

Abstract
Purpose
To review pharmacokinetic and pharmacodynamic characteristics of antibodies that bind to soluble ligands within the framework of calcitonin gene-related peptide antibodies.
Overview
Calcitonin gene-related peptide has been implicated in the pathophysiology of migraine. Galcanezumab is an antibody that binds to the ligand calcitonin gene-related peptide. Other antibodies that target calcitonin gene-related peptide include eptinezumab and fremanezumab. To understand how antibodies can affect the extent and duration of free ligand concentrations, it is important to consider the dose and pharmacokinetics of an antibody, and the kinetics of the ligand and antibody–ligand complex. Insights regarding the pharmacokinetic/pharmacodynamic properties of galcanezumab as a probe antibody drug and calcitonin gene-related peptide as its binding ligand regarding its clinical outcomes are provided.
Discussion
Antibodies are administered parenterally because oral absorption is limited by gastrointestinal degradation and inefficient diffusion through the epithelium. The systemic absorption of antibodies following intramuscular or subcutaneous administration most likely occurs via convective transport through lymphatic vessels into blood. The majority of antibody elimination occurs via intracellular catabolism into peptides and amino acids following endocytosis. Binding of ligand to an antibody reduces the free ligand that is available to interact with the receptor and efficacy is driven by the magnitude and duration of the reduction in free ligand concentration. A galcanezumab pharmacokinetic/pharmacodynamic model shows that galcanezumab decreases free calcitonin gene-related peptide concentrations in a dose- and time-dependent manner and continues to suppress free calcitonin gene-related peptide with repeated dosing. The model provides evidence for a mechanistic linkage to galcanezumab therapeutic effects for the preventive treatment of migraine.
Keywords: Migraine, CGRP, pharmacokinetics, pharmacodynamics, antibody, galcanezumab
Introduction
Migraine is a chronic, recurrent neurological disorder characterized by attacks of severe pain and associated symptoms, such as nausea, photophobia, and phonophobia, with significant unmet medical need. Although the pathophysiology of migraine is still under evaluation, many data indicate a crucial role of calcitonin gene-related peptide (CGRP) (1). CGRP is a 37-amino acid neuropeptide and a member of the calcitonin family, which includes calcitonin, adrenomedullin, amylin, and intermedin (2). It is expressed in both central and peripheral nervous systems, especially in dorsal root and trigeminal ganglions and their projections (3). Mammals express two isoforms, α-CGRP and β-CGRP, encoded by two distinct genes (4). The α-CGRP and β-CGRP are differentially regulated, differ in pharmacological profile, and have distinct physiological functions (5). During a migraine attack, CGRP concentrations increase with rapid turnover of effect (6–9). Galcanezumab is a monoclonal antibody (mAb) that binds to the soluble ligand, CGRP, and is approved for the preventive treatment of migraine (10). Fremanezumab, approved for the preventive treatment of migraine (11) and eptinezumab (12–14), currently in Phase 3 development, are other monoclonal antibodies that bind to CGRP. Of note, erenumab is a mAb that binds to the CGRP receptor and is approved for the preventive treatment of migraine (15). See Table 1 for a summary of the monoclonal antibodies that modulate CGRP.
Table 1.
Monoclonal antibodies that modulate CGRP.
| Generic name | Sponsor | Patient population | Target | Route | Dose (mg) | IgG Type | t1/2 (days) | Tmax |
|---|---|---|---|---|---|---|---|---|
| Eptinezumaba (ALD403) | Alder Biopharmaceuticals | Episodic and chronic migraine | CGRP-α CGRP-β | IV | 30 mg QTLY 100 mg QTLY 300 mg QTLY | IgG 1 | 26 | 3 hours |
| Erenumabb (AMG 334) | Novartis and Amgen | Episodic and chronic migraine | CGRP receptor | SC | 70 mg QM 140 mg QM | IgG 2 | 28 | 6 days |
| Fremanezumabb (TEV-48125) | Teva Pharmaceutical Industries | Episodic and chronic migraine | CGRP-α CGRP-β | SC | 225 mg QM with 675 mg LD | IgG 2 | 32 | 5 days |
| Galcanezumabb (LY2951742) | Eli Lilly and Company | Episodic and chronic migraine | CGRP-α CGRP-β | SC | 240 mg LD followed by 120 mg QM | IgG 4 | 27 | 5 days |
CGRP: calcitonin gene-related peptide; IV: intravenous; SC: subcutaneous; QM: once monthly; LD: loading dose; QTLY: quarterly; t1/2: half-life; Tmax: time of maximum observed drug concentration.
Currently in Phase 3 trials.
Approved by the FDA.
Pharmacokinetic (PK) and pharmacodynamic (PD) analyses are essential components of drug discovery and development. Pharmacokinetics is the study of the time-course of drug absorption, distribution, metabolism, and excretion, and these processes are applied to enable safe and effective therapeutic management of drugs in patients. Population PK principles are applied to understand the sources and correlates of variability in drug concentrations in a population of patients, which could have important implications for clinical dosing. Results from population PK analyses have been included in all drug labels from antibodies currently on the market.
Pharmacodynamics is the relationship between the measured drug concentration and the resulting effect, including the time course and intensity of therapeutic and adverse effects. A PD endpoint can be a clinical endpoint or biomarker. The concentration of a biomarker may be increased or decreased as an indicator of a pharmacological response to a therapeutic intervention.
Antibody drugs represent one of the fastest growing areas of drug development, and they often exhibit PK and PD properties that are more complex than those typically associated with small-molecule drugs (16). Antibody drugs that bind soluble ligands make up a large proportion of antibody therapeutics. As such, kinetics of both the antibody and the ligand are important for informing the pharmacological characteristics of the antibody drug. Pharmacokinetic/pharmacodynamic models that describe the in vivo interaction of an antibody and a soluble ligand provide unique characterizations and insights and can provide a framework for predicting the time course of antibody exposure and ligand response following different dose regimens (17–19). Models can support dose regimen selection during drug development and regulatory review.
This manuscript will provide a review of PK and PD characteristics of antibodies that bind to soluble ligands within a framework to CGRP antibodies. A fundamental understanding of modulation of CGRP following antibody administration is required to comprehend the pharmacology of antibodies that bind CGRP. As such, an evaluation of the PK/PD properties of galcanezumab as a probe antibody drug and CGRP as its binding ligand provides insights regarding their clinical interaction and therapeutic outcome.
Antibody nomenclature and structure
An antibody, also known as an immunoglobulin (Ig), is produced mainly by plasma cells that are used by the immune system to identify and neutralize pathogens such as bacteria and viruses. There are five Ig isotypes, namely Ig alpha (IgA), Ig delta (IgD), Ig epsilon (IgE), Ig gamma (IgG), and Ig mu (IgM), with different structural characteristics including molecular weight and valence (20). The predominant isotype comprising 70–85% of the Ig in human serum is IgG and this has, on average, the highest serum concentration of ∼10 mg/mL and the longest half-life (t1/2) of ∼23 days (21).
The majority of antibodies on the market and in drug development are of the IgG isotype and are mAb preparations. These preparations are derived from a cloned population of cells and have a unique structure, affinity, and specificity, which is in contrast to polyclonal antibodies that have a distribution of affinity and specificity resulting from antigen stimulation of genetically distinct cells (22). Humanized antibodies have the suffix “-zumab” and the amino acid sequence is ∼90% derived from a human DNA sequence, whereas human antibodies have the suffix “-umab” and 100% of the amino acid sequence is derived from a human DNA sequence. IgG are further divided into four subclasses based on the structure of their heavy chains: IgG1, IgG2, IgG3, and IgG4. Structural differences among IgG heavy chains can lead to differences in binding to cell-associated receptors (20). The literature is limited regarding IgG subclass and its effect on PK/PD of monoclonal antibodies, and studies in this area are warranted to address this topic. The antibodies in Table 1 that modulate CGRP are of the IgG isotype and are either humanized or human antibodies.
Pharmacokinetics
Delivery and absorption
Most small-molecule drugs are delivered orally, undergoing absorption in the gastrointestinal (GI) tract followed by first-pass liver metabolism before entering the peripheral circulation. Antibodies are administered via the IV, SC, intramuscular (IM), or ophthalmic route because oral administration is limited by GI degradation and inefficient diffusion or convection through the epithelium. Most antibodies are administered by the IV route because it allows for the antibody to be delivered rapidly with complete availability to the systemic circulation and can be given in larger volumes and doses compared to the IM or SC route. However, the IV route may not be as convenient as IM or SC administration, as it requires patient visits to hospitals or clinics to receive the medication. As IM and SC dosing allows for self-administration, these routes are useful when effective treatment requires maintenance or chronic dosing. A limitation of IM and SC dosing is that large doses may not be feasible due to the solubility of IgG (∼100–150 mg/mL) and because larger injection volumes tend to cause more injection site pain. For these reasons, multiple injections must be given to achieve higher doses. The antibodies in Table 1 that modulate CGRP administered by the SC route are galcanezumab, fremanezumab, and erenumab. The dose in a 1-mL injection is 120 mg for galcanezumab (10) and 70 mg or 140 mg for erenumab (15), and for fremanezumab (11) the dose in a 1.5-mL injection is 225 mg. Multiple injections of these antibodies are administered to achieve higher doses.
Although antibody absorption is limited after oral administration, IgG may be available to the systemic circulation by crossing epithelial cells via paracellular transport or via receptor-mediated processes. In neonates of several non-human species (e.g. mice, rats, dogs), IgG is efficiently absorbed following oral administration (23) during the first few weeks postpartum. This GI absorption, observed only in neonates, allows for transmission of IgG from mother's milk, and is due to transport of IgG by the Brambell receptor (i.e. FcRn) (24). FcRn is located in many tissues in adults such as endothelial cells of kidneys, liver, lungs, hepatocytes, intestinal macrophages, peripheral blood monocytes, and dendritic cells (25–27). Although FcRn is located in the GI epithelium, it appears that IgG is absorbed minimally in humans (28). FcRn functions within systemic tissues to protect IgG from degradation, and the receptor may be an important determinant of tissue disposition (see Distribution and Elimination sections) (23).
The systemic absorption of an antibody following IM or SC administration most likely occurs via convective transport of antibody through lymphatic vessels into blood (Figure 1). Convective transport occurs as a result of fluid movement from the interstitial space and blood (29–31). Additional determinants of convective transport of antibody include osmotic pressure gradients and the nature of paracellular pores. The lymph fluid drains slowly into the systemic circulation, and antibody absorption following IM or SC administration can occur for days, resulting in maximum antibody concentrations in serum ∼1 to 2 weeks post administration. For galcanezumab, the time to maximum concentration is 5 days (10). For fremanezumab, after single SC administrations of 225 mg, 675 mg, and 900 mg fremanezumab, median time to maximum concentrations was 5 to 7 days (11). Following a single subcutaneous dose of 70 mg or 140 mg erenumab administered to healthy adults, median peak serum concentrations were attained in approximately 6 days (15). The time to maximum concentration of IV-administered eptinezumab is about 3 hours (32). The SC or IM bioavailability of antibodies typically range from 50–100%, but may be dose-dependent, and the extent is determined by presystemic catabolism and systemic absorption. Erenumab's SC bioavailability is reported to be 82% (15), and may be dependent on rates of proteolysis, antibody endocytosis, and recycling with FcRn.
Figure 1.

General pathways for IgG absorption, distribution and elimination.
Distribution
Movement of antibodies from the blood into the interstitial fluid of tissues is dependent on their limited ability to cross cell membranes. Extravasation of antibodies may occur by paracellular or transcellular movement by means of convective processes, diffusion, or endocytosis (33). Due to their large molecular weight and polarity, it is unlikely that significant extravasation occurs with antibodies by transcellular diffusion (33). The main processes in which endocytosis of antibodies can occur are phagocytosis, fluid-phase pinocytosis, and receptor-mediated endocytosis (34). Virtually all cells in the body have the ability to take up proteins and other macromolecules, including antibodies, from the surrounding fluid space via fluid-phase pinocytosis. Receptor-mediated endocytosis may occur following binding of antibodies to cell surface antigens or Fcy receptors, which are present on a variety of cells including monocytes, macrophages, B-lymphocytes, and platelets, and contribute to the movement of antibodies from blood to tissues (33).
Convective transport through paracellular pores is considered to be the primary mechanism of antibody extravasation in tissues (35). Convective transport is driven by the hydrostatic pressure gradient between blood and tissues and is dependent on the gradient of osmotic pressure and the diameter and tortuosity of paracellular pores. Convection also plays a role in the distribution of antibodies within the interstitial fluid and facilitates the transport of antibodies out of the tissue as the interstitial fluid enters the lymphatic capillaries (Figure 1). Given that the paracellular pores of the lymphatic capillaries are much larger than those in the vascular endothelium, it is assumed that the convective clearance of antibodies from the tissue is much more efficient than the process of convective extravasation, thereby maintaining relatively low monoclonal antibody concentrations in the interstitial fluid (35). Distribution of antibodies within tissues may be hindered by high-affinity monoclonal antibody binding to tissue proteins, which creates a barrier to mAb distribution in tissues following extravasation (36–39).
The limited distribution of monoclonal antibodies to the brain has primarily been explained by tight junctions in the brain vascular endothelium, and plasma-to-brain concentration ratios are typically in the range of 500:1 (40). The role of FcRn in limiting IgG exposure in the brain has been the focus of several investigations, and conflicting data have been reported (40–42). Galcanezumab shows limited distribution into the central nervous system and cerebrospinal fluid (43). For other monoclonal antibodies that bind CGRP, no data exists to our knowledge. While the central penetration of galcanezumab is low, a central effect cannot be definitively ruled out. For many neurodegenerative diseases, monoclonal antibodies are being developed suggesting that they can act centrally to mediate their effects (44–46).
It has been hypothesized that a migraine attack could potentially disrupt the blood-brain barrier (BBB), allowing a mAb entry into the brain. However, it has been shown experimentally that there is no increase in BBB permeability during an aura phase of migraine and there is no disruption of the BBB during a glyceryl trinitrate-triggered migraine attack (47,48).
Elimination
Small molecule drugs are typically metabolized by Phase 1 or 2 enzyme systems in the GI tract and liver after oral administration and eliminated from the body by renal and biliary pathways (49). For monoclonal antibodies, elimination occurs by excretion and catabolism. The majority of mAb elimination occurs via intracellular catabolism, following fluid-phase or receptor-mediated endocytosis. The general pathway for IgG elimination after SC administration is shown in Figure 1. The contribution of different organs to mAb catabolism is estimated to be 33% for liver, 24% for skin, 16% for muscle, and 12% for intestine (50). An mAb is catabolized into peptides and amino acids primarily by the reticuloendothelial system, which consists of phagocytic cells such as macrophages and monocytes (51). Cells in the body have the ability to take up proteins or macromolecules from the surrounding fluid space via fluid phase endocytosis, from which the formed endosome is delivered to a lysosome where its content undergoes enzymatic degradation. A significant portion of IgG is rescued from lysosomal catabolism by FcRn (23,52,53). Within the acidified environment of the early endosome, IgG binds tightly to FcRn. The IgG–FcRn complexes are not delivered to the lysosome for catabolism but rather are sorted to the cell surface for fusion with the cell membrane. The receptor shows virtually no affinity for IgG at physiological pH and, upon fusion of the sorting vesicle with the cell membrane, IgG dissociates from the receptor and is released into blood or interstitial fluid. This phenomenon may explain the longer elimination t1/2 of most IgGs of 3 to 4 weeks compared to IgA, IgD, IgE, and IgM, whose elimination t1/2 range from 0.5 to 1 week. Although IgG1, IgG2, and IgG4 subclasses exhibit elimination t1/2 of around 3 to 4 weeks, the elimination t1/2 of IgG3 is only 7 days, which is attributed to differences in binding to FcRn across IgG subclasses (54).
Receptor-meditated endocytosis is a process whereby the antibody Fab domain binds with target epitopes on cell surfaces, which triggers internalization and subsequent lysosomal degradation of the mAb–receptor complex (55). This process can lead to target-mediated disposition, where the interaction of the mAb and its receptor contributes to the kinetics of mAb elimination, as well as distribution (56). This form of elimination can be capacity-limited because of a finite expression of the receptor. Higher doses of mAb can saturate cell membrane targets responsible for elimination, leading to greater-than-proportional increases in mAb serum concentration relative to the dose administered, known as nonlinear PK. As an example, erenumab exhibits nonlinear PK as a result of binding to the CGRP receptor (57). For antibodies that bind to soluble ligands, target-mediated elimination may also occur (58,59). Galcanezumab exhibits linear PK and exposure increases proportionally with doses between 1 and 600 mg (10). For fremanezumab, dose proportionality, based on population PK, was observed between 225 mg to 900 mg (11). It is worthy to note that antibodies can target soluble ligands and cell membrane-bound receptors (60).
The liver and kidney are involved predominantly in the metabolism and elimination of small molecule drugs and can lead to changes in PK, possibly affecting the efficacy and toxicity profile in patients with renal and hepatic impairment. Although the liver and kidneys may contribute to catabolism of antibodies, in general, renal or hepatic impairment does not affect mAb elimination (61,62). The large size of a mAb (150 kDa) prevents efficient filtration through the glomerulus and mAb PK is expected to be unchanged by renal impairment, but specific cases do exist where mAb PK is affected (63,64). Antibody fragments (non-intact mAb) of lower molecular weight (less than 60 kDa) filtered by the glomerulus can be influenced by renal impairment (65,66). Other organs including skin, muscle, and intestine also contribute to mAb degradation, thereby limiting major influences to mAb elimination due to liver injury or insufficiency. Dose adjustment of a mAb is usually not warranted in patients with renal or hepatic impairment (67). No dedicated clinical studies were conducted to evaluate the effect of hepatic impairment or renal impairment on the PK of monoclonal antibodies that modulate CGRP. Population PK analysis of integrated data from the galcanezumab clinical studies revealed that creatinine clearance did not affect the PK of galcanezumab in patients with mild or moderate renal impairment (10). For fremanezumab, a population PK analysis of integrated data from the fremanezumab clinical studies did not reveal a difference in the PK of fremanezumab in patients with mild hepatic impairment, relative to those with normal hepatic function (11). For erenumab, population PK analysis of integrated data from the erenumab clinical studies did not reveal a difference in the PK of erenumab in patients with mild or moderate renal impairment relative to those with normal renal function (15). For galcanezumab, population PK analysis revealed that bilirubin, an indirect marker of hepatic function, did not influence the clearance of galcanezumab (10).
Immunogenicity
Therapeutic antibodies may be viewed by the body as foreign and activate immune responses that lead to anti-drug antibody (ADA) development that can bind to the therapeutic antibody. Many factors can influence the likelihood of ADA formation, including manufacturing process, antibody structure (i.e. derived from rodent, chimeric, humanized, human), dose, dosing frequency, route of administration (68–70), and patient factors such as immune status or genetics. The effect that ADA could have on the PK of an antibody is complex and difficult to predict, but this interaction can influence the elimination of the antibody by either decreasing or increasing the t1/2. Some ADA may have competitive binding activity, preventing the binding of the ligand to the antibody (71,72). Overall, these changes may alter the efficacy and toxicity profile of the antibody.
Assays for immunogenicity are difficult to develop and verify, and a comparison of observed incidences of ADA formation across various detection assays is complicated by potential differences in assay sensitivity. It is expected that therapeutic antibodies will show some immunogenicity, and even completely human therapeutic antibodies have unique idiotypes and posttranslational modifications or impurities associated with the manufacturing process that may trigger an immune response (72–75).
The immunogenicity of galcanezumab (76,77), erenumab (78), and fremanezumab (79) have been reported elsewhere from clinical trials. The drug labels for these three drugs state similarly that, although the immunogenicity profiles do not demonstrate an impact of ADA development on efficacy or safety, the available data are too limited to make definitive conclusions (10,11,15). In addition, ADA development does not demonstrate an impact on the PK of galcanezumab (10).
Demographics
Age, body size/weight, sex, and ethnicity are demographic factors commonly examined using population PK analyses to understand their influence on drug exposure, efficacy, and safety to help guide clinical dosing regimens. Age may affect lymph flow rate and endocytosis rate, leading to reduced absorption, distribution, and/or elimination of a mAb (62). Pharmacokinetic studies show mixed results regarding the effect of age (80–84), but limited data exist in the young and elderly populations (85). Body weight is a commonly identified factor that influences mAb PK, which is not surprising based on the theory of allometry and PK principles (86). Body weight-adjusted dosing can reduce variability in PK, but the degree to which the variability is affected and the therapeutic window of the mAb are important to consider for rational dose recommendations. The PK of galcanezumab were not affected by age, sex, race, or subtypes of migraine spectrum (episodic or chronic migraine), based on a population PK analysis. Body weight has no clinically relevant effect on the PK of galcanezumab (10). For fremanezumab, a population PK analysis assessing effects of age, race, sex, and weight was conducted, and no dose adjustments are recommended (11). The PK of erenumab were not affected by age, gender, race, or subtypes of migraine spectrum (episodic or chronic migraine) based on population PK analysis (15).
Pharmacodynamics
Considerations for antibodies that bind soluble ligands
Attention to the PD effects of antibodies via the characterization of the interaction of the antibody and the soluble ligand is important, given that the soluble ligand is the active agent that elicits the pharmacological effect. When antibody is administered, the premise is that the ligand will bind to it and the free ligand concentration that is available to interact with the ligand receptor will be reduced. Theoretically, efficacy is driven by the magnitude and duration of the reduction in free ligand concentration (87). Galcanezumab, eptinezumab, and fremanezumab are antibodies that bind to the soluble ligand CGRP (Table 1). Galcanezumab and fremanezumab are approved in the US for the preventive treatment of migraine, and eptinezumab is currently in Phase 3 development.
During drug development of antibodies, measurements of total (free + bound) or free ligand concentrations have the potential to help understand the kinetics of ligand inhibition, their correlation to clinical outcomes, and use as disease biomarkers. The time course of ligand inhibition may help define dosing regimens that sustain the required pharmacological activity of the antibody for effective treatment. Unfortunately, many bioanalytical challenges exist that make measurement of free ligand concentrations uncertain, or not practical/possible at times (88). Soluble ligands often have low concentrations and rapid elimination, and as assays are needed to measure the reduction of the free ligand after antibody administration, the sensitivity requirements for the assay may not be achievable. Furthermore, the antibody–ligand complex formed after antibody administration may have a t1/2 that is longer than the free ligand, leading to an accumulation of the antibody–ligand complex (89). As such, the complex has a much greater concentration than the free ligand, and this increases the difficulty in accurately determining free ligand concentration by the assay. Although approaches have been taken to mitigate the challenge of measuring free ligand concentration, it is generally felt that overestimation of free ligand concentration remains a concern (90). For the aforementioned reasons, assays that measure total (free + bound) ligand concentrations are most common. Total CGRP, rather than free CGRP, was measured following galcanezumab administration as described below in A PK/PD evaluation of galcanezumab clinical effects on CGRP.
Mechanistic PK/PD models have been used successfully in recent years to characterize relationships between mAb PK and the time course of mAb effects, and to enhance understanding of concentration–effect relationships between the mAb and the suppression of target ligand (58,91,92). In cases of low concentrations and rapid turnover of ligand, PK/PD models that characterize the interaction of the mAb and the ligand can be a valuable tool to mitigate the bioanalytical challenges associated with reliable free ligand assays, and facilitate an understanding of the time course of free ligand suppression by using the measured antibody–ligand complex and mAb concentrations (17–19). These models may provide unique characterizations and insights, and have the potential to support safe and effective dose regimens during drug development, regulatory review, approval, and labeling (88,93).
A PK/PD evaluation of galcanezumab clinical effects on CGRP
Galcanezumab is a humanized mAb that binds to the soluble ligand CGRP. In patients with migraine, CGRP is elevated (6–8,94–96) and may function as a biomarker for migraine diagnosis (97). In patients with episodic and chronic migraine, galcanezumab was demonstrated to be a safe and effective preventive treatment (76,77,98–100). In addition, galcanezumab was shown to be a promising preventive treatment for patients with cluster headache (101).
An objective during development of galcanezumab was to characterize the interaction between galcanezumab and CGRP, and to infer the effect of galcanezumab on free CGRP concentrations using a PK/PD model-based approach. To support this objective, 174 subjects (48% male, 52% female, aged 19–65 years) were given a SC administration of a single dose of 240 mg galcanezumab or 300 mg galcanezumab (102), and blood samples were collected pre-dose and up to approximately 20 weeks post-dose to measure concentrations of total CGRP (18 samples per subject; 8.5 mL per sample) and galcanezumab (18 samples per subject; 2.5 mL per sample). Galcanezumab serum concentrations were measured using a validated enzyme-linked immunosorbent assay (ELISA) and total CGRP plasma concentrations were measured using a galcanezumab-tolerant electrochemiluminescent assay, as reported previously (99). A bioanalytical assay to measure free CGRP was not used because free ligand assay results can have significant error in the presence of ligand bound to antibody, leading to an inability to accurately distinguish and measure concentrations of free and bound ligand. See previous section Pharmacodynamics, considerations for antibodies that bind soluble ligands, for additional details regarding challenges associated with ligand assays.
The interaction between galcanezumab and CGRP was characterized with the PK/PD antibody-ligand interaction model shown in Figure 2 using non-linear mixed effects modeling software (NONMEM, Version 7.3). A detailed review of the properties and assumptions of a general PK/PD antibody-ligand interaction model can be found elsewhere (103). The model herein is described by several PK and PD parameters that govern the interaction between galcanezumab and CGRP, and definitions of these parameters are found in the footnotes under Figure 2.
Figure 2.
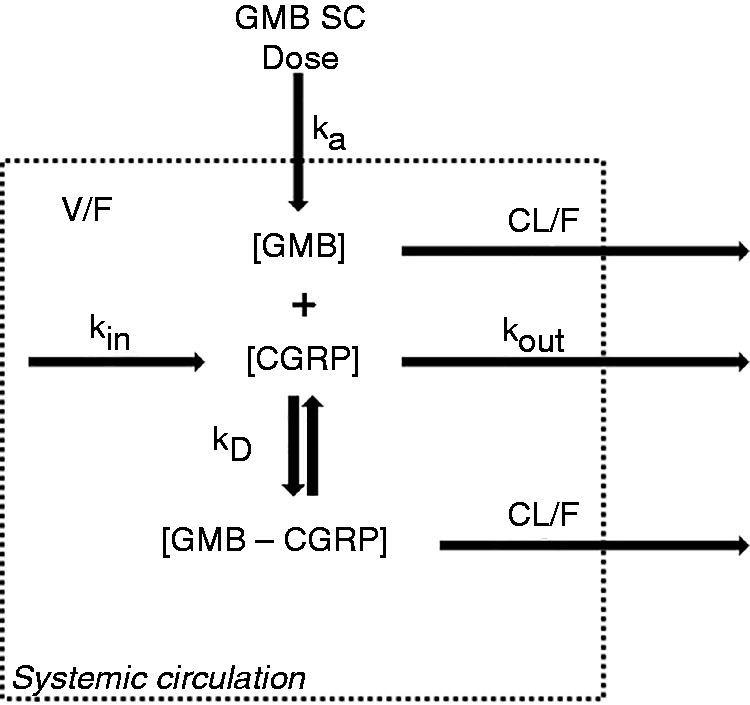
Schematic of the galcanezumab–CGRP interaction.
[CGRP]: calcitonin gene-related peptide concentration; CL/F: apparent clearance; [GMB]: galcanezumab concentration; [GMB–CGRP]: GMB–CGRP complex concentration; ka: rate of absorption of galcanezumab from the SC tissue to the systemic circulation; kD: in vivo equilibrium constant governing galcanezumab and CGRP binding; kin: zero-order production rate constant of CGRP; kout: first-order elimination rate constant of CGRP; SC = subcutaneous; V/F: apparent volume of distribution of galcanezumab.
Note: Parameter values estimated by the PK/PD model: ka = 0.02 h−1; V/F = 6.5 L; CL/F = 0.008 L/h; kin = 0.005 ng*h/mL; kout = 0.17 h−1; kD = 1.3 µM.
The time course of total galcanezumab, total (bound + free) CGRP, and free CGRP concentrations following a single administration of 300 mg galcanezumab is shown in Figure 3. Galcanezumab concentrations rise and reach peak levels ∼5 days post-dose and decline thereafter with a t1/2 of ∼27 days. Administration of galcanezumab leads to slow increases in total CGRP concentrations with a plateau maintained at 4–8 weeks after dosing. Free CGRP has a faster elimination compared to the galcanezumab-CGRP complex (GMB-CGRP), and when CGRP becomes bound to galcanezumab and takes on the slow distribution and clearance properties of galcanezumab, there is an increase in total CGRP concentrations. The binding of CGRP to galcanezumab prevents the normal processes involved in the rapid CGRP clearance, such as diffusion to sites of catabolism and proteolysis. The initial slow rise of total CGRP concentrations is a reflection of the accumulation or production of GMB-CGRP in the body.
Figure 3.

Time-course of total galcanezumab, total CGRP, and free CGRP following a single dose of 300 mg galcanezumab. Lines denote model-based predictions, circles denote observed galcanezumab concentrations at 300 mg, and triangles denote observed total CGRP concentrations at 300 mg. Galcanezumab concentration (nM) shown in terms of galcanezumab binding sites.
The model assumes that the free CGRP concentration, which is dependent on the ratio of kin to kout (kin/kout; see Figure 2 legend), is in equilibrium at baseline. In the model, increasing kin is predicted to increase the free CGRP concentration. The authors acknowledge that free CGRP is elevated during a migraine attack, but elevations in CGRP during a migraine attack do not affect the model. The premise of galcanezumab as a preventive treatment for migraine is to have a sufficient concentration of galcanezumab that will inhibit CGRP activity even when there is a spike in free CGRP during a migraine attack. The model predicts a rapid decrease of free CGRP concentrations from predose within 1 day after 300 mg galcanezumab was administered, with a slow return to predose free CGRP concentrations (Figure 2). Interestingly, the predicted effect of galcanezumab on free CGRP diminishes even in the presence of high galcanezumab concentrations. These data are supportive evidence to address a common misconception that if high antibody concentrations are present (i.e. excess of antibody relative to the ligand) then the ligand should be completely bound to the antibody.
Figure 4 illustrates the results from a simulation using the developed model to show the predicted effect of galcanezumab on free CGRP relative to baseline when galcanezumab is administered monthly for 5 months at doses of 5, 50, 120, 240, and 300 mg, and 240 mg as a loading dose followed by 120 mg monthly. Table 2 provides a descriptive summary of the key parameters. After the initial administration, the maximum reduction of free CGRP from baseline ranged from 39% at 5 mg to a near plateau of 97% at 240 mg and 300 mg. The reduction at trough (1 month after the first galcanezumab administration) ranged from 0% at 5 mg to 70% at 300 mg. These findings suggest that administration of galcanezumab leads to a dose- and time-dependent reduction in free CGRP. Higher galcanezumab doses provide greater maximum reduction of free CGRP and suppress free CGRP to a greater extent than lower doses during the first month after the initial dose. Repeated monthly dosing of galcanezumab to steady state continued to suppress free CGRP ranging from 6% at 5 mg to 80% at 300 mg.
Figure 4.

Simulation of the percent change of free CGRP from baseline following monthly administration of galcanezumab.
Table 2.
Predicted decrease in free CGRP from baseline following administration of galcanezumab.
| First dose administration |
Steady state administration |
||
|---|---|---|---|
| Dose (mg) | Maximum effect (%) | Effect at trough (%) | Average effect (%) |
| 5 | 39 | 0 | 6 |
| 50 | 86 | 12 | 39 |
| 120 | 94 | 41 | 61 |
| 240 | 97 | 64 | 76 |
| 240/120* | 97 | 64 | 61 |
| 300 | 97 | 70 | 80 |
Loading dose of 240 mg followed by 120 mg monthly.
Clinical perspectives and inferences
Skljarevski et al. reported on the dose dependency of 5, 50, 120 and 300 mg galcanezumab in a Phase 2 trial for the preventive treatment of migraine (98). In that study, both 120 mg and 300 mg demonstrated a statistically significant difference in the change from baseline in the number of migraine headache days compared to placebo during the overall 3-month treatment period, but 5 mg and 50 mg did not. The PK/PD simulation herein demonstrated that the average steady-state reduction in free CGRP was only 6% for 5 mg and 39% at 50 mg, but 61% at 120 mg and 80% at 300 mg (Table 2). The overall lack of efficacy over the 3-month treatment phase at 5 mg and 50 mg may be due to the lower effects of galcanezumab on free CGRP. An important consideration for the treatment of patients with migraine is to provide early onset of effect. Our analyses indicate that the reduction of free CGRP at trough (1 month after first galcanezumab administration) was 0% at 5 mg and only 12% at 50 mg, suggesting that these doses do not sustain appreciable target binding throughout the initial dosing interval. As such, initial doses greater than 120 mg may be preferable for achieving and sustaining higher target engagement.
In subsequent Phase 3 trials in patients with episodic (76) or chronic (100) migraine, galcanezumab was administered at doses of 240 mg monthly and 120 mg monthly with an initial loading dose of 240 mg (240 mg/120 mg). Both dose regimens had a similar and statistically significant difference in the overall change from baseline in the number of migraine headache days compared to placebo during the overall 6-month treatment period, the primary endpoint. Our modeling analysis indicates that the average steady state decrease in free CGRP at 120 mg and 240 mg monthly was 61% and 76%, respectively, thus informing the degree of target engagement that was associated with efficacy. Furthermore, a 240-mg loading dose achieved a near-maximum reduction of free CGRP (97%) within the first day and continued to suppress free CGRP at trough (1 month after first galcanezumab administration) by 64%. A 240-mg loading dose provides a 23% greater decrease of free CGRP at trough compared to 120 mg (41%; Table 2). These data show that the 240-mg loading dose achieves by 1 month the same degree of target engagement that was observed at steady state and associated with clinical efficacy.
Summary
The PK and PD properties of an antibody are complex and differ from small molecule drugs. To understand how antibodies can affect the extent and duration of free ligand concentrations, the dose and PK of the antibody, and the kinetics of the ligand and the antibody–ligand complex, are important factors to consider. The PK/PD model developed for galcanezumab is a tool that serves as a mechanistic explanation based on free CGRP for galcanezumab therapeutic effects and was used to help justify dose regimens in clinical efficacy trials.
Article highlights
The antibodies for the preventive treatment of migraine being developed and on the market are of the IgG isotype, having consistency in structure, being mAb preparations, and either fully human or humanized. Antibodies are administered parenterally because absorption by oral administration is limited by GI degradation and inefficient diffusion or convection through the epithelium. Absorption of antibodies following SC administration involves convection and subsequent uptake into the lymphatic system, usually leading to a slower absorption and a delayed time to maximum antibody serum concentrations compared to small molecules administered orally.
The majority of antibody elimination occurs via intracellular catabolism into peptides and amino acids following endocytosis. The large size of an antibody (150 kDa) prevents efficient filtration through the glomerulus of the kidney and many organs can contribute to antibody catabolism; therefore, major influences on antibody PK are unlikely in patients with renal or hepatic impairment. Production of ADA may increase or decrease the clearance of the antibody from the body or inhibit the ligand binding to the antibody, thus affecting efficacy. Comparison of incidence of ADA across products may be misleading because immunogenicity results are highly dependent on the assay methodology used.
Antibody drugs that bind soluble ligands make up a large proportion of antibody therapeutics. The premise is that binding of ligand to the antibody reduces the free ligand that is available to interact with the receptor and efficacy is driven by the magnitude and duration of the reduction in free ligand concentration. A galcanezumab PK/PD model was developed to provide insights regarding the effects of galcanezumab serum concentrations on total and free CGRP concentrations. The model provides evidence for a mechanistic linkage to galcanezumab therapeutic effects for the preventive treatment of migraine.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors are employees of Eli Lilly, which has developed galcanezumab.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was sponsored and funded by Eli Lilly and Company, Indianapolis, IN, USA.
References
- 1.Tepper SJ. History and review of anti-calcitonin gene-related peptide (CGRP) therapies: From translational research to treatment. Headache 2018; 58: 238–275. [DOI] [PubMed] [Google Scholar]
- 2.Brain SD, Williams TJ, Tippins JR, et al. Calcitonin gene-related peptide is a potent vasodilator. Nature 1985; 313: 54–56. [DOI] [PubMed] [Google Scholar]
- 3.McCarthy PW, Lawson SN. Cell type and conduction velocity of rat primary sensory neurons with calcitonin gene-related peptide-like immunoreactivity. Neuroscience 1990; 34: 623–632. [DOI] [PubMed] [Google Scholar]
- 4.Steenbergh PH, Hoppener JW, Zandberg J, et al. A second human calcitonin/CGRP gene. FEBS Lett 1985; 183: 403–407. [DOI] [PubMed] [Google Scholar]
- 5.Mulderry PK, Ghatei MA, Spokes RA, et al. Differential expression of alpha-CGRP and beta-CGRP by primary sensory neurons and enteric autonomic neurons of the rat. Neuroscience 1988; 25: 195–205. [DOI] [PubMed] [Google Scholar]
- 6.Fusayasu E, Kowa H, Takeshima T, et al. Increased plasma substance P and CGRP levels, and high ACE activity in migraineurs during headache-free periods. Pain 2007; 128: 209–214. [DOI] [PubMed] [Google Scholar]
- 7.Sarchielli P, Alberti A, Codini M, et al. Nitric oxide metabolites, prostaglandins and trigeminal vasoactive peptides in internal jugular vein blood during spontaneous migraine attacks. Cephalalgia 2000; 20: 907–918. [DOI] [PubMed] [Google Scholar]
- 8.Juhasz G, Zsombok T, Modos EA, et al. NO-induced migraine attack: Strong increase in plasma calcitonin gene-related peptide (CGRP) concentration and negative correlation with platelet serotonin release. Pain 2003; 106: 461–470. [DOI] [PubMed] [Google Scholar]
- 9.Kraenzlin ME, Ch'ng JL, Mulderry PK, et al. Infusion of a novel peptide, calcitonin gene-related peptide (CGRP) in man. Pharmacokinetics and effects on gastric acid secretion and on gastrointestinal hormones. Regul Pept 1985; 10: 189–197. [DOI] [PubMed] [Google Scholar]
- 10.Eli Lilly and Company. Emgality: Highlights of prescribing information, https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761063s000lbl.pdf (2018, accessed 29 November 2018).
- 11.Teva Pharmaceuticals. AJOVY: Highlights of prescribing information, https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761089s000lbl.pdf (2018, accessed 29 November 2018).
- 12.Giamberardino MA, Affaitati G, Curto M, et al. Anti-CGRP monoclonal antibodies in migraine: Current perspectives. Intern Emerg Med 2016; 11: 1045–1057. [DOI] [PubMed] [Google Scholar]
- 13.Mitsikostas DD, Reuter U. Calcitonin gene-related peptide monoclonal antibodies for migraine prevention: Comparisons across randomized controlled studies. Curr Opin Neurol 2017; 30: 272–280. [DOI] [PubMed] [Google Scholar]
- 14.Raffaelli B, Reuter U. The biology of monoclonal antibodies: Focus on calcitonin gene-related peptide for prophylactic migraine therapy. Neurotherapeutics 2018; 15: 324–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amgen and Novartis. Aimovig: Highlights of prescribing information, https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761077s001lbl.pdf (2019, accessed 15 March 2019).
- 16.Mould DR, Sweeney KR. The pharmacokinetics and pharmacodynamics of monoclonal antibodies – mechanistic modeling applied to drug development. Curr Opin Drug Discov Devel 2007; 10: 84–96. [PubMed] [Google Scholar]
- 17.Fetterly GJ, Aras U, Meholick PD, et al. Utilizing pharmacokinetics/pharmacodynamics modeling to simultaneously examine free CCL2, total CCL2 and carlumab (CNTO 888) concentration time data. J Clin Pharmacol 2013; 53: 1020–1027. [DOI] [PubMed] [Google Scholar]
- 18.Wang W, Wang X, Doddareddy R, et al. Mechanistic pharmacokinetic/target engagement/pharmacodynamic (PK/TE/PD) modeling in deciphering interplay between a monoclonal antibody and its soluble target in cynomolgus monkeys. AAPS J 2014; 16: 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiao JJ, Krzyzanski W, Wang YM, et al. Pharmacokinetics of anti-hepcidin monoclonal antibody Ab 12B9m and hepcidin in cynomolgus monkeys. AAPS J 2010; 12: 646–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frazer JK, Capra JD. Immunoglobulins: Structure and function. In: Paul WE. (eds). Fundamental Immunology, 4th edn Philadelphia, PA: Lippincott-Raven, 1999, pp. 37–74. . [Google Scholar]
- 21.Llewelyn MB, Hawkins RE, Russell SJ. Discovery of antibodies. BMJ 1992; 305: 1269–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975; 256: 495–497. [DOI] [PubMed] [Google Scholar]
- 23.Junghans RP. Finally! The Brambell receptor (FcRB). Mediator of transmission of immunity and protection from catabolism for IgG. Immunol Res 1997; 16: 29–57. [DOI] [PubMed] [Google Scholar]
- 24.Waldmann TA, Jones EA. The role of cell-surface receptors in the transport and catabolism of immunoglobulins. Ciba Found Symp 1972; 9: 5–23. [DOI] [PubMed] [Google Scholar]
- 25.Zhu X, Meng G, Dickinson BL, et al. MHC class I-related neonatal Fc receptor for IgG is functionally expressed in monocytes, intestinal macrophages, and dendritic cells. J Immunol 2001; 166: 3266–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aarli A, Matre R, Thunold S. IgG Fc receptors on epithelial cells of distal tubuli and on endothelial cells in human kidney. Int Arch Allergy Appl Immunol 1991; 95: 64–69. [DOI] [PubMed] [Google Scholar]
- 27.Spiekermann GM, Finn PW, Ward ES, et al. Receptor-mediated immunoglobulin G transport across mucosal barriers in adult life: Functional expression of FcRn in the mammalian lung. J Exp Med 2002; 196: 303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blum PM, Phelps DL, Ank BJ, et al. Survival of oral human immune serum globulin in the gastrointestinal tract of low birth weight infants. Pediatr Res 1981; 15: 1256–1260. [DOI] [PubMed] [Google Scholar]
- 29.Covell DG, Barbet J, Holton OD, et al. Pharmacokinetics of monoclonal immunoglobulin G1, F(ab′)2, and Fab′ in mice. Cancer Res 1986; 46: 3969–3978. [PubMed] [Google Scholar]
- 30.Baxter LT, Zhu H, Mackensen DG, et al. Physiologically based pharmacokinetic model for specific and nonspecific monoclonal antibodies and fragments in normal tissues and human tumor xenografts in nude mice. Cancer Res 1994; 54: 1517–1528. [PubMed] [Google Scholar]
- 31.Flessner MF, Lofthouse J, Zakaria el R. In vivo diffusion of immunoglobulin G in muscle: Effects of binding, solute exclusion, and lymphatic removal. Am J Physiol 1997; 273: H2783–H2793. [DOI] [PubMed] [Google Scholar]
- 32.Baker B, Hodsman P and Smith J. PK & PD supporting a single dose, placebo-controlled randomized ascending dose study of ALD403, a humanized anti-calcitonin gene-related peptide (CGRP) monoclonal antibody administered IV or SC, https://www.alderbio.com/wp-content/uploads/2014/04/ALD403-IHC-Poster-Baker-Smith-29-April-2015-for-Jim.pdf (2015, accessed 5 December 2018).
- 33.Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci 2004; 93: 2645–2668. [DOI] [PubMed] [Google Scholar]
- 34.Kuester K, Kloft C. Pharmacokinetics of monoclonal antibodies. In: Meibohm B. (eds). Pharmacokinetics and pharmacodynamics of biotech drugs: Principles and case studies in drug development, Weinheim, Germany: Wiley-VCH, 2006, pp. 45–92. . [Google Scholar]
- 35.Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther 2008; 84: 548–558. [DOI] [PubMed] [Google Scholar]
- 36.Juweid M, Neumann R, Paik C, et al. Micropharmacology of monoclonal antibodies in solid tumors: Direct experimental evidence for a binding site barrier. Cancer Res 1992; 52: 5144–5153. [PubMed] [Google Scholar]
- 37.Blumenthal RD, Fand I, Sharkey RM, et al. The effect of antibody protein dose on the uniformity of tumor distribution of radioantibodies: An autoradiographic study. Cancer Immunol Immunother 1991; 33: 351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Langmuir VK, Mendonca HL, Woo DV. Comparisons between two monoclonal antibodies that bind to the same antigen but have differing affinities: Uptake kinetics and 125I-antibody therapy efficacy in multicell spheroids. Cancer Res 1992; 52: 4728–4734. [PubMed] [Google Scholar]
- 39.Adams GP, Schier R, McCall AM, et al. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res 2001; 61: 4750–4755. [PubMed] [Google Scholar]
- 40.Garg A, Balthasar JP. Investigation of the influence of FcRn on the distribution of IgG to the brain. AAPS J 2009; 11: 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Pardridge WM. Mediated efflux of IgG molecules from brain to blood across the blood-brain barrier. J Neuroimmunol 2001; 114: 168–172. [DOI] [PubMed] [Google Scholar]
- 42.Abuqayyas L, Balthasar JP. Investigation of the role of FcgammaR and FcRn in mAb distribution to the brain. Mol Pharm 2013; 10: 1505–1513. [DOI] [PubMed] [Google Scholar]
- 43.Johnson MP, Ellis BB, Maren DL, et al. Peripheral and central nervous system distribution of the CGRP neutralizing antibody 125I-LY2951742 in male rats. Neurology 2016; 86: S26.007. [DOI] [PubMed]
- 44.Yu YJ, Watts RJ. Developing therapeutic antibodies for neurodegenerative disease. Neurotherapeutics 2013; 10: 459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finke JM, Banks WA. Modulators of IgG penetration through the blood-brain barrier: Implications for Alzheimer's disease immunotherapy. Hum Antibodies 2017; 25: 131–146. [DOI] [PubMed] [Google Scholar]
- 46.Webster CI, Caram-Salas N, Haqqani AS, et al. Brain penetration, target engagement, and disposition of the blood-brain barrier-crossing bispecific antibody antagonist of metabotropic glutamate receptor type 1. FASEB J 2016; 30: 1927–1940. [DOI] [PubMed] [Google Scholar]
- 47.Hougaard A, Amin FM, Christensen CE, et al. Increased brainstem perfusion, but no blood-brain barrier disruption, during attacks of migraine with aura. Brain 2017; 140: 1633–1642. [DOI] [PubMed] [Google Scholar]
- 48.Schankin CJ, Maniyar FH, Seo Y, et al. Ictal lack of binding to brain parenchyma suggests integrity of the blood-brain barrier for 11C-dihydroergotamine during glyceryl trinitrate-induced migraine. Brain 2016; 139: 1994–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wohlrab J. Pharmacokinetic characteristics of therapeutic antibodies. J Dtsch Dermatol Ges 2015; 13: 530–534. [DOI] [PubMed] [Google Scholar]
- 50.Ferri N, Bellosta S, Baldessin L, et al. Pharmacokinetics interactions of monoclonal antibodies. Pharmacol Res 2016; 111: 592–599. [DOI] [PubMed] [Google Scholar]
- 51.Keizer RJ, Huitema AD, Schellens JH, et al. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet 2010; 49: 493–507. [DOI] [PubMed] [Google Scholar]
- 52.Brambell FW. The transmission of immunity from mother to young and the catabolism of immunoglobulins. Lancet 1966; 2: 1087–1093. [DOI] [PubMed] [Google Scholar]
- 53.Ober RJ, Martinez C, Vaccaro C, et al. Visualizing the site and dynamics of IgG salvage by the MHC class I-related receptor, FcRn. J Immunol 2004; 172: 2021–2029. [DOI] [PubMed] [Google Scholar]
- 54.Tabrizi MA, Tseng CM, Roskos LK. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov Today 2006; 11: 81–88. [DOI] [PubMed] [Google Scholar]
- 55.Mahmood I, Green MD. Pharmacokinetic and pharmacodynamic considerations in the development of therapeutic proteins. Clin Pharmacokinet 2005; 44: 331–347. [DOI] [PubMed] [Google Scholar]
- 56.Mager DE. Target-mediated drug disposition and dynamics. Biochem Pharmacol 2006; 72: 1–10. [DOI] [PubMed] [Google Scholar]
- 57.Vu T, Ma P, Chen JS, et al. Pharmacokinetic-pharmacodynamic relationship of erenumab (AMG 334) and capsaicin-induced dermal blood flow in healthy and migraine subjects. Pharm Res 2017; 34: 1784–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marathe A, Peterson MC, Mager DE. Integrated cellular bone homeostasis model for denosumab pharmacodynamics in multiple myeloma patients. J Pharmacol Exp Ther 2008; 326: 555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tabrizi M, Raskos LK. Exposure-response relationships for therapeutic biologic products. In: Meibohm B. (eds). Pharmacokinetics and pharmacodynamics of biotech drugs: principles and case studies in drug development, Weinheim, Germany: Wiley-VCH, 2006, pp. 295–330. [Google Scholar]
- 60.Kuang B, King L, Wang HF. Therapeutic monoclonal antibody concentration monitoring: free or total? Bioanalysis 2010; 2: 1125–1140. [DOI] [PubMed] [Google Scholar]
- 61.Meibohm B, Zhou H. Characterizing the impact of renal impairment on the clinical pharmacology of biologics. J Clin Pharmacol 2012; 52: 54S–62S. [DOI] [PubMed] [Google Scholar]
- 62.Mould DR, Meibohm B. Drug development of therapeutic monoclonal antibodies. BioDrugs 2016; 30: 275–293. [DOI] [PubMed] [Google Scholar]
- 63.Roberts BV, Susano I, Gipson DS, et al. Contribution of renal and non-renal clearance on increased total clearance of adalimumab in glomerular disease. J Clin Pharmacol 2013; 53: 919–924. [DOI] [PubMed] [Google Scholar]
- 64.Struemper H, Chen C, Cai W. Population pharmacokinetics of belimumab following intravenous administration in patients with systemic lupus erythematosus. J Clin Pharmacol 2013; 53: 711–720. [DOI] [PubMed] [Google Scholar]
- 65.Zhang Y, Yao Z, Kaila N, et al. Pharmacokinetics of ranibizumab after intravitreal administration in patients with retinal vein occlusion or diabetic macular edema. Ophthalmology 2014; 121: 2237–2246. [DOI] [PubMed] [Google Scholar]
- 66.Glund S, Stangier J, van Ryn J, et al. Effect of age and renal function on idarucizumab pharmacokinetics and idarucizumab-mediated reversal of dabigatran anticoagulant activity in a randomized, double-blind, crossover phase Ib study. Clin Pharmacokinet 2017; 56: 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Berdeja J, Jagannath S, Zonder J, et al. Pharmacokinetics and safety of elotuzumab combined with lenalidomide and dexamethasone in patients with multiple myeloma and various levels of renal impairment: Results of a phase Ib study. Clin Lymphoma Myeloma Leuk 2016; 16: 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hwang WY, Foote J. Immunogenicity of engineered antibodies. Methods 2005; 36: 3–10. [DOI] [PubMed] [Google Scholar]
- 69.Moss AC, Brinks V, Carpenter JF. Review article: Immunogenicity of anti-TNF biologics in IBD – the role of patient, product and prescriber factors. Aliment Pharmacol Ther 2013; 38: 1188–1197. [DOI] [PubMed] [Google Scholar]
- 70.Shankar G, Shores E, Wagner C, et al. Scientific and regulatory considerations on the immunogenicity of biologics. Trends Biotechnol 2006; 24: 274–280. [DOI] [PubMed] [Google Scholar]
- 71.Shankar G, Arkin S, Cocea L, et al. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptides-harmonized terminology and tactical recommendations. AAPS J 2014; 16: 658–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schellekens H. Factors influencing the immunogenicity of therapeutic proteins. Nephrol Dial Transplant 2005; 20(Suppl 6): vi3–9. . [DOI] [PubMed] [Google Scholar]
- 73.Xu Z, Davis HM, Zhou H. Rational development and utilization of antibody-based therapeutic proteins in pediatrics. Pharmacol Ther 2013; 137: 225–247. [DOI] [PubMed] [Google Scholar]
- 74.Chen X, Hickling T, Kraynov E, et al. A mathematical model of the effect of immunogenicity on therapeutic protein pharmacokinetics. AAPS J 2013; 15: 1141–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kimball JA, Norman DJ, Shield CF, et al. The OKT3 Antibody Response Study: A multicentre study of human anti-mouse antibody (HAMA) production following OKT3 use in solid organ transplantation. Transpl Immunol 1995; 3: 212–221. [DOI] [PubMed] [Google Scholar]
- 76.Stauffer VL, Dodick DW, Zhang Q, et al. Evaluation of galcanezumab for the prevention of episodic migraine: The EVOLVE-1 randomized clinical trial. JAMA Neurol 2018; 75: 1080–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Skljarevski V, Matharu M, Millen BA, et al. Efficacy and safety of galcanezumab for the prevention of episodic migraine: Results of the EVOLVE-2 Phase 3 randomized controlled clinical trial. Cephalalgia 2018; 38: 1442–1454. [DOI] [PubMed] [Google Scholar]
- 78.Vargas B, Starling A, Silberstein S, et al. Erenumab immunogenicity: A pooled analysis of Phase 2 and Phase 3 migraine prevention clinical trials (P4.098). Neurology 2018; 90: P4.098. . [Google Scholar]
- 79.Cohen-Barak O, Weiss S, Rasamoelisolo M, et al. A phase 1 study to assess the pharmacokinetics, safety, and tolerability of fremanezumab doses (225 mg, 675 mg and 900 mg) in Japanese and Caucasian healthy subjects. Cephalalgia 2018; 38: 1960–1971. [DOI] [PubMed] [Google Scholar]
- 80.Sun YN, Lu JF, Joshi A, et al. Population pharmacokinetics of efalizumab (humanized monoclonal anti-CD11a antibody) following long-term subcutaneous weekly dosing in psoriasis subjects. J Clin Pharmacol 2005; 45: 468–476. [DOI] [PubMed] [Google Scholar]
- 81.Ma P, Yang BB, Wang YM, et al. Population pharmacokinetic analysis of panitumumab in patients with advanced solid tumors. J Clin Pharmacol 2009; 49: 1142–1156. [DOI] [PubMed] [Google Scholar]
- 82.Muralidharan KK, Kuesters G, Plavina T, et al. Population pharmacokinetics and target engagement of natalizumab in patients with multiple sclerosis. J Clin Pharmacol 2017; 57: 1017–1030. [DOI] [PubMed] [Google Scholar]
- 83.Long A, Chigutsa E, Wallin J. Population pharmacokinetics of necitumumab in cancer patients. Clin Pharmacokinet 2017; 56: 505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang Y, Wei X, Bajaj G, et al. Challenges and considerations for development of therapeutic proteins in pediatric patients. J Clin Pharmacol 2015; 55: S103–S115. [DOI] [PubMed] [Google Scholar]
- 85.Edlund H, Melin J, Parra-Guillen ZP, et al. Pharmacokinetics and pharmacokinetic-pharmacodynamic relationships of monoclonal antibodies in children. Clin Pharmacokinet 2015; 54: 35–80. [DOI] [PubMed] [Google Scholar]
- 86.Boxenbaum H. Interspecies scaling, allometry, physiological time, and the ground plan of pharmacokinetics. J Pharmacokinet Biopharm 1982; 10: 201–227. [DOI] [PubMed] [Google Scholar]
- 87.Lowe PJ, Georgiou P, Canvin J. Revision of omalizumab dosing table for dosing every 4 instead of 2 weeks for specific ranges of bodyweight and baseline IgE. Regul Toxicol Pharmacol 2015; 71: 68–77. [DOI] [PubMed] [Google Scholar]
- 88.Lee JW, Kelley M, King LE, et al. Bioanalytical approaches to quantify “total” and “free” therapeutic antibodies and their targets: Technical challenges and PK/PD applications over the course of drug development. AAPS J 2011; 13: 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chames P, Van Regenmortel M, Weiss E, et al. Therapeutic antibodies: Successes, limitations and hopes for the future. Br J Pharmacol 2009; 157: 220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zheng S, McIntosh T, Wang W. Utility of free and total target measurements as target engagement and efficacy biomarkers in biotherapeutic development – opportunities and challenges. J Clin Pharmacol 2015; 55: S75–S84. [DOI] [PubMed] [Google Scholar]
- 91.Hayashi N, Tsukamoto Y, Sallas WM, et al. A mechanism-based binding model for the population pharmacokinetics and pharmacodynamics of omalizumab. Br J Clin Pharmacol 2007; 63: 548–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lowe PJ, Tannenbaum S, Gautier A, et al. Relationship between omalizumab pharmacokinetics, IgE pharmacodynamics and symptoms in patients with severe persistent allergic (IgE-mediated) asthma. Br J Clin Pharmacol 2009; 68: 61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tabrizi M, Bornstein GG, Suria H. Biodistribution mechanisms of therapeutic monoclonal antibodies in health and disease. AAPS J 2010; 12: 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Goadsby PJ, Edvinsson L. The trigeminovascular system and migraine: Studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol 1993; 33: 48–56. [DOI] [PubMed] [Google Scholar]
- 95.Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol 1990; 28: 183–187. [DOI] [PubMed] [Google Scholar]
- 96.Ho TW, Edvinsson L, Goadsby PJ. CGRP and its receptors provide new insights into migraine pathophysiology. Nat Rev Neurol 2010; 6: 573–582. [DOI] [PubMed] [Google Scholar]
- 97.Cernuda-Morollon E, Larrosa D, Ramon C, et al. Interictal increase of CGRP levels in peripheral blood as a biomarker for chronic migraine. Neurology 2013; 81: 1191–1196. [DOI] [PubMed] [Google Scholar]
- 98.Skljarevski V, Oakes TM, Zhang Q, et al. Effect of different doses of galcanezumab vs placebo for episodic migraine prevention: A randomized clinical trial. JAMA Neurol 2018; 75: 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Oakes TMM, Skljarevski V, Zhang Q, et al. Safety of galcanezumab in patients with episodic migraine: A randomized placebo-controlled dose-ranging Phase 2b study. Cephalalgia 2018; 38: 1015–1025. [DOI] [PubMed] [Google Scholar]
- 100.Detke HC, Goadsby PJ, Wang S, et al. Galcanezumab in chronic migraine: The randomized, double-blind, placebo-controlled REGAIN study. Neurology 2018; 90: e2211–e2221. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Martinez JM, Goadsby P, Dodick D, et al. Study CGAL: A Phase 3 placebo-controlled study of galcanezumab in patients with episodic cluster headache: Results from the 8-week double-blind treatment phase. Presented at: 60th Annual Scientific Meeting American Headache Society, San Fransisco, CA, USA, 28 June–1 July 2018; Wiley, pp.1287–1337.
- 102.Kielbasa W, Williams D, Coutant D, et al. Tolerability, pharmacokinetics, and pharmacodynamics of galcanezumab in healthy subjects following a subcutaneous administration of a lyophilized formulation or a solution formulation. Presented at: American Headache Society, Annual Scientific Meeting, Boston, MA, USA, 8–11 June 2017.
- 103.Davda JP, Hansen RJ. Properties of a general PK/PD model of antibody-ligand interactions for therapeutic antibodies that bind to soluble endogenous targets. MAbs 2010; 2: 576–588. [DOI] [PMC free article] [PubMed] [Google Scholar]