

Abstract
Aims
Rituximab is an anti‐CD20 monoclonal antibody approved in non‐Hodgkin lymphoma (NHL). This study aimed to assess the relationship between antigen mass and nonlinear pharmacokinetics of rituximab in NHL patients.
Methods
In a retrospective cohort of 25 NHL patients treated with rituximab, antigen mass was assessed at baseline by measuring metabolic tumour volume (MTV) by positron emission tomography. Rituximab pharmacokinetics was described using a semimechanistic 2‐compartment model including a latent target antigen. Rituximab target‐mediated elimination was described as irreversible binding between rituximab and it target. Histology (follicular or diffuse large B‐cell lymphomas), initial MTV and body weight were tested as covariates on pharmacokinetic parameters.
Results
The model allowed a satisfactory description of rituximab serum concentrations. Target‐mediated elimination was maximum at the beginning of treatment and became negligible towards the end of follow‐up. The second‐order elimination of rituximab due to target binding and complex elimination increased with baseline MTV. Central volume of distribution increased with body weight (P = .022) and baseline MTV (P = .005).
Conclusions
This study quantified for the first time the target‐mediated elimination of rituximab in NHL patients and confirmed rituximab retention by antigen mass.
Keywords: metabolic tumour volume, non‐Hodgkin lymphoma, pharmacokinetics, rituximab, target‐mediated drug disposition
What is already known on the subject
The pharmacokinetics of rituximab was described in previous works in non‐Hodgkin lymphoma (NHL), chronic lymphocytic leukaemia and rheumatoid arthritis using 2‐compartment models.
Antigen mass was found to influence rituximab pharmacokinetics in all of these diseases.
Nonlinear elimination of rituximab due to target antigen (CD20) has only been detected in chronic lymphocytic leukaemia so far.
What this study adds
Target‐mediated drug disposition modelling was used to describe rituximab pharmacokinetics in NHL for the first time.
This study showed a nonlinear elimination of rituximab in NHL due to target antigen in NHL for the first time.
This study confirms retention of rituximab by tumour burden.
1. INTRODUCTION
Rituximab, a chimaeric IgG1 therapeutic monoclonal antibody binding CD20 antigen, is approved in 2 B‐cell malignancies, non‐Hodgkin lymphomas (NHL) and chronic lymphocytic leukaemia (CLL). The pharmacokinetics of rituximab were described in 12 pharmacokinetic modelling studies in NHL,1, 2, 3, 4, 5, 6, 7 CLL8, 9 and rheumatoid arthritis.10, 11 and in patients under plasmapherisis.12 Some of these studies suggested that rituximab pharmacokinetics might be influenced by antigen mass (i.e. the amount of membrane and circulating CD20 available for rituximab binding). Indeed, higher CD20 expression in CLL,9 larger metabolic tumour volume (MTV) in lymphomas7 and higher CD19+ count in rheumatoid arthritis10 were associated with lower rituximab concentrations. Rituximab pharmacokinetics were reported to be linear in lymphomas1, 2, 3, 4, 5, 7 and rheumatoid arthritis,10, 11 despite the influence of antigen mass on either rituximab clearance10 or volume of distribution.7 Nonlinear pharmacokinetics were reported in a murine model of lymphoma expressing human CD20, where rituximab clearance increased with tumour volume.13 In CLL, rituximab was shown to exhibit a time‐dependent elimination rate,8 and a target‐mediated elimination related with antigen mass.9 Nonlinear pharmacokinetics of therapeutic antibodies are usually described using target‐mediated drug disposition (TMDD) models14, 15 that provide a description of joint kinetics of the antibody, its target antigen and the immune complex. It is known that the onset of nonlinearity occurs for even higher antibody concentrations if antigen mass is high.16 Thus, the absence of nonlinearity in rheumatoid arthritis may be explained by low CD20 burden compared to CLL, where CD20 amount is much higher. In NHL, nonlinear pharmacokinetics was not reported, despite clear influence of CD20 levels on rituximab concentrations17 and of tumour volume on rituximab volume of distribution.7 Therefore, the presence or absence of nonlinear elimination kinetics of rituximab in NHL has not yet been demonstrated. The objective of this study was to detect, if they exists, nonlinear kinetics of rituximab in NHL.
2. METHODS
2.1. Patient cohort and study design
This study was conducted using the data from a retrospective cohort of 25 routine practice patients treated in the Tours University Hospital (Tours, France) between May 2006 and October 2010. This study was approved by the local ethics committee. As part of the routine therapeutic drug monitoring of rituximab, blood samples are drawn to measure rituximab trough concentrations and individual results are interpreted and sent to the prescriber and discussed during clinic–biological rounds. As for a similar previous study,10 the samples were therefore not collected specifically for this study; pharmacokinetic modelling was performed retrospectively.
Patients were treated with rituximab 375 mg/m2 doses every 2 or 3 weeks. A dose‐dense regimen consisted in supplemental rituximab infusions at days 1 and 4 after the first rituximab infusion.
2.2. Rituximab concentration measurements
Blood samples were collected immediately before (trough) and 2 hours after (peak) an infusion of rituximab at several visits. Rituximab concentrations were determined using a validated enzyme‐linked immunosorbent assay technique derived from Blasco et al.1 Limit of detection, lower and upper limits of quantitation of the assay were 0.061, 0.20 and 9.0 mg/L, respectively. Coefficients of variation from intraday variability were 8.4, 5.4 and 6.4% and from interday variability were 4.6, 6.8 and 5.3% for 0.2, 3.0 and 7.0 quality controls, respectively.10
2.3. Determination of tumour volume
In this study, MTV was used as surrogate of antigen mass and was determined at baseline. In this cohort, positron emission tomography/computed tomography with 2‐deoxy‐2‐fluorine‐18‐fluoro‐D‐glucose images were computed on image contouring software Dosisoft (Planet Onco, Dosisoft, France). A volume of interest was set around each lesion (node or organ involvement). The intensity threshold for the contouring was made using Nestle et al.’s method.18 This method have taken into account the background noise intensity and mean intensity of the lesion: Ithreshold = β. Iaverage + Inoise with β = 0.15, Iaverage for a volume determined with 70% of I maximum and Inoise the mean intensity for a volume set in a 1‐cm section around lesional volume. Bone marrow involvement was not accounted. Spleen was considered as involved if there was focal uptake or diffuse uptake >150% of the liver background.
2.4. Pharmacokinetic modelling
Pharmacokinetic analysis of rituximab concentration–time data was performed using population approach with the nonlinear mixed‐effects software MONOLIX suite 2018 (Lixoft, Orsay, France). Many iterations were performed to reach the best stochastic approximation of the expectation–maximization convergence (K1 = 1000, K2 = 300, where K1 and K2 are the iteration kernels 1 and 2 in MONOLIX). Two Markov chains were used. Fisher information matrix and likelihood were computed using stochastic approximation and importance sampling, respectively.
2.4.1. Structural pharmacokinetic model design
The objective was the detection of nonlinear pharmacokinetics, if present. Therefore, the first step was the development of a linear pharmacokinetic model. One‐ and 2‐compartment models were tested, with first‐order elimination and transfer rate constants and with volume and clearance parameterization. On the basis of the best linear model (Model 1), the following nonlinear pharmacokinetic models were tested (Figure 1):
-
‐
Model 2: a clearance exponentially decreasing with time, as already done in CLL for rituximab8 and obinutuzumab, another anti‐CD20 therapeutic antibody.19
-
‐
Model 3: a Michaelis–Menten elimination;
-
‐
Model 4: irreversible binding of rituximab to CD20, as already used for rituximab in CLL.9 This model was developed on the basis of a target turnover with zero‐ and first‐order antigen mass input (kin) and endogenous output (kout), respectively. Rituximab target‐mediated elimination was described using a second‐order target‐mediated elimination rate constant kdeg, as usually used in TMDD models.15 Target antigen was described as a latent variable (L), which corresponds to CD20 antigen interacting with rituximab, notably blood and extra‐blood B‐cells and tumour cells. This variable may also correspond to other (unknown) factors that could cause pharmacokinetic nonlinearity.
-
‐
Model 5: In the case of nonidentifiability of kin and kout, the initial amount of latent target (L0) was estimated.
Figure 1.
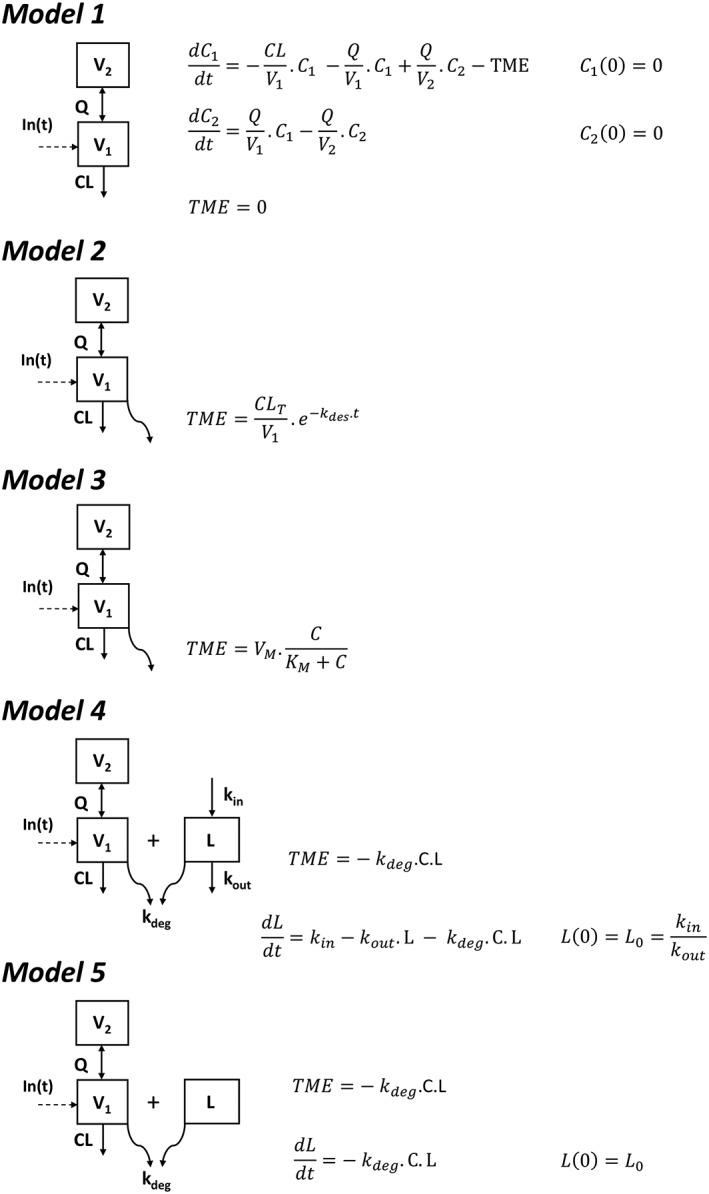
Pharmacokinetic 2‐compartment models used to describe rituximab pharmacokinetics: linear model with no target‐mediated elimination (model 1), with time‐dependent elimination rate (model 2), with Michaelis–Menten elimination (model 3), with irreversible binding of rituximab to latent target and target turnover (model 4), and irreversible binding of rituximab to latent target with no description of target turnover (model 5). C and L are rituximab concentrations and latent target amount, respectively; V1 and V2 are central and peripheral volumes, respectively; CL and Q are systemic and intercompartment clearances, respectively; TME is target‐mediated elimination; CLT and kdes are initial target‐mediated clearance and its time‐decrease rate constant, respectively; VM and KM are maximum rate and Michaelis constant, respectively; kin, kout and kdeg are latent target input and output and elimination due to its binding on rituximab, respectively
2.4.2. Interindividual and residual model design
Interindividual variability was described by an exponential model, defined by θi = θTV. exp (ηi), where θ is structural parameter of subject i, θTV is typical value of parameter and ηi is individual deviation of subject i from typical value, with mean 0 and interindividual variance ω2. Interindividual variances that could not be estimated properly were fixed to 0. The residual variability was described using a proportional model, defined as Cij,obs = Cij,pred. (1 + εij,prop), where Cij,obs and Cij,pred are observed and model‐fitted concentrations, respectively, for subject i and observation j, and εij,prop is proportional residual error with mean 0 and variance σprop 2.
2.4.3. Covariate analysis
Since 25 patients were studied, only a limited number of individual sources of variability could be tested. Baseline body weight (BW), MTV and histology (follicular [FL] or diffuse large B‐cell [DLBCL] lymphomas) were tested as covariates. The association of structural parameters and BW was tested as a power function: ln (θBW) = ln (θpop) + βBW. ln (BW/med (BW)). Histology was coded with FL as reference: ln (θDLBCL) = ln (θFL) + βDLBCL, where θDLBCL and θFL are values of θ for DLBCL and FL, respectively, and βDLBCL is the parameter which provides the value of θ for DLBCL.
The association of MTV was tested as power function, multiplicative, θMTV = θpop βMTV.MTV, exponential, θMTV = θpop. Exp (βMTV.MTV). In addition, we attempted to estimate structural parameters were estimated by unit of MTV (cm3), θMTV = θpop. MTV, leading to θMTV in (unit.cm−3). This strategy avoids the estimation of a covariate coefficient. Covariate selection was based on the likelihood ratio test (LRT), where the difference in objective function value (OFV, −2.ln likelihood) between 2 nested models is assumed to follow a χ2 distribution with 1 degree of freedom. The effect of potential covariates on pharmacokinetic parameters was tested using a backward stepwise selection process. In the univariate step, covariates showing a significant association with pharmacokinetic parameters (P < .10) where kept in the complete model. In the multivariate step, covariates for which P < .05 were retained in the final model.
2.4.4. Model evaluation
Comparison between structural, interindividual and residual models was made using OFV or Akaike's information criterion (AIC), defined as AIC = OFV + 2.p, where p is the number of estimated model parameters. Models were evaluated graphically using goodness‐of‐fit diagnostic plots: observed vs population (PRED) and individual‐PRED fitted concentrations; population and individual weighted residuals vs PRED and individual‐PRED, respectively. Visual predictive checks and normalized prediction distribution errors (NPDE) were also performed by simulating 1000 replicates using both fixed and random effect final parameters.
2.4.5. Model‐based simulations
The typical parameters of the final pharmacokinetic model were used to simulate typical rituximab concentration–time profiles for several MTV values (from 10 to 5000 m3) of 2 dosing regimens:
-
‐
Every 3 weeks: 4 doses of rituximab 375 mg/m2 dose every 3 weeks;
-
‐
Dose‐dense: 6 doses of 375 mg/m2 rituximab at day 0, 1 and 4, and every 2 weeks.
2.4.6. Nomenclature of target and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY,20 and are permanently achieved in the Concise Guide to PHARMACOLOGY 2017/2018.21
3. RESULTS
3.1. Patients
Table 1 summarizes patient characteristics. A total of 134 rituximab serum concentrations were available for the 25 patients included in this study; for each patient, median (range) number of samples was 5 (2–10). Among the 25 patients, 13 and 12 had FL or DLBCL, respectively. Fourteen and 13 patients were treated with rituximab every 3 or every 2 weeks, respectively, and 6 DLBCL patients had dose‐dense dosing regimen (Table 1). Median (range) baseline MTV was 600 (7–6217) cm3 and was not significantly different between FL and DLBCL patients (Mann–Whitney test, P = .24). Median (range) infusion duration were 6.5 (5.0–10.5) hours and 2.5 (1.5–7.0) hours for first and following infusions, respectively.
Table 1.
Summary of patient characteristics
| Characteristics | Patients (n = 25) |
|---|---|
| Women, n (%) | 9 (36) |
| Age, y (range) | 61 (31–77) |
| BW, kg (range) | 67 (50–114) |
| BSA, m 2 (range) | 1.8 (1.5–2.2) |
| MTV, cm 3 (range) | 600 (7–6217) |
| Histology, n (%) | |
| Follicular grade 1 | 9 (36) |
| Follicular grade 2 | 4 (16) |
| Diffuse large B‐cell | 12 (48) |
| Ann‐Arbor stage | |
| I | 3 (12) |
| II | 3 (12) |
| III | 8 (32) |
| IV | 11 (44) |
| Chemotherapy, n (%) | |
| RCHOP 21 | 13 (52) |
| RCHOP 14 | 11(44) |
| RDHAP 21 | 1 (4) |
| Dose‐dense, n (%) | 6 (24%) |
BW: body weight; BSA: body surface area; MTV: metabolic tumour volume; RCHOP, rituximab, cyclophosphamide, hydroxy‐doxorubicine, vincristine, prednisone; RDHAP, rituximab, dexamethasone, high‐dose cytarabine, cisplatin; 14 and 21, intercourse duration (days).
3.2. Pharmacokinetic analysis
3.2.1. Structural, interindividual and residual models
The (linear) 2‐compartment model with first‐order transfer and elimination rates (AIC = 1400.81) was better than a 1‐compartment model (model 1, AIC = 1464.12). The 2‐compartment models with time‐dependent elimination rate (model 2, AIC = 1400.43), and with Michaelis–Mentel elimination (model 3, AIC = 1405.76) did not improve model performance of the linear 2‐compartment model. The semimechanistic model with second‐order target‐mediated elimination and turnover of latent target allowed a slight improvement of data description (model 4, AIC = 1387.78), with kout being poorly estimated. The semimechanistic model with second‐order target‐mediated elimination without target turnover (model 5, AIC = 1378.11), led to the best description of data. Indeed, only the initial condition of latent target amount (L0) was identifiable. Therefore, model was selected as a base model. Interindividual variances of V2, Q and kdeg were not identifiable and were therefore set to 0. Best error model was proportional.
3.2.2. Covariate model
In the univariate analysis, V1 was found to be associated with both body size, expressed as BW, and MTV coded as exponential; CL was associated with BW; The initial amount of latent target (L0) was associated with MTV. Histology was associated with none of tested covariates. Among several coding strategies (additive, multiplicative and power models) for coding the association of MTV with L0, the multiplicative model led to the greatest reduction in AIC (−19.66) compared to power (−18.91) or exponential (−10.31) coding strategies compared to base model with no influence of MTV on L0. In addition, estimating L0 by unit of MTV (in nmol.cm−3) led to similar reduction of −2LL as the multiplicative model (−21.66 vs −21.33) but higher reduction of AIC (−21.33 vs −19.66) Thus, the strategy of L0 estimation in unit of MTV was chosen because it led to an economy of parameters to be estimated. The association of MTV with L0 shows that variable L is partly explained by MTV. The variations of L and MTV should therefore be positively correlated.
In the final model, V1 was influenced by BW (LRT = 5.2, P = .022) and MTV (LRT = 7.9, P = .005).
3.2.3. Model evaluation
Structural parameters were estimated with good accuracy, while interindividual and residual parameters were estimated with acceptable accuracy (Table 2). Plots of predicted vs observed concentrations showed satisfactory fitting of pharmacokinetic data by the model, with no bias for typical parameter or individual parameter estimates (relative bias <5%). Diagnostic plots of residuals and NPDE showed Gaussian residuals, which is confirm by a non‐significant Kolmogorov–Smirnov test for individual residuals (KS = 0.07493, P = .0601) and NPDE (KS = 0.05337, P = .16). Visual predictive checks showed a good adequacy between observed and predicted concentrations. Overall, our model presents no bias or model misspecification (Figure 2).
Table 2.
Population pharmacokinetic parameter estimates
| Parameter (unit) | Estimate | RSE (%) |
|---|---|---|
| V1 (L) | 2.9 | 8 |
| CL (L/day) | 0.23 | 10 |
| MTV on V1 | 0.00012 | 36 |
| BW on V1 | 0.49 | 45 |
| V2 (L) | 5.0 | 9 |
| Q (L/day) | 1.8 | 28 |
| L0 (nmol.cm−3) | 3.1 | 18 |
| Kdeg (cm3.nmol−1.day−1) | 0.000079 | 17 |
| ωV1 | 0.19 | 28 |
| ωCL | 0.42 | 19 |
| ωL0 | 0.35 | 34 |
| σprop | 0.26 | 8 |
RSE, relative standard error; V1, V2: central and peripheral volumes of distribution; CL, Q: systemic and intercompartment clearances; MTV: metabolic tumour volume; BW: body weight, L0, initial latent target amount available for rituximab by unit of MTV; Kdeg, second‐order target‐mediated elimination rate; ωV1, ωCL, ωL0: interindividual standard deviations of V1, CL and L0, respectively; σprop: residual proportional error.
Figure 2.
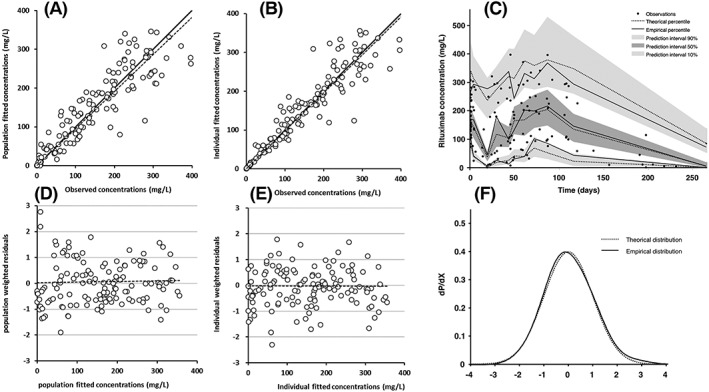
Diagnostic plots of the final pharmacokinetic model: (A) observed vs population model‐predicted rituximab concentrations and (B) observed vs individual model‐predicted rituximab concentrations; open circles are fitted vs observed concentrations, bold and dashed lines are first bisector and fitted trend line, respectively. (C) Prediction‐corrected visual predictive check; observed concentrations (black circles), theoretical (dashed lines) and empirical (continuous lines) percentiles (from bottom to top: 10%, 50% and 90% percentiles) and prediction interval (from bottom to top: 10%, 50% and 90% prediction intervals); time zero is the first even for each patient, i.e. start of the first rituximab infusion. (D) Population weighted residuals vs population predicted rituximab concentrations; (E) individual weighted residuals vs individual predicted rituximab concentrations; dashed line is fitted trend line. (F) Normalized prediction distribution errors (dotted) vs gaussian law (continuous)
3.2.4. Model‐based simulations
Four doses every 3 weeks: For MTV values ranging from 10 to 2000 cm3, latent target amount (L) was decreased to null for all simulated MTV values, leading to negligible target‐mediated elimination; T½‐β ranged from 25.1 to 27.2 days. For MTV = 5000 cm3, L and thus target‐mediated elimination were not null, leading to a terminal elimination half‐live (T½‐β = 10 days) shorter than for other MTV values.
Dose‐dense: For MTV values ranging from 10 to 5000 cm3, L and target‐mediated elimination were decreased to null for all simulated MTV values; T½‐β increased from 25.1 to 31.5 days (Figure 3).
Figure 3.
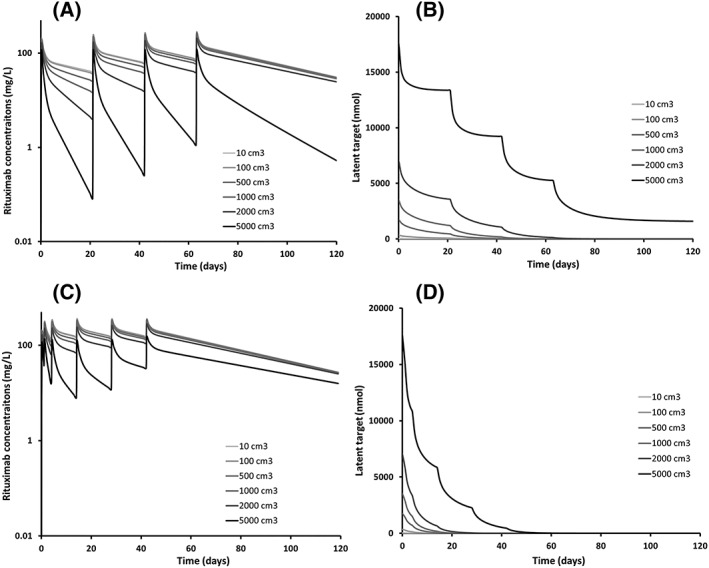
Simulated typical profiles of rituximab concentration–time (left) and latent target amount (right) for increasing metabolic tumour volumes (10–5000 cm3) for 4 doses of 375 mg/m2 rituximab every 3 weeks (top) and every 2 weeks (bottom)
Independently from kdeg value, the time to reach TMDD disappearance depends only on rituximab concentrations, but the remote value of L depends not only on initial antigen amount, but also on rituximab concentrations.
4. DISCUSSION
To our knowledge, this study is the first to detect a target‐mediated elimination of rituximab in patients treated for NHL. A semimechanistic model including target‐mediated elimination fitted satisfactorily rituximab serum concentrations measured in our patients. Model 5, which included a latent variable (L), allowed the best description of rituximab serum concentrations. The influence of MTV on L is described as baseline value of L (L0) being expressed in unit of MTV. The strong association of MTV with L0 (R2 = 0.59), suggests that 59% of L0 variability is explained by tumour volume. Thus, L should mainly correspond to CD20 antigen interacting with rituximab, notably blood and extra‐blood B‐cells and tumour cells. Rituximab pharmacokinetics was not significantly different between FL and DLBCL.
The pharmacokinetics of rituximab were described using 2 compartment models in 12 previous studies (Table 3), including 7 in lymphomas, 2 in CLL and 2 in rheumatoid arthritis. Among these studies, the influence of antigen mass on pharmacokinetic parameters was assessed and quantified in only 2 studies, both in diffuse large B‐cell lymphoma.6, 7 Notably, Rozman et al. reported a decrease in rituximab clearance with time, the rate of decrease being higher in progressive disease patients. A decrease in rituximab clearance with time was also reported in CLL8 and rheumatoid arthritis10 patients.
Table 3.
Pharmacokinetic studies of rituximab using pharmacokinetic modelling
| Study | Year | Disease | Nonlinear elimination | Antigen mass | V1 (L) | CL (L/day) | T½‐β (days) |
|---|---|---|---|---|---|---|---|
| Regazzi | 2005 | FL | – | – | 2.98 | 0.208 | 22.4 |
| Blasco | 2008 | DLBCL | – | – | 1.77 | 0.117 | 94.1 |
| Muller | 2012 | DLBCL | – | – | 3.88 | 0.226 | 53.9 |
| Rozman | 2017 | DLBCL | Time‐varying | Disease progression on kdes | 4.62 | 0.252 | 40.3 |
| Tout | 2017 | DLBCL | – | Tumour volume on V1 and V2 | 6.4 | 0.55 | 12.8 |
| Gota | 2016 | DLBCL | – | – | 0.95 | 0.141 | 11.2 |
| Candelaria | 2018 | DLBCL | – | – | 3.19 | 0.3 | 21.2 |
| Li | 2012 | CLL | Time‐varying | – | 4.15 | 0.171 | 26.7 |
| Tout | 2016 | CLL | TMDD | Circulating CD20 on kdeg | 3.08 | 0.137 | 31.3 |
| Ng | 2006 | RA | – | – | 2.98 | 0.257 | 20.2 |
| Lioger | 2017 | RA | Time‐varying | CD19 count on k10 | 4 | 0.44 | 18.5 |
| Puisset | 2013 | Plasma‐pheresis | – | – | 2.48 | 0.15936 | 22.8 |
All pharmacokinetic models were bicompartmental.
FL: follicular lymphoma; DLBCL, diffuse large B‐cell lymphoma; CLL: chronic lymphocytic leukaemia; RA, rheumatoid arthritis; TMDD, target‐mediated drug disposition; V1, V2: central and peripheral volumes of distribution; CL: clearance; k10: first‐order elimination rate constant; kdes: time‐varying elimination rate; kdeg: second‐order target‐mediated elimination rate; T½‐β: elimination half‐life.
Time‐decreasing clearance assumes a strict decrease of target antigen during treatment. Our irreversible binding TMDD model should allow a semimechanistic description of target‐mediated elimination, as done for rituximab in our previous study in CLL patients.9 Target‐mediated drug disposition is frequently reported for monoclonal antibodies.15, 22, 23 It is due to the formation of antibody–target complexes, which are eliminated. Therefore, this elimination depends not only on antibody concentrations, but also on target antigen amount. Our TMDD model assumes irreversible binding of rituximab on the target antigen,15 where target‐mediated elimination is described using second‐order elimination rate, depending on both rituximab concentrations and latent antigen amount. As well as in our previous study,9 the irreversible binding TMDD model showed better descriptive performance than time‐varying clearance model (difference in AIC = 22.32).
The estimation of L0 in the present study on NHL was at the same order of magnitude as in our previous study in CLL patients (1250 nmol vs 1850 nmol, respectively), even if somewhat LOWER. This suggests a lower antigen mass and/or a lower access to the target cells in lymphoma than in CLL. However, target‐mediated elimination rate (kdeg) is half in the present study compared to CLL patients (0.000079 vs 0.000176 nmol−1.day−1, respectively), which suggests a lower turnover, a lower avidity of rituximab for target cells, and/or lower elimination rate of complexes in NHL than in CLL patients.9 Simulations confirmed that high tumour volume (≥5000 cm3) would necessitate higher doses of rituximab to obtain the disappearance of tumour, which is in accordance with our previous findings in DLBCL.7 In addition, dose‐dense induction protocol may be useful to accelerate elimination of tumour.
In addition, we showed a significant increase in V1 with MTV. The increase in V1 is associated with an increased terminal elimination half‐life. In a previous study of rituximab pharmacokinetics in DLBCL patients, we have reported this phenomenon,7 which may be explained by reversible interactions of rituximab with its target (target‐mediated retention).15 An association of increased V1 and terminal elimination half‐life with higher antigen mass was also reported for trastuzumab.24 Antibody retention by antigen may be explained, at least in part, by low antibody‐target complex elimination rate. The fact that both target‐mediated elimination and retention were observed in the present study suggest that antigen target is present in several tissues and rituximab‐target complexes in these tissues may be cleared with different rates.
Our study has nevertheless limitations. First, MTV was available only at baseline, which prevented us to describe the variations of latent target amount with tumour burden. As a consequence, it was not possible to verify that latent target amount decreasing toward zero was linked to an extinction of tumour. Second, the elimination rate constant of latent target (kout) was not identifiable, which prevented us from using model 5. Two explanations are possible: (i) no nonlinear terminal elimination shape was visible; and (ii) there was a low rate of target output independent from rituximab treatment. Of note, in our previous study in CLL patients, the identifiability of kout was associated with a nonlinear elimination shape. Third, we were not able to estimate the interindividual variability of kdeg. It was therefore impossible to test a possible association of this parameter with MTV. Fourth, our cohort included both follicular lymphoma and DLBCL patients. No significant association of disease (FL or DLBCL) with pharmacokinetic parameters was detected but this should be considered with caution as absence of significance may be due to the relative small number of patients of our cohort.
In conclusion, this is the first study reporting nonlinearity of rituximab pharmacokinetics in NHL patients. By describing this nonlinearity using a semimechanistic model, a clear relationship between nonlinear elimination and antigen mass, assessed by MTV, was shown. In addition, this study confirms the retention of rituximab by the target‐antigen. Our simplified TMDD model with irreversible binding of mAb to latent target may be extended to other antibodies and/or diseases.
COMPETING INTERESTS
D.T. reports grants from Boehringer Ingelheim and Amgen, outside the submitted work. G.P. reports grants from Novartis, grants from Roche Pharma, grants from Sanofi‐Genzyme, grants from Chugai, grants from Pfizer, outside the submitted work. E.G. reports personal fees from ROCHE, during the conduct of the study; personal fees from GILEAD, personal fees from FRESENIUS KABI, grants from NOVARTIS, outside the submitted work. H.M., Y.V., C.P.‐A., F.A. and P.C. have nothing to disclose.
CONTRIBUTORS
D.T., H.M., P.C., G.P., E.G., conceived and desgined the experiments. H.M., Y.V., C.P.‐A., F.A., P.C. and E.G., performed the experiments. D.T., H.M. and G.P., analyzed the data. D.T., H.M., G.P. and E.G., wrote the paper. All authors approved the manuscript.
Ternant D, Monjanel H, Venel Y, et al. Nonlinear pharmacokinetics of rituximab in non‐Hodgkin lymphomas: A pilot study. Br J Clin Pharmacol. 2019;85:2002–2010. 10.1111/bcp.13991
Ethical approval: This study was approved by local ethics committee. As part of the routine therapeutic drug monitoring of rituximab, blood samples are drawn to measure rituximab trough concentrations and individual results are interpreted and sent to the prescriber and discussed during clinic‐biological rounds.
The authors confirm that Prof. Emmanuel Gyan was the principal investigator of this study.
REFERENCES
- 1. Blasco H, Chatelut E, de Bretagne IB, Congy‐Jolivet N, Le Guellec C. Pharmacokinetics of rituximab associated with CHOP chemotherapy in B‐cell non‐Hodgkin lymphoma. Fundam Clin Pharmacol. 2009;23(5):601–608. [DOI] [PubMed] [Google Scholar]
- 2. Candelaria M, Gonzalez D, Fernandez Gomez FJ, et al. Comparative assessment of pharmacokinetics, and pharmacodynamics between RTXM83, a rituximab biosimilar, and rituximab in diffuse large B‐cell lymphoma patients: a population PK model approach. Cancer Chemother Pharmacol. 2018;81(3):515–527. [DOI] [PubMed] [Google Scholar]
- 3. Gota V, Karanam A, Rath S, et al. Population pharmacokinetics of Reditux, a biosimilar rRituximab, in diffuse large B‐cell lymphoma. Cancer Chemother Pharmacol. 2016;78(2):353–359. [DOI] [PubMed] [Google Scholar]
- 4. Muller C, Murawski N, Wiesen MH, et al. The role of sex and weight on rituximab clearance and serum elimination half‐life in elderly patients with DLBCL. Blood. 2012;119(14):3276–3284. [DOI] [PubMed] [Google Scholar]
- 5. Regazzi MB, Iacona I, Avanzini MA, et al. Pharmacokinetic behavior of rituximab: a study of different schedules of administration for heterogeneous clinical settings. Ther Drug Monit. 2005;27(6):785–792. [DOI] [PubMed] [Google Scholar]
- 6. Rozman S, Grabnar I, Novakovic S, Mrhar A, Jezersek Novakovic B. Population pharmacokinetics of rituximab in patients with diffuse large B‐cell lymphoma and association with clinical outcome. Br J Clin Pharmacol. 2017;83(8):1782–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tout M, Casasnovas O, Meignan M, et al. Rituximab exposure is influenced by baseline metabolic tumor volume and predicts outcome of DLBCL patients: a Lymphoma Study Association report. Blood. 2017;129(19):2616–2623. [DOI] [PubMed] [Google Scholar]
- 8. Li J, Zhi J, Wenger M, et al. Population pharmacokinetics of rituximab in patients with chronic lymphocytic leukemia. J Clin Pharmacol. 2012;52(12):1918–1926. [DOI] [PubMed] [Google Scholar]
- 9. Tout M, Gagez AL, Lepretre S, et al. Influence of FCGR3A‐158V/F genotype and baseline CD20 antigen count on target‐mediated elimination of rituximab in patients with chronic lymphocytic leukemia: a study of FILO Group. Clin Pharmacokinet. 2016;2016:25. [DOI] [PubMed] [Google Scholar]
- 10. Lioger B, Edupuganti SR, Mulleman D, et al. Antigenic burden and serum IgG concentrations influence rituximab pharmacokinetics in rheumatoid arthritis patients. Br J Clin Pharmacol. 2017;83(8):1773–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ng CM, Bruno R, Combs D, Davies B. Population pharmacokinetics of rituximab (anti‐CD20 monoclonal antibody) in rheumatoid arthritis patients during a phase II clinical trial. J Clin Pharmacol. 2005;45(7):792–801. [DOI] [PubMed] [Google Scholar]
- 12. Puisset F, White‐Koning M, Kamar N, et al. Population pharmacokinetics of rituximab with or without plasmapheresis in kidney patients with antibody‐mediated disease. Br J Clin Pharmacol. 2013;76(5):734–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dayde D, Ternant D, Ohresser M, et al. Tumor burden influences exposure and response to rituximab: pharmacokinetic‐pharmacodynamic modeling using a syngeneic bioluminescent murine model expressing human CD20. Blood. 2009;113(16):3765–3772. [DOI] [PubMed] [Google Scholar]
- 14. Gibiansky L, Gibiansky E. Target‐mediated drug disposition model: approximations, identifiability of model parameters and applications to the population pharmacokinetic‐pharmacodynamic modeling of biologics. Expert Opin Drug Metab Toxicol. 2009;5(7):803–812. [DOI] [PubMed] [Google Scholar]
- 15. Ternant D, Azzopardi N, Raoul W, Bejan‐Angoulvant T, Paintaud G. Influence of antigen mass on the pharmacokinetics of therapeutic antibodies in humans. Clin Pharmacokinet. 2018;2018:018–0680. [DOI] [PubMed] [Google Scholar]
- 16. Stein AM, Peletier LA. Predicting the onset of nonlinear pharmacokinetics. CPT Pharmacometrics Syst Pharmacol. 2018;7(10):670–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Berinstein NL, Grillo‐Lopez AJ, White CA, et al. Association of serum rituximab (IDEC‐C2B8) concentration and anti‐tumor response in the treatment of recurrent low‐grade or follicular non‐Hodgkin's lymphoma. Ann Oncol. 1998;9(9):995–1001. [DOI] [PubMed] [Google Scholar]
- 18. Nestle U, Kremp S, Schaefer‐Schuler A, et al. Comparison of different methods for delineation of 18F‐FDG PET‐positive tissue for target volume definition in radiotherapy of patients with non‐small cell lung cancer. J Nucl Med. 2005;46(8):1342–1348. [PubMed] [Google Scholar]
- 19. Gibiansky E, Gibiansky L, Carlile DJ, Jamois C, Buchheit V, Frey N. Population pharmacokinetics of obinutuzumab (GA101) in chronic lymphocytic leukemia (CLL) and non‐Hodgkin's lymphoma and exposure‐response in CLL. CPT Pharmacometrics Syst Pharmacol. 2014;2014:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res. 2018;46(D1):D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alexander SPH, Kelly E, Marrion NV, et al. The Concise Guide to PHARMACOLOGY 2017/18: Overview. Br J Pharmacol. 2017;174(Suppl 1):S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dirks NL, Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(10):633–659. [DOI] [PubMed] [Google Scholar]
- 23. Dostalek M, Gardner I, Gurbaxani BM, Rose RH, Chetty M. Pharmacokinetics, pharmacodynamics and physiologically‐based pharmacokinetic modelling of monoclonal antibodies. Clin Pharmacokinet. 2013;52(2):83–124. [DOI] [PubMed] [Google Scholar]
- 24. Bruno R, Washington CB, Lu JF, Lieberman G, Banken L, Klein P. Population pharmacokinetics of trastuzumab in patients with HER2+ metastatic breast cancer. Cancer Chemother Pharmacol. 2005;56(4):361–369. [DOI] [PubMed] [Google Scholar]