

Abstract
We set out to investigate the genetic adaptations of the marine fungus Paradendryphiella salina CBS112865 for degradation of brown macroalgae. We performed whole genome and transcriptome sequencing and shotgun proteomic analysis of the secretome of P. salina grown on three species of brown algae and under carbon limitation. Genome comparison with closely related terrestrial fungi revealed that P. salina had a similar but reduced CAZyme profile relative to the terrestrial fungi except for the presence of three putative alginate lyases from Polysaccharide Lyase (PL) family 7 and a putative PL8 with similarity to ascomycete chondroitin AC lyases. Phylogenetic and homology analyses place the PL7 sequences amongst mannuronic acid specific PL7 proteins from marine bacteria. Recombinant expression, purification and characterization of one of the PL7 genes confirmed the specificity. Proteomic analysis of the P. salina secretome when growing on brown algae, revealed the PL7 and PL8 enzymes abundantly secreted together with enzymes necessary for degradation of laminarin, cellulose, lipids and peptides. Our findings indicate that the basic CAZyme repertoire of saprobic and plant pathogenic ascomycetes, with the addition of PL7 alginate lyases, provide P. salina with sufficient enzymatic capabilities to degrade several types of brown algae polysaccharides.
Subject terms: Microbiology, Fungi, Proteomics, Molecular ecology
Introduction
Knowledge of the diversity and distribution of marine fungi is increasing in step with improvements in culture-based and molecular identification techniques and increased research efforts1–9. Numerous fungal species have been isolated from brown algae1 and observations and enzyme screenings have shown them to be able to grow on macroalgal substrates while producing enzymes that catalyze degradation of algal polysaccharides2–5, however, only few marine fungi have been genome sequenced and even fewer studied in combination with culture-based analyses of enzymatic degradation of marine macroalgae (seaweeds)6.
The observed association of the fungus Paradendryphiella salina (Ascomycota) to brown algae dates back to 1916 when the fungus was first described as Cercospora salina. Its ecological mode and habitat were described as saprophytic on seaweeds7. Several reports investigating enzyme secretion and carbon utilization have confirmed the ability of P. salina to utilize alginate, laminarin and cellulose from brown algae8,9. Based on this we identified P. salina as an interesting candidate for studying potential adaptations of its enzyme repertoire to the breakdown of brown algae polysaccharides.
Brown macroalgae (Phaeophyceae) are a large and diverse class of marine macrophytes found abundantly in the seas of the southern and northern hemisphere10. The cell wall of brown algae consists mainly of alginate, the most abundant polysaccharide found in the outermost cell wall layer11, fucose-containing sulfated polysaccharides (FCSPs) and cellulose in a 3:1:1 ratio10. Presence of mixed linkage glucan (MLG)12 and arabinogalactans linked to proteins have also been reported13. Carbon storage in brown algae consists mainly of laminarin, whilst trehalose and mannitol appear to function more as osmotic stress regulators14.
As a part of our quest to annotate enzymes for the biorefining of brown algae, this study was undertaken to elucidate the enzymatic repertoire utilized by a known saprophytic marine fungus in the breakdown of brown algae and to examine the degree of enzyme overlap shared by this fungus with its terrestrial counterparts. By using a combination of genomics, culture-based methods and shotgun proteomics we show that P. salina secretes enzymes capable of catalyzing degradation of a range of polysaccharides found in brown macroalgae, notably also alginate. Our study also indicates that alginate lyase is the only additional enzyme adaptation necessary for allowing P. salina to degrade brown algae, when compared to its terrestrial relatives.
Methods
Brown macroalgae species
Ascophyllum nodosum was harvested along the coastline of Norway (European Protein, Bække, Denmark). Fucus serratus was harvested in the Kattegat Sea (Dansk TANG, Nykøbing Sj., Denmark). Saccharina latissima was harvested along the coastline of the Faroe Islands (Ocean Rainforest, Kaldbak, Faroe Islands). All three species were obtained in dry form and milled to particle sizes under 1 mm.
Fungal strain and culture conditions
Paradendryphiella salina CBS112865 (G.K. Sutherland) Woudenb. & Crous, isolated from F. serratus in the North Sea, was obtained from CBS-KNAW culture collection (www.westerdijkinstitute.nl/collections/) and maintained on 2% oatmeal agar. Spores were added in a final concentration of 105 spores/ml. The base medium used for all fermentations was prepared with 0.10% (w/v) glucose and otherwise as previously described15. The three species of powdered brown algae were autoclaved (121 °C, 20 min.) before being added to the base medium at 2% w/v. The carbon limited medium consisted solely of the base medium. All fungal fermentations were carried out in five replicates (23 °C, 180 rpm, 14 days) in standard 250 ml Erlenmeyer flasks. All fermentations were routinely checked for contamination by microscopy. One of the replicates in the P. salina fermentation of A. nodosum failed due to an inoculation error, thus only four yielded results.
Genome and transcriptome sequencing and assembly
DNA was extracted from P. salina mycelium growing in liquid YPD medium for 4 days using a Qiagen (Venlo, Holland) DNeasy plant minikit. RNA was extracted from mycelium growing in liquid base medium with 2% S. latissima for 7 days using a Qiagen RNeasy minikit. mRNA was purified by polyA capture, fragmented and converted to double-stranded cDNA. The “dUTP method” was used to generate strand-specific mRNA-seq libraries16. Paired-end sequence reads (125 bp) were generated using an Illumina HiSeq 2500. FASTQ sequence files were generated using Illumina Casava (1.8.3) for DNA and bcl2fastq2 (2.18) for RNA. The draft genome was assembled using the CLC genomics workbench (9.5.1). Misassemblies and nucleotide disagreement between the Illumina data and the contig sequences were corrected with Pilon17 (1.20). The contigs were linked into scaffolds using SSPACE Premium scaffolder (2.3)18 and the gapped regions within the scaffolds were partially closed in an automated manner using GapFiller (1.10)19. The transcriptome was assembled by performing genome alignment using Tophat2 (2.1.1)20. The alignment was used to guide a de novo assembly of the RNA reads using Trinity (2.4.0)21.
Gene prediction and annotation
Genome-wide gene prediction was accomplished by creating a training set using AUGUSTUS (2.7) (http://bioinf.uni-greifswald.de/webaugustus/)22 using the assembled transcripts as hints. The predicted protein sequences were annotated using HMMer323 with dbCAN24 models using an E-value threshold of 10−5, InterProScan 525 and BLASTp26 (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Phobius27 and Euk-mPLoc (2.0)28 (http://www.csbio.sjtu.edu.cn/bioinf/euk-multi-2/) were used for subcellular localization and transmembrane domain prediction. For functional predictions of CAZymes, HotPep29 and CUPP30 were used. Substrate specificities of CAZymes were inferred by manual inspection of CAZy (www.cazy.org)31 and the BRENDA database (www.brenda-enzymes.org)32. Another version of HotPep was used for prediction of protease families33 (https://sourceforge.net/projects/hotpep-protease/). For all accepted annotations at family level, at least two different methods had to agree. Sulfatase annotation consisted of BLASTp searching sulfatase hits found with InterProScan in the SulfAtlas34 database (http://abims.sb-roscoff.fr/sulfatlas/) with E-value 0 as threshold.
Phylogenetic analysis
Phylogenetic analyses were performed by aligning the amino acid sequences of the putative catalytic domains with Mafft35 which were manually inspected in CLC main workbench (8). Maximum likelihood analyses were performed with RaxML blackbox36 using WAG as substitution matrix and otherwise default parameters at the CIPRESS server (www.phylo.org/)37. RaxML reached the MRE-based Bootstopping criterion after 252 replicates.
Genome comparison
Genome protein sequences were downloaded from Genbank38. Local BLASTp searches of protein sequences were executed in CLC main workbench (version 8) with default parameters. Venn analysis and diagram development of CAZymes were performed online (http://bioinformatics.psb.ugent.be/webtools/Venn/).
Carbohydrate monomer composition
Carbohydrate monomer composition was determined by HPAEC-PAD analysis following a two-step sulfuric acid hydrolysis as previously described39.
Shotgun proteomic analysis of fungal secretomes
Protein precipitation was performed as previously described40. The precipitated proteins were diluted in digestion buffer, consisting of 10% Acetonitrile and 50 mM HEPES buffer pH 8.5. 10 μg of protein from each sample was reduced, alkylated and in-solution digested with trypsin (Sigma-Aldrich, St. Louise, MI, US) and LysC (Wako, Osaka, Japan) and desalted on C18 filters (Thermo Fisher Scientific, Rockford, USA). 1 μg from each sample was analysed by Liquid Chromatography-tandem Mass Spectrometry (LC-MS/MS) as previously described40. Protein identification was performed using the open-source software MaxQuant (1.6.3.4)41 and Perseus42 as previously described40. A hit was only considered if present in three out of five biological replicates. The iBAQ values were normalized across the samples by multiplying them with factors calculated from the original protein concentration in the supernatants (Supplementary Table S1) before being averaged across the replicates and used for further analyses.
Enzyme assays of supernatants
The AZurine CrossLinked (AZCL) assay was carried out in 96-well microtiter plates as suggested by the manufactor. Each of the 12 selected AZCL substrates (Supplementary Table S2) (Megazyme, Bray, Ireland) was prepared by adding 0.01% w/v of substrate to 0.1% agarose dissolved in 0.05 M Britten-Robinson (BR) buffer43 (pH 5). Each well contained 200 μl of substrate and 100 μl of supernatant. The plates were incubated (30 °C, 45 h), centrifuged (15 min. 3000 g) and 100 μl of reaction was transferred to a new plate and quantified by measuring A590.
Alginate lyase activity was assayed on 1.5 mg/ml sodium alginate from F. vesiculosus (Sigma) mixed with 10 mM BR buffer (pH 5). The reactions were carried out in 96-well microtiter plates by mixing 20 μl of sample with 180 μl of substrate and incubating (30 °C, 120 min.) Degradation of substrates was monitored by measuring A235.
Fucoidanase activity was assayed on fucoidan extracted from Fucus evanescens and Fucus vesiculosus using Carbohydrate–Polyacrylamide Gel Electrophoresis (C-PAGE) as previously described44.
Genes, cloning, expression and purification of PL7 alginate lyase
An open reading frame encoding PsAlg7A (ENA acc. LR536815) was identified in P. salina. The gene sequence was truncated to exclude the 22 amino acids long predicted signal peptide. The codon optimized gene including a C-terminal His-tag (Supplementary Table S3) for Pichia pastoris was cloned into pPICZαA (GenScript, Piscataway, NJ, USA). The resulting construct was transformed into E. coli strain DH5α and selected on low salt LB agar plates with 25 μg ml−1 zeocin and propagated in low salt LB medium with 25 μg ml−1 zeocin (Invitrogen, Carlsbad, CA, USA). The pPICZαA-PsAgl7A construct was linearized by PmeI (New England BioLabs, Ipswich, MA, USA), transformed into P. pastoris X-33 by electroporation (Micropulser; Bio-Rad, Hercules, CA, USA), and selected (30 °C, 3 days) on yeast peptone dextrose plates with 100 μg ml−1 zeocin. Transformants were grown in a 5 L Sartorius Biostat Aplus fermenter via glycerol fed batch fermentation at 30 °C for 24 hours. Then, a methanol fed-batch phase was initiated to induce expression and secretion of the enzyme. After 96 hours of methanol induction at 20 °C. The cell-free supernatant was concentrated and buffer-exchanged into 50 mM Tris, 500 mM NaCl, 20 mM imidazole pH 7.5 by ultrafiltration using the cross-flow filter reactor equipped with a 10 kDa cutoff membrane (Millipore, Sartorius, Goettingen, Germany). The supernatant was applied to a 5 ml HisTrap FF HP column (GE Healthcare Uppsala, Sweden) equilibrated with 50 mM Tris, 500 mM NaCl, 20 mM imidazole pH 7.5 and eluted (2 ml.min−1) by a linear 20–500 mM imidazole gradient (30 CV). Fractions containing the target enzymes were pooled, concentrated (Viaspin (10 kDa), Goettingen, Germany), Sartorius) and applied to a Hiload 16/60 Superdex G75 column (GE Healthcare) equilibrated with 10 mM NaOAc, 150 mM NaCl, pH 6 (0.5 ml.min−1). Fractions containing pure target enzymes were concentrated (Viaspin (10 kDa), Sartorius) and stored at 4 °C. The purity was checked on 12% SDS-PAGE gels. The theoretical molar extinction coefficient and size were calculated using ProtParam (http://web.expasy.org/protparam) to 30035 M−1 cm−1 and 25.445 kDa. Protein concentrations were determined by A280 using the theoretically obtained molar extinction coefficients.
Enzyme kinetics assays
Reactions on 0.1875 mg ml−1 of PsAlg7A were set up in triplicates in a 96 well quartz plate. Alginate (Sigma), polyguluronic acid (larger than 5 kDa) (Carbosynth, Compton, UK), and polymannuronic acid (larger than 5 kDa) (Carbosynth) substrates were prepared in 20 mM Britten-Robinson, 200 mM NaCl, pH 5 buffer. Substrate concentrations ranging from 0.025 to 1.5 mg ml−1 were assayed. To determine enzyme kinetics, the averaged initial velocities (linear range over 10 minute runs) in milli-absorbance units (mAU) at A235 were calculated to mM per seconds of 4-deoxy-4,5-unsaturated mono-uronates from measurement of double bonds at absorbance of 235 nm caused by lyase induced β-elimination, versus the substrate concentrations using the extinction coefficient of 6150 M−1 cm−145,46.
Results
Genome properties and comparison
The draft genome of P. salina was assembled to a size of 27.4Mbp with an average GC content of 52%, a N50 of 32 K and an average scaffold size of 15062 bp. The assembled transcriptome contained 21394 transcripts with an average size of 1789bp and a N50 of 2792 (Supplementary Table S4). For gene prediction both the genome and transcripts were submitted to the AUGUSTUS server, which predicted 9281 proteins in the genome (Supplementary File S1). The transcripts were used as is, in a tBLASTn search towards the genome predicted proteins and were found to match approximately 8600 of predicted proteins after filtering (identity >75%, HSP length >200 bp) (Supplementary File S2). The combined annotations from CUPP, HotPep, dbCAN and InterProScan predicted putative domains for 8184 proteins of which 448 contained CAZyme domains. CUPP and HotPep annotated putative functions for 323 of the putative CAZymes (Supplementary File S2).
In order to elucidate P. salina potential genetic macroalgae specific specializations, a genome comparison of a terrestrial fungus belonging to the same family was undertaken. Candidates were found by BLASTn (megablast)47 of the ribosomal barcode from P. salina (accession MH873443.1) against the The Whole Genome (WGS) blast database with restriction to Pleosporaceae (taxid:28556). The first hit was the terrestrial plant pathogen Stemphylium lycopersici at E-value 0.0, 100% query coverage and 98% identity (Supplementary Table S5). The result correlated with the reported close taxonomic relationship of P. salina and Stemphylium species48. The protein sequences from the S. lycopersici genome (accession ASM119154v1)49 were subjected to the same annotation analyses as P. salina and used for comparisons (Supplementary File S3). The P. salina and S. lycopersici cross genome BLASTp showed a 98.3% agreement between the annotations of CAZymes and an average pairwise identity of 88% (±10). Of the 9282 predicted P. salina proteins, 7581 showed 75% identity or higher to S. lycopersici proteins, 461 proteins fell under the E-value threshold of these, and 274 remained unannotated (Supplementary File S3). The CAZyme annotations were binned according to broad substrate specificities and compared. The two fungi showed similar substrate profiles, however, P. salina generally had reduced numbers of CAZYme encoding genes in most categories related to polysaccharides found mainly in the primary plant-cell walls of terrestrial plants, the most significant being hemicellulose, pectin, β-galactan and α-arabinan. Chondroitin AC lyase (chondroitin sulfate) and alginate lyase were unique to P. salina (Fig. 1).
Figure 1.

Comparative distribution of CAZymes in P. salina and its terrestrial relative S. lycopersici, binned according to substrate specificities. CAZy domains were annotated with dbCAN HMM models and CUPP. Substrate designations were inferred by using the predicted functions from CUPP (Supplementary Files S2 and S3).
Protein sequences from two additional fungal plant pathogens, Bipolaris maydis (Ascomycota) (accession GCF_000354255.1)50 and Alternaria alternate (Ascomycota) (accession GCF_001642055.1)51, found amongst the top BLASTn results were included in a broad CAZyme comparison, to broaden the taxonomic variance. The CAZyme profiles showed high similarity with respect to families and paralogue copies within them (Fig. 2) (Supplementary Table S6). Compared to the three terrestrial fungal plant pathogens, P. salina harbored 25% fewer CAZyme domains than S. lycopersici and B. maydis and 35% fewer than A. alternata. Approximately the same relationship applied for the putative sugar transporters (Table 1).
Figure 2.
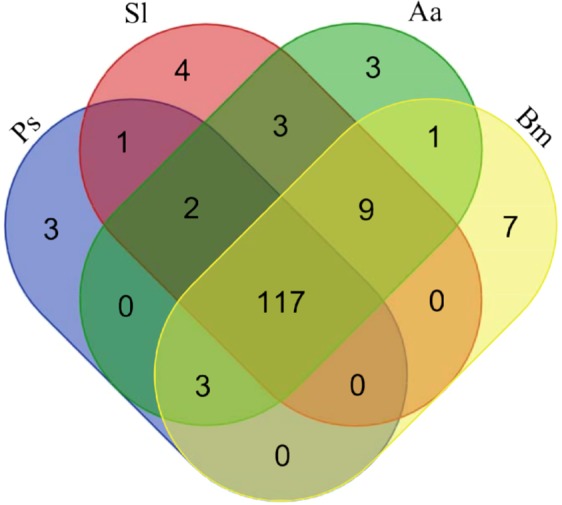
Venn diagram of shared CAZyme domain families between P. salina (Ps), and the terrestrial fungi: S. lycopersici (Sl), A. alternata (Aa) and B. maydis (Bm). The domains were annotated using dbCAN HMM models and CUPP (Supplementary Table S6). The three unique families in P. salina (indicated in the blue zone) were PL7, PL8 and CBM24.
Table 1.
Summary of annotated CAZymes and potential sugar transporters in P. salina (Ps), S. lycopersici (Sl), B. maydis (Bm) and A. alternata (Aa).
| Organism | Auxiliary Activities | Carbohydrate Esterases | Polysaccharide Lyases | Glycosyl- Transferases | Glycoside Hydrolases | Total CAZymes | Major Facilitator (PF07690) | Sugar (and other) transporter (PF00083) | Total transporters |
|---|---|---|---|---|---|---|---|---|---|
| Ps | 86 | 55 | 22 | 209 | 82 | 454 | 157 | 73 | 230 |
| Sl | 136 | 80 | 23 | 278 | 99 | 616 | 205 | 91 | 296 |
| Bm | 141 | 77 | 16 | 274 | 99 | 607 | 219 | 78 | 297 |
| Aa | 167 | 95 | 26 | 303 | 108 | 699 | 256 | 104 | 360 |
CAZymes were annotated using dbCAN HMM models and CUPP and putative transporter genes were annotated using PFAM HMM models.
Unique to P. salina were three PL7 alginate lyases, in addition to a PL8 protein with similarity to ascomycete chondroitin AC lyases, and a CBM24 appended to a GH18 domain (Fig. 2). Family GH18 harbors enzymes such as chitinases and CBM24 has been reported to bind to α-1,3-mutan, a mixed linkage glucan from Streptococcus sp.52.
Brown algae sugar monomer composition
Three brown algae species A. nodosum, F. serratus and S. latissima were selected representatives of the orders Fucales and Laminariales, commonly found in the North Sea. To assess the content and variation of sugars in the selected species, we performed monomer composition analysis by acid hydrolysis with subsequent HPAEC-PAD of products. The analysis showed the combined content of guluronic and mannuronic acids, which constitute alginate, to be as high as 30–40% in all three species of algae (Fig. 3). Fucose content, assumed to mainly stem from FCSPs, varied significantly across the three species, with A. nodosum containing almost 10%. The majority of the glucose is assumed to come from laminarin and cellulose of which F. serratus contained almost double the amount compared to A. nodosum. Mannitol content was relatively the same for all the species at approximately seven percent. The total sugar content averaged around 62% of the dry weight (Fig. 3).
Figure 3.
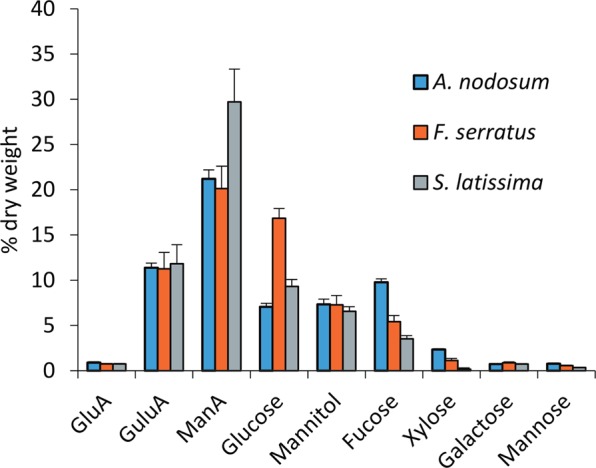
HPAEC-PAD based analysis of monomer sugars in three species of brown algae. GluA (glucuronic acid), GuluA (guluronic acid), ManA (mannuronic acid). The samples were analyzed in triplicates.
Enzymes relevant for brown algae cell wall degradation in the P. salina genome
Based on the combination of assumed substrate specificities of the CAZymes and the monomer analysis of the brown algae, it was possible to identify enzymes in the P. salina genome relevant for the degradation of all major types of polysaccharides in brown algae except FCSPs (Table 2).
Table 2.
Overview of putative CAZymes found in the P. salina genome relevant for degrading polysaccharides in brown algae. Family and functional annotation was performed using dbCAN and CUPP (Supplementary File S2).
| Substrate | Enzyme activity | EC no. | CAZyme families | ||||
|---|---|---|---|---|---|---|---|
| Alginate | Mannuronate-specific alginate lyase | 4.2.2.3 | 3 PL7 | ||||
| Cellulose | Lytic Polysaccharide MonoOxygenase (LPMO) | — | 25 AA9 | ||||
| Cellobiose dehydrogenase (acceptor) | 1.1.99.18 | 2 AA3 | |||||
| Endo-β-1,4-glucanase | 3.2.1.4 | 2 GH5 | 2 GH45 | 1 GH12 | |||
| β-1,4-cellobiohydrolase (reducing end) | 3.2.1.176 | 3 GH7 | |||||
| β-1,4-cellobiohydrolase (non-reducing end) | 3.2.1.91 | 2 GH6 | |||||
| β-glucosidase | 3.2.1.21 | 3 GH1 | 9 GH3 | ||||
| β-1,3-glucan | Endo-β-1,3-glucosidase | 3.2.1.39 | 4 GH16 | 2 GH17 | 1 GH64 | 2 GH81 | 3 GH128 |
| Exo-β-1,3-glucanase | 3.2.1.58 | 4 GH5 | 3 GH55 | ||||
| Endo-β-1,6-glucanase | 3.2.1.75 | 2 GH5 | |||||
| Exo-β-1,3/1,6-glucanase | 3.2.1.- | 2 GH131 | |||||
| β-glucosidase | 3.2.1.21 | 3 GH1 | 9 GH3 | ||||
| β-1,3-glucosidase | 3.2.1.- | 1 GH132 | |||||
| Mixed linkage glucan | Endo-β-1,3(4)-glucanase | 3.2.1.6 | 2 GH16 | ||||
| Endo-β-1,3-glucosidase | 3.2.1.39 | 2 GH17 | |||||
| Arabinogalactan | β-1,3-galactosidase | 3.2.1.145 | 1 GH43 | ||||
| Endo-β-1,6-galactosidase | 3.2.1.164 | 2 GH5 | |||||
| β-galactosidase | 3.2.1.23 | 3 GH2 | 2 GH35 | ||||
| Trehalose | α,α-trehalase | 3.2.1.28 | 2 GH37 | 1 GH65 | |||
The predicted PL7 genes were found on three different contigs and interestingly none of them contained introns. The GC percentage did not significantly vary from the contigs they were found in or the average GC content of the genome (Supplementary File S4). The top ten hits of the BLASTp analysis revealed marine bacterial sequences with the highest identities at a maximum of 30% similarity and no fungal hits despite several fungal sequences being listed in CAZy (Supplementary Table S7). Furthermore, the fungal PL7 members in CAZy all belong to subfamily four, but the dbCAN result of the three P. salina sequences yielded no such hit (Supplementary File S2). To expand on this, the sequences were aligned with PL7 domain sequences derived from CAZy and analyzed in a maximum likelihood phylogenetic analysis (Fig. 4 and Supplementary Fig. S1). The three P. salina PL7 sequences clustered together in a clade dominated by marine proteobacteria and a single sequence from the red seaweed Pyropia yezoensis, all without any subfamily classifications. All characterized members of this clade have been characterized as β-mannuronate specific lyases53–56 (EC 4.2.2.3) (Fig. 4). Recently three putative PL7 proteins were annotated in a marine Calcarisporium sp. KF525 (Basidiomycota)57 and it would have been interesting to include these sequences in the phylogenetic analysis, however no sequence information from this genome was available at the time of this study.
Figure 4.

Maximum likelihood phylogenetic analysis of selected CAZy-listed PL7 protein sequences, including the three sequences from P. salina found in this study. The species names are indicated along with accession numbers of corresponding PL7 sequences. Branch numbers indicate bootstrap values. The tree scale bar indicates substitution changes per site. A *indicate EC numbers of characterized members listed in CAZy. Sequences belonging to PL7 subfamily 4 are indicated in the same column. The tree was pruned from a larger phylogenetic tree containing all CAZy listed sequences (Supplementary Fig. S1).
We found putative genes for the complete degradation of cellulose in P. salina (Table 2). Additionally, a total number of 25 putative AA9 Lytic Polysaccharide MonoOxygenases (LPMOs) genes were found. The AA9 LPMOs have been reported to act mainly on cellulose which is the broad category chosen for them in this study, but there have also been reports of specificity towards hemicellulose58 and xyloglucan59.
Three different sulfatases were annotated: an aryl S1_6 N-acetylglucosamine-6-sulfatase (EC 3.1.6.14) (g8134.t1), a S1_12 choline-sulfatase (EC 3.1.6.6) (g7726.t1) and a S3 alkyl sulfatase (g9014.t1). Only the S1_6 sulfatase of these three was predicted for extracellular secretion. Five sulfate transporter proteins (PF00916, PF01740) were also found: one sulfate permease 1 (2.A.53.1.1) (g6037.t1), three sulfate permease 2 proteins (2.A.53.1.2) (g1349.t1, g2846.t1, g5922.t1) and a putative sulfate transporter (2.A.53.1.11) (g1670.t1) (Supplementary File S2).
The P. salina genome contains enzymes relevant for degradation of algal carbon storage compounds
A wide variety of putative β-1,3-glucanases were identified in the P. salina genome (Table 2). It is also likely that some of the numerous putative GH1 and GH3 β-glucosidases identified are active on β-1,3 oligosaccharides catalyzing the release of glucose.
We found several enzymes belonging to the mannitol II degradation pathway. A mannitol dehydrogenase (EC 1.1.1.255) (g460.t1), which converts d-mannitol into d-mannose, which is further converted to d-mannose-6-phosphate by hexokinases and then to d-fructose-6-phosphate by mannose-6-phosphate-isomerase before entering the glycolysis II pathway60. Two such putative isomerases and multiple hexokinases were found. The presence of one mannitol-1-phosphate 5-dehydrogenase (EC 1.1.1.17) (g1637.t1) belonging to the mannitol I degradation pathway indicates P. salina has its own mannitol metabolism61 (Supplementary File S2).
Three putative trehalase genes (EC 3.2.1.28) were found in P. salina: one GH65 with a CBM32 and a signal peptide appended and two GH37, the latter also containing a signal peptide but no CBM (Table 2). The presence of three GT20 genes, two α, α-trehalose-phosphate synthases (EC 2.4.1.15) and one trehalose-phosphatase (EC 3.1.3.12) (Supplementary File S2) indicates that P. salina also has its own trehalose pathway.
Oxidative enzymes
A total of 16 putative peroxidase genes were found: four AA2 peroxidases, five catalases, three haem peroxidases, one glutathione peroxidase, two chloroperoxidases and a bromoperoxidase/chloroperoxidase gene (Supplementary File S2). In a comprehensive search for vanadium-dependent haloperoxidases (VHPO’s), the genome was searched for all PAP2 domains (PF01569). Nine sequences were found which were subsequently aligned with the sequences used by Fournier and colleagues62 and analyzed with maximum likelihood analysis. The phylogenetic tree showed that one PAP2 sequence (g2873.t1) was positioned with vanadium-dependent bromoperoxidases (VBPOs) from brown algae and another sequence along with vanadium-dependent chloroperoxidases from fungi (VCPOs) (g3610.t1). The remaining PAP2 sequences were positioned with putative acid phosphatases (Supplementary Fig. S2).
Other putative oxidative enzymes found included eight laccases, numerous AA7 sugar oxidases, AA3 oxidoreductases and five tannases (Supplementary File S2).
P. salina produces active enzymes under both carbon limitation and fermentation of brown algae
We tested the fermentation supernatants on a variety of substrates found in plant, fungal and brown algal cell walls. Overall α-amylase activity was equally strong in all fermentations regardless of substrate (Fig. 5a). In the carbon limited fermentation, amylase activity was observed very soon after day two together with protease activity (Supplementary Fig. S3). Endo-β-1,3-glucanase activity was also observed in all fermentations, but at significantly higher levels in two of the brown algae fermentations (Fig. 5a). Unique to the brown algae fermentations were endo-β-1,4-glucanase, endo-β-1,4-mannanase, endo-1,4-xylanase, mixed linkage endo-β-glucanase (Fig. 5a) and alginate lyase activity (Fig. 5b). No fucoidanase activities were observed in the C-PAGE (Supplementary Fig. S4).
Figure 5.

Enzyme activities in the fermentation supernatants. The supernatants of P. salina after 14 days of incubation on brown algae and under carbon starvation were assayed on a variety of substrates at pH 5 and 30 °C. (a) AZCL assay of endo-lytic activities after 45 hours of incubation. (b) Alginate lyase activity after two hours of incubation. Five biological replicates for each type of fermentation were analyzed.
Characterization of PsAlg7A confirmed the preference for mannuronic acid
To confirm the in silico prediction of the mannuronic acid specificity (EC 4.2.2.3) of the PL7 lyases we chose the most abundantly secreted PL7 (PsAlg7A) (g8132.t1) for recombinant expression in P. pastoris. The recombinant PsAlg7A had an approx. size of 25 kDa according to the SDS-PAGE (Supplementary Fig. S5, lane 2), which corresponds with the predicted molecular mass. PsAlg7A displayed a higher kcat for polymannuronic acid than for alginate or polyguluronic acid and a lower Km for polymannuronic acid compared to polyguluronic acid (Table 3, Fig. 6), confirming the in silico predicted specificity for PsAlg7A.
Table 3.
PsAlg7A kinetic parameters on alginate, polymannuronic acid and polyguluronic acid. Values ± denotes the standard error.
| Substrate | Vmax (μM/s) | Km (mM) | Kcat (s−1) | Kcat/Km (mM−1 s−1) |
|---|---|---|---|---|
| Alginate | 0.49 ± 0.03 | 0.68 ± 0.12 | 0.66 ± 0.04 | 0.97 |
| polymannuronic acid | 2.5 ± 0.15 | 2.8 ± 0.41 | 3.4 ± 0.18 | 1.2 |
| polyguluronic acid | 1.9 ± 0.38 | 19 ± 5.4 | 2.1 ± 0.33 | 0.11 |
Figure 6.

Michaelis-Menten plots of PsAlg7A. Evolution of initial velocities measured as the formation of double bonds for increasing substrate concentrations of (a) alginate, (b) polymannuronic acid and (c) polyguluronic acid.
The secreted proteome of P. salina reveal similarities and differences in enzyme profiles depending on which algal strain is used as substrate
Maxquant identified 662 proteins from P. salina across the fermentations (Supplementary File S2).
The relative quantity analysis of the secretomes of the four fermentations revealed different profiles for each of the four types (Fig. 7). As expected, based on the enzyme activities, CAZymes were represented two to three times higher in the fermentations with algae, both in relative distribution (Fig. 6b–d), but also in quantities (Supplementary File S2) compared to the carbon limited fermentation which favored proteases and other proteins (Fig. 7a).
Figure 7.

Relative composition of the secreted proteomes of P. salina incubated on three species of brown algae and under carbon starvation. (a) Carbon limited, (b) F. serratus, (c) S. latissima, (d) A. nodosum. The compositions were derived from LC-MS/MS calculated iBAQ values (Supplementary File S2). Proteins representing less than one percent were accumulated under the “other” category.
Highly abundant in all fermentations were several α-glucosidases (EC 3.2.1.20), a starch specific AA13 LPMO with an appended CBM20 and a GH13 α-amylase (EC 3.2.1.1) also with a CBM20 appended; the latter enzyme was most likely constitutively expressed and responsible for the observed amylase activity (Fig. 5a). Starch-like polysaccharides or the genes necessary for their synthesis have not been found in brown algae.
The fermentations with algae contained significantly higher levels of PL7 alginate lyases (5–16%), as was expected based on the observed alginate lyase activity (Fig. 5b), but surprisingly also of the PL8 lyase that constituted 4–12% of the observed proteins in the fermentations with algae (Fig. 7).
All three algal P. salina fermentations contained high amounts of cellulases, in particular high in the S. latissima fermentation where cellulases constituted 41% of the total protein content (Fig. 7d). This was due to two reducing end acting GH7 β-1,4-cellobiohydrolases (EC 3.2.1.176) (g4260.t1, g793.t1) that constituted approximately 35% of the proteome. Together with a non-reducing end-acting GH6 β-1,4-cellobiohydrolase (EC 3.2.1.91) (g468.t1) and a GH5 endo-1,4-β-glucanase (EC 3.2.1.4) (g5427.t1) (Supplementary File S2), these were most likely linked to the observed cellulase activity (Fig. 5a).
The majority of CAZymes in the carbon limited fermentation consisted of β-1,3-glucanases, also known as laminarinases63, from nine different families (Supplementary File S2). The two most abundant enzymes were putative endo-β-1,3-glucosidases (EC 3.2.1.39): a GH17 (g2064.t1) and a GH55 (g36.t1). These were also the most abundant in the fermentations with algae (Supplementary File S2) and most likely linked to the observed endo-β-1,3-glucanase activity (Fig. 5a).
The P. salina fermentations with A. nodosum diverged from the other fermentations with high percentages of AA7 glucose-oxidases, AA3-oxidoreductases and a putative gluconolactonase. Together, these enzymes constituted as much as 47% of the secretome (Fig. 7b). The essential activities for gluconic acid production in Aspergillus niger were found to consist of an AA3 glucose-oxidase (EC 1.1.3.4), a catalase (EC 1.11.1.6) and a gluconolactonase (EC 3.1.1.17)64.
Of other noteworthy observations was a GH11 endo-β-1,4-xylanase, which was upregulated in the F. serratus fermentation (Supplementary File S2). This enzyme was most likely responsible for the activity observed on arabinoxylan and xylan (Fig. 5a). Additionally, two putative pectate lyases (EC 4.2.2.2), a PL3 and a PL1 were observed solely in the fermentations with algae (Supplementary file S2).
Proteins upregulated in the fermentations with algae other than CAZymes included abundant proteases/peptidases, a highly abundant lipase and a catalase (Supplementary File S2). One of the most abundant single proteins in all the P. salina fermentations was a 193aa long unknown protein with homology to allergen-like proteins in other fungi (g5788.t1) (Fig. 7).
Discussion
The annotation analysis of the P. salina genome revealed a repertoire of putative enzymes, which theoretically degrade the majority of the polysaccharides in brown algae, including alginate. The P. salina genome harbored 150–250 fewer CAZymes and 60–130 fewer sugar transporters compared to its close terrestrial relatives (Table 1). However, in spite of the lower numbers of CAZymes, P. salina still harbors CAZymes relevant for degradation of most of the major types of terrestrial polysaccharides, e.g. cellulases, pectinases, hemicellulases and starch-degrading enzymes.
The proteomic and genomic analysis of the CAZymes of P. salina strongly suggests that presence of the PL7 alginate lyases is one of the most significant adaptations of P. salina to thrive on brown macro-algae. The absence of alginate lyase families in the terrestrial relatives, the absence of introns in the genes and the phylogenetic placement among bacterial sequences could indicate possible horizontal gene transfers from marine bacteria, granting P. salina the ability to degrade alginate. This hypothesis however, is difficult to confirm and the GC content analysis of the genes and the low sequence identity to all other PL7 proteins provided no further evidence. The gene loss of the PL7 genes in the terrestrial fungi is also a possibility, but even more difficult to confirm. No alginate lyases from other enzyme families were found in P. salina, thus it is reasonable to conclude that one of these genes is identical to a poly-specific β-1,4-mannuronide lyase (EC 4.2.2.3) isolated from the supernatant of a different strain, P. salina IFO3213965. However, this study did not produce the sequence of the protein and therefore no family classification or homology to other enzymes was provided. Our phylogenetic analysis of the PL7 sequences from P. salina indicated them to be mannuronic acid specific, which we confirmed by performing enzyme kinetics on the purified PsAlg7A, which also showed a low activity towards guluronic acids, which seems to be a common occurrence in the PL7 family53,66. Considering that the carbohydrate analysis of the algae used in this study showed mannuronic acid contents to be approximately two-fold higher than guluronic acid contents in all three brown algae species, it is plausible that the specificities of the enzymes would mirror this composition. To our knowledge PsAlg7A is the first recombinantly expressed and characterized PL7 from a marine fungus. The combined data from the activity assays and proteomic analysis confirmed that the PL7 proteins were abundantly present and active in the fermentations with brown algae. Our study thus indicates that P. salina is capable of degrading the alginate in the brown algae cell wall, thereby exposing other polysaccharides for enzymatic degradation. Such polysaccharides may for example be cellulose microfibrils, which P. salina appears to be able to degrade due to the numerous putative cellulases in its genome and corroborated by their active presence in the fermentation supernatants. This is not surprising considering that cellulases are quite common enzymes found in many if not all plant cell wall degrading fungi as well as in fungi not typically associated with cellulose degradation33. Cellulase activity and quantity were significantly higher in the algal fermentations compared to the carbon limited fermentation (Fig. 5a). Yet the presence of minor amounts of cellulase activity in the carbon limited fermentation suggests these enzymes are either constitutively expressed in small amounts or induced by starvation.
The carbon storage compounds in brown algae all have similar roles and structures in fungi. There is for example a partial structural overlap between the β-1,3-glucan found in fungal cell walls67 and the brown algal laminarin. The only differences between these compounds are the terminal d-mannitol residues in the algal M-series laminarin14. Filamentous fungi utilize β-1,3-glucanases in the synthesis, remodeling and recycling of the β-1,3-glucan in their cell wall, but also in connection with mycoparatism68. Our data indicates that the same putative β-1,3-glucan active enzyme, which is expressed by P. salina when carbon limited, is significantly upregulated when P. salina grows on brown algae.
Mannitol is an abundant sugar found in both fungi60 and brown algae14 and mostly related to carbon storage and stress tolerance. P. salina has been shown to utilize mannitol as sole carbon source8. This indicates that P. salina can metabolize extracellular sources of mannitol. We found genes belonging to both the mannitol I and II degradation pathways, which indicates that P. salina is capable of both synthesis and metabolization. Similarly, trehalose is found in both fungi69 and brown algae14. Fungi are known to utilize both cytosolic and extracellular sources of trehalose70, and our data suggests that this is the case in P. salina as well.
The numerous catalases and peroxidases may allow P. salina to cope with defense-related oxidative bursts in the form of hydrogen peroxide from brown algae. Such responses have been observed in Laminaria digitata where oligo-guluronates from alginate degradation was observed to elicit oxidative defenses in the algae71. The VHPOs may play a similar role72 during attack by the fungus by remobilizing halides from the algae or playing a role in hyphal invasion of the algal cell wall, as has been hypothesized for the many terrestrial fungi where a similar enzyme has been found73. Reactive oxygen species (ROS), including hydrogen peroxide, play several critical roles in terrestrial plants during plant-fungal interactions74 and fungi have numerous coping mechanisms in place for dealing with ROS75,76. Recently, it has been observed that LPMOs are effectively activated by hydrogen peroxide prior to the degradation of substrates like cellulose77. The numerous AA9 LPMOs found in the P. salina genome and in the algal fermentations suggest that they play an important role in the degradation of the brown algal cell. It is possible that the fungal LPMOs in synergy with peroxidases could provide an ingenious way to use the algal/plant defense against itself while speeding up the degradation of polysaccharides like cellulose and hemicellulose. Extensive additional experiments would need to be performed to confirm these hypotheses. Unfortunately this stretches beyond the scope of this study.
The identification of a putative PL8 chondroitin AC lyase gene in P. salina without any equivalent genes in the compared terrestrial fungi and the fact that this enzyme was highly abundant in the algal fermentations raises interesting questions regarding the role and substrate specificity of the lyase. Chondroitin sulfate belongs to the glycosaminoglycan (GAG) family of linear heteropolysaccharides attached covalently to proteins forming proteoglycans78. These types of proteoglycans are almost exclusively associated with animal tissues and cells79 and to our knowledge have never been found in algae or fungi. Two putative PL8 proteins from Trametes versicolor F21a (Basidiomycota) were previously found by proteomic analysis when the fungus fermented microalga, one of which significantly upregulated80.
Aside from the numerous β-glucan active enzymes, the P. salina genome like that of S. lycopersici contains abundant hemicellulolytic and pectinolytic enzymes. As many as 36 putative pectinase genes were annotated in the genome suggesting that P. salina effectively degrades pectin. The shared sequence identities with S. lycopersici pectinases, like many of the other CAZymes in the P. salina genome, indicate commonalities between marine and terrestrial sources of pectin. Pectinases indeed seem to be a common occurrence in marine ascomycetes6,81 and they can even be induced by terrestrial sources of pectin82. In the case of marine bacteria, various horizontal gene transfers of whole pectinolytic loci from terrestrial bacteria have been observed, which might indicate an evolutionary pressure towards the metabolization of marine pectin, said to stem from seagrasses and marine diatoms83,84. The two putative pectate lyases found solely in the P. salina fermentations with brown algae raises an interesting question regarding their substrate specificities. Similarly, in the fermentation with F. serratus as substrate, the observation of endo-xylanase activity on xylan and arabinoxylan linked to the presence of a GH11 endo-xylanase is slightly perplexing. The proposed model of the brown algal cell wall incorporates short-chained hemi-cellulose molecules85. However, to our knowledge no polysaccharide with a xylan backbone has been identified in brown algae. Like the pectinases, the large number of annotated hemicellulases in the P. salina genome is most likely relevant for seagrasses86 and various terrestrial sources washed out to sea.
Generally, the enzymatic capabilities of P. salina suggest that this fungus is capable of degrading most components of brown algae, seagrasses and terrestrial plant material, which is indicative of a broad and saprobic lifestyle with plant pathogenic traits. Based on the high percentage of shared CAZymes with its plant-pathogenic family members and the acquired alginate lyases, it seems plausible that P. salina belongs to the proposed group of marine fungi that were secondary colonizers from the terrestrial to the marine environment6,87.
This study provides detailed insights into the adaptations and brown algae enzymatic degradation pattern in a marine fungus. Our genome analysis indicates that the repertoire of enzymes found in P. salina is highly similar to that of terrestrial saprobic and plant pathogenic fungi from the same family. With this enzymatic repertoire and an acquired ability to degrade alginate, P. salina is capable of degradation and metabolization of most major polysaccharides from brown algae.
Supplementary information
Acknowledgements
We would like to thank European Protein for providing the A. nodosum sample and we acknowledge the proteomic core facility at DTU Bioengineering for performing the final Mass Spectrometry analyses. This work was funded by the European Commission H2020 Bio-based Industries Joint Consortium via the Macro Cascade Project, BBI Grant no. 720755, as well as by the Technical University of Denmark.
Author Contributions
B.P. and L.L. conceived the study. B.P. conducted the majority of the experiments assisted by C.W., F.H., M.V. and N.K. Data analysis was performed by B.P. with contributions by L.L., C.W. and A.M. regarding interpretation and supervision of the project. The paper was written by B.P., C.W., A.M. and L.L. All authors reviewed the manuscript.
Data Availability
The draft genome and transcriptome assemblies, along with the cDNA paired-end reads were submitted to the European Nucleotide Archive (ENA) (www.ebi.ac.uk/ena) under the study accession number PRJEB30354. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD012152 (www.proteomexchange.org/). All other data generated or analyzed during this study are included in this published article and its Supplementary Information files.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-48823-9.
References
- 1.Zuccaro A, et al. Detection and Identification of Fungi Intimately Associated with the Brown Seaweed Fucus serratus. Appl. Environ. Microbiol. 2008;74:931–941. doi: 10.1128/AEM.01158-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cunliffe M, Hollingsworth A, Bain C, Sharma V, Taylor JD. Algal polysaccharide utilisation by saprotrophic planktonic marine fungi. Fungal Ecol. 2017;30:135–138. doi: 10.1016/j.funeco.2017.08.009. [DOI] [Google Scholar]
- 3.Gomaa M, Hifney AF, Fawzy MA, Issa AA, Abdel-Gawad KM. Biodegradation of Palisada perforata (Rhodophyceae) and Sargassum sp. (Phaeophyceae) biomass by crude enzyme preparations from algicolous fungi. J. Appl. Phycol. 2015;27:2395–2404. doi: 10.1007/s10811-014-0517-x. [DOI] [Google Scholar]
- 4.Wang Y, Barth D, Tamminen A, Wiebe MG. Growth of marine fungi on polymeric substrates. BMC Biotechnol. 2016;16:3. doi: 10.1186/s12896-016-0233-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balabanova LA, et al. Polysaccharide-Degrading Activity in Marine and Terrestrial Strains of Mycelial Fungi. Russ. J. Bioorganic Chem. 2018;44:431–437. doi: 10.1134/S1068162018040039. [DOI] [Google Scholar]
- 6.Balabanova, L., Slepchenko, L., Son, O. & Tekutyeva, L. Biotechnology Potential of Marine Fungi Degrading Plant and Algae Polymeric Substrates. Front. Microbiol. 9 (2018). [DOI] [PMC free article] [PubMed]
- 7.Sutherland G. K. Marine fungi imperfecti. New Phytol. 1916;15:35–48. doi: 10.1111/j.1469-8137.1916.tb07201.x. [DOI] [Google Scholar]
- 8.dela Cruz TEE, Schulz BE, Kubicek CP, Druzhinina IS. Carbon source utilization by the marine Dendryphiella species D. arenaria and D. salina. FEMS Microbiol. Ecol. 2006;58:343–353. doi: 10.1111/j.1574-6941.2006.00184.x. [DOI] [PubMed] [Google Scholar]
- 9.dela Cruz TE, Wagner S, Schulz B. Physiological responses of marine Dendryphiella species from different geographical locations. Mycol. Prog. 2006;5:108. doi: 10.1007/s11557-006-0504-y. [DOI] [Google Scholar]
- 10.Gurvan M, Thierry T, Delphine S, Mark CJ, Bernard K. The cell wall polysaccharide metabolism of the brown alga Ectocarpus siliculosus. Insights into the evolution of extracellular matrix polysaccharides in Eukaryotes. New Phytol. 2010;188:82–97. doi: 10.1111/j.1469-8137.2010.03374.x. [DOI] [PubMed] [Google Scholar]
- 11.Deniaud-Bouët E, Hardouin K, Potin P, Kloareg B, Hervé C. A review about brown algal cell walls and fucose-containing sulfated polysaccharides: Cell wall context, biomedical properties and key research challenges. Carbohydr. Polym. 2017;175:395–408. doi: 10.1016/j.carbpol.2017.07.082. [DOI] [PubMed] [Google Scholar]
- 12.Salmeán AA, et al. Insoluble (1 → 3), (1 → 4)-β-D-glucan is a component of cell walls in brown algae (Phaeophyceae) and is masked by alginates in tissues. Sci. Rep. 2017;7:2880. doi: 10.1038/s41598-017-03081-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hervé Cécile, et al. Arabinogalactan proteins have deep roots in eukaryotes: identification of genes and epitopes in brown algae and their role in Fucus serratus embryo development. New Phytol. 2015;209:1428–1441. doi: 10.1111/nph.13786. [DOI] [PubMed] [Google Scholar]
- 14.Michel G, Tonon T, Scornet D, Cock JM, Kloareg B. Central and storage carbon metabolism of the brown alga Ectocarpus siliculosus: insights into the origin and evolution of storage carbohydrates in Eukaryotes. New Phytol. 2010;188:67–81. doi: 10.1111/j.1469-8137.2010.03345.x. [DOI] [PubMed] [Google Scholar]
- 15.Ramada MHS, Steindorff AS, Bloch C, Ulhoa CJ. Secretome analysis of the mycoparasitic fungus Trichoderma harzianum ALL 42 cultivated in different media supplemented with Fusarium solani cell wall or glucose. Proteomics. 2016;16:477–490. doi: 10.1002/pmic.201400546. [DOI] [PubMed] [Google Scholar]
- 16.Levin JZ, et al. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat. Methods. 2010;7:709–715. doi: 10.1038/nmeth.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walker BJ, et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. Plos One. 2014;9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27:578–579. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- 19.Boetzer M, Pirovano W. Toward almost closed genomes with GapFiller. Genome Biol. 2012;13:R56. doi: 10.1186/gb-2012-13-6-r56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grabherr MG, et al. Nat. Biotechnol. 2011. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data; pp. 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stanke M, Morgenstern B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005;33:W465–W467. doi: 10.1093/nar/gki458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eddy Sean R. Accelerated Profile HMM Searches. PLoS Computational Biology. 2011;7(10):e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang L, et al. DbCAN-seq: A database of carbohydrate-active enzyme (CAZyme) sequence and annotation. Nucleic Acids Res. 2018;46:D516–D521. doi: 10.1093/nar/gkx894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones P, et al. InterProScan 5: genome-scale protein function classification. Bioinforma. Oxf. Engl. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Käll L, Krogh A, Sonnhammer ELL. Advantages of combined transmembrane topology and signal peptide prediction—the Phobius web server. Nucleic Acids Res. 2007;35:W429–W432. doi: 10.1093/nar/gkm256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chou, K. & Shen, H. A. New Method for Predicting the Subcellular Localization of Eukaryotic Proteins with Both Single and Multiple Sites: Euk-mPLoc 2.0. [DOI] [PMC free article] [PubMed]
- 29.Busk PK, Pilgaard B, Lezyk MJ, Meyer AS, Lange L. Homology to peptide pattern for annotation of carbohydrate-active enzymes and prediction of function. BMC Bioinformatics. 2017;18:214. doi: 10.1186/s12859-017-1625-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barrett K, Lange L. Peptide-based functional annotation of carbohydrate-active enzymes by conserved unique peptide patterns (CUPP) Biotechnol. Biofuels. 2019;12:102. doi: 10.1186/s13068-019-1436-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeske Lisa, Placzek Sandra, Schomburg Ida, Chang Antje, Schomburg Dietmar. BRENDA in 2019: a European ELIXIR core data resource. Nucleic Acids Research. 2018;47(D1):D542–D549. doi: 10.1093/nar/gky1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Busk PK, Lange M, Pilgaard B, Lange L. Several Genes Encoding Enzymes with the Same Activity Are Necessary for Aerobic Fungal Degradation of Cellulose in Nature. Plos One. 2014;9:e114138. doi: 10.1371/journal.pone.0114138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barbeyron Tristan, Brillet-Guéguen Loraine, Carré Wilfrid, Carrière Cathelène, Caron Christophe, Czjzek Mirjam, Hoebeke Mark, Michel Gurvan. Matching the Diversity of Sulfated Biomolecules: Creation of a Classification Database for Sulfatases Reflecting Their Substrate Specificity. PLOS ONE. 2016;11(10):e0164846. doi: 10.1371/journal.pone.0164846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Katoh, K., Rozewicki, J. & Yamada, K. D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform., 10.1093/bib/bbx108 (2017). [DOI] [PMC free article] [PubMed]
- 36.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinforma. Oxf. Engl. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 37.Miller, M. A., Pfeiffer, W. & Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In 2010 Gateway Computing Environments Workshop (GCE) 1–8, 10.1109/GCE.2010.5676129 (IEEE, 2010).
- 38.Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2016;44:D67–D72. doi: 10.1093/nar/gkv1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rhein-Knudsen N, Ale MT, Ajalloueian F, Meyer AS. Characterization of alginates from Ghanaian brown seaweeds: Sargassum spp. and Padina spp. Food Hydrocoll. 2017;71:236–244. doi: 10.1016/j.foodhyd.2017.05.016. [DOI] [Google Scholar]
- 40.Huang Y, Busk PK, Herbst F-A, Lange L. Genome and secretome analyses provide insights into keratin decomposition by novel proteases from the non-pathogenic fungus Onygena corvina. Appl. Microbiol. Biotechnol. 2015;99:9635–9649. doi: 10.1007/s00253-015-6805-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tyanova S, Temu T, Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016;11:2301–2319. doi: 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- 42.Tyanova S, et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 43.Britton HTS, Robinson RA. CXCVIII.—Universal buffer solutions and the dissociation constant of veronal. J. Chem. Soc. Resumed. 1931;0:1456–1462. doi: 10.1039/JR9310001456. [DOI] [Google Scholar]
- 44.Cao H, et al. Novel Enzyme Actions for Sulphated Galactofucan Depolymerisation and a New Engineering Strategy for Molecular Stabilisation of Fucoidan Degrading Enzymes. Mar. Drugs. 2018;16:422. doi: 10.3390/md16110422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Swift SM, Hudgens JW, Heselpoth RD, Bales PM, Nelson DC. Characterization of AlgMsp, an Alginate Lyase from Microbulbifer sp. 6532A. Plos One. 2014;9:e112939. doi: 10.1371/journal.pone.0112939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matsubara Y, Iwasaki K, Muramatsu T. Action of poly (alpha-Lguluronate) lyase from Corynebacterium sp. ALY-1 strain on saturated oligoguluronates. Biosci. Biotechnol. Biochem. 1998;62:1055–1060. doi: 10.1271/bbb.62.1055. [DOI] [PubMed] [Google Scholar]
- 47.Morgulis A, et al. Database indexing for production MegaBLAST searches. Bioinformatics. 2008;24:1757–1764. doi: 10.1093/bioinformatics/btn322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woudenberg JHC, Groenewald JZ, Binder M, Crous PW. Alternaria redefined. Stud. Mycol. 2013;75:171–212. doi: 10.3114/sim0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Franco, M. E. E., López, S., Medina, R., Saparrat, M. C. N. & Balatti, P. Draft Genome Sequence and Gene Annotation of Stemphylium lycopersici Strain CIDEFI-216. Genome Announc. 3 (2015). [DOI] [PMC free article] [PubMed]
- 50.Ohm, R. A. et al. Diverse Lifestyles and Strategies of Plant Pathogenesis Encoded in the Genomes of Eighteen Dothideomycetes Fungi. PLoS Pathog. 8 (2012). [DOI] [PMC free article] [PubMed]
- 51.Zeiner Carolyn A., Purvine Samuel O., Zink Erika M., Paša-Tolić Ljiljana, Chaput Dominique L., Haridas Sajeet, Wu Si, LaButti Kurt, Grigoriev Igor V., Henrissat Bernard, Santelli Cara M., Hansel Colleen M. Comparative Analysis of Secretome Profiles of Manganese(II)-Oxidizing Ascomycete Fungi. PLOS ONE. 2016;11(7):e0157844. doi: 10.1371/journal.pone.0157844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fuglsang CC, et al. Biochemical analysis of recombinant fungal mutanases. A new family of alpha1,3-glucanases with novel carbohydrate-binding domains. J. Biol. Chem. 2000;275:2009–2018. doi: 10.1074/jbc.275.3.2009. [DOI] [PubMed] [Google Scholar]
- 53.Inoue A, et al. Characterization of an Eukaryotic PL-7 Alginate Lyase in the Marine Red Alga Pyropia yezoensis. Curr. Biotechnol. 2015;4:240–248. doi: 10.2174/2211550104666150915210434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Malissard M, et al. Sequence of a gene encoding a (poly ManA) alginate lyase active on Pseudomonas aeruginosa alginate. FEMS Microbiol. Lett. 1993;110:101–106. doi: 10.1111/j.1574-6968.1993.tb06302.x. [DOI] [PubMed] [Google Scholar]
- 55.Kawamoto H, et al. Cloning and Sequencing Analysis of Alginate Lyase Genes from the Marine Bacterium Vibrio sp. O2. Mar. Biotechnol. 2006;8:481–490. doi: 10.1007/s10126-005-6157-z. [DOI] [PubMed] [Google Scholar]
- 56.Inoue A, Anraku M, Nakagawa S, Ojima T. Discovery of a Novel Alginate Lyase from Nitratiruptor sp. SB155-2 Thriving at Deep-sea Hydrothermal Vents and Identification of the Residues Responsible for Its Heat Stability. J. Biol. Chem. 2016;291:15551–15563. doi: 10.1074/jbc.M115.713230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kumar, A. et al. Genome Sequencing and analyses of Two Marine Fungi from the North Sea Unraveled a Plethora of Novel Biosynthetic Gene Clusters. Sci. Rep. 8 (2018). [DOI] [PMC free article] [PubMed]
- 58.Agger JW, et al. Discovery of LPMO activity on hemicelluloses shows the importance of oxidative processes in plant cell wall degradation. Proc. Natl. Acad. Sci. 2014;111:6287–6292. doi: 10.1073/pnas.1323629111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kojima Y, et al. A Lytic Polysaccharide Monooxygenase with Broad Xyloglucan Specificity from the Brown-Rot Fungus Gloeophyllum trabeum and Its Action on Cellulose-Xyloglucan Complexes. Appl. Environ. Microbiol. 2016;82:6557–6572. doi: 10.1128/AEM.01768-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meena, M., Prasad, V., Zehra, A., Gupta, V. K. & Upadhyay, R. S. Mannitol metabolism during pathogenic fungal–host interactions under stressed conditions. Front. Microbiol. 6 (2015). [DOI] [PMC free article] [PubMed]
- 61.Calmes, B. et al. Role of mannitol metabolism in the pathogenicity of the necrotrophic fungus Alternaria brassicicola. Front. Plant Sci. 4 (2013). [DOI] [PMC free article] [PubMed]
- 62.Fournier J-B, et al. The Vanadium Iodoperoxidase from the Marine Flavobacteriaceae Species Zobellia galactanivorans Reveals Novel Molecular and Evolutionary Features of Halide Specificity in the Vanadium Haloperoxidase Enzyme Family. Appl. Environ. Microbiol. 2014;80:7561–7573. doi: 10.1128/AEM.02430-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nlelsen CO. Laminarinases in Soil and Litter Invertebrates. Nature. 1963;199:1001. doi: 10.1038/1991001a0. [DOI] [Google Scholar]
- 64.Witteveen CFB, et al. Induction of glucose oxidase, catalase, and lactonase in Aspergillus niger. Curr. Genet. 1993;24:408–416. doi: 10.1007/BF00351849. [DOI] [PubMed] [Google Scholar]
- 65.Shimokawa T, et al. Purification and Characterization of Extracellular Poly(β-D-1,4-mannuronide) Lyase from Dendryphiella salina IFO 32139. Biosci. Biotechnol. Biochem. 1997;61:636–640. doi: 10.1271/bbb.61.636. [DOI] [PubMed] [Google Scholar]
- 66.Zhu B, Ni F, Ning L, Sun Y, Yao Z. Cloning and characterization of a new pH-stable alginate lyase with high salt tolerance from marine Vibrio sp. NJ-04. Int. J. Biol. Macromol. 2018;115:1063–1070. doi: 10.1016/j.ijbiomac.2018.04.108. [DOI] [PubMed] [Google Scholar]
- 67.Fesel PH, Zuccaro A. β-glucan: Crucial component of the fungal cell wall and elusive MAMP in plants. Fungal Genet. Biol. 2016;90:53–60. doi: 10.1016/j.fgb.2015.12.004. [DOI] [PubMed] [Google Scholar]
- 68.Gruber S, Seidl-Seiboth V. Self versus non-self: fungal cell wall degradation in Trichoderma. Microbiology. 2012;158:26–34. doi: 10.1099/mic.0.052613-0. [DOI] [PubMed] [Google Scholar]
- 69.Elbein AD, Pan YT, Pastuszak I, Carroll D. New insights on trehalose: a multifunctional molecule. Glycobiology. 2003;13:17R–27R. doi: 10.1093/glycob/cwg047. [DOI] [PubMed] [Google Scholar]
- 70.Jorge JA, Polizeli M, de LTM, Thevelein JM, Terenzi HF. Trehalases and trehalose hydrolysis in fungi. FEMS Microbiol. Lett. 1997;154:165–171. doi: 10.1111/j.1574-6968.1997.tb12639.x. [DOI] [PubMed] [Google Scholar]
- 71.Küpper FC, Kloareg B, Guern J, Potin P. Oligoguluronates Elicit an Oxidative Burst in the Brown Algal Kelp Laminaria digitata. Plant Physiol. 2001;125:278–291. doi: 10.1104/pp.125.1.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Leblanc C, et al. Iodine transfers in the coastal marine environment: the key role of brown algae and of their vanadium-dependent haloperoxidases. Biochimie. 2006;88:1773–1785. doi: 10.1016/j.biochi.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 73.Barnett P, et al. Isolation, Characterization, and Primary Structure of the Vanadium Chloroperoxidase from the Fungus Embellisia didymospora. J. Biol. Chem. 1998;273:23381–23387. doi: 10.1074/jbc.273.36.23381. [DOI] [PubMed] [Google Scholar]
- 74.Camejo D, Guzmán-Cedeño Á, Moreno A. Reactive oxygen species, essential molecules, during plant–pathogen interactions. Plant Physiol. Biochem. 2016;103:10–23. doi: 10.1016/j.plaphy.2016.02.035. [DOI] [PubMed] [Google Scholar]
- 75.Angelova MB, Pashova SB, Spasova BK, Vassilev SV, Slokoska LS. Oxidative stress response of filamentous fungi induced by hydrogen peroxide and paraquat. Mycol. Res. 2005;109:150–158. doi: 10.1017/S0953756204001352. [DOI] [PubMed] [Google Scholar]
- 76.Li Q, McNeil B, Harvey LM. Adaptive response to oxidative stress in the filamentous fungus Aspergillus niger B1-D. Free Radic. Biol. Med. 2008;44:394–402. doi: 10.1016/j.freeradbiomed.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 77.Bissaro B, et al. Oxidative cleavage of polysaccharides by monocopper enzymes depends on H2O2. Nat. Chem. Biol. 2017;13:1123–1128. doi: 10.1038/nchembio.2470. [DOI] [PubMed] [Google Scholar]
- 78.Wang W, Wang J, Li F. Hyaluronidase and Chondroitinase. Adv. Exp. Med. Biol. 2017;925:75–87. doi: 10.1007/5584_2016_54. [DOI] [PubMed] [Google Scholar]
- 79.Lindahl, U., Couchman, J., Kimata, K. & Esko, J. D. Proteoglycans and Sulfated Glycosaminoglycans. In Essentials of Glycobiology (eds Varki, A. et al.) (Cold Spring Harbor Laboratory Press, 2015).
- 80.Gao, X. et al. Proteomic analysis reveals large amounts of decomposition enzymes and major metabolic pathways involved in algicidal process of Trametes versicolor F21a. Sci. Rep. 7 (2017). [DOI] [PMC free article] [PubMed]
- 81.Kumar A, et al. De Novo Assembly and Genome Analyses of the Marine-Derived Scopulariopsis brevicaulis Strain LF580 Unravels Life-Style Traits and Anticancerous Scopularide Biosynthetic Gene Cluster. Plos One. 2015;10:e0140398. doi: 10.1371/journal.pone.0140398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Poveda, G., Gil-Durán, C., Vaca, I., Levicán, G. & Chávez, R. Cold-active pectinolytic activity produced by filamentous fungi associated with Antarctic marine sponges. Biol. Res. 51 (2018). [DOI] [PMC free article] [PubMed]
- 83.Hehemann J-H, et al. Aquatic adaptation of a laterally acquired pectin degradation pathway in marine gammaproteobacteria. Environ. Microbiol. 2017;19:2320–2333. doi: 10.1111/1462-2920.13726. [DOI] [PubMed] [Google Scholar]
- 84.Hobbs, J. K., Hettle, A. G., Vickers, C. & Boraston, A. B. Biochemical Reconstruction of a Metabolic Pathway From a Marine Bacterium Reveals its Mechanism of Pectin Depolymerization. Appl. Environ. Microbiol., 10.1128/AEM.02114-18 (2018). [DOI] [PMC free article] [PubMed]
- 85.Deniaud-Bouët E, et al. Chemical and enzymatic fractionation of cell walls from Fucales: insights into the structure of the extracellular matrix of brown algae. Ann. Bot. 2014;114:1203–1216. doi: 10.1093/aob/mcu096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Davies P, Morvan C, Sire O, Baley C. Structure and properties of fibres from sea-grass (Zostera marina) J. Mater. Sci. 2007;42:4850–4857. doi: 10.1007/s10853-006-0546-1. [DOI] [Google Scholar]
- 87.Richards TA, Jones MDM, Leonard G, Bass D. Marine Fungi: Their Ecology and Molecular Diversity. Annu. Rev. Mar. Sci. 2011;4:495–522. doi: 10.1146/annurev-marine-120710-100802. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The draft genome and transcriptome assemblies, along with the cDNA paired-end reads were submitted to the European Nucleotide Archive (ENA) (www.ebi.ac.uk/ena) under the study accession number PRJEB30354. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD012152 (www.proteomexchange.org/). All other data generated or analyzed during this study are included in this published article and its Supplementary Information files.