdown cells did not have an effect (Figures 1H and S1K). In adherent ECHDC2 knockdown PEO1 and OVCAR4 cells, proliferation was reduced in one of the two shRNA compared with shCtrl (Figures 1H and S1K). In suspension culture, both ACADVL and ECHDC2 knockdown in PEO1 and OVCAR4 cells significantly decreased cell number compared with shCtrl (Figures 1I and S1L). These results provide confidence that the CRISPR/Cas9 screen identified suspension-related genes and highlight the importance of two metabolic enzymes.
Global Metabolomics Confirms Metabolic Alterations in Anoikis-Resistant Cells
Relative levels of metabolites were examined in adherent versus suspension cultured PEO1 cells via global non-targeted metabolomics, which is a powerful mass-spectrometry-based approach to assess the metabolic profile in a non-biased fashion (Figure 2A and Table S1). Notably, 15 of the 35 detected FA metabolites were significantly (p < 0.05) enriched in cells grown in suspension cells (Figure 2B). In an independent cell line (OVARY1847, TP53-mutated, BRCA wild-type) global non-targeted metabolomics demonstrated 19 of 35 detected FA metabolites enriched in cells grown in suspension compared with adherent cells (Figure S2A and Table S1). Enriched FA between PEO1 and OVARY1847 significantly overlapped, highlighting that the accumulation of FA in anoikis-escaped cells is not cell-line dependent (Figure 2C). We observed differences between PEO1 and OVARY1847 cells, particularly in acyl-carnitine metabolites. For example, two short-chain acyl-carnitine metabolites (propionyl and butanoyl carnitine) are decreased only in PEO1, whereas three medium chain are increased in PEO1 against five in OVARY1847 (from hexanoyl-L-carnitine to O-tetradecanoyl-carnitine). These data suggest that the rate-limiting step of FA β-oxidation mediated by carnitine palmityltransferase is differently regulated in PEO1 suspension compared with OVARY1847 suspension. Both PEO1 and OVARY1847 cells in suspension are enriched for long- and very-long-chain fatty acids, specifically poly-unsaturated FA (e.g., linoleic acid) compared with adherent cells. In PEO1 and OVARY1847 cells grown in suspension, linoleic acid was enriched when compared with adherent cells, which was validated in an independent experiment (Figures 2D and S2B). These data suggest that cells grown in suspension accumulate PUFAs as a potential metabolic adaptation to resist anoikis.
Figure 2.

HGSOC Cells Cultured in Suspension Are Enriched for Fatty Acids and Fatty Acid Metabolites
HGSOC cells (PEO1 and OVARY1847) were cultured in adherent and suspension cultures for 48 h. Cells were collected and used for global non-targeted metabolomics.
(A) Metabolites grouped based on pathway. The number of significant (p < 0.05) metabolites indicated with percentage (%) of pathway. See Table S1 for a full list of metabolites.
(B) Fatty acids and fatty acids metabolites in PEO1 cells grown in adherent (black bars) or suspension (gray bars) conditions. ∗p value < 0.05.
(C) Comparison of differentially enriched fatty acids and fatty acid metabolites in PEO1 versus OVARY1847.
(D) An example of poly-unsaturated fatty acids enriched in PEO1 and OVARY1847 suspension cells.
(E) Global non-targeted metabolomics analysis of shCtrl PEO1 cells cultured in adherent versus suspension. Left Panel, metabolite counts graphed as a scatterplot. x axis, shCtrl adherent and y axis, shCtrl suspension. Right Panel, x axis, Log2-Fold change of metabolites and y axis, p value.
(F) Metabolomics analysis of shCtrl PEO1 cells cultured in suspension versus shACADVL cells cultured in suspension. Left Panel, metabolite counts graphed as a scatterplot. x axis, shCtrl suspension and y axis, shACADVL suspension. Right Panel, x axis, Log2-Fold change of metabolites and y axis, p value.
(G) Metabolomics analysis of shCtrl PEO1 cells cultured in suspension versus shECHDC2 cells cultured in suspension. Left Panel, metabolite counts graphed as a scatterplot. x axis, shCtrl suspension and y axis, shECHDC2 suspension. Right Panel, x axis, Log2-Fold change of metabolites and y axis, p value. Red dotted lines indicated p value threshold. Statistical test = unpaired t test. Error bars = S.E.M.
See also Figure S2.
In the CRISPR/Cas9 screen analysis, two of the top hits were involved in FA metabolism, ACADVL and ECHDC2. Loss of either enzyme reduces the viability of cells grown in suspension culture (Figure 1I). ACADVL promotes β-oxidation of FA and ECHDC2 contributes to FA biosynthesis, thus these enzymes function at different aspects of FA metabolism. We therefore wanted to assess the metabolic profile of suspension cells following the knockdown of either ACADVL or ECHDC2. Relative levels of metabolites were examined in adherent versus suspension cultured shCtrl PEO1 and shACADVL or shECHDC2 cells via global non-targeted metabolomics (Table S2). We confirmed that PUFAs are enriched in shCtrl suspension cells compared with shCtrl adherent cells (Figure 2E). ACADVL or ECHDC2 knockdown (Figure 1G) cells cultured in suspension led to the significant depletion of PUFAs in suspension cells (Figures 2F and 2G). Notably, ACADVL or ECHDC2 knockdown cells cultured in suspension displayed similar metabolic profiles (Figures S2C and S2D). Taken together, these data indicate that ACADVL or ECHDC2 has distinct functions in FA metabolism, and loss of either enzyme leads to the depletion of PUFAs in suspension cultured cells.
Anoikis Resistance Associated Transcriptome
The CRISPR/Cas9 KO screen interrogated the loss of individual genes and the ability of HGSOC cells to remain viable in suspension. In non-transduced PEO1 cells a sub-population of cells survive under forced suspension culture. Therefore, to assess transcriptional adaptations that allow for suspension survival and to cross-reference genes identified in CRISPR/Cas9 KO screen, we examined the transcriptome of PEO1 cells that survive under suspension culture conditions for 10 days. Principal component analysis and hierarchal clustering showed that the “Adherent” and “Suspension” samples grouped (Figures S3A and S3B). Comparing the transcriptome of the adherent to suspension cells, we identified 804 differentially expressed genes (p < 0.0001; Figure 3A and Table S3). We found 718 upregulated and 86 downregulated genes associated with suspension culture. Although not statistically significant the CRISPR/Cas9 screen hits ACADVL and ECHDC2 were upregulated 1.14X and 1.86X, respectively. Notably, the most significant differentially expressed gene was MUC16 (Log2 FC = 5.29, False Discovery Rate = 1.85 × 10−29), which was upregulated in cells in suspension culture. MUC16 is known as cancer antigen 125 (CA125), which is routinely used as a biomarker in the clinic to monitor HGSOC progression.
Figure 3.

Suspension-Dependent Transcriptomic Changes Are Associated with Metastatic HGSOC
(A) PEO1 cells were grown in adherent or suspension cultures for 10 days. RNA was extracted from cells and utilized for RNA-sequencing. Scatterplot of Fold Change (Log2, x axis) comparing p value (Log10, y axis). Red dots are 804 differentially regulated genes (p < 0.0001).
(B) Workflow on combining RNA-Seq and CRISPR/Cas9 datasets.
(C) Overlapped genes identified from CRISPR/Cas9 and RNA-seq analyses before thresholding directionality of gRNA.
(D) Number of gRNA for 108 genes.
(E) Scatterplot of the gRNAs associated with 2,206 genes identified from CRISPR/Cas9 criteria. Fold change of most significant gRNA (x axis) compared with adjusted p value (y axis). Red dots (3 gRNAs) and blue dots (2 gRNA) indicate 108 genes that overlapped with RNA-seq.
(F) Protein-protein interaction (STRING) analysis of 108 genes. Disconnected nodes are hidden. Line thickness correlates to confidence of protein-protein interaction.
(G) Overlap analysis of 108 genes with differentially expressed genes in primary (n = 166) versus metastatic (n = 75) ovarian adenocarcinoma from the Bittner Ovarian Cancer Dataset.
(H) Overlap analysis of 108 genes with differentially expressed genes in primary (n = 189) versus metastatic (n = 54) ovarian adenocarcinoma from the Tothill Ovarian Cancer Dataset.
(I) Overlap analysis of common genes identified in (G) and (H).
(J) CRABP2 expression in metastatic versus primary HGSOC tumors from G and H, and PEO1 cells adherent versus suspension cells. p Values = unpaired t test. Error bars = S.E.M.
See also Figure S3.
KEGG pathway analysis of the 804 genes differentially regulated in anoikis-resistant cells revealed enriched pathways including focal adhesion, ECM-receptor interaction, TGFβ-signaling, cell adhesion molecules, and arachidonic acid metabolism (Table 2). KEGG pathway enrichment for arachidonic acid (a PUFA) metabolism highlights an overlap between the CRISPR/Cas9 screen, metabolomics, and RNA-sequencing analysis. Gene ontology for biological processes related to the 804 genes showed significant enrichment for tissue development, regulation of cell proliferation, and positive regulation of response to stimulus (Table 2). Gene ontology for the molecular function related to the 804 genes showed enrichment for receptor binding and activity, signal transducer activity, and transmembrane transporter activity (Table 2). The combined –omics approaches indicate a role of PUFA metabolism in conveying a survival advantage for cells cultured in suspension. Independent of the metabolomics, we further investigated the overlap between the CRISPR/Cas9 screen and RNA-seq analysis.
Table 2.
Pathway Analysis of Significant Genes Identified with RNA-Sequencing
| Gene Set Name | # Genes in Gene Set (K) | # Genes in Overlap (k) | k/K | p Value | FDR Q-Value |
|---|---|---|---|---|---|
| KEGG | |||||
| Pathways in cancer | 328 | 26 | 7.9% | 7.97 × 10−11 | 1.48 × 10−8 |
| Focal adhesion | 201 | 17 | 8.5% | 5.76 × 10−8 | 5.36 × 10−6 |
| Dilated cardiomyopathy | 92 | 11 | 12.0% | 4.06 × 10−7 | 2.52 × 10−5 |
| ECM-receptor interaction | 84 | 10 | 11.9% | 1.42 × 10−6 | 5.48 × 10−5 |
| Hypertrophic cardiomyopathy (HCM) | 85 | 10 | 11.8% | 1.59 × 10−6 | 5.48 × 10−5 |
| TGF-beta signaling pathway | 86 | 10 | 11.6% | 1.77 × 10−6 | 5.48 × 10−5 |
| Complement and coagulation cascades | 69 | 9 | 13.0% | 2.20 × 10−6 | 5.85 × 10−5 |
| Cell adhesion molecules (CAMs) | 134 | 12 | 9.0% | 2.79 × 10−6 | 6.48 × 10−5 |
| Regulation of actin cytoskeleton | 216 | 15 | 6.9% | 4.09 × 10−6 | 8.46 × 10−5 |
| Arachidonic acid metabolism | 58 | 8 | 13.8% | 5.28 × 10−6 | 9.82 × 10−5 |
| GO Biological Processes | |||||
| Tissue development | 1,518 | 138 | 9.1% | 1.12 × 10−60 | 4.98 × 10−57 |
| Regulation of multicellular organismal development | 1,672 | 138 | 8.3% | 1.45 × 10−55 | 3.21 × 10−52 |
| Positive regulation of developmental process | 1,142 | 103 | 9.0% | 1.23 × 10−44 | 1.82 × 10−41 |
| Epithelium development | 945 | 94 | 10.0% | 2.79 × 10−44 | 3.09 × 10−41 |
| Regulation of cell proliferation | 1,496 | 116 | 7.8% | 9.54 × 10−44 | 8.46 × 10−41 |
| Regulation of anatomical structure morphogenesis | 1,021 | 94 | 9.2% | 1.87 × 10−41 | 1.38 × 10−38 |
| Regulation of cell differentiation | 1,492 | 111 | 7.4% | 3.89 × 10−40 | 2.46 × 10−37 |
| Positive regulation of multicellular organismal process | 1,395 | 106 | 7.6% | 4.00 × 10−39 | 1.99 × 10−36 |
| Positive regulation of response to stimulus | 1,929 | 125 | 6.5% | 4.05 × 10−39 | 1.99 × 10−36 |
| Neurogenesis | 1,402 | 106 | 7.6% | 6.26 × 10−39 | 2.78 × 10−36 |
| GO Molecular Function | |||||
| Receptor binding | 1,476 | 107 | 7.3% | 1.16 × 10−37 | 1.05 × 10−34 |
| Molecular function regulator | 1,353 | 82 | 6.1% | 9.58 × 10−24 | 4.32 × 10−21 |
| Receptor activity | 1,649 | 90 | 5.5% | 7.09 × 10−23 | 2.13 × 10−20 |
| Signal transducer activity | 1,731 | 90 | 5.2% | 1.82 × 10−21 | 4.10 × 10−19 |
| Transporter activity | 1,276 | 74 | 5.8% | 2.11 × 10−20 | 3.80 × 10−18 |
| Transmembrane transporter activity | 997 | 63 | 6.3% | 2.50 × 10−19 | 3.75 × 10−17 |
| Enzyme binding | 1,737 | 86 | 5.0% | 3.28 × 10−19 | 4.22 × 10−17 |
| Sequence specific DNA binding | 1,037 | 61 | 5.9% | 2.84 × 10−17 | 3.20 × 10−15 |
| Calcium ion binding | 697 | 49 | 7.0% | 4.22 × 10−17 | 4.22 × 10−15 |
| RNA polymerase II transcription factor activity sequence specific DNA binding | 629 | 46 | 7.3% | 8.93 × 10−17 | 8.05 × 10−15 |
Related to Figure 3.
CRISPR/Cas9 Screen Combined with Transcriptome
Genes identified in the CRISPR/Cas9 screen (2,206 genes) were compared with the RNA-seq differentially expressed genes (804 genes) (Figure 3B), which showed a significant overlap of 108 genes (Hypergeometric test, p = 2.55 × 10−6; Figures 3C and Table S4). Overall, within the 108 genes that overlapped within the datasets, 96 genes had two gRNAs and 12 genes had three gRNAs (Figure 3D). Within the 108 genes, we did not detect genes involved in FA metabolism, highlighting potential differences in essential (CRISPR/Cas9) and sufficient (RNA-seq) genes. These 108 genes had gRNAs that were distributed between being enriched or depleted in suspension cultures conditions (Figure 3E), and notably 10 of 12 genes with the three-gRNA genes were significantly upregulated in suspension (Table S4). KEGG pathway analysis of these 108 genes revealed enriched pathways including pathways in cancer, dorsoventral axis formation (Notch signaling), and TGFβ signaling (Table 3). Gene ontology for biological processes related to the 108 genes showed significant enrichment for tissue development, regulation of cell differentiation, and positive regulation of developmental process (Table 3). Gene ontology for molecular function related to the 108 genes showed enrichment of receptor binding and activity, and transporter activity (Table 3). Protein-protein network interaction analysis on the 108 genes was conducted (Szklarczyk et al., 2017). Protein-protein network analysis and kMeans clustering identified three distinct protein-protein interaction clusters: NOTCH, TGFβ, and ERBB receptor signaling, receptor-ligand interactions, and intracellular signaling (Figure 3F). Although FA metabolic pathways were not enriched in the 108 CRISPR/RNA-seq gene set, NOTCH, TGFβ, and ERBB signaling regulate FA uptake, activate transcription of FA metabolism genes, and promote FA synthesis, respectively (Bian et al., 2015, Jabs et al., 2018, Soukupova et al., 2017). The high degree of concordance between the two (CRISPR/Cas9 screen and RNA-seq) independent genomic datasets and the previously described pathways provide confidence in the findings.
Table 3.
Pathway Analysis of Significant Genes Common between CRISPR/Cas9 Screen and RNA-Sequencing
| Gene Set Name | # Genes in Gene Set (K) | # Genes in Overlap (k) | k/K | p Value | FDR Q-Value |
|---|---|---|---|---|---|
| KEGG | |||||
| Pathways in cancer | 328 | 8 | 2.4% | 8.79 × 10−7 | 1.63 × 10−4 |
| Dorsoventral axis formation | 25 | 3 | 12.0% | 2.50 × 10−5 | 2.32 × 10−3 |
| TGF beta signaling pathway | 86 | 4 | 4.7% | 4.56 × 10−5 | 2.83 × 10−3 |
| Taste transduction | 52 | 3 | 5.8% | 2.30 × 10−4 | 1.01 × 10−2 |
| Basal cell carcinoma | 55 | 3 | 5.5% | 2.71 × 10−4 | 1.01 × 10−2 |
| P53 signaling pathway | 69 | 3 | 4.4% | 5.29 × 10−4 | 1.53 × 10−2 |
| Melanoma | 71 | 3 | 4.2% | 5.75 × 10−4 | 1.53 × 10−2 |
| Calcium signaling pathway | 178 | 4 | 2.3% | 7.40 × 10−4 | 1.72 × 10−2 |
| GO Biological Processes | |||||
| Circulatory system development | 788 | 19 | 2.4% | 1.75 × 10−14 | 7.76 × 10−11 |
| Regulation of multicellular organismal development | 1,672 | 22 | 1.3% | 2.11 × 10−11 | 3.37 × 10−8 |
| Regulation of cellular component movement | 771 | 16 | 2.1% | 2.28 × 10−11 | 3.37 × 10−8 |
| Positive regulation of developmental process | 1,142 | 18 | 1.6% | 1.02 × 10−10 | 1.13 × 10−7 |
| Regulation of cell differentiation | 1,492 | 20 | 1.3% | 1.42 × 10−10 | 1.26 × 10−7 |
| Tissue development | 1,518 | 20 | 1.3% | 1.92 × 10−10 | 1.42 × 10−7 |
| Epithelium development | 945 | 16 | 1.7% | 4.43 × 10−10 | 2.81 × 10−7 |
| Anatomical structure formation involved in morphogenesis | 957 | 16 | 1.7% | 5.31 × 10−10 | 2.93 × 10−7 |
| Regulation of transport | 1,804 | 21 | 1.2% | 5.95 × 10−10 | 2.93 × 10−7 |
| Blood vessel morphogenesis | 364 | 11 | 3.0% | 7.66 × 10−10 | 3.31 × 10−7 |
| GO Molecular Function | |||||
| Receptor binding | 1,476 | 19 | 1.3% | 8.53 × 10−10 | 7.68 × 10−7 |
| Receptor activity | 1,649 | 17 | 1.0% | 1.79 × 10−7 | 8.05 × 10−5 |
| Transporter activity | 1,276 | 14 | 1.1% | 1.17 × 10−6 | 3.52 × 10−4 |
| S100 protein binding | 13 | 3 | 23.1% | 3.17 × 10−6 | 6.27 × 10−4 |
| Kinase activity | 842 | 11 | 1.3% | 3.48 × 10−6 | 6.27 × 10−4 |
| Metal ion transmembrane transporter activity | 417 | 8 | 1.9% | 5.18 × 10−6 | 7.18 × 10−4 |
| Cation channel activity | 298 | 7 | 2.4% | 5.58 × 10−6 | 7.18 × 10−4 |
| Gated channel activity | 325 | 7 | 2.2% | 9.80 × 10−6 | 1.10 × 10−3 |
| Protein kinase activity | 640 | 9 | 1.4% | 1.58 × 10−5 | 1.38 × 10−3 |
| Transferase activity transferring phosphorus containing groups | 992 | 11 | 1.1% | 1.61 × 10−5 | 1.38 × 10−3 |
Related to Figure 3.
Clinical Relevance
Anoikis escape during ovarian cancer dissemination is a part of the metastatic process; therefore, we next examined the 108 top identified genes against published ovarian cancer datasets that evaluated primary and metastatic (disseminated) disease. The Bittner Ovarian Dataset (GEO: GSE2109) with human genome microarray examined 241 patients with ovarian carcinoma and included both primary tumors (n = 166) and metastatic tumors (n = 75). Comparing primary and metastatic tumors, 5,104 genes were differentially regulated and significantly 45 of our 108 top genes overlapped (Figure 3G, p = 0.0002). The Tothill Ovarian Cohort (GEO: GSE9899) examined 243 patients with ovarian carcinoma including both primary tumors (n = 189) and metastatic tumors (n = 54). Comparing primary versus metastatic tumors, 7,466 genes were differentially regulated and significantly 52 of 108 top hits overlapped (Figure 3H, p = 0.022). On examination of the level of congruency between the genes that overlapped within the metastatic signatures of both datasets we observed that 29 genes overlapped, representing a significant (p = 0.003) enrichment (Figure 3I and Table S5). Examination of expression directionality within these 29 genes found 10 genes that shared directionality between all of the datasets. For example, Cellular Retinoic Acid Binding Protein 2 (CRABP2) was upregulated in all of the datasets (Figure 3J) and is a biomarker for ovarian cancer (Toyama et al., 2012). Furthermore, we examined the expression of the 108 genes in matched primary tumor, ascites tumor cells, and metastatic tumors from five patients with ovarian cancer (GEO: GSE73064) (Gao et al., 2019). Of the 108 genes, there were 41 genes that were expressed in a similar direction (Table S6, Columns D-I). These data highlight that the 108 genes identified through –omics approaches are relevant and relatable to disseminated and metastatic ovarian cancer clinical specimens.
Validation and Ovarian Cancer Relevance
To validate a subset of the 108 genes identified in the RNA-seq and CRISPR/Cas9 comparisons, The Protein Atlas database (Uhlen et al., 2010, Uhlen et al., 2015) was used to examine tissue and pathologic expression of the genes (Table S6, Columns J-K). We identified genes that are predominantly expressed in ovarian cancer compared with 16 other cancer types. Furthermore, we cross-referenced the 108 genes to differentially regulated genes in a dataset that examined fallopian tube epithelium (FTE) cells in adherent and suspension (GEO: GSE51220 [Lawrenson et al., 2013]) (Table S6, Columns L-M). Lastly, expression of the 108 genes was examined in a dataset that compared normal FTE with high-grade serous carcinoma; 46 of 108 were significantly differentially regulated (GEO: GSE10971 [Tone et al., 2008]) (Table S6, Columns N-O). Based on the Protein Atlas and FTE datasets we selected 13 genes to confirm their role in anoikis escape (Figure 4A). Of 13 genes, 12 shared directionality between FTE and PEO1 cells grown in suspension, with the exception of Retinoblastoma Binding Protein 8 (RBBP8), which was downregulated in FTE cells. In the PEO1 RNA-seq analysis, all 13 genes were differentially upregulated in suspension and we validated the expression of these genes (Figure 4B). In contrast, OVCAR4 grown under adherent and suspension conditions displayed a slightly different expression profile. In OVCAR4, 8 of 13 genes were differentially expressed in suspension compared with adherent cells (Figure S4A). The gRNA profile for each gene demonstrated that all had gRNA depletion when cells were cultured in suspension, suggesting these genes are required for anchorage-independent cell survival (Figure 4C).
Figure 4.

Validation of CRISPR/Cas9 and RNA-seq in HGSOC Cells Reveals an Autophagy Effector
(A) Cross-referencing 108 genes with Protein Atlas and a publicly available dataset examining FTE grown in suspension (GEO: GSE51220). Thirteen genes were selected that are predominantly expressed in ovarian cancer and were significantly changed in FTE cells grown in suspension.
(B) PEO1 cells were grown in adherent and suspension settings for 7 days. RNA was extracted from cells and used for RT-qPCR against indicated genes. Internal control = 18S. Statistical test = two-sided unpaired t test.
(C) All target gRNA counts (y axis) for each of the 13 selected genes in adherent and suspension. Connecting line demonstrate the direction of abundance between adherent and suspension.
(D) Control shRNA and pooled shRNA against the 13 selected genes were transduced into PEO1 cells. RNA was extracted and used for RT-qPCR against indicated gene. 18S = internal control. Statistical test = ANOVA.
(E) shControl and pooled shRNA cells were cultured in adherent conditions for 1 and 7 days. On day 1 and 7 a CyQuant assay examined double-stranded DNA content as a surrogate for cell number. Intensity of CyQuant at Day 7/Day 1 graphed.
(F) Same as (E), but examined cells grown in suspension settings.
(G) Percent change between Adherent and Suspension growth shown in (E) and (F).
(H) shCtrl and shULK1 PEO1 and OVCAR4 cells grown in adherent (Adh) or suspension (Sus) for 48 h. RNA was extracted from cells and used for RT-qPCR against ULK1. Internal control = 18S. Statistical test = two-sided unpaired t test.
(I) Same as H, collected protein and immunoblotted against ULK1, p62, and LC3-II. Loading control = Actin. Values underneath blots are densitometry analysis of p62 (top) and LC3-II (bottom) expression normalized to Actin and shCtrl Adh.
(J) OVCAR4 shCtrl and shULK1 cells cultured in adherent (Adh) and suspension (Sus) culture for 48 h. Caspase activity was measured with Caspase-Glo assay. Assays in panels E-G performed in quintuplicate in at least two independent experiments. Statistical test = ANOVA.
∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Error bars = S.E.M. See also Figure S4.
These 13 genes potentially contribute to HGSOC cell survival in suspension, so we assessed cell viability following the knock down of each gene. Two independent shRNAs for each gene were pooled together, and PEO1 and OVCAR4 cells were transduced with the control shRNA or pooled shRNA. Gene knockdown was evaluated and confirmed with quantitative RT-PCR (RT-qPCR) (Figures 4D and S4B). In both PEO1 and OVCAR4 cells, 10 of 13 genes were significantly downregulated (Figures 4D and S4B). In PEO1 and OVCAR4 knockdown cells, we assessed growth in adherent and suspension settings. Double-stranded DNA content was measured as a surrogate for cell viability. In PEO1, 9 of 10 knockdowns had reduced viability in suspension compared with adherent (Figures 4E–4G). Knockdown of LGR6 was the exception, which showed increased cell viability in suspension compared with cells grown in adherent conditions (Figure 4G). In OVCAR4, only the 10 genes with significant knockdown resulted in decrease anchorage-independent cell growth, highlighting a direct correlation between the level of gene knockdown and suspension growth (Figures S4C and S4D). Taken together, most of the 13 genes selected for validation were confirmed to be important in maintaining cell viability when cells were cultured in suspension.
In both PEO1 and OVCAR4 cell lines, knockdown of ULK1 showed the most significant loss of cell viability in cells cultured in suspension over cells in adherent conditions. We next assessed whether suspension induced autophagy. Autophagy was assessed with a tandem fluorescently tagged LC3 (mCherry/GFP). Upon autophagosome fusion with the lysosome, acidification quenches GFP, but not mCherry. The percent mCherry-positive and GFP-negative (mCherry+/GFP-) cells is a functional autophagy indicator. In PEO1 and OVCAR8 cultured in suspension, there was an average increase of 6.8% and 15.4% mCherry+/GFP- cells compared with adherent cells, respectively (Figures S4E and S4F). Although OVCAR4 adherent cells had a high basal level of mCherry+/GFP- cells (57.4%), suspension culture actually reduced the frequency of mCherry+/GFP- cells by 9.5%, suggesting that, although autophagy is differentially regulated in suspension culture, there are cell-line-dependent effects that require further investigation.
We next evaluated ULK1-dependent autophagy by evaluating expression of ULK1, sequestosome 1 (SQSTM1/p62) depletion, and accumulation of lipidated microtubule-associated protein light chain 3 (LC3-II). To assess the role of ULK1 we utilized shULK1 PEO1, OVCAR4, and OVCAR8 cells cultured in suspension. We confirmed the loss of ULK1 protein expression in the three cell lines (Figure S4G). In shCtrl PEO1, OVCAR4, and OVCAR8, both ULK1 mRNA and protein expression were elevated upon suspension culture (Figures 4H–4I and S4H). In shCtrl and shULK1 PEO1 cells cultured in suspension, p62 and LC3 expression remained mostly unchanged (Figure 4I). In contrast, shCtrl OVCAR4 and OVCAR8 suspension cells had depletion of p62 and loss of ULK1 attenuated p62 depletion (Figures 4I and S4F). Furthermore, ULK1 knockdown enhanced forced suspension-induced apoptosis measured via caspase activity (Figure 4J). ULK1 and ULK2 are functionally redundant in the initiation of the pro-survival autophagic response; ULK2 was not identified in either the CRISPR/Cas9 or RNA-seq analysis. Also, ULK1 knockdown in PEO1, OVCAR8, and OVCAR4 cell lines did not promote a ULK2 compensatory response (Figure S4J). Taken together, suspension culture promotes autophagy and ULK1 is upregulated upon forced suspension and contributes to cell survival potentially by promoting an autophagic response.
Given anoikis resistance and dissemination are related to metastasis and a worse prognosis, we determined whether any of the 108 genes, individually or combined could predict survival. As an example, we examined the relationship between ULK1 expression in primary ovarian adenocarcinoma samples and recurrence at 3 years (No recurrence, n = 23 and Recurrence, n = 96) and found that increased ULK1 expression significantly correlated with recurrence (Figure 5A). We also cross-referenced the individual 108 genes to The Cancer Genome Atlas for ovarian serous cystadenocarcinoma (TCGA, Nature) to determine if mRNA expression correlated to overall survival (Table S6, Column P). “High” and “Low” expression was determined by the median Z score. The expression of eight genes correlated with poor overall survival (Figure 5B) (2011). We developed a gene signature by combining the 108 genes and examined the ovarian serous cystadenocarcinoma (TCGA, Nature) dataset and found that high expression of the top 108 genes significantly (p = 0.0015) predicted a worse overall survival (Figure 5C). One caveat is the risk of overfitting the model due to the high number of genes in the candidate list. Therefore, we tested whether our 108 genes would perform better at predicting survival than 108 random genes. We randomly selected genes to serve as permuted gene signatures and used these random genes to evaluate survival. Our 108 gene signature performs significantly better than the permuted models. Taken together, in ovarian cancer we confirmed that the genes identified significantly correlated to ovarian cancer clinical outcomes including metastasis, recurrence, and survival.
Figure 5.

ULK1 Predicts HGSOC Recurrence and Survival and the 108 Gene Signature Predicts Overall Survival
(A) Examination of ULK1 expression in primary ovarian adenocarcinoma that had no recurrence (n = 23) or recurrence (n = 96) after 3 years. Statistical test = two-sided unpaired t test. Error bars = S.E.M.
(B) A median expression was determined from each indicated gene to distinguish patient tumors with high expression (High, gray line) versus low expression (Low, dark line). KM curves were generated. TCGA (Ovarian cancer, Cancer Genome Atlas Research Network, 2011) dataset was used. Statistical test = Wilcoxon rank sum.
(C) For the TCGA Ovarian Cystadenocarcinoma dataset, 103 genes were mapped to their associated probe set or if the reference dataset was a list of genes, then expression data were pulled out based on matching gene names. Of the candidate genes, there was a varying number of probe sets present on all array platforms. Number of probe sets used indicated above KM curves. For each dataset, modeling survival and generating a KM curve on the candidate gene signature score. The dichotomous description of gene signature score (high or low gene signature score) was assigned. p Values were taken from the log likelihood statistic from the Cox proportional hazard models. For verification, we permuted a subset of random expression and random outcome (time, vital status) values, and broke the relationship between expression and outcome. This was done 1,000 times, and for each permuted dataset we modeled survival as described for the original analysis and generated a distribution of log likelihood statistics. Note: not all genes were identified in each dataset.
Looking beyond Ovarian Cancer
Anoikis escape is required for metastasis in most carcinomas, meaning they resist programmed cell death upon detachment from the ECM. The genes identified in this study could potentially contribute to our understanding of disease progression for multiple carcinomas. The 108 gene signature developed above and the random gene signature were tested against TCGA datasets for a variety of epithelial-derived cancers. Owing to the filtering step, there was not a static number of genes used in every analysis. For most datasets not all 108 genes were retained, but for most datasets at least 100 of the 108 genes were retained and used in the respective analysis. Except for colorectal cancer, our 108 gene set significantly predicted survival better than a random 108 genes (Table S7). All of the Kaplan-Meier (KM) curves show good separation of survival based on the gene signature scores (survival estimates for high scores are in gray and low scores are in black), except the curve for TCGA-COAD-colorectal cancer, which shows curve intersections (Figure 6). Overall, the 108 genes identified through the genome-wide approaches possibly playing a role in HGSOC progression and their expression predicts survival in a variety of carcinomas.
Figure 6.

108 Gene Signature Predicts Overall Survival in Several Epithelial-Derived Cancers
For the TCGA datasets, 108 gene expression data were pulled out based on matching gene names in the hg38 annotation. For each dataset, modeling survival and generating a KM curve on the candidate gene signature score. The dichotomous description of gene signature score (high or low gene signature score) was assigned. p Values were taken from the log likelihood statistic from the Cox proportional hazard models. For verification, we permuted a subset of random expression and random outcome (time, vital status) values and broke the relationship between expression and outcome. This was done 1,000 times, and for each permuted dataset we modeled survival as described for the original analysis and generated a distribution of log likelihood statistics. Note: not all genes were identified in each dataset and the number of genes used to generate KM curves indicated above graphs.
Discussion
Up to 80% of HGSOC is diagnosed at advanced stage and is characterized by extensive peritoneal metastases and development of ascites. HGSOC cells do not undergo a classical metastatic program mainly because of the disease etiology and progression. Most HGSOC are derived from transformed fallopian tube epithelial (FTE) cells on the fallopian tube fimbriae. Transformed FTE form STIC lesions that are histologically characterized by a mutated p53 signature. Completely independent of vasculature or lymphatic invasion, malignant cells within STIC lesions will detach from the ECM, exfoliate into the peritoneum, escape anoikis, and form distant disease. In this report, a combination of genome-wide approaches (CRISPR/Cas9 screen and RNA-seq) in conjunction with non-targeted metabolomics were utilized to evaluate the anchorage-independent transcriptional reprogramming HGSOC cells undergo to escape anoikis and exhibit anchorage independence. Each of the –omic approaches alone provides sufficient biological insight, and there is a significant amount of information gained from each technique individually. However, integration of all of the datasets provides a deeper and more comprehensive understanding of the biology. We identified both known and novel effectors of anoikis and discovered that these effectors predicted overall survival of patients with a variety of carcinomas.
The loss of ECM attachment activates and represses both inside-out (i.e., activation of ligand binding) and outside-in (i.e., ligand binding to a receptor) signaling. NOTCH, TGFβ, and ERBB signaling are well-established pathways that drive anoikis resistance. The CRISPR/Cas9 screen and RNA-seq analyses detected NOTCH (NOTCH1/3), TGFβ (LTBP1/THBS1), and ERBB (ERBB3/4) signaling pathways, which highlights the strength and confidence of these approaches. NOTCH3 is altered (amplified/overexpressed) in 17% of HGSOC cases, and NOTCH3 alterations correlated to poor overall survival (Hu et al., 2014). TGFβ signaling is similarly linked to anoikis resistance through several mechanisms including promoting an epithelial-to-mesenchymal transition, activation of Akt pathway, and increased SMAD-dependent transcriptional activation (Cieply et al., 2012, Horowitz et al., 2007, Munoz et al., 2008). Latent Transforming Growth Factor β Binding Protein 1 (LTBP1) is a latent ligand for the TGFβ receptor, and upon Thrombospondin-1 (THBS1)-mediated cleavage it promotes Akt and Erk activation (reviewed in Xu et al., 2012). Notably, THBS1 was also a top hit in our dataset; however, in suspended cells the THBS1 transcript was significantly downregulated and THBS1 gRNAs were depleted suggesting a regulatory role in anoikis. We observed that ERBB3 and ERBB4 receptor tyrosine kinases were upregulated in cells grown under forced suspension. ERBB3 is amplified or overexpressed in greater than 50% of HGSOC cases (Cerami et al., 2012, Gao et al., 2013). Pardeep et al. reported that ERBB3 is important for HGSOC dissemination to the omentum and ERBB3 knockdown reduced tumor burden. In contrast, in vitro knockdown of ERBB3 failed to inhibit HGSOC cell proliferation suggesting a regulatory role of the tumor microenvironment or a context-dependent function (Pradeep et al., 2014). The role of ERRB4 in HGSOC is complex. ERRB4 is expressed in 89%–96% of HGSOC tumors, is associated with chemotherapy refractory disease, and has at least three isoforms that are detectable in multiple cellular compartments (Davies et al., 2014, Gilmour et al., 2001). In PEO1 cells, targeting ERRB4 unexpectedly stimulated cell proliferation (Gilmour et al., 2001), highlighting that both ERBB3 and ERBB4 regulate proliferation and dissemination in a context-dependent manner. Our findings are consistent with previous reports that NOTCH, TGFβ, and ERBB signaling are important regulators of anoikis.
Another top hit, Unc-51 like autophagy activating kinase 1 (ULK1) is a serine/threonine kinase that under nutrient deprivation promotes autophagy in an AMP-activated protein kinase (AMPK)-dependent fashion (Egan et al., 2015). ULK1 is expressed in ∼40% of HGSOC and is correlated with progressive disease (Cerami et al., 2012, Gao et al., 2013). Consistently, we found ULK1 expression predicts HGSOC recurrence and overall survival. SBI-0206965 is a small molecule ULK1 inhibitor that, in models of lung and renal clear cell carcinoma, showed anti-cancer effects through inhibition of autophagy and the pentose phosphate pathway (Egan et al., 2015, Lu et al., 2018). In the HGSOC cell lines, forced suspension elevated ULK1 expression and knockdown of ULK1 inhibited cell viability. Notably, forced suspension induced autophagy in two of three cell lines, suggesting an autophagy-independent ULK1 function. For instance, ULK1 is key for stress/metabolic-induced clearance of damaged mitochondria (Kundu et al., 2008). Also, in adipocytes ULK1 functions to regulate FA oxidation and FA metabolism (Ro et al., 2013). Identification of ULK1 highlights the relationship between autophagy, anoikis, and metabolism.
In light of the metabolomics data demonstrating accumulation of PUFAs in suspension cells, NOTCH, TGFβ, ERBB, and ULK1 signaling have all been linked to increased FA metabolism. In cardiac endothelial cells, NOTCH signaling is an important regulator for FA transport and angiogenesis (Jabs et al., 2018). In hepatocellular carcinoma, TGFβ signaling is associated with increased FA oxidation and increased expression of Peroxisome proliferator-activated receptor gamma, a regulator of FA and glucose metabolism (Soukupova et al., 2017). ERBB signaling contributes to FA synthase by promoting increased expression of FASN, a known regulator of HGSOC dissemination (Grunt et al., 2009, Jiang et al., 2014). Loss of ULK1 in adipocytes reduces FA β-oxidation (Ro et al., 2013). The NOTCH, TGFβ, ERBB signaling pathways were highly enriched in our studies, suggesting these pathways are promoting ovarian cancer dissemination in part through promoting FA metabolism. In both ovarian and colon cancer, inhibition of a critical mitochondrial FA transporter, carnitine palmityltransferase I (CPT1), significantly promoted anchorage-independent cell death (Shao et al., 2016, Wang et al., 2018). Clinically applicable drugs (e.g., Etomoxir) have been developed to inhibit FA metabolism and CPT1 for non-oncogenic indications, suggesting that repurposing these drugs might a useful way to exploit a metabolic vulnerability to reduce HGSOC dissemination.
Two of the top CRISPR/Cas9 screen hits, ACADVL and ECHDC2, are associated with long-chain and very-long-chain FA metabolism and biosynthesis, respectively. Seemingly, these enzymes function on opposite ends of FA biology. Upon knock down of ACADVL or ECHDC2 cells, followed by metabolomics analysis, the suspension-induced PUFA enrichment was significantly attenuated. Similar to most cellular process, FA metabolism maintains energy and signaling equilibrium within cells. In the CRISPR/Cas9 screen, we expect that the loss of ACADVL or ECHDC2 was a mutually exclusive event. In suspension culture, ACADVL or ECHDC2 potentially regulates this equilibrium and loss of these effectors disrupts the equilibrium, exacerbates cellular stress, and reduces cell viability.
In conclusion, we uncovered novel regulators of HGSOC growth in suspension, and potentially, anoikis escape, through a CRISPR/Cas9 gene knockout screen, global metabolomics, and RNA-seq approaches. This project is unique in its use of new, unbiased technology to identify drivers of a critical aspect of ovarian cancer dissemination. We confirmed our top hits in a secondary cell line model and extensively cross-referenced them through several comprehensive and complex HGSOC datasets. The data presented here provide a more complete understanding of pathways that facilitate HGSOC dissemination and ovarian cancer progression and will stimulate development of novel therapeutics.
Limitations of the Study
Most of our primary analyses were performed in PEO1 cells (TP53-mutant and BRCA2-mutant). We performed validations in several secondary HGSOC cell lines with differing mutational backgrounds, including OVCAR4 (TP53-mutant, BRCA1/2-wild-type), OVCAR8, and OVARY1847 (both TP53-mutant, BRCA1/2-wild-type, ERBB2-mutant, and KRAS-mutant). However, we cannot make predictions for every scenario, as certain mutations that we have not examined may result in different behavior in suspension. For patients, their individual mutational background may significantly affect mechanisms of anoikis resistance. Cross-referencing our gene lists with publicly available ovarian cancer datasets gives us confidence that our findings have clinical importance, but new targets will need to be examined in vivo before ultimate significance can be determined. This is especially true of immunocompetent animal models and human patients, in which transformed cells must not only survive in suspension, but must also evade immune surveillance within the peritoneal cavity. We cannot make predictions for immune evasion based only on the in vitro data of the current study.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We would like to acknowledge Dr. Andrew P. Bradford and Alexandra McMellen for critical reading of this manuscript. This work was supported by the National Institutes of Health (B.G.B., R00CA194318; J.M.E., R01CA117907 and R01GM120109), the Department of Defense OCRP (B.G.B., OC170228), as well as the American Cancer Society (L.J.W., ACS-IRG and B.T.S., ACS-IRG, 16-184-56), the University of Colorado Department of Obstetrics and Gynecology Academic Enrichment Fund (AEF), RNA Biosciences Initiative (J.K.R.), and the University of Colorado Cancer Center Support Grant (P30CA046934).
Author Contributions
L.J.W., Z.L.W., and B.G.B. conceived of the project and experiments. L.J.W., Z.L.W., T.M.Y., M.J., S.W.B., and B.G.B. designed and performed experiments. L.J.W., Z.L.W., L.Q., T.M.Y., B.T.S., K.D.S., S.K., V.F.-R., H.S., L.A.V., C.M.C., H.K., J.M.E., J.K.R., and B.G.B. synthesized and analyzed data and aided in interpretation. L.J.W., Z.L.W., and B.G.B. wrote the manuscript. All authors read and revised the manuscript and have read and approved of this version of the manuscript.
Declaration of Interests
The authors declare no competing financial interests.
Published: September 27, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.07.049.
Supplemental Information
CmpdID = compound ID. HMDB = human metabolome database.
CmpdID = compound ID. HMDB = human metabolome database.
TPM = transcripts per million. FDR = false discovery rate. FC = fold change.
FC = fold change. gRNA = guide RNA.
FC = fold change. Sus = suspension. Adh = adherent. Mets = metastasis.
GSE73064 = primary high-grade serous tumors compared with metastatic (ascites and distant sites) tumors. Protein Atlas - Ovarian cancer mRNA (Yes is greater than 0 Fragments Per Kilobase of transcript per Million (FPKM) /No is 0 Fragments Per Kilobase of transcript per Million). Protein Atlas – Ovarian cancer protein (Yes is greater than 0% of patients). GSE51220 = mRNA expression from fallopian tube epithelial (FTE) cells cultured in 2D and 3D conditions. FC = fold change. GSE10971 = mRNA expression in FTE (n = 24) compared with high grade serous carcinoma (HGS). TCGA (Nature, 2011) – Kaplan Meier analysis based on gene expression (z-score > 0) and p-value calculated by Wilcoxon Rank Sum.
Using indicated TCGA datasets calculated p-values for 108 gene signature or random 108 genes across cancer types.
References
- Bahnson B.J., Anderson V.E., Petsko G.A. Structural mechanism of enoyl-CoA hydratase: three atoms from a single water are added in either an E1cb stepwise or concerted fashion. Biochemistry. 2002;41:2621–2629. doi: 10.1021/bi015844p. [DOI] [PubMed] [Google Scholar]
- Berek J.S., Hacker N.F. Wolters Kluwer Health/Lippincott Williams & Wilkins; 2015. Berek & Hacker's Gynecologic Oncology. [Google Scholar]
- Bian Y., Yu Y., Wang S., Li L. Up-regulation of fatty acid synthase induced by EGFR/ERK activation promotes tumor growth in pancreatic cancer. Biochem. Biophys. Res. Commun. 2015;463:612–617. doi: 10.1016/j.bbrc.2015.05.108. [DOI] [PubMed] [Google Scholar]
- Brown C.W., Brodsky A.S., Freiman R.N. Notch3 overexpression promotes anoikis resistance in epithelial ovarian cancer via upregulation of COL4A2. Mol. Cancer Res. 2015;13:78–85. doi: 10.1158/1541-7786.MCR-14-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieply B., Riley P.T., Pifer P.M., Widmeyer J., Addison J.B., Ivanov A.V., Denvir J., Frisch S.M. Suppression of the epithelial-mesenchymal transition by Grainyhead-like-2. Cancer Res. 2012;72:2440–2453. doi: 10.1158/0008-5472.CAN-11-4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies S., Holmes A., Lomo L., Steinkamp M.P., Kang H., Muller C.Y., Wilson B.S. High incidence of ErbB3, ErbB4, and MET expression in ovarian cancer. Int. J. Gynecol. Pathol. 2014;33:402–410. doi: 10.1097/PGP.0000000000000081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Steen S., Bulten J., Van De Vijver K.K., Van Kuppevelt T.H., Massuger L. Changes in the extracellular matrix are associated with the development of serous tubal intraepithelial carcinoma into high-grade serous Carcinoma. Int. J. Gynecol. Cancer. 2017;27:1072–1081. doi: 10.1097/IGC.0000000000000933. [DOI] [PubMed] [Google Scholar]
- Egan D.F., Chun M.G., Vamos M., Zou H., Rong J., Miller C.J., Lou H.J., Raveendra-Panickar D., Yang C.C., Sheffler D.J. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol. Cell. 2015;59:285–297. doi: 10.1016/j.molcel.2015.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q., Yang Z., Xu S., Li X., Yang X., Jin P., Liu Y., Zhou X., Zhang T., Gong C. Heterotypic CAF-tumor spheroids promote early peritoneal metastasis of ovarian cancer. J. Exp. Med. 2019;216:688–703. doi: 10.1084/jem.20180765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour L.M., Macleod K.G., Mccaig A., Gullick W.J., Smyth J.F., Langdon S.P. Expression of erbB-4/HER-4 growth factor receptor isoforms in ovarian cancer. Cancer Res. 2001;61:2169–2176. [PubMed] [Google Scholar]
- Grunt T.W., Wagner R., Grusch M., Berger W., Singer C.F., Marian B., Zielinski C.C., Lupu R. Interaction between fatty acid synthase- and ErbB-systems in ovarian cancer cells. Biochem. Biophys. Res. Commun. 2009;385:454–459. doi: 10.1016/j.bbrc.2009.05.085. [DOI] [PubMed] [Google Scholar]
- Horowitz J.C., Rogers D.S., Sharma V., Vittal R., White E.S., Cui Z., Thannickal V.J. Combinatorial activation of FAK and AKT by transforming growth factor-beta1 confers an anoikis-resistant phenotype to myofibroblasts. Cell Signal. 2007;19:761–771. doi: 10.1016/j.cellsig.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W., Liu T., Ivan C., Sun Y., Huang J., Mangala L.S., Miyake T., Dalton H.J., Pradeep S., Rupaimoole R. Notch3 pathway alterations in ovarian cancer. Cancer Res. 2014;74:3282–3293. doi: 10.1158/0008-5472.CAN-13-2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabs M., Rose A.J., Lehmann L.H., Taylor J., Moll I., Sijmonsma T.P., Herberich S.E., Sauer S.W., Poschet G., Federico G. Inhibition of endothelial notch signaling impairs fatty acid transport and leads to metabolic and vascular remodeling of the adult heart. Circulation. 2018;137:2592–2608. doi: 10.1161/CIRCULATIONAHA.117.029733. [DOI] [PubMed] [Google Scholar]
- Jayson G.C., Kohn E.C., Kitchener H.C., Ledermann J.A. Ovarian cancer. Lancet. 2014;384:1376–1388. doi: 10.1016/S0140-6736(13)62146-7. [DOI] [PubMed] [Google Scholar]
- Jiang L., Wang H., Li J., Fang X., Pan H., Yuan X., Zhang P. Up-regulated FASN expression promotes transcoelomic metastasis of ovarian cancer cell through epithelial-mesenchymal transition. Int. J. Mol. Sci. 2014;15:11539–11554. doi: 10.3390/ijms150711539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Kim B., Song Y.S. Ascites modulates cancer cell behavior, contributing to tumor heterogeneity in ovarian cancer. Cancer Sci. 2016;107:1173–1178. doi: 10.1111/cas.12987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu M., Lindsten T., Yang C.Y., Wu J., Zhao F., Zhang J., Selak M.A., Ney P.A., Thompson C.B. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112:1493–1502. doi: 10.1182/blood-2008-02-137398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz D.M., Rinaldo P., Rhead W.J., Tian L., Millington D.S., Vockley J., Hamm D.A., Brix A.E., Lindsey J.R., Pinkert C.A. Targeted disruption of mouse long-chain acyl-CoA dehydrogenase gene reveals crucial roles for fatty acid oxidation. Proc. Natl. Acad. Sci. U S A. 1998;95:15592–15597. doi: 10.1073/pnas.95.26.15592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrenson K., Notaridou M., Lee N., Benjamin E., Jacobs I.J., Jones C., Gayther S.A. In vitro three-dimensional modeling of fallopian tube secretory epithelial cells. BMC Cell Biol. 2013;14:43. doi: 10.1186/1471-2121-14-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y., Miron A., Drapkin R., Nucci M.R., Medeiros F., Saleemuddin A., Garber J., Birch C., Mou H., Gordon R.W. A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J. Pathol. 2007;211:26–35. doi: 10.1002/path.2091. [DOI] [PubMed] [Google Scholar]
- Lengyel E., Burdette J.E., Kenny H.A., Matei D., Pilrose J., Haluska P., Nephew K.P., Hales D.B., Stack M.S. Epithelial ovarian cancer experimental models. Oncogene. 2014;33:3619–3633. doi: 10.1038/onc.2013.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Metzinger M.N., Lewellen K.A., Cripps S.N., Carey K.D., Harper E.I., Shi Z., Tarwater L., Grisoli A., Lee E. Obesity contributes to ovarian cancer metastatic success through increased lipogenesis, enhanced vascularity, and decreased infiltration of M1 macrophages. Cancer Res. 2015;75:5046–5057. doi: 10.1158/0008-5472.CAN-15-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J., Zhu L., Zheng L.P., Cui Q., Zhu H.H., Zhao H., Shen Z.J., Dong H.Y., Chen S.S., Wu W.Z., Tan J.M. Overexpression of ULK1 represents a potential diagnostic marker for clear cell renal carcinoma and the Antitumor effects of SBI-0206965. EBioMedicine. 2018;34:85–93. doi: 10.1016/j.ebiom.2018.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda F., Mannion D., Liu S., Zheng Y., Mangala L.S., Redondo C., Herrero-Gonzalez S., Xu R., Taylor C., Chedom D.F. Salt-inducible kinase 2 couples ovarian cancer cell metabolism with survival at the adipocyte-rich metastatic niche. Cancer Cell. 2016;30:273–289. doi: 10.1016/j.ccell.2016.06.020. [DOI] [PubMed] [Google Scholar]
- Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010;22:132–139. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Munoz N.M., Baek J.Y., Grady W.M. TGF-beta has paradoxical and context dependent effects on proliferation and anoikis in human colorectal cancer cell lines. Growth Factors. 2008;26:254–262. doi: 10.1080/08977190802291667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piek J.M., Van Diest P.J., Zweemer R.P., Jansen J.W., Poort-Keesom R.J., Menko F.H., Gille J.J., Jongsma A.P., Pals G. Dysplastic changes in prophylactically removed Fallopian tubes of women predisposed to developing ovarian cancer. J. Pathol. 2001;195:451–456. doi: 10.1002/path.1000. [DOI] [PubMed] [Google Scholar]
- Pradeep S., Kim S.W., Wu S.Y., Nishimura M., Chaluvally-Raghavan P., Miyake T., Pecot C.V., Kim S.J., Choi H.J., Bischoff F.Z. Hematogenous metastasis of ovarian cancer: rethinking mode of spread. Cancer Cell. 2014;26:77–91. doi: 10.1016/j.ccr.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ro S.H., Jung C.H., Hahn W.S., Xu X., Kim Y.M., Yun Y.S., Park J.M., Kim K.H., Seo M., Ha T.Y. Distinct functions of Ulk1 and Ulk2 in the regulation of lipid metabolism in adipocytes. Autophagy. 2013;9:2103–2114. doi: 10.4161/auto.26563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H., Mohamed E.M., Xu G.G., Waters M., Jing K., Ma Y., Zhang Y., Spiegel S., Idowu M.O., Fang X. Carnitine palmitoyltransferase 1A functions to repress FoxO transcription factors to allow cell cycle progression in ovarian cancer. Oncotarget. 2016;7:3832–3846. doi: 10.18632/oncotarget.6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukupova J., Malfettone A., Hyrossova P., Hernandez-Alvarez M.I., Penuelas-Haro I., Bertran E., Junza A., Capellades J., Giannelli G., Yanes O. Role of the transforming growth factor-beta in regulating hepatocellular carcinoma oxidative metabolism. Sci. Rep. 2017;7:12486. doi: 10.1038/s41598-017-12837-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D., Morris J.H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N.T., Roth A., Bork P. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:D362–D368. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tone A.A., Begley H., Sharma M., Murphy J., Rosen B., Brown T.J., Shaw P.A. Gene expression profiles of luteal phase fallopian tube epithelium from BRCA mutation carriers resemble high-grade serous carcinoma. Clin. Cancer Res. 2008;14:4067–4078. doi: 10.1158/1078-0432.CCR-07-4959. [DOI] [PubMed] [Google Scholar]
- Toyama A., Suzuki A., Shimada T., Aoki C., Aoki Y., Umino Y., Nakamura Y., Aoki D., Sato T.A. Proteomic characterization of ovarian cancers identifying annexin-A4, phosphoserine aminotransferase, cellular retinoic acid-binding protein 2, and serpin B5 as histology-specific biomarkers. Cancer Sci. 2012;103:747–755. doi: 10.1111/j.1349-7006.2012.02224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M., Oksvold P., Fagerberg L., Lundberg E., Jonasson K., Forsberg M., Zwahlen M., Kampf C., Wester K., Hober S. Towards a knowledge-based human protein Atlas. Nat. Biotechnol. 2010;28:1248–1250. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- Uhlen M., Fagerberg L., Hallstrom B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Wang Y.N., Zeng Z.L., Lu J., Wang Y., Liu Z.X., He M.M., Zhao Q., Wang Z.X., Li T., Lu Y.X. CPT1A-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis. Oncogene. 2018;37:6025–6040. doi: 10.1038/s41388-018-0384-z. [DOI] [PubMed] [Google Scholar]
- Wheeler L.J., Watson Z.L., Qamar L., Yamamoto T.M., Post M.D., Berning A.A., Spillman M.A., Behbakht K., Bitler B.G. CBX2 identified as driver of anoikis escape and dissemination in high grade serous ovarian cancer. Oncogenesis. 2018;7:92. doi: 10.1038/s41389-018-0103-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P., Liu J., Derynck R. Post-translational regulation of TGF-beta receptor and Smad signaling. FEBS Lett. 2012;586:1871–1884. doi: 10.1016/j.febslet.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CmpdID = compound ID. HMDB = human metabolome database.
CmpdID = compound ID. HMDB = human metabolome database.
TPM = transcripts per million. FDR = false discovery rate. FC = fold change.
FC = fold change. gRNA = guide RNA.
FC = fold change. Sus = suspension. Adh = adherent. Mets = metastasis.
GSE73064 = primary high-grade serous tumors compared with metastatic (ascites and distant sites) tumors. Protein Atlas - Ovarian cancer mRNA (Yes is greater than 0 Fragments Per Kilobase of transcript per Million (FPKM) /No is 0 Fragments Per Kilobase of transcript per Million). Protein Atlas – Ovarian cancer protein (Yes is greater than 0% of patients). GSE51220 = mRNA expression from fallopian tube epithelial (FTE) cells cultured in 2D and 3D conditions. FC = fold change. GSE10971 = mRNA expression in FTE (n = 24) compared with high grade serous carcinoma (HGS). TCGA (Nature, 2011) – Kaplan Meier analysis based on gene expression (z-score > 0) and p-value calculated by Wilcoxon Rank Sum.
Using indicated TCGA datasets calculated p-values for 108 gene signature or random 108 genes across cancer types.