

Summary:
Pyroptosis is an inflammatory cell death program initiated by supramolecular organizing centers known as inflammasomes. In a recent issue of Science, Rühl et al. challenge the paradigm that inflammasome signaling necessitates pyroptosis by demonstrating ESCRTIII-dependent membrane repair can delay or prevent gasdermin D mediated cell death.
Inflammasome signaling connects the recognition of intracellular perturbations to activation of the inflammatory caspases. A major function of inflammasome signaling and subsequent caspase activation is the cleavage of pro-form IL-1 family cytokines such as IL-1β and IL-18 into their bioactive forms in the cytosol. These cytokines are secreted in an unconventional manner, as they lack N-terminal signal sequences necessary for entry into the vesicle-mediated biosynthetic pathway. Downstream of inflammasome activation, the lytic program of cell death, pyroptosis, has long been a favored mechanism of bioactive IL-1 family release into the extracellular space where these cytokines can signal through their respective receptors. In addition to cleaving IL-1 family cytokines, active inflammatory caspases cleave the latent cytosolic protein gasdermin D (GSDMD) (He et al., 2015; Kayagaki et al., 2015; Shi et al., 2015a). Cleavage of GSDMD releases the pore-forming N terminal fragment from the autoinhibitory C terminal fragment, allowing the N terminus of GSDMD to oligomerize and perforate the plasma membrane (Aglietti et al., 2016; Ding et al., 2016; Liu et al., 2016; Sborgi et al., 2016). These pores mediate IL-1 secretion and cell lysis (Evavold et al., 2017; Heilig et al., 2017; Kayagaki et al., 2015; Shi et al., 2015a). In a recent issue of Science, Rühl et al. reveal that a calcium-dependent membrane repair response, mediated by ESCRTIII components, antagonizes the execution phase of pyroptosis (Rühl et al., 2018). This ESCRTIII-mediated repair likely maintains membrane integrity by removal of GSDMD pores and thus prevents ultimate osmotic lysis of highly perforated cells during pyroptosis (Figure 1).
Figure 1: GSDMD pores activate ESCRT-III dependent membrane repair.
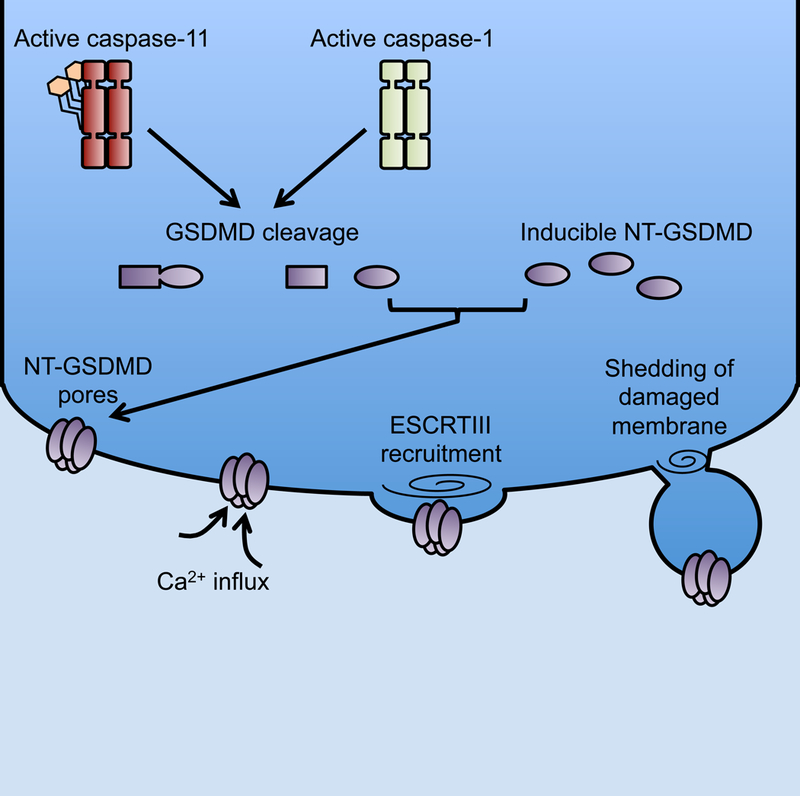
Active inflammatory caspases cleave GSDMD into N and C terminal fragments. The N terminal fragment (NT-GSDMD) binds the inner leaflet of the plasma membrane and oligomerizes into a large pore. NT-GSDMD expression allows for the flux of calcium (Ca2+) ions likely from the hypercalcemic extracellular space into the hypocalcemic cytosol. Ca2+ flux serves as a signal to recruit ESCRT-III machinery to the site of membrane damage. ESCRT-III filament oligomerization allows for shedding of damaged membrane through bleb formation.
One mechanism of inflammasome activation occurs upon recognition of bacterial lipopolysaccharide (LPS) in the cytosol (Kayagaki et al., 2011; 2013; Shi et al., 2015b). The CARD domain of caspase-11 binds LPS leading to the activation of its latent enzymatic activity (Shi et al., 2015b). Active caspase-11 then cleaves GSDMD resulting in plasma membrane perforation (Kayagaki et al., 2015; Shi et al., 2015a). Previous work illustrated that caspase-11 dependent potassium efflux occurs prior to assembly of the NLRP3 inflammasome (Rühl and Broz, 2015). Caspase-11 activation cleaves GSDMD after cytosolic delivery of LPS, and activation of the NLRP3 inflammasome under these conditions requires cleavage of GSDMD (Kayagaki et al., 2015; Shi et al., 2015a). Based on these combined data, it is likely that GSDMD mediated potassium efflux is placed upstream of NLRP3 and caspase-1 dependent IL-1β cleavage. As premature rupture of the plasma membrane would result in the release of primarily pro-form IL-1 family members, the skewed presence of cleaved IL-1β in culture supernatants invokes a model whereby GSDMD is not instantaneously lytic. Mechanisms that may prevent cell lysis during the critical time of NLRP3 inflammasome mediated cleavage of IL-1β have not been identified until now (Rühl et al., 2018).
Rühl et al. illustrate that in addition to potassium efflux, cells flux calcium in a GSDMD and caspase-11 dependent manner in response to intracellular delivery of bacterial LPS using primary and immortalized bone marrow derived macrophages. Careful chelation of calcium through co-treatment of fast acting BAPTA-AM and a drug export inhibitor was sufficient to increase cell lysis after LPS transfection in macrophages. These results suggest calcium dependent processes antagonize cell death after caspase-11 activation. Calcium flux is an evolutionarily conserved trigger for plasma membrane repair through exocytosis of vesicles such as lysosomes, mobilization of annexins, and recruitment of ESCRT machinery to sites of membrane injury (Andrews et al., 2014; Cooper and McNeil, 2015). ESCRTIII machinery has been implicated in the membrane repair response to light induced injury, bacterial pore forming toxins, and the host-derived MLKL pore (Gong et al., 2017b; Jimenez et al., 2014). Consistent with data on other membrane damaging agents, Rühl et al. demonstrate recruitment of the ESCRT protein CHMP4 to the plasma membrane after expression of pore forming N terminal fragment of GSDMD. This recruitment was impaired in the presence of calcium chelators, reinforcing a role for GSDMD mediated calcium flux in triggering repair of these pores. In the necroptosis pathway, which is a caspase-independent process of lytic cell death, the pores formed by MLKL mediate membrane permeabilization and phosphatidylserine (PS) externalization prior to full rupture of the plasma membrane (Gong et al., 2017b; 2017a). Rühl et al also notice membrane permeabilization and PS externalization prior to cell lysis through monitoring calcium flux with Fluo-8, incorporation of the normally membrane impermeable DNA intercalating dye propidium iodide, and surface staining with the PS binding protein annexin V.
To solidify their claims that ESRCTIII machinery negatively regulates the pyroptotic cell fate, the authors characterize cells that overexpress the ESCRT associated protein VPS4A or a dominant negative VPS4A with the single amino acid change E228Q. One expectation would be that wild-type VPS4A provides a survival benefit to the cell by increasing reparative capacity, whereas the dominant negative variant will inhibit membrane repair responses (Jimenez et al., 2014). Directly comparing these two altered cell states after LPS transfection and caspase-11 activation, the authors report an increase in membrane permeability and cell lysis in VPS4A E228Q cells. This increased permeability and cell lysis proved independent of the NLPR3 inflammasome. As deficiency in ESCRTIII machinery initiates spontaneous necroptotic cell death (Gong et al., 2017a; 2017b), the authors performed knockdown experiments in the context of RIPK3 deficient immortalized macrophages that are genetically unable to undergo necroptosis. Knockdown of several known ESCRTIII proteins with siRNA transfection also results in increased cell lysis after activation of caspase-11.
Intriguingly, ESCRTIII-dependent membrane repair negatively regulates cell lysis and inflammasome-mediated cytokine release during Salmonella typhimurium infection. In addition, synthetic caspase-1 activation by a chemically dimerizable caspase-1 variant in HEK293T cells revealed similar cellular responses. These results suggest that GSDMD-dependent membrane permeability is a conserved signal among the inflammasome pathways that recruit ESCRTIII machinery to the plasma membrane for the purpose of delaying or preventing lysis.
There are two implications of these findings. First, this study adds GSDMD pores to the list of membrane disruptions that can be repaired by the ESCRTIII machinery. The common use of ESCRTIII in membrane repair processes is explained by the common use of calcium fluxes as a signal that initiates these events. A major question that arises from these studies is whether other macrophage proteins regulate GSDMD pore residence in the plasma membrane. Whereas ESCRTIII controls the removal of these pores, a host protein that promotes GSDMD pore insertion in the membrane is yet to be defined. Perhaps the process of membrane insertion and removal will emerge as a new regulatory step in the path to pyroptosis. The second implication of these findings relates to several recent studies demonstrating that pyroptosis is not a necessary consequence of inflammasome signaling (Chen et al., 2014; Gaidt et al., 2016; Wolf et al., 2016; Zanoni et al., 2016; 2017). Indeed, macrophages, dendritic cells and neutrophils can utilize inflammasome-dependent processes to activate GSDMD pore formation and remain viable (Evavold et al., 2017; Heilig et al., 2017). Under these conditions, the pores formed at the plasma membrane do not promote pyroptosis, but rather promote the secretion of IL-1 through these pores. The ability of living cells to add IL-1 to the repertoire of cytokines they secrete is notable, as this cytokine has been considered to only be released upon cell lysis. Consequently, cells that secrete cytokines from the cytosol and vesicle mediated secretory pathway are considered to have achieved a hyperactive state, which coincides with an enhanced ability to stimulate adaptive immunity (Sanchez et al., 2017; Zanoni et al., 2016). The mechanisms that permit GSDMD to either promote pyroptosis or cell hyperactivation are unclear, but hyperactive cells display evidence of lower amounts of GSDMD pores than pyroptotic cells (Evavold et al., 2017).
One hypothesis based on this recent report is that hyperactive cells are able to survive GSDMD pores at the plasma membrane through rapid removal by ESCRTIII membrane shedding. A prediction based on this hypothesis would be that inhibition of membrane repair proteins, such as the ESCRTIII machinery, or chelation of calcium would convert hyperactive cells into pyroptotic cells due to increased plasma membrane occupancy of lytic GSDMD pores leading to osmotic lysis. Moreover, if ESCRTIII mediated membrane repair is a common response to host-derived GSDMD pores, one would expect that cell free culture supernatants would contain GSDMD, as has been reported (Liu et al., 2016). According to the known role of ESCRTIII proteins at the plasma membrane in the budding of viral particles and extracellular vesicles (Jimenez et al., 2014; Van Engelenburg et al., 2014), this pool of GSDMD is likely associated with shed plasma membrane.
References:
- Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, and Dueber EC (2016). GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. U.S.a 113, 7858–7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews NW, Almeida PE, and Corrotte M (2014). Damage control: cellular mechanisms of plasma membrane repair. Trends in Cell Biology 24, 734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KW, Groß CJ, Sotomayor FV, Stacey KJ, Tschopp J, Sweet MJ, and Schroder K (2014). The neutrophil NLRC4 inflammasome selectively promotes IL-1β maturation without pyroptosis during acute Salmonella challenge. CellReports 8, 570–582. [DOI] [PubMed] [Google Scholar]
- Cooper ST, and McNeil PL (2015). Membrane Repair: Mechanisms and Pathophysiology. Physiol. Rev 95, 1205–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang D-C, and Shao F (2016). Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116. [DOI] [PubMed] [Google Scholar]
- Evavold CL, Ruan J, Tan Y, Xia S, Wu H, and Kagan JC (2017). The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity [DOI] [PMC free article] [PubMed]
- Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, Robertson AAB, Cooper MA, Graf T, and Hornung V (2016). Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity 44, 833–846. [DOI] [PubMed] [Google Scholar]
- Gong Y-N, Guy C, Crawford JC, and Green DR (2017a). Biological events and molecular signaling following MLKL activation during necroptosis. Cell Cycle 16, 1748–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y-N, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, Linkermann A, and Green DR (2017b). ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell 169, 286–300.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W-T, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang Z-H, Zhong C-Q, and Han J (2015). Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res 25, 1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, and Broz P (2017). The Gasdermin-D pore acts as a conduit for IL-1β secretion in mice. Eur. J. Immunol [DOI] [PubMed]
- Jimenez AJ, Maiuri P, Lafaurie-Janvore J, Divoux S, Piel M, and Perez F (2014). ESCRT machinery is required for plasma membrane repair. Science 343, 1247136. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszyński A, et al. (2013). Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, and Lieberman J (2016). Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rühl S, and Broz P (2015). Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur. J. Immunol 45, 2927–2936. [DOI] [PubMed] [Google Scholar]
- Rühl S, Shkarina K, Demarco B, Heilig R, Santos JC, and Broz P (2018). ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362, 956–960. [DOI] [PubMed] [Google Scholar]
- Sanchez M, Kolar SL, Müller S, Reyes CN, Wolf AJ, Ogawa C, Singhania R, De Carvalho DD, Arditi M, Underhill DM, et al. (2017). O-Acetylation of Peptidoglycan Limits Helper T Cell Priming and Permits Staphylococcus aureus Reinfection. Cell Host and Microbe [DOI] [PMC free article] [PubMed]
- Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, Farady CJ, Müller DJ, Broz P, and Hiller S (2016). GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. Embo J 35, 1766–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F (2015a). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, and Shao F (2015b). Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192. [DOI] [PubMed] [Google Scholar]
- Van Engelenburg SB, Shtengel G, Sengupta P, Waki K, Jarnik M, Ablan SD, Freed EO, Hess HF, and Lippincott-Schwartz J (2014). Distribution of ESCRT machinery at HIV assembly sites reveals virus scaffolding of ESCRT subunits. Science 343, 653–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML, Cho HC, Popescu NI, Coggeshall KM, Arditi M, et al. (2016). Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell 166, 624–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, Donado CA, Shao F, Wu H, Springstead JR, et al. (2016). An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 352, 1232–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoni I, Tan Y, Di Gioia M, Springstead JR, and Kagan JC (2017). By Capturing Inflammatory Lipids Released from Dying Cells, the Receptor CD14 Induces Inflammasome-Dependent Phagocyte Hyperactivation. Immunity 47, 697–709.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]