

Abstract
This method describes the chemoenzymatic synthesis of several nucleotide sugars, which are essential substrates in the biosynthesis of prokaryotic N- and O-linked glycoproteins. Protein glycosylation is now known to be widespread in prokaryotes and proceeds via sequential action of several enzymes, utilizing both common and modified prokaryote-specific sugar nucleotides. The latter, which include UDP-hexoses such as UDP-diNAc-bacillosamine (UDP-diNAcBac), UDP-diNAcAlt and UDP-2,3-diNAcManA, are also important components of other bacterial and archaeal glycoconjugates. The ready availability of these “high-value” intermediates will enable courses of study into inhibitor screening, glycoconjugate biosynthesis pathway discovery, and unnatural carbohydrate incorporation toward metabolic engineering.
Keywords: Chemoenzymatic synthesis; glycoconjugate biosynthesis; nucleotide-activated carbohydrates; 2,4-di-N-acetyl-bacillosamine; pseudaminic acid; 2,3-di-N-acetyl-glucuronic acid
1. Introduction
Prokaryote-specific carbohydrates are important constituents of bacterial and archaeal glycoconjugates. In the past two decades, the accelerated pace of genome sequencing, complemented by bioinformatic analysis, has led to identification of key genes associated with prokaryotic glycoconjugate biosynthesis. Exploiting this extensive resource of information, bioanalytical tools originally developed for eukaryotic glycan analysis (Balonova, Hernychova & Bilkova 2009), including glycan arrays (Hsu & Mahal 2006), high-sensitivity mass spectrometry (Hitchen & Dell 2006; Reid, Stupak, Szymanski & Li 2009; Scott & Cordwell 2015), and high-resolution NMR analysis (Leeflang, Faber, Erbel & Vliegenthart 2000; Young et al. 2002; McCallum, Shaw & Creuzenet 2013) have provided a definition of some of the structures of unusual, highly-modified saccharides, which are embedded within prokaryotic glycans. Selected examples of prokaryotic glycosyl donors and some of the corresponding known bacterial sources are illustrated in Figure 1. Modified sugars are generally integrated into complex glycans using activated glycosyl donors such as UDP-hexoses, including pyranoses like UDP-diNAc-bacillosamine (UDP-diNAcBac) (Morrison & Imperiali 2014; Sharon 2007), furanoses such as UDP-galactofuranose (UDP-Galfur) (Koplin, Brisson & Whitfield 1997), GDP-heptoses such as GDP-6-deoxy-altrose (McCallum, Shaw & Creuzenet 2013), and cytidine monophosphate nonulosonic acids like CMP-pseudaminic acid (CMP-Pse) (Schoenhofen, McNally, Brisson & Logan 2006). In some instances, like with diNAcBac, different nucleotide donors may also be employed, even for the same carbohydrate, which may provide a mechanism for regulation of biosynthetic pathways using common precursor sugars (Schoenhofen, Vinogradov, Whitfield, Brisson & Logan 2009). Some of the many interesting glycosyl donors that are utilized by pathogenic bacteria are illustrated in Figure 1.
Fig. 1.

Structures of selected prokaryote-specific glycosyl donors. Shown are donors from Pseudomonas aeruginosa (P. aeruginosa); Bordatella pertussis, (B. pertussis); Campylobacter jejuni, (C. jejuni); Vibrio cholera, (V. cholera); Mycobacterium tuberculosis, (M. tuberculosis); Klebsiella pneumoniae, (K. pneumonia); Helicobacter pylori, (H. pylori); and Legionella pneumoniae, (L. pneumonia).
Herein we focus on selected carbohydrate components of bacterial and archaeal glycoproteins. However, while the methods presented are discussed in the context of bacterial and archaeal protein glycosylation, these same building blocks are also valuable reagents in the study of other classes of complex glycoconjugates. Protein glycosylation, once believed to be a unique modification of eukaryotic proteins, it is now known to be widespread in prokaryotes (Schmidt, Riley & Benz 2003; Messner 2009; Nothaft & Szymanski 2010) and differentiated, in part, by the inclusion of a rich diversity of highly-derivitized carbohydrates. For example, the N- and O-linked glycoproteins of Gram-negative pathogens Campylobacter jejuni and Neisseria gonorrhoeae feature 2,4-di-N-acetylbacillosamine (diNAcBac) at the reducing end of the transferred glycan. (Glover, Weerapana, Chen & Imperiali 2006) (Hartley et al. 2011) Additionally, highly modified hexoses such as 2,3-diacetamido-glucuronic acid (diNAcGlcA) are represented in N-linked glycans that feature in the S-layer glycoproteins of Methanococcus voltae (Voisin et al. 2005). These modified hexose donors are biosynthesized from common sugar nucleotides such as UDP-GlcNAc via the sequential action of several enzymes. The nonulosonic acid donors, such as CMP-pseudaminic acid, are also assembled from common NDP-sugar precursors via the intermediacy of modified hexosamines following an analogous pathway to sialic acid biosynthesis (Schoenhofen, Vinogradov, Whitfield, Brisson & Logan 2009). Pseudaminic acid and related nonulosonic acids such as legionaminic acid are critical O-linked modifications of the Fla proteins and are essential for flagellar assembly (Schirm et al. 2003) for example in Helicobacter pylori and C. jejuni. The structures of the protein-linked glycans incorporating these unusual NDP-sugar precursors are illustrated in Figure 2.
Fig. 2.
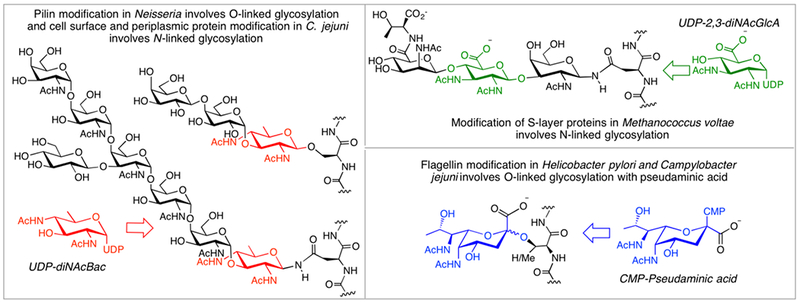
N- and O-linked prokaryotic glycans featuring modified carbohydrate building blocks.
A major impediment to studies on the biological and physical roles of modified carbohydrates in prokaryotic glycoconjugates centers on the availability of chemically-defined carbohydrate reagents. In particular, there is a need for nucleotide-activated sugars that can be used: 1) to assess the activities of specific phosphoglycosyl and glycosyl transferases involved in glycoconjugate assembly, 2) as ligands for biophysical and structural analyses, and 3) as precursors for the assembly of complex glycans and glycoconjugates for detailed biological studies. If readily available in milligram quantities, the nucleotide-activated sugar reagents can be immediately deployed to investigate specificity of enzymes in glycoconjugate biosynthesis pathways, in assays for inhibitor screening and as intermediates that can be advanced to more elaborate glycoconjugates through biosynthetic pathways.
The chemical synthesis of highly-modified carbohydrates is demanding and relies on specialized expertise in carbohydrate chemistry. For example, targets can be prepared via asymmetric synthetic methods or by using common carbohydrates as building blocks from the chiral pool. An additional challenge, with respect to the preparation of useful reagents for studying aspects of glycan assembly, is that the saccharides must be appropriately modified as activated nucleotide derivatives, thus requiring additional synthetic manipulation. This method will focus on prokaryote-specific carbohydrates in glycans in the structures of prokaryotic N- and O-linked glycoproteins, specifically the chemoenzymatic synthesis of biologically-relevant carbohydrate building blocks prepared as the biologically-relevant activated nucleotide derivatives. Applying the alternative chemoenzymatic approach using a combination of biotransformation together with simple chemical steps maximizes efficiency and provides milligram-quantities of chemically-defined nucleotide-activated sugars.
2. Equipment and consumables
Equipment
VWR Scientific 1500E Incubator set to 37 °C
Innova 4330 refrigerated incubator shaker
Beckman Allegra 6R centrifuge
Beckman L-80 Optima Ultracentrifuge
Sonics VibraCell sonicator
Gel casting equipment and electrophoresis chambers for SDS-PAGE
BioRad PowerPac 300 and GelDoc XR+ Imaging System for SDS-PAGE
Shimadzu UV-2401PC UV-Vis Spectrophotometer
Waters 600 Delta HPLC pump with Waters 2487 Dual Absorbance Detector
Äktaprime plus FPLC chromatography system (GE Healthcare Life Sciences)
Labconco Freezone 2.5 lyophilizer
BioTek Synergy H1 Hybrid Reader
P/ACE MDQ capillary electrophoresis system with UV detector (Beckman Coulter)
Consumables
Chromatography/separation: Poly-Prep and Econo-Prep chromatography columns (Bio-Rad); Hi-Trap Q 5 mL columns; Normal-phase (NP) Varian Microsorb HPLC column; Synergi C18 Hydro preparatory RP-HPLC column (4 µM, 80 Å, 250 × 21.2 mm; Phenomenex); YMC-Pack-PVA-SIL-NP NP-HPLC column 250 mm x 4.6 mm (YMC); 6 mL Supelclean ENVI-Carb Solid Phase Extraction (SPE) tubes; Amicon Ultra-15 centrifugal filter units with 5 kDa and 10 kDa MWCO (EMD Millipore); SiliaFlash P60 silica gel (Silicycle) thin-layer chromatography (TLC) plates: 25 × 75 mm, MF254 glass-backed (AgelaTech).
Plasmids: pET-24a(+) (EMD Biosciences)
Cells: BL21-CodonPlus(DE3) RIL competent cells and DH5α competent cells (Agilent)
Plasmid purification kit: Wizard Plus SV Minipreps (Promega)
Resins: Ni-NTA agarose resin (Thermo Scientific), Glutathione-S-transferase resin (GST resin) (Sigma-Alrich)
Stains: ninhydrin, p-anisaldehyde, ceric ammonium molybdate (Sigma-Aldrich)
Substrates and coenzymes: UDP-GlcNAc, NAD+/NADP+, Acetyl Coenzyme A (AcCoA), Pyridoxal phosphate (PLP), L-Glutamic acid (L-Glu) (Sigma-Aldrich)
Reagent and chemicals: Isopropyl β-D-1-thiogalactopyranoside (IPTG); Adams’ catalyst (PtO2); dithiothreitol (DTT) (S)-tol-BINAP RuCl2; bis-(2-cyanoethyl)-N,N-diisopropyl-phosphoramidite; acetic anhydride; silver (I) acetate (all from Sigma-Aldrich); n-dodecyl-β-D-maltoside (DDM) (Anatrace)
Enzymes: lysozyme from chicken egg white (Research Products International Corp.); DNAseI (New England Biolabs)
Antibiotics: Chloramphenicol (CAM) (Sigma-Aldrich), Kanamycin (KAN) (Teknova)
Media: Lysogeny Broth (LB) powder (Teknova)
Buffers: HEPES, triethyl ammonium bicarbonate (TEAB) (Sigma-Aldrich)
Antibodies: T7•Tag® Antibody (EMD Millipore); Tetra·His Antibody (Qiagen)
Dialysis: Slide-a-Lyzer dialysis cassettes (Thermo Scientific), SnakeSkin Dialysis Tubing (34 mm dry flat width, Thermo Scientific)
BCA protein assay (Thermo Fisher)
Protease Inhibitor Cocktail Set III, EDTA-free (Calbiochem)
Radiochemicals: [3H]-AcCoA, [14C]-AcCoA, UDP-[3H-C6]GlcNAc (American Radiolabeled Chemicals, Inc.)
Note: All sugars are D-hexoses unless designated otherwise.
3.1. Discussion
di-N-Acetylbacillosamine (diNAcBac), shown in Figure 3 as an activated uridine diphosphate derivative (Sharon & Jeanloz 1960; Sharon 2007), is a member of a class of 2,4-diacetamido-2,4,6-trideoxyhexoses (DATDHs). DATDHs feature in the N- and O- linked glycoproteins and lipopolysaccharides of many microorganisms (Maki & Renkonen 2004), including the human pathogens N. meningitides (Stimson, Virji, Makepeace, Dell & Morris 1995), N. gonorrhoeae (Hegge et al. 2004; Aas, Vik, Vedde, Koomey & Egge-Jacobsen 2007), P. aeruginosa (Singh, Singh, Hogan & Feingold 1990; Castric, Cassels & Carlson 2001), V. cholera (Chowdhury et al. 1991), and C. jejuni (Young et al. 2002). A UDP-diNAcBac diastereomer with inverted stereochemistry at C4 and C5 (termed UDP-diNAcAlt, see Section 4) is also a key intermediate in the biosynthesis of CMP-Pse, a monosaccharide which is a component of the O-linked glycan that decorates flagellar proteins in H. pylori and features in the lipopolysaccharide (LPS) of P. aeruginosa. Thus, chemoenzymatic synthesis of these and other DATDHs is broadly applicable toward a variety of studies in many bacterial species.
Fig. 3.

Biosynthetic pathway of UDP-diNAcBacillosamine in C. jejuni.
In Campylobacter jejuni, biosynthesis of UDP-diNAcBac (Sharon & Jeanloz 1960; Sharon 2007) glycosyl donors proceeds via three enzymatic steps, shown in Figure 3, starting from the common UDP-GlcNAc precursor. First, membrane-associated enzyme PglF carries out an oxidoreductase-coupled dehydration, followed by the UDP-ketosugar product undergoing pyridoxamine-dependent transamination to the installed ketone at C4 by soluble protein PglE. Finally, an acetyl CoA-dependent acetylation of the C4 amino group by acetyltransferase PglD results in the final product. While this triad of enzymes (dehydratase, aminotransferase, and acetyltransferase) is both prevalent and unique to many prokaryotic pathogens, there has not been widespread focus on inhibitor development efforts against them, no doubt in part due to limited access to their cognate substrates.
In response to this need, our laboratory has developed chemoenzymatic methodology capable of routinely yielding tens of milligrams of useful DATDH glycosyl donors. This section outlines the synthesis of the first three intermediates in the C. jejuni pgl pathway, the third of which comprises the reducing sugar of the N-linked heptasaccharide of this organism and the corresponding sugar in the O-linked heptasaccharide in selected Neisseria sp. (Fig. 2). Synthesis is achieved through the use of three enzymes from two homologous pathways: NAD+-dependent dehydratase PglF from C. jejuni, aminotransferase PglC from N. gonorrhoeae and acetyltransferase PglD from C. jejuni. PglC, essential for the biosynthesis of O-linked glycoproteins in Neisseria, has been shown to be significantly more efficient in catalyzing the conversion of the UDP-4-ketosugar to the UDP-4-aminosugar than the C. jejuni homolog (Morrison 2014). As a result, it has become the preferred enzyme in our laboratory for accessing the UDP-4-aminosugar intermediate. Unlike PseB, discussed in the Section 4, PglF has no associated C-5 epimerase activity when acting on UDP-GlcNAc (Morrison & Imperiali 2014).Thus, this hexose retains an energetically-favored conformation due to the “all equatorial” gluco stereochemistry and therefore is not susceptible to non-enzymatic epimerization. This stability allows for the products of PglF and PglC to be isolated individually, a stark difference from the synthesis of UDP-6-deoxy-4-amino-2-NAc-L-Alt, outlined in Section 4.2, which benefits from a coupled one-pot approach toward this unique intermediate. In addition, immobilization of enzymes on affinity purification resin, as is done with the GST-PglFΔ1–130 construct, allows a greater percentage of the enzyme to be catalytically active during synthesis than when employed in solution (Morrison 2014; Olivier, Chen, Behr & Imperiali 2006), significantly improving workflow and ease of use.
Chemoenzymatic syntheses of UDP-diNAcBac and the two precursor glycosyl donors enable several distinct avenues for fundamental studies and development of small molecule inhibitors. As UDP-diNAcBac is the essential reducing sugar for bacterial N- and O-glycosylation pathways in several organisms, it is a valuable reagent for further elucidation of these pathways (Nothaft & Szymanski 2010; Hartley et al. 2011; Glover, Weerapana, Chen & Imperiali 2006; Hartley, Schneggenburger & Imperiali 2013). The semi-preparative scale of synthesis is ideal for carrying out high-throughput screens of small molecule inhibitors of Pgl enzymes, as we have recently shown with PglD (De Schutter, Morrison, Morrison, Ciulli & Imperiali 2017). The incorporation of radioisotopes into UDP-diNAcBac for biochemical characterization purposes can easily be achieved through the use of the [14C]-labeled [14C]acetyl CoA using a chemoenzymatic approach (Hartley, Schneggenburger & Imperiali 2013) and also enables other derivatizations such as the introduction of a C-4-N-azidoacetyl group using a chemical approach for click chemistry (Lukose, Whitworth, Guan & Imperiali 2015) and subsequent applications to install fluorophore and biotin labels (Musial-Siwek, Jaffee & Imperiali 2016). Regardless of any variations the user incorporates, the method described in this method, with its ease of use and convenient scale, has proved a superior alternative in our experience to multistep and labor-intensive synthetic routes (Glover, Weerapana, Chen & Imperiali 2006).
3.2. Procedure
Buffers and Solvents:
Lysogeny Broth (LB): Dissolve 25 g LB to 1 L of water and autoclave to sterilize before use.
Resuspension buffer: 100 mM NaCl, 25 mM Tris-HCl, pH 7.5
Working buffer: 100 mM NaCl, 50 mM HEPES, pH 7.4
TLC solvent: 5:1:3:1 n-propanol/EtOAc/H2O/25% ammonium hydroxide
TLC stains: Ninhydrin - 1.5 g of ninhydrin is dissolved in 100 mL n-butanol. To this, 3 mL of acetic acid is added dropwise. Store at 4 ºC. When used, heat to develop.; p-Anisaldehyde - 15 mL of acetic acid is added to 350 mL ice-cold ethanol. To this, add 50 mL of concentrated sulfuric acid dropwise, over ice, with stirring over one hour. Store at 4 ºC under foil. When used, heat to develop.
FPLC Solvent A: sterile-filtered Milli-Q-purified water
FPLC Solvent B: 0.9 M ammonium bicarbonate
3.2.1. Expression of PglF (GST-PglFΔ1–130) from C. jejuni, PglC from N. gonorrhoeae, and PglD from C. jejuni
Protocol:
Amplify plasmid of choice in DH5α using standard procedures and purify plasmid DNA using Miniprep kit.
Transform plasmid into freshly-competent BL21 (DE3) RIL cells, growing transformants on LB-agar selection media containing corresponding vector selection antibiotic (see Table 1) and 30 μg/mL CAM.
Choose a single colony and use it to inoculate a starter culture: 100 mL of LB media with antibiotics. Shake overnight at 37 ºC.
Use 10 mL of starter culture to inoculate 1 L of LB with antibiotics until an optical density at 600 nm of 0.5–0.8 is reached.
Induce each culture with IPTG to a final concentration of 1 mM to begin induction. Grow at 16 °C overnight with shaking.
Pellet cultures by centrifugation at 3,200 x g for 15 min and discard the supernatant.
Bring all cell pellets up in 40 mL resuspension buffer and transfer to a 50 mL centrifugation tube. Centrifuge the samples once more.
Decant the supernatant and store pellets immediately at −80 °C until needed. Pellet storage time should not exceed 3 months for optimal enzyme activity
Table 1.
Plasmids used in this manuscript.
| Construct | Vector | Resistance | Reference | Addgene ID |
|---|---|---|---|---|
| GST-PglFΔ1–130 (Cj) | pGEX4T-2 | Carb, 50 μg/mL | Olivier et al., 2006 | 89708 |
| His8-TEV-PglC (Ng) | modified pET30b(+) | Kan, 30 μμg/mL | Hartley et al., 2011 | 89709 |
| T7-PglD-His6 (Cj) | pET24a(+) | Kan, 30 μg/mL | Morrison et al., 2010 | 89710 |
| PseB-His6 (Cj) | pET-24a(+) | Kan, 30 μg/mL | this report | 89723 |
| His8-TEV-PseC (Cj) | modified pET30b(+) | Kan, 30 μg/mL | Hartley et al., 2011 | 89724 |
| His8-TEV-PseH (Cj) | modified pET30b(+) | Kan, 30 μg/mL | Hartley et al., 2011 | 89725 |
| T7-WbpB-His6 (Pa) | pET-24a(+) | Kan, 30 μg/mL | Larkin et al., 2009 | 89711 |
| T7-WbpE-His6 (Pa) | pET-24a(+) | Kan, 30 μg/mL | Larkin et al., 2009 | 89712 |
| T7-WbpD-His6 (Pa) | pET-24a(+) | Kan, 30 μg/mL | Larkin et al., 2009 | 89713 |
| T7-AglK-His6 (Mv) | pET-24a(+) | Kan, 30 μg/mL | Larkin et al., 2013 | 89714 |
| T7-AglC-His6 (Mv) | pET-24a(+) | Kan, 30 μg/mL | Larkin et al., 2013 | 89726 |
Notes:
Two antibiotics are always needed; one to select BL-21 RIL cells (CAM) and one to select and maintain the plasmid (see Table 1). These two antibiotics are added to LB anytime it is used.
3.2.2. Chemoenzymatic Synthesis of UDP-4-ketosugar
Chemoenzymatic synthesis proceeds simply by the addition of UDP-GlcNAc and NAD+ to immobilized GST-PglFΔ1–130. Kinetic characterization of this enzyme has shown it to be the rate-limiting step in this enzyme triad in C. jejuni and related species. The GST-PglFΔ1–130 construct truncates the protein and removes the transmembrane domains, enabling higher yields from protein purification while retaining efficient activity.
Protocol:
Thaw a 1-L pellet of BL21 overexpressing GST-PglF130 and thoroughly resuspend in 40 mL ice-cold working buffer containing 0.5 mg/mL lysozyme, 0.32 μL DNase I and 8 μL protease inhibitor cocktail. Tumble this solution at 4 °C for 30 min.
Sonicate the slurry for 90 s at 50% amplitude while on ice, allowing the slurry to rest on ice for 5 min afterwards. Repeat twice more. The thick cell slurry becomes noticeably clarified and homogenous after sonication.
Transfer the lysate to ultracentrifuge tubes and spin at 145,000 x g for 1 h at 4 °C.
Transfer the clarified lysate to a clean 50 mL centrifuge tube containing 4 mL GST resin that has been previously washed in working buffer. Add NAD+ to a final concentration of 500 μM and gently rock the slurry at 4 °C for 4 h.
Transfer the slurry to a chromatography column and elute the solution from the column via gravity.
Wash the column with 8 CV of working buffer to remove excess cofactor and non-specific proteins to waste. Immobilized GST-PglF130 should be used immediately for best results.
To synthesize UDP-4-ketosugar, dissolve 100 mg of UDP-GlcNAc in 10 mL working buffer and add to the column containing GST-PglFΔ1–130 on GST resin.
Using a plastic scoopula, very gently loosen the resin until free flowing. Cap the column and incubate the resin at RT with gentle rocking for 48 h.
Monitor the reaction during this time by thin layer chromatography (TLC) in TLC solvent. Spot the reaction on TLC plates and allow to fully dry. Once dry, run the TLC in TLC solvent and visualize by short and long wavelength UV, along with p-anisaldehyde stain, to reveal the conversion of UDP-GlcNAc to the lower-running UDP-4-ketosugar product. Spotting TLC plates with UDP-GlcNAc, along with a co-spot of starting material and the reaction mixture, is essential for best results.
Once the reaction is complete, collect the column flow-through containing the product in a 50 mL centrifuge tube and wash the column with 3 CV of working buffer.
Combine all flow-through fractions in a 50 mL centrifuge tube and heat the solution in a 60 °C water bath for 1 h to precipitate any unwanted soluble protein. This results in a slightly opaque solution.
Centrifuge the solution at 3,200 x g for 30 minutes and filter the supernatant through a 3,000 MWCO centrifuge filter. This additional filtration is to ensure the removal of any remaining protein from the reaction mixture, but may be omitted. In either case, the solution was taken to the next step immediately without further purification. Alternatively, this solution may be flash-frozen in 1 mL aliquots in liquid nitrogen and stored at −20 ºC for up to 6 months.
3.2.3. Chemoenzymatic Synthesis of UDP-4-aminosugar
Immobilized His8-TEV-PglC from N. gonorrhoeae is employed in the synthesis of UDP-4-aminosugar from UDP-4-ketosugar. Importantly, when approximately 60 mg of enzyme is used, an upper limit of 50 mg UDP-4-ketosugar for 100% turnover has been observed. As such, the amount of enzyme in the following protocol has been scaled appropriately to allow all material from Section 3.2.2 to be carried through to UDP-4-aminosugar.
Protocol:
To prepare immobilized PglC, thaw a 2-L pellet of BL21 overexpressing His8-TEV-PglC and thoroughly resuspend in 40 mL ice-cold working buffer containing 0.5 mg/mL lysozyme, 0.32 μL DNase I and 8 μL protease inhibitor cocktail. Tumble this solution at 4 °C for 30 min.
Sonicate the slurry for 90 s at 50% amplitude while on ice, allowing the slurry to rest on ice for 5 min afterwards. Repeat twice more.
Transfer the solution to ultracentrifuge tubes and spin at 145,000 x g for 1 h at 4 ºC.
Transfer the clarified lysate to a clean 50 mL centrifuge tube containing pyridoxal phosphate (PLP) at a final concentration of 500 μM and 5 mL Ni-NTA resin (previously thoroughly washed with water). Invert the lysate, resin and PLP gently at 4 ºC for 4 h.
Transfer the bright yellow slurry to a chromatography column and elute the solution from the column via gravity.
Wash the resin with an additional 50 mL (10 CV) of ice-cold working buffer with 30 mM imidazole to remove excess PLP and non-specific proteins to waste.
To synthesize UDP-4-amino-sugar, pass 3 CV of ice-cold working buffer with no imidazole through the column.
With the stopcock closed, add 1 batch of UDP-4-ketosugar (in ~12 mL working buffer) to the column with very careful stirring of the settled resin with a plastic scoopula to loosen.
Add L-glutamate to a final concentration of 25 mM and tumble the resin gently at room temperature for 72 h.
Monitor the reaction during this time by TLC in TLC solvent. Spot the reaction on TLC plates and allow to fully dry. Once dry, run the TLC in TLC solvent and visualize by short and long wavelength UV, along with ninhydrin stain, to reveal the conversion of UDP-ketosugar to the higher-running UDP-4-aminosugar product, which stains pink with ninhydrin. Although heat is required for this stain to reveal compounds on the plate, slow heating will reduce the amount of background coloration and improve visualization.
Once the reaction is complete, collect the column flow-through containing the product and wash the column with 3 CV of working buffer.
Combine all flow-through fractions in a 50 mL centrifuge tube and heat the solution in a 60 °C water bath for 1 h to precipitate any unwanted soluble protein. This results in a slightly opaque solution.
Centrifuge the solution at 3,200 x g for 30 minutes and filter the supernatant through a 3,000 MWCO centrifuge filter.
- To purify crude UDP-4-aminosugar, two Hi-Trap Q columns connected in sequence are used for FPLC purification. The method is as follows:
- Sample: 1 mL aliquot of crude reaction mixture
- Flow rate: 1.5 mL/min
- Column is equilibrated for at least 25 mL in 1% Solvent B
- Detection wavelength is 280 nm
- Gradient: 1–50% Solvent B over 80 mL
- 3 mL fractions are collected for optimal resolution
- Column is washed in 20 mL 100% Solvent B, followed by 30 mL 100% Solvent A.
TLC fractions to identify peaks containing UDP-4-aminosugar (representative trace shown in Figure 4) and combine in 50 mL centrifuge tubes.
Freeze the sample and lyophilize to yield a white amorphous solid. This material (>30 mg) should be stored as a lyophilized powder at −20 °C.
Fig. 4.
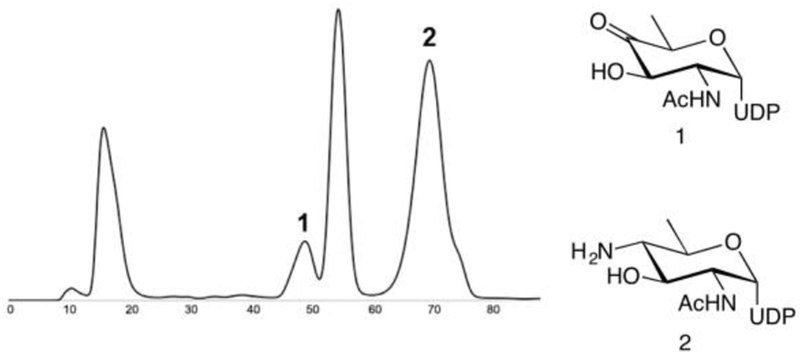
FPLC profile (280 nm, 1.5 mL/min, y-axis in volume) from chemoenzymatic synthesis of UDP-4-aminosugar.
3.2.4. Synthetic acetylation toward UDP-diNAcBac
The chemical acetylation of the UDP-4-amino-sugar has been reported previously (Glaze, Watson, Young & Tanner 2008). We have found this method, carried out under anhydrous conditions, to be an expedient route to UDP-diNAcBac. Furthermore, for large-scale preparations, the chemical strategy using acetic anhydride is far less costly that the application of the enzymatic method utilizing AcCoA.
Protocol:
Add 5 mg of previously purified and lyophilized UDP-4-aminosugar to a 15 mL round-bottom flask flame-dried under vacuum and charge with N2 and stir bar.
Add anhydrous MeOH (2.5 mL) via syringe under N2, followed by 140 µL Ac2O. Maintain the vessel under positive N2 pressure via balloon or manifold.
Allow the reaction to stir for 12 h. Monitor by TLC run in TLC solvent, staining with ninhydrin solution to observe the disappearance of starting material (staining pink) and the appearance of a higher-running UV-active spot for the product (staining brown).
Once complete, concentrate the solution under reduced pressure.
Dissolve the residue in water, freeze, and lyophilize to a yellow amorphous solid.
Take up the lyophilized material in 5 mL H2O and purify via the same method described in Section 3.2.3 for the FPLC purification of UDP-4-aminosugar, yielding UDP-diNAcBac as a fine white powder.
Starting from 100 mg UDP-GlcNAc in Section 3.2.2, >30 mg of UDP-diNAcBac can be easily obtained via this procedure.
3.2.5. Purification of PglD from C. jejuni and Chemoenzymatic Synthesis of UDP-diNAcBac
UDP-diNAcBac can also be produced using recombinantly-expressed and purified acetyl transferase PglD. This method is advantageous for the preparation of isotopically-labeled UDP-diNAcBac product, which must be carried out on a small scale with costly radiolabeled reagent.
Protocol:
Thaw a 1-L cell pellet of BL-21 expressing T7-PglD-His6 and resuspend in ice-cold working buffer containing 30 mM imidazole.
Lyse and clarify the cell slurry via sonication and centrifugation as described in Section 3.2.2.
Treat the clarified supernatant with 2 mL of previously washed Ni-NTA resin in a 50 mL centrifuge tube and tumble for 3 h at 4 °C.
Pass the resin slurry through a chromatography column to retain PglD via the C-terminal His6-tag and elute other soluble proteins.
Wash the column with 20 CV of working buffer with 30 mM imidazole.
Elute PglD with working buffer containing 300 mM imidazole in 2 mL fractions.
Analyze the contents of each fraction, the pellet, and the flow-through via 10% SDS-PAGE, with Coomassie staining, for the 21.4 kD protein of interest.
Combine pure fractions and dialyze against 2 L ice-cold working buffer at 4 °C using a 10 kD Slide-A-Lyzer cassette of an appropriate volume.
Concentrate further to ~10 mg/mL using a 10 kDa MWCO centrifugal filter device, measuring protein concentration by UV absorption at 280 nm.
Determine the final protein concentration by BCA assay and aliquot protein immediately into microcentrifuge tubes. Snap-freeze these tubes in N2 for storage at −80 ºC. Thawing and handling of any aliquots after initial freezing should be done over ice to prevent protein precipitation.
To synthesize UDP-diNAcBac, add 10 mg UDP-4-aminosugar and 14 mg AcCoA to 6 mg PglD in 5 mL of working buffer in a 15-mL centrifuge tube.
Incubate the reaction mixture at 37 °C for 12 h.
Monitor the reaction progress by TLC as outlined in Section 3.2.4.
Remove enzyme from the reaction crude by protein precipitation and filtration in Section 3.2.2.
This reaction mixture can now be purified via FPLC in 1 mL aliquots as described above in Section 3.2.3. Alternatively, the filtrate can be aliquoted into microcentrifuge tubes, flash-frozen in liquid nitrogen and stored at −20 °C for 6 months for later purification.
Notes:
This procedure can be carried out on various scales to accommodate synthesis with [3H]-AcCoA or [14C]-AcCoA to afford radiolabeled UDP-diNAcBac for activity assays (Hartley, Schneggenburger & Imperiali 2013; Lukose, Whitworth, Guan & Imperiali 2015)
4. Chemoenzymatic Preparation of UDP-diNAcAlt with PseB, PseC, and PseH
4.1. Discussion
Pseudaminic acid (5,7-diacetamido-3,5,7,9-tetradeoxy-L-glycero-α-L-manno-nonulosonic acid) from the class of nonulosonic acids was first identified by the Kochetkov lab in 1984 in lipopolysaccharides (LPS) of Pseudomonas aeruginosa and Shigella boydii (Knirel et al. 1984). Pseudaminic acid has been implicated in the pathogenesis of Gram-negative bacteria including C. jejuni and H. pylori, where it features as an important modification of the FlaA and FlaB proteins (Schirm et al. 2003; Thibault et al. 2001). As these glycoproteins are essential for flagellar function, inhibition of glycosylation results in loss of bacterial motility and a reduction in virulence (Guerry et al. 2006). For example, studies with enzyme mutants that are defective in pseudaminic acid biosynthesis (including PseH) in C. jejuni demonstrated that a disruption of these genes resulted in the reduction of flagellar filaments, hook structures and non-motile phenotypes (Guerry et al. 2006). Our research interests lie in the characterization of the enzymes in the pseudaminic acid and related pathways as potential targets for inhibition for the development of tools to understand the roles of modified carbohydrates in pathogeneses and as potential lead compounds as antivirulence agents to address infectious diseases.
The formation of CMP-pseudaminic acid proceeds through several unusual sugar derivatives, as illustrated in Figure 5. The pathway begins with the common sugar nucleotide UDP-GlcNAc, with the first three enzymes (PseB, PseC, and PseH) proceeding analogously to PglF, PglE, and PglD from the C. jejuni pgl pathway, with the exception that PseB includes C-5 epimerase activity. In this case, because the sugar that is produced is less stable, the UDP-keto-sugar product can rapidly epimerize at C-5 to the corresponding C. jejuni Pgl pathway UDP-4-ketosugar intermediate. Therefore, the application of a PseB/PseC-coupled reaction to immediately channel the labile product is essential (Schoenhofen et al. 2006). In the CMP-Pse pathway, PseH-catalyzed C-4 amine acylation is followed by UDP hydrolysis by PseG, condensation with phosphoenol pyruvate (PEP) by PseI, and reaction with CTP by PseF to form CMP-Pse. A peptidyl pseudaminyltransferase then transfers pseudaminic acid to serine or threonine resulting in O-linked protein glycosylation.
Fig. 5.
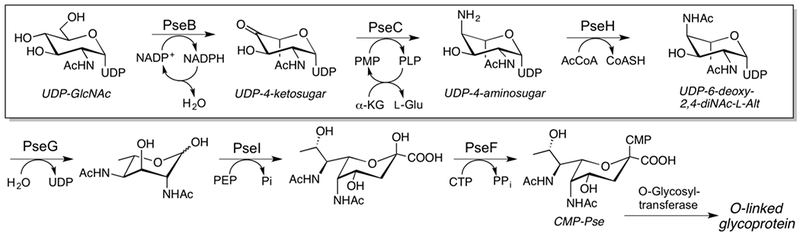
CMP-pseudaminic acid biosynthetic pathway. Steps covered in this method are boxed.
Two flagellin proteins in C. jejuni and H. pylori (FlaA and FlaB), which are glycosylated in this fashion, have been identified as playing a role in pathogenesis. FlaA isolated from C. jejuni is pseudaminylated at 19 serine or threonine residues, and 8 of these sites have been shown to make a significant contribution to the motility and autoagglutination of the bacteria (Guerry et al. 2006; Ewing, Andreishcheva & Guerry 2009). Similarly, FlaA in H. pylori has been found to contain 7 glycosylation sites in the central core region of the protein as well (Schirm et al. 2003). However, much yet remains to be understood about the structure and mechanism of pseudaminyltransferases involved in these processes.
Chemical syntheses of pseudaminic acid, starting with N-acetyl glucosamine (Lee, Kubota, Ishiwata & Ito 2011) or sialic acid (Williams, Corcilius, Kiefel & Payne 2016; Zunk, Williams, Carter & Kiefel 2014) have been reported. However, while these syntheses offer access to multiple analogs and the opportunity for selective customization, they proceed through some difficult/air sensitive or low-yielding steps, requiring several purifications and considerable protecting group manipulation. Alternatively, chemoenzymatic synthesis can afford efficient conversion of substrate to product, in which several enzymes can often be applied in tandem (Schoenhofen, McNally, Brisson & Logan 2006), provided that a thorough understanding of the enzymes, substrates, and cofactors is known. A chemoenzymatic approach also provides access to the biosynthetic intermediates of the pseudaminic acid pathway, and the opportunity to study substrate-protein interactions through protein crystallography. This structural insight is in turn valuable for inhibition and mechanistic analyses. In the interest of studying the acetyltransferase PseH as a potential inhibition target, we have established efficient methodology for the expression and purification of C. jejuni PseB, PseC, and PseH. We have outlined here the use of PseB and PseC in the synthesis of PseH substrate UDP-4-amino-2-NAcAlt and a kinetic analysis of PseH. Together these studies establish the foundation for future inhibitor development of PseH.
4.2. Procedure
Buffers and Solvents:
Cell lysis and Ni-NTA chromatography buffers: 50 mM sodium phosphate pH 7.3, 400 mM NaCl, 5 mM β-mercaptoethanol (β-ME), with 10, 100, 300 or 500 mM imidazole
Dialysis buffer: 25 mM sodium phosphate pH 7.3, 50 mM NaCl
Assay buffer: 50 mM HEPES pH 7.4
NP-HPLC isocratic elution buffer: 50 mM ammonium formate pH 4.2
ENVI-Carb SPE wash buffers: 10 mM and 50 mM triethylammonium acetate
4.2.1. Enzyme Expression and Purification
Protocol:
To express PseB, transform E. coli BL21 RIL competent cells with a pET-24a(+) plasmid containing the pseB gene. For expression of PseC and PseH, modified pET-30a(+) plasmids with an N-terminal His8-tag and a TEV protease site and the pseC and pseH genes are used (see Table 1).
Inoculate 8 mL of LB media containing 30 μg/mL KAN and 30 μg/mL CAM with transformants and incubate with shaking overnight at 37 °C.
Inoculate 1 L of LB media containing 30 μg/mL KAN and 30 μg/mL CAM with the overnight culture of cells. Allow the cells to grow at 37 °C while shaking until an OD600 of ~0.6 is reached.
Cool the culture to 16 °C, induce with 0.5 mM IPTG, and incubate for 18 h with shaking.
Harvest the cells by centrifugation (3200 x g for 30 min) and store at −80 °C until needed.
Carry out each cell lysis and purification step at 4 °C. Resuspend the cell pellet in 40 mL of 10 mM imidazole lysis buffer, and tumble with 4 μL protease inhibitor cocktail and 20 mg lysozyme for 15 minutes. For PseC, add 500 µM PLP to this buffer.
Add 10 µg/mL DNaseI, and lyse by sonication by pulsing two times for 90 seconds each at 40% amplitude.
Clear the lysate by centrifugation (100,000 x g for 50 min) and add to 3 mL of Ni-NTA resin in a chromatography column at a rate of 1 mL/min. Wash the resin with 10 CV of 10 mM imidazole lysis buffer, followed sequentially by 40 mM, 70 mM, and 100 mM imidazole lysis buffers.
Elute the protein of interest with 300 mM imidazole lysis buffer and pool fractions containing the purified protein (as analyzed by SDS-PAGE) and dialyze with dialysis buffer overnight with 10 kD MWCO dialysis tubing. Calculate protein concentration based upon the predicted extinction coefficients (generated by http://web.expasy.org/protparam/) at λ = 280 nm.
For PseH, add 500 µL of a 100 μM His-tagged TEV protease (commercially available from ThermoFisher Scientific or can be prepared in-house) and 5 mM DTT to protein in dialysis buffer at 4°C. Stationary incubation at 4°C resulted in no precipitation, and complete removal of His-tag.
For PseH, purify the cleaved protein away from the tag by Ni-NTA chromatography by collecting the flow-through, and determine purity by running a 12% SDS-PAGE protein gel.
Notes:
Initially the TEV protease reaction was subject to tumbling overnight, however, this resulted in complete protein precipitation therefore stationary incubation is preferred.
For optimal enzyme activity, carry out all steps at 4°C in as few days as possible, and either use immediately for chemoenzymatic reactions or snap-freeze aliquoted samples with liquid nitrogen and store at −80°C. For optimum activity, use flash-frozen proteins within 3–4 months.
4.2.2. Chemoenzymatic synthesis of UDP-6-deoxy-4-amino-2-NAc-L-Alt
Protocol:
Make a pyridoxal 5’-phosphate (PLP) stock with 100 mM NaOH solution, as it is sparingly soluble near to neutral pH.
Prepare 2 µM PseB-His6 and 2 µM His7-PseC in 10 mL dialysis buffer containing 5 mM UDP-GlcNAc disodium salt (0.033 g), 20 mM monosodium glutamate, 50 µM PLP, and 250 µM NADP+, and incubate overnight at 37°C.
Monitor reaction completion by analytical NP-HPLC with an isocratic eluent comprising 50 mM ammonium formate pH 4.2, as shown in Figure 6.
Remove reagent enzymes with a 10 kDa MWCO centrifugal filter device, and concentrate the filtrate by lyophilization.
Resuspend the reaction mixture in 1 mL H2O, and load onto an ENVI-Carb SPE tube that has been conditioned with 2 mL MeOH and 2 mL H2O. The desired UDP-4-amino-2-NAc-L-Alt elutes immediately with water, followed by any of the corresponding 6-deoxy-4-keto intermediate, while UDP-GlcNAc elutes only with 10–50 mM triethylammonium acetate (TEAA) buffer (Barnes et al. 2016).
Quantify UDP-6-deoxy-4-amino-2-NAc-L-Alt obtained using the molar extinction coefficient of UDP (ε262 = 9890 M−1 cm−1): 0.018 g, 58% yield.
Fig. 6.
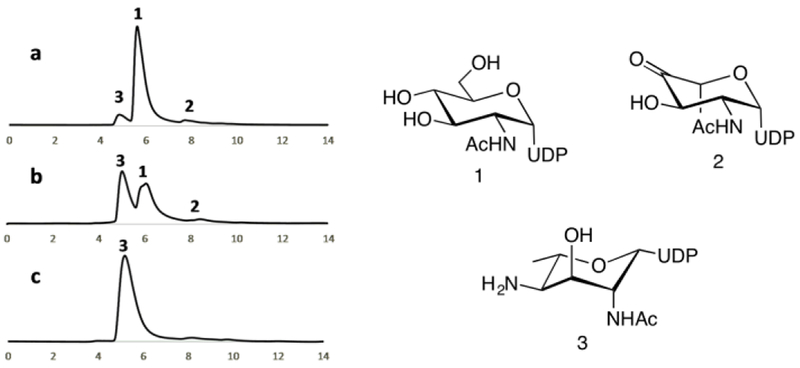
NP-HPLC profiles (260 nm, 1 mL/min, y-axis in volume) from chemoenzymatic synthesis of UDP-6-deoxy-4-amino-2-NAc-L-Alt. a) Reaction at t = 0; b) Reaction progress; c) Purified UDP-6-deoxy-4-amino-2-NAc-L-Alt (3).
Notes:
While PLP has been employed at higher concentrations, excess PLP is troublesome and can interfere with both protein quantification and absorbance-dependent assays. A screen of PLP concentrations from 1 mM to 50 µM showed minimal differences in product conversion and therefore the lowest concentration of PLP is adequate.
PseB and PseC must be coupled in a single reaction to avoid the rapid epimerization to the UDP-4-amino-2-NAcBac diastereomer as described in the discussion.
4.2.3. Synthetic acetylation toward UDP-6-deoxy-2,4-diNAc-L-Alt
Protocol:
As modified from (Liu & Tanner 2006), add acetic anhydride (0.026 mL, 0.272 mmol) and AgOAc (6.5 mg, 0.039 mmol) to a round-bottomed flask containing 0.5 mL anhydrous methanol with crushed, activated 3 Å molecular sieves, and stir under N2 for 1 hr.
Dissolve lyophilized UDP-6-deoxy-4-amino-2-NAc-L-Alt (4.0 mg, 0.0068 mmol) in 0.5 mL anhydrous methanol, add to the reaction flask, and stir for 12 hours.
Monitor reaction completion by TLC (5:1:3:1 n-propanol/EtOAc/H2O/25% ammonium hydroxide; Rf = 0.23) and quantify by A260 UV absorption after separation by isocratic HPLC elution.
Notes:
Complete consumption of starting material and conversion to UDP-6-deoxy-2,4-diNAc-L-Alt is achieved with the described anhydrous conditions, in comparison to only ~60% conversion under less stringent conditions and without molecular sieves.
The synthetic acetylation of UDP-6-deoxy-4-amino-2-NAc-L-Alt is advantageous because it is higher yielding, does not require costly AcCoA reagent and can be carried out on larger scales.
4.2.4. Chemoenzymatic acetylation toward UDP-6-deoxy-2,4-diNAc-L-Alt with PseH
Chemoenzymatic acetylation of UDP-6-deoxy-4-amino-2-NAc-L-Alt has been described in a multi-enzyme format, using either the first six or first three enzymes in the pseudaminic acid biosynthetic pathway (Schoenhofen, McNally, Brisson & Logan 2006) (Fig. 3). Because of the multienzyme format previously applied, kinetic characterization of the acetyltransferase PseH was not previously reported. Therefore, we carried out steady-state kinetic analysis of PseH using 5,5’-dithiobis(2-nitrobenzoic acid) (Ellman’s reagent) to detect CoASH release through generation of the TNB2- chromophore (Ellman 1959), in a continuous fashion, as described previously (Hartley et al. 2011; De Schutter, Morrison, Morrison, Ciulli & Imperiali 2017).
Protocol:
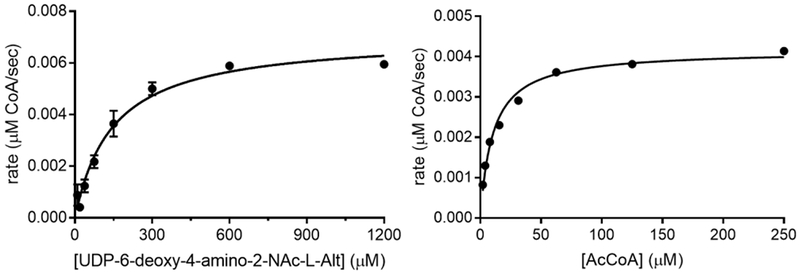
1. To a flat-bottom, black, 96-well plate (Grenier) add HEPES buffer, 5 mM MgCl2, 0.05% BSA, 0.001% Triton X-100, 1 mM DTNB, and 10 nM PseH, with substrate concentrations of AcCoA and UDP-6-deoxy-4-amino-2-NAc-L-Alt varied separately, with the other substrate at saturation. Use saturated concentrations of 250µM AcCoA, and 1200µM UDP-6-deoxy-4-amino-2-NAc-L-Alt. Final volume: 100 µL.
2. Run reactions in duplicate and measure initial rates in the linear portion of the reaction curve over a 5 min time period at 30°C. A background control reaction in the absence of UDP-6-deoxy-4-amino-2-NAc-L-Alt is subtracted from each reaction rate.3. Graph and analyze kinetic constants with GraphPad Prism 7 software.
Notes:
Quantify PseH concentration (ε280 = 17795 M−1 cm−1), UDP-sugar (ε262 = 9890 M−1 cm−1), AcCoA (ε260 = 16,400 M−1 cm−1) by UV absorption.
Make DTNB solution fresh before each use, and store AcCoA solution at −20°C for best results.
The reactions followed Michaelis-Menten kinetics with values of kcat = 0.70 ± 0.03 s−1, Km = 147 ± 21 µM, and kcat/Km= 4.8 M−1 s−1 with respect to the UDP-6-deoxy-4-amino-2-NAc-L-Alt substrate; and values of kcat = 0.42 ± 0.1 s−1, Km = 15.9 ± 0.8 µM, and kcat/Km = 173.7 M−1 s−1 with respect to AcCoA.
5. Chemoenzymatic Preparation of UDP-2,3-diNAcGlcA with WbpB, WbpE, and WbpD
5.1. Discussion
Two of the most intensively studied archaeal N-linked glycosylation pathways are found in the marine methanogen Methanococcus voltae and the closely related M. maripaludis (VanDyke et al. 2009; Chaban, Voisin, Kelly, Logan & Jarrell 2006). M. voltae is known to generate N-linked glycoproteins with a unique trisaccharide composed of NAc(6Thr)ManA-β1,4–2,3-diNAcGlcA-β1,3-GlcNAc (Fig. 2), where 2,3-diNAcGlcA is 2,3-diacetamido-2,3-dideoxy-D-glucuronic acid. Both the M. voltae and M. maripaludis pathways require UDP-2,3-diNAcGlcA as the donor for the second sugar in the N-linked glycan structures found in the archaeal flagellins and S-layer glycoproteins of these organisms (Voisin et al. 2005; Kelly et al. 2006). While the enzymes that are responsible for glycoprotein assembly have been biochemically characterized in M. voltae (Larkin, Chang, Whitworth & Imperiali 2013), the complete set of genes responsible for the biosynthesis of the glycosyl donor substrates in M. voltae has not been fully annotated, preventing the enzymatic synthesis of UDP-2,3-diNAcGlcA, which is required for investigation of oligosaccharyl transferase activity in this organism.
The chemical synthesis of UDP-2,3-diNAcGlcA from GlcNAc is a laborious process which entails seventeen steps and results in a low final yield (Rejzek et al. 2009). To address this, and provide valuable material for enzymatic studies, our group has applied chemoenzymatic synthesis towards mulitmilligram quantities of this material by exploiting the WbpB, WbpE, and WbpD enzymes involved in the Pseudomonas aeruginosa PAO1 lipopolysaccharide biosynthetic pathway (Fig. 8) (Larkin & Imperiali 2009). The coupled reaction of WbpB and WbpE, a dehydrogenase and aminotransferase, uses a unique NAD+ recycling mechanism to convert UDP-2-NAcGlcA to UDP-2-NAc-3-NH2GlcA. WbpD then catalyzes the acetylation of UDP-2-NAc-3-NH2GlcA to produce UDP-2,3-diNAcGlcA. In this section, we present the simple procedure for the preparation of UDP-2-NAcGlcA via oxidation of UDP-GlcNAc, followed by the enzymatic conversion of UDP-2-NAcGlcA to UDP-2,3-diNAcGlcA, the rare nucleotide sugar that is critical for studies of lipopolysaccharide assembly and oligosaccharyl transferase activity in methanogenic archaea (Larkin, Chang, Whitworth & Imperiali 2013).
Fig. 8.

Biosynthetic pathway of UDP-2,3-diNAcManA, a lipopolysaccharide subunit in P. aeruginosa PAO1. UDP-2,3-diNAcGlcA is boxed.
5.2. Procedure
Buffers:
Cell lysis buffer: 50 mM HEPES, pH 8.0, 300 mM NaCl, 10 mM imidazole
Ni-NTA wash buffer: 50 mM HEPES, pH 8.0, 300 mM NaCl, 25 mM imidazole
Ni-NTA elution buffer: 50 mM HEPES, pH 8.0, 300 mM NaCl, 250 mM imidazole
Dialysis buffer: 50 mM HEPES, pH 8.0, 100 mM NaCl
FPLC Solvent A: sterile-filtered Milli-Q water
FPLC Solvent B: 0.9 M NH4HCO3
RP-HPLC buffer: 50 mM TEAB, pH 7.1
CE running buffer: 25 mM sodium tetraborate, pH 9.5
5.2.1. Enzyme expression and purification
Protocol:
Transform E. coli BL21-CodonPlus(DE3) RIL competent cells with pET-24a(+) plasmids carrying the desired genes using KAN and CAM as selection markers for expression as proteins with N-terminal T7 and C-terminal His6 tags (see Table 1.)
Inoculate transformants into a 5 mL starter culture in LB, supplemented with KAN (50 μg/mL) and CAM (30 μg/mL), and incubate at 37 °C overnight.
Inoculate 1 L of LB media with 5 ml of starter culture and incubate at 37 °C while shaking until an OD600 of 0.6–0.8 is reached. Lower the temperature to 16 °C and induce with 1 mM IPTG. After 16 h, harvest the cells by centrifugation (5,000 x g for 15 min) and store at −80 °C until needed.
Resuspend fresh or frozen cell pellets from 1 L of culture in 50 mL of cold cell lysis buffer. Carry out each lysis and purification step at 4 °C. Lyse cells by sonication on ice (50% amplitude, 1 s pulses, 3 × 90 s). Clear the cell lysate of all cellular debris by centrifugation (145,000 x g for 1 h). Incubate the supernatant with Ni-NTA agarose resin (2 mL) for 2 h with gentle rocking and then pour into a chromatography column.
Wash the resin with 25 ml Ni-NTA wash buffer and elute protein with 20 ml of Ni-NTA elution buffer. Combine the fractions containing the eluted protein, as determined by SDS-PAGE, and dialyze (10 kDa MWCO) overnight at 4 °C.
Remove any precipitate by filtration and add glycerol to a final concentration of 25%. Store purified WbpB and WbpE at −20 °C and WbpD at 4 °C. Protein purity is measured by SDS-PAGE and Western blot analysis for T7 and His tags. Protein concentration is calculated based upon the predicted extinction coefficients (generated by http://web.expasy.org/protparam/) at λ = 280 nm.
5.2.2. Synthesis and purification of UDP-GlcNAcA
WbpA, the first enzyme in the biosynthetic pathway (Fig. 8), catalyzes the oxidation of UDP-GlcNAc to UDP-2-NAcGlcA (Miller et al. 2004). However, the overall substrate conversion yield is only modest. Alternately, UDP-2-NAcGlcA can be prepared via platinum-catalyzed oxidation of UDP-GlcNAc to the corresponding uronic acid according to the method of Field and coworkers (Rejzek, Mukhopadhyay, Wenzel, Lam & Field 2007).
Protocol:
Activate Adams’ catalyst (PtO2) by adding 10 mg in 1 mL water under H2 for 2 h and then flush with N2.
Transfer the catalyst to a three-necked round bottom flask containing a stirring mixture of UDP-GlcNAc (100 mg, 0.153 mmol) and NaHCO3 (25.8 mg, 0.307 mmol) in water (5 mL). Insert a gas bubbler (medium grade) into the side neck, and reflux the reaction for 72 h with streaming O2. Add three more aliquots of activated catalyst (10 mg) over the course of the reaction.
Remove the Pt catalyst by filtration and purify the UDP-2-NAcGlcA product away from remaining starting material via FPLC on 2 × 5 mL HiTrap Q FF anion exchange columns connected in sequence, monitoring at 260 nm, eluting with a linear gradient of 0–50% Solvent B over 250 mL.
Combine fractions containing UDP-2-NAcGlcA, lyophilize, and resuspend in water for further purification using a preparatory RP-HPLC column for removal of salt and impurities.
Equilibrate RP-HPLC column with RP-HPLC buffer prior to loading sample. Elute UDP-2-NAcGlcA with a gradient of 0–50% acetonitrile in RP-HPLC buffer over 30 min, monitoring at 228 and 260 nm.
Quantify the UDP-2-NAcGlcA product using the molar extinction coefficient of uridine at 262 nm (10,000 M−1cm−1) and characterize by ESI-MS. 1H and 13C NMR spectroscopy data are in agreement with published values (Rejzek, Mukhopadhyay, Wenzel, Lam & Field 2007).
Notes:
More than one round of lyophilization may be needed to fully remove the volatile NH4HCO3 and TEAB buffers.
5.2.3. Synthesis and purification of UDP-2-NAc-3-NH2GlcA by the coupled reaction of WbpB and WbpE
The coupled WbpB and WbpE reaction utilizes a method of NAD+ recycling that requires the concomitant reduction of α-ketoglutarate and oxidation of NADH by WbpB, while L-glutamate serves as the amino-transferase donor substrate for WbpE. The dehydrogenation and aminotransferase reactions convert UDP-2-NAcGlcA to UDP-2-NAc-3-NH2-GlcA (Larkin & Imperiali 2009).
Protocol:
To synthesize UDP-2-NAc-3-NH2GlcA, add 4.5 mg each of WbpB and WbpE, 0.75 mM UDP-2-NAcGlcA, 0.2 mM NAD+, 25 mM L-glutamate, 0.1 mM PLP, 2.5 mM DTT, and 2 mM MgCl2 in a total volume of 50 mL 50 mM HEPES pH 8.0.
Incubate the reaction at 30 °C for 24 h and monitor the progress of the reaction by capillary electrophoresis (CE) (see section 5.2.5.)
Remove protein from the mixture by filtration with a 10 kDa centrifugation filter unit and lyophilize the resulting filtrate. Resuspend the crude mixture in water and load onto a pre-equilibrated preparatory RP-HPLC column. Elute the UDP-2-NAc-3-NH2GlcA product with a linear gradient of 0–50% acetonitrile over 65 min, monitoring at 228 and 260 nm. Lyophilize to remove TEAB.
Quantify the UDP-2-NAc-3-NH2GlcA product using the molar extinction coefficient of uridine at 262 nm (10,000 M−1cm−1) and characterize by ESI-MS as well as 1H and 13C NMR spectroscopy (Larkin & Imperiali 2009).
5.2.4. Acetylation with WbpD toward of UDP-2,3-diNAcGlcA
WbpD catalyzes the acetyl-CoA-dependent acetylation of UDP-2-NAc-3-NH2GlcA to give UDP-2,3-diNAcGlcA.
Protocol:
To synthesize UDP-2,3-diNAcGlcA, 1.5 mg of WbpD enzyme, 0.75 mM UDP-2-NAc-3-NH2GlcA, and 0.75 mM AcCoA are solubilized in a final volume of 7 mL 50 mM HEPES pH 7.0.
Incubate the reaction at 30 °C for at least 2 h prior to CE analysis (see section 5.2.5.) Filter, lyophilize, and purify the crude reaction mixture as described in Section 5.2.3.
Characterize the UDP-2,3-diNAcGlcA product is characterized by ESI-MS, 1H and 13C NMR spectroscopy analysis (Larkin & Imperiali 2009).
Notes:
Radiolabeled UDP-2-[3H]3-diNAcGlcA can be prepared by incubation of UDP-2-NAc-3-NH2GlcA (1 mM) with [3H]-AcCoA (20 Ci/mmol), 50 mM HEPES (pH 7.0), and WbpD (0.5 mg) in a reaction volume of 1 mL at 30 °C for 5 min, followed by a chase with unlabeled AcCoA (0.75 mM) for 4 h.
5.2.5. Analysis of reaction products by capillary electrophoresis
Capillary electrophoresis (CE) is used to monitor reaction progress and to identify final products (Fig. 9). A bare silica capillary (75 µm x 80 cm) is utilized with detection at 72 cm.
Fig. 9.

Capillary electrophoresis time course analysis of the WbpD reaction. (1) AcCoA; (2) CoASH; (3) UDP-2,3-diNAcGlcA; (4) UDP-2-NAc-3-NH2GlcA. The peak labeled x represents an impurity present in the AcCoA starting material (Larkin & Imperiali 2009).
Protocol:
Prepare samples by filtration (5 kDa MWCO) and dilute (2x) with water.
Prior to each run, condition the capillary sequentially with 0.1 M NaOH, water, and CE running buffer for 2 minutes.
Introduce samples to the capillary by pressure injection for 15 seconds at 30 mbar and perform separation at 22 kV and monitored by UV absorbance at 254 nm.
Carry out manual peak integration using Beckman 32 Karat software suite.
6. Isolation and synthesis of polyprenol-phosphate carrier Dol-P
6.1. Discussion
Biosynthesis of N-linked glycoproteins in nature begins with the stepwise assembly of an oligosaccharide onto a membrane-bound polyprenol phosphate-linked carrier, followed by the flipping of the glycan across the membrane, and subsequent transfer to protein. The oligosaccharyl transferase (OTase) is the key enzyme of this pathway and catalyzes the en bloc transfer of the oligosaccharide onto asparagine residues of nascent or fully folded proteins found within an N-X-S/T sequon, where X can be any amino acid except proline. Archaeal N-linked glycosylation bears similarities to both the bacterial and eukaryotic pathways. As in bacteria, archaea display a tremendous variety of N-linked glycans and the OTase is a single integral membrane protein. Similar to eukaryotes, N-linked glycoproteins are an important feature of secretory proteins, appearing in S-layer and flagellar proteins (Calo, Kaminski & Eichler 2010). Also, as in eukaryotes, the polyprenol carrier is a dolichol rather than the α-isoprene unit unsaturated polyprenols, which are characteristic of bacteria. Dolichols are distinguished from undecaprenols by the presence of a saturated α-isoprene unit (Hartley & Imperiali 2012). These interesting attributes make the archaeal pathway an attractive system for detailed biochemical characterization.
Both Dol-P and Dol-PP-linked glycosyl donor substrates have been implicated in archaeal glycan assembly pathways (Jarrell et al. 2014). AglB, the OTase in the M. voltae pathway, has been demonstrated to specifically utilize Dol-P-GlcNAc-β-(1–3)-2,3-diNAcGlcA as its glycan donor substrate to efficiently generate N-linked glycopeptides (Larkin, Chang, Whitworth & Imperiali 2013). This study was only made possible by the availability of the polyprenol phosphate carrier and UDP-2,3-diNAcGlcA (see Section 5) required for the generation of the appropriate polyprenol phosphate-linked glycan substrate. In this section, we present the isolation, purification, and phosphorylation of polyprenols, as well as the enzymatic synthesis of Dol-P-GlcNAc-β-(1–3)-2,3-diNAcGlcA, which has been applied to studies of the archaeal oligosaccharyl transferase activity (Larkin, Chang, Whitworth & Imperiali 2013).
6.2. Procedure
Buffers and solvents:
Cell lysis buffer: 50 mM HEPES, pH 7.5, 300 mM NaCl, 10 mM imidazole
Membrane resuspension buffer: 50 mM HEPES, pH 7.5, 150 mM NaCl, 10 mM imidazole
Ni-NTA wash buffer: 50 mM HEPES, pH 7.5, 150 mM NaCl, 25 mM imidazole, 0.05% DDM
Ni-NTA elution buffer: 50 mM HEPES, pH 7.5, 150 mM NaCl, 250 mM imidazole, 0.05% DDM
Dialysis buffer: 50 mM HEPES, pH 7.5, 150 mM NaCl
NP-HPLC Solvent A: 4:1 CHCl3/MeOH
NP-HPLC Solvent B: 10:10:3 CHCl3/MeOH/2 M NH4OAc
Pure Solvent Upper Phase (PSUP): 15 mL CHCl3, 240 mL MeOH, 1.83 g KCl in 235 mL water
TLC Solvent: 65:25:4:0.5 CHCl3/MeOH/H2O/conc. NH4OH
TLC stain: Ceric ammonium molybdate - Add 5 g cerium sulfate and 25 g of ammonium molybdate to 450 mL water. To this, add 50 mL concentrated sulfuric acid dropwise over one hour. Filter as needed.
6.2.1. Synthesis of (S)-dolichols
Polyprenols are first extracted from the leaves of Rhus typhina (staghorn sumac) (Swiezewska et al. 1994), which affords a distribution of C50–65 isomers. The isolated polyprenols are further fractionated to obtain a 5:1 mixture of C55:C60 linear polyprenols. The purified polyprenols are then subject to regioselective asymmetric hydrogenation to yield the (C55–60) (S)-dolichols according to the procedure of Sowa and coworkers (Wu, Beauchamps, Laquidara & Sowa 2012). The (S)-dolichols are derived from plant sources and therefore include three E-isoprene units, in contrast to the non-plant derived linear polyprenols that include two E-isoprene units and an additional Z-isoprene unit.
Protocol:
Dissolve a sample of linear polyprenols (C55–60 5:1, 0.08 g, 0.10 mmol) in toluene and concentrate by rotary evaporation.
Add (S)-tol-BINAP RuCl2 (p-cymene) (0.01 g, 0.01 mmol) and KOH (0.001 g, 0.02 mmol) to the polyprenols and dry under vacuum. Backfill N2 into the flask and reestablish the vacuum for 1 h.
In a separate flask, add n-propanol (anhydrous, 10 mL) to activated 4 Å molecular sieves and sparge with N2 for 30 min. Transfer the sparged n-propanol to the flask containing polyprenols, (S)-tol-BINAP RuCl2 and KOH, and immediately purge the flask of atmosphere and backfill with N2. Repeat this process three times, then add a dry stir bar and allow the reaction to proceed for 8 h under N2.
Monitor the reaction by NMR analysis (Larkin, Chang, Whitworth & Imperiali 2013). Upon completion, add silica gel to the mixture, and flash silica gel column chromatography (toluene:EtOAc, 49:1, v/v) affords the desired (S)-dolichols (5:1 C55–60 mixture) as a colorless oil with typical yields of approximately 90%.
6.2.2. Synthesis of (S)-dolichol phosphate
Dolichol phosphate is prepared essentially as described by Branch and coworkers (Branch, Burton & Moss 1999).
Protocol:
Dissolve a sample of short (S)-dolichols (C55–60, 0.02 g, 0.003 mmol) in THF (anhydrous, 1 mL) under argon, then add tetrazole (0.006 g, 0.008 mmol) and bis-(2-cyanoethyl)-N,N-diisopropylphosphoramidite (0.03 g, 0.009 mmol) and stir the solution for 2 h.
Cool the solution to −30 °C and add 30% hydrogen peroxide (0.2 mL). After stirring for 10 min, warm the mixture to room temperature.
Rapidly dilute the reaction with Na2SO3 (10%, 5 mL) and extract with EtOAc (3 × 10 mL). Wash the combined organic extracts with NaHCO3 (5%, 2 mL), brine (5 mL), and MgSO4, then concentrate by rotary evaporation under vacuum.
Dissolve the resulting oil in MeOH (anhydrous, 5 mL), introduce a stoichiometric equivalent of NaOMe, and stir the mixture for 2 d.
Carry out flash column chromatography on silica with a gradient of EtOAc to EtOAc:MeOH (5:2) to isolate the desired Dol-P with typical yield of 70%.
Characterize the material 1H and 31P NMR analysis to verify the correct product (Larkin, Chang, Whitworth & Imperiali 2013). Dol-P quantification is carried out using a standard phosphate quantification protocol (Chen, Toribara & Warner 1956).
Note: If preferred, dolichol phosphate and other polyprenol phosphates can also be prepared via a chemoenzymatic approach using the promiscuous diacylglycerol kinase (DAGK) from Streptococcus mutans (Hartley, Larkin & Imperiali 2008 ). This approach is particularly valuable for the preparation of [32P]- and [33P]-labeled polyprenol phosphates.
6.2.3. Dol-P-GlcNAc-β-(1–3)-2,3-diNAcGlcA synthesis with AglK and AglC
In the first membrane-committed step of the M. voltae pathway, AglK is a specific Dol-P-GlcNAc synthase that utilizes UDP-GlcNAc and (C55–60) Dol-P to produce α-linked Dol-P-GlcNAc. AglC is a UDP-2,3-diNAcGlcA glycosyltransferase that converts the AglK product, Dol-P-GlcNAc, to Dol-P-GlcNAc-β-(1–3)-2,3-diNAcGlcA. The expression and purification of AglK and AglC, as well as preparative AglK and AglC reactions to generate Dol-P-GlcNAc-β-(1–3)-2,3-diNAcGlcA are presented here (Larkin, Chang, Whitworth & Imperiali 2013).
Protocol:
Transform plasmids for AglK and AglC into E. coli BL21-CodonPlus(DE3) RIL competent cells with KAN and CAM as selection markers for expression as proteins with N-terminal T7 and C-terminal His6 tags (see Table 1.)
Inoculate 1 L of LB media, supplemented with KAN (50 μg/mL) and CAM (30 μg/mL), with 5 mL of starter culture and incubate at 37 °C with shaking until an OD600 of 0.6–0.8 is obtained. Lower the temperature to 16 °C and induce with 1 mM IPTG.
After 16 h, harvest the cells by centrifugation (5,000 x g for 15 min) and store at −80 °C until needed.
Perform all cell lysis and protein purification steps at 4 °C. Resuspend cell pellets 50 mL of lysis buffer and 5 μL protease inhibitor cocktail, then lyse cells by sonication on ice (50% amplitude, 1 s pulses, 3 × 90 s).
To prepare cell membrane fractions, remove cellular debris from the cell lysate by centrifugation (6,000 x g for 30 min), saving the supernatant. Then pellet the membranes (145,000 x g for 1 h) and discard the supernatant. Resuspend the membrane pellet in 2 mL of membrane resuspension buffer and store at −80 °C.
To purify AglK and AglC proteins away from crude membrane fractions, solubilize the membranes for 1 h in membrane resuspension buffer supplemented with 1% n-dodecyl-β-D-maltoside (DDM) and centrifuge (145,000 x g for 1 h).
Incubate the supernatant with Ni-NTA agarose resin (1 mL) for 3 h with gentle rocking and then pour into a chromatography column. Wash the resin with 25 ml of Ni-NTA wash buffer, and elute the proteins with 20 ml of Ni-NTA elution buffer. Collect the fractions containing protein, as determined by SDS-PAGE, and dialyze (10 kDa MWCO) overnight at 4 °C.
Quantify AglK and AglC concentrations by UV-Vis absorption spectroscopy, using the predicted extinction coefficients (generated by http://web.expasy.org/protparam/) at λ = 280 nm.
To synthesize Dol-P-GlcNAc, resuspend dried (S)-Dol-P (200 nmol) in DMSO (30 μL) by vortexing, followed by the addition of 0.715% DDM (70 μL), 0.5 M HEPES, pH 7.5 (100 μL), 1 M MgCl2 (10 μL), 100 mM DTT (20 μL), UDP-GlcNAc (250 nmol), and AglK (0.5 μM), along with water for a final volume of 1 mL. Incubate the reaction at 25 °C for 3 h. Quench the reaction by dilution into 2:1 CHCl3:MeOH (12 ml), followed by the addition of 3 mL pure solvent upper phase (PSUP) (Folch, Lees & Sloane Stanley 1957). Extract the organic layer three times with PSUP (3 mL) before drying the Dol-P-GlcNAc product under streaming N2.
To synthesize Dol-P-GlcNAc-β-(1–3)-2,3-diNAcGlcA, resuspend dried Dol-P-GlcNAc (50 nmol) in DMSO (15 μL) by vortexing, followed by the addition of 0.715% DDM (35 μL), 0.5 M HEPES, pH 7.5 (50 μL), 1 M MgCl2 (5 μL), 100 mM DTT (10 μL), UDP-2,3-diNAcGlcA (50 nmol), and AglC (0.5 μM), along with water for a final volume of 0.5 mL. Incubate the reaction at 25 °C for 3 h. Quench and extract as described in Step 9 for the AglK reaction. Dry the Dol-P-GlcNAc-β-(1–3)-2,3-diNAcGlcA product under streaming N2.
Purify the crude Dol-P-glycans products using a normal phase Varian Microsorb HPLC column, separating over 21–28% NP-HPLC Solvent B.
Monitor elution of Dol-P-saccharides by running TLC plates in TLC solvent and staining with CAM or by scintillation counting for the radiolabeled products.
Notes:
UDP-[3H]GlcNAc can be used to generate radiolabeled Dol-P-[3H]GlcNAc to monitor the reaction progress by liquid scintillation counting analysis and to have radiolabeled substrate for future studies.
UDP-2-[3H]3-diNAcGlcA can be used to generate radiolabeled Dol-P-GlcNAc-β-(1–3)-2-[3H]3-diNAcGlcA to monitor the reaction progress and to have radiolabeled donor substrate for OTase investigations.
To scale up either the AglK or AglC reaction, keep the nmol of starting Dol-P or Dol-P-GlcNAc constant and increase the number of reactions instead of increasing the volume in one reaction.
Conclusions
A major obstacle to investigating the biosynthetic pathways and biological roles of prokaryotic glycoconjugates is the limited availability chemically-defined carbohydrate reagents, including nucleotide- and polyprenol phosphate-activated glycosyl donors. This issue is exacerbated by the densely-modified nature of key carbohydrate building blocks that feature in prokaryotes, which render total synthesis approaches challenging for all but the experienced synthetic chemist. To address this current need and enable studies in prokaryotic glycobiology, we have established straightforward chemoenzymatic approaches for milligram-scale preparation and purification of 8 different prokaryote-specific monosaccharides donors, which are activated as their biologically-relevant nucleotide derivatives. These chemically well-defined derivatives include intermediates from three different pathways. We also report the synthesis of a (S)-dolichol phosphate acceptor and the resulting polyprenol phosphate-linked disaccharide Dol-P-GlcNAc-β(1–3)-2,3-diNAcGlcA to illustrate the utility of the unusual UDP-sugar derivatives in defining an alternate pathway for protein glycosylation in archaea.
Prokaryote-specific carbohydrates are recurring features of diverse glycoconjugates in bacteria and archaea. Therefore, each established and validated chemoenzymatic synthesis can be useful for many researchers even though their focus may be on a different organism or pathway. In this context, we have shown that it can be advantageous to apply homologous enzymes from different pathways to gain the advantage of better catalytic properties and enzyme stability for specific steps. This indeed proved to be key to the development of an optimized protocol for the preparation of >100 mg of UDP-4-amino-2-acetamido-Bac for high throughput inhibitor screening, where C. jejuni and N. gonorrhoeae enzymes, from N- and O-linked protein glycosylation pathways respectively, were applied in sequence. Similarly, it proved advantageous in the study of M. voltae protein glycosylation to apply four of the five enzymes from a well-annotated P. aeruginosa pathway, rather than the less-defined M. voltae enzymes, to prepare a critical UDP-sugar that was essential for unraveling the fascinating details of the archaeal pathway. It is also valuable to implement simple chemical alternatives for selected transformations, such as a PtO2-catalyzed oxidation for converting UDP-GlcNAc to the C-6 carboxylate derivative instead of WbpA, and, chemical N-acetylation instead of the PglD- and PseH-catalyzed processes, which require a costly AcCoA cosubstrate. Nevertheless, in some cases, for example for the introduction of isotopic labeling ([3H] or [14C]) into N-acetamido-sugars, the corresponding enzymatic transformation may be preferable as it is more efficient at small scale and radiolabeled precursors are readily available.
In the application of sequential enzymatic processes, we also demonstrate that it is can be critical to couple sequential transformations when particular intermediates exhibit chemical lability, as observed with the facile epimerization of the UDP-ketosugar intermediate in the H. pylori pse pathway. In this context we also found it advantageous to apply enzymes in an immobilized format, for example using existing GST- and His-coexpression tags to simplify reaction work up and minimize delays associated with handling. Optimization of coexpression tags is also valuable. For example, in the case of the C. jejuni PglF, the expression and stability of a truncation variant, in which the non-essential membrane-associated domains were replaced by GST (in GST-PglFΔ1–130), provided considerable advantages in the chemoenzymatic synthesis of UDP-Bac derivatives.
In establishing these methods, we have defined several useful principles that can be applied to the chemoenzymatic syntheses of other high-value carbohydrate reagents. These practical aspect of the approaches will be critical as the needs of the community expand in concert with the growing appreciation of the key biological roles of prokaryotic glycoconjugates.
Fig. 7.
Steady State kinetics for C. jejuni acetyltransferase PseH.
Acknowledgements
We gratefully acknowledge support from the NIH (GM-097241) for our work on prokaryote-specific carbohydrates. We also thank Drs. Angelyn Larkin, Garrett Whitworth and Michael J. Morrison for their contributions towards the establishment of these methods.
References
- Aas FE, Vik A, Vedde J, Koomey M, & Egge-Jacobsen W (2007). Neisseria gonorrhoeae O-linked pilin glycosylation: Functional analyses define both the biosynthetic pathway and glycan structure, Molecular Microbiology, 65(3), 607–24. 10.1111/j.1365-2958.2007.05806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balonova L, Hernychova L, & Bilkova Z (2009). Bioanalytical tools for the discovery of eukaryotic glycoproteins applied to the analysis of bacterial glycoproteins, Expert Reviews Proteomics, 6(1), 75–85. 10.1586/14789450.6.1.75. [DOI] [PubMed] [Google Scholar]
- Barnes J, Tian L, Loftis J, Hiznay J, Comhair S, Lauer M, & Dwek R (2016). Isolation and analysis of sugar nucleotides using solid phase extraction and fluorophore assisted carbohydrate electrophoresis, MethodsX, 3, 251–60. 10.1016/j.mex.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branch CL, Burton G, & Moss SF (1999). An expedient synthesis of allylic polyprenyl phosphates, Synthetic Communications, 29(15), 2639–44. doi: <Go to ISI>://000081112800013 [Google Scholar]
- Calo D, Kaminski L, & Eichler J (2010). Protein glycosylation in archaea: Sweet and extreme, Glycobiology, 20(9), 1065–76. 10.1093/glycob/cwq055. [DOI] [PubMed] [Google Scholar]
- Castric P, Cassels FJ, & Carlson RW (2001). Structural characterization of Pseudomonas aeruginosa 1244 pilin glycan, Journal of Biological Chemistry, 276, 26479–85. [DOI] [PubMed] [Google Scholar]
- Chaban B, Voisin S, Kelly J, Logan SM, & Jarrell KF (2006). Identification of the genes involved in the biosynthesis and attachment of Methanococcus voltae N-linked glycosylation pathways in archaea, Molecular Microbiology, 61, 259–68. [DOI] [PubMed] [Google Scholar]
- Chen PS, Toribara TY, & Warner H (1956). Microdetermination of phosphorus, Analytical Chemistry, 28, 1756–57. [Google Scholar]
- Chowdhury TA, Jansson PE, Lindberg B, J. L, Gustafsson B, & Holme T (1991). Structural studies of the Vibrio cholera O:3 O-antigen polysaccharide, Carbohydrate Research, 215(303–14. [DOI] [PubMed] [Google Scholar]
- De Schutter JW, Morrison JP, Morrison MJ, Ciulli A, & Imperiali B (2017). Targeting bacillosamine biosynthesis in bacterial pathogens: Development of inhibitors to a bacterial amino-sugar acetyltransferase from Campylobacter jejuni, Journal of Medicinal Chemistry. 10.1021/acs.jmedchem.6b01869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellman GL (1959). Tissue sulfhydryl groups, Archives of Biochemistry and Biophysics, 82(1), 70–7. doi: https://www.ncbi.nlm.nih.gov/pubmed/13650640. [DOI] [PubMed] [Google Scholar]
- Ewing CP, Andreishcheva E, & Guerry P (2009). Functional characterization of flagellin glycosylation in Campylobacter jejuni 81–176, Journal of Bacteriology, 191(22), 7086–93. 10.1128/JB.00378-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Lees M, & Sloane Stanley GH (1957). A simple method for the isolation and purification of total lipides from animal tissues, Journal of Biological Chemistry, 226(1), 497–509. doi: https://www.ncbi.nlm.nih.gov/pubmed/13428781. [PubMed] [Google Scholar]
- Glaze PA, Watson DC, Young NM, & Tanner ME (2008). Biosynthesis of CMP-N,N’-diacetyllegionaminic acid from UDP-N,N’-diacetylbacillosamine in Legionella pneumophila, Biochemistry, 47(10), 3272–82. 10.1021/bi702364s. [DOI] [PubMed] [Google Scholar]
- Glover JK, Weerapana E, Chen MM, & Imperiali B (2006). Direct biochemical evidence for the utilization of UDP-bacillosamine by pglc, an essential glycosyl-1-phosphate-transferase in the C. jejuni N-linked glycosylation pathway, Biochemistry, 45, 5343–50. [DOI] [PubMed] [Google Scholar]
- Guerry P, Ewing CP, Schirm M, Lorenzo M, Kelly J, Pattarini D, … Logan S (2006). Changes in flagellin glycosylation affect campylobacter autoagglutination and virulence, Molecular Microbiology, 60(2), 299–311. 10.1111/j.1365-2958.2006.05100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley MD, & Imperiali B (2012). At the membrane frontier: A prospectus on the remarkable evolutionary conservation of polyprenols and polyprenyl-phosphates, Arch Biochem Biophys, 517(2), 83–97. 10.1016/j.abb.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley MD, Morrison MJ, Aas FE, Borud B, Koomey M, & Imperiali B (2011). Biochemical characterization of the O-linked glycosylation pathway in neisseria gonorrhoeae responsible for biosynthesis of protein glycans containing N,N’-diacetylbacillosamine, Biochemistry, 50(22), 4936–48. 10.1021/bi2003372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley MD, Schneggenburger PE, & Imperiali B (2013). Lipid bilayer nanodisc platform for investigating polyprenol-dependent enzyme interactions and activities, Proceedings of the National Academy of Sciences USA, 110(52), 20863–70. 10.1073/pnas.1320852110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley MH, Larkin A, & Imperiali B (2008. ). Chemoenzymatic synthesis of polyprenyl phosphates, Bioorganic and Medicinal Chemistry, 16, 5149–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegge FT, Hitchen PG, Aas FE, Kristiansen H, Lovold C, Egge-Jacobsen W, … Koomey M (2004). Unique modifications with phosphocholine and phosphoethanolamine define alternate antigenic forms of Neisseria gonorrhoeae type iv pili, Proceedings of the National Academy Sciences USA, 101(29), 10798–803. doi: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15249686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchen PG, & Dell A (2006). Bacterial glycoproteomics, Microbiology, 152(Pt 6), 1575–80. 10.1099/mic.0.28859-0. [DOI] [PubMed] [Google Scholar]
- Hsu KL, & Mahal LK (2006). A lectin microarray approach for the rapid analysis of bacterial glycans, Nature Protocols, 1(2), 543–9. 10.1038/nprot.2006.76. [DOI] [PubMed] [Google Scholar]
- Jarrell KF, Ding Y, Meyer BH, Albers SV, Kaminski L, & Eichler J (2014). N-linked glycosylation in archaea: A structural, functional, and genetic analysis, Microbiology Molecular Biology Reviews, 78(2), 304–41. 10.1128/MMBR.00052-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly J, Jarrell H, Millar L, Tessier L, Fiori LM, Lau PC, … Szymanski CM (2006). Biosynthesis of the N-linked glycan in Campylobacter jejuni and addition onto protein through block transfer, Journal of Bacteriology, 188, 2427–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knirel YA, Vinogradov EV, L’Vov V L, Kocharova NA, Shashkov AS, Dmitriev BA, & Kochetkov NK (1984). Sialic acids of a new type from the lipopolysaccharides of Pseudomonas aeruginosa and shigella boydii, Carbohydrate Research, 133(2), C5–8. doi: https://www.ncbi.nlm.nih.gov/pubmed/6437679. [DOI] [PubMed] [Google Scholar]
- Koplin R, Brisson JR, & Whitfield C (1997). Udp-galactofuranose precursor required for formation of the lipopolysaccharide O antigen of klebsiella pneumoniae serotype o1 is synthesized by the product of the rfbdkpo1 gene, Journal of Biological Chemistry, 272(7), 4121–8. doi: https://www.ncbi.nlm.nih.gov/pubmed/9020123. [DOI] [PubMed] [Google Scholar]
- Larkin A, Chang MM, Whitworth GE, & Imperiali B (2013). Biochemical evidence for an alternate pathway in N-linked glycoprotein biosynthesis, Nature Chemical Biology, 9(6), 367–73. 10.1038/nchembio.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin A, & Imperiali B (2009). Biosynthesis of UDP-GlcNac(3NAc)A by WbpB, WbpE, and WbpD: Enzymes in the wbp pathway responsible for O-antigen assembly in Pseudomonas aeruginosa pao1, Biochemistry, 48(23), 5446–55. 10.1021/bi900186u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YJ, Kubota A, Ishiwata A, & Ito Y (2011). Synthesis of pseudaminic acid, a unique nonulopyranoside derived from pathogenic bacteria through 6-deoxy-AltdiNAc, Tetrahedron Letters, 52(3), 418–21. 10.1016/j.tetlet.2010.11.078. [DOI] [Google Scholar]
- Leeflang BR, Faber EJ, Erbel P, & Vliegenthart JF (2000). Structure elucidation of glycoprotein glycans and of polysaccharides by nmr spectroscopy, Journal of Biotechnology, 77(1), 115–22. doi: https://www.ncbi.nlm.nih.gov/pubmed/10674218. [DOI] [PubMed] [Google Scholar]
- Liu F, & Tanner ME (2006). PseG of pseudaminic acid biosynthesis: A UDP-sugar hydrolase as a masked glycosyltransferase, Journal of Biological Chemistry, 281(30), 20902–9. 10.1074/jbc.M602972200. [DOI] [PubMed] [Google Scholar]
- Lukose V, Whitworth G, Guan Z, & Imperiali B (2015). Chemoenzymatic assembly of bacterial glycoconjugates for site-specific orthogonal labeling, Journal of the Americal Chemical Society, 137(39), 12446–9. 10.1021/jacs.5b07146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki M, & Renkonen R (2004). Biosynthesis of 6-deoxyhexose glycans in bacteria, Glycobiology, 14(3), 1r–15r. doi: <Go to ISI>://000189140600001. [DOI] [PubMed] [Google Scholar]
- McCallum M, Shaw GS, & Creuzenet C (2013). Comparison of predicted epimerases and reductases of the Campylobacter jejuni D-altro- and L-gluco-heptose synthesis pathways, Journal of Biological Chemistry, 288(27), 19569–80. 10.1074/jbc.M113.468066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messner P (2009). Prokaryotic protein glycosylation is rapidly expanding from “curiosity” to “ubiquity”, ChemBioChem, 10(13), 2151–4. 10.1002/cbic.200900388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller WL, Wenzel CQ, Daniels C, Larocque S, Brisson JR, & Lam JS (2004). Biochemical characterization of WbpA, a UDP-n-acetyl-d-glucosamine 6-dehydrogenase involved in O-antigen biosynthesis in Pseudomonas aeruginosa pao1, Journal of Biological Chemistry, 279, 37551–58. [DOI] [PubMed] [Google Scholar]
- Morrison MJ PhD Thesis 2014. ‘Elucidation of the pathways responsible for the biosynthesis of UDP-N,N’-diacetyl bacillosamine in bacterial pathogens’, Massachusetts Institute of Technology. [Google Scholar]
- Morrison MJ, & Imperiali B (2014). The renaissance of bacillosamine and its derivatives: Pathway characterization and implications in pathogenicity, Biochemistry, 53(4), 624–38. 10.1021/bi401546r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musial-Siwek M, Jaffee MB, & Imperiali B (2016). Probing polytopic membrane protein-substrate interactions by luminescence resonance energy transfer, Journal of the American Chemical Society, 138(11), 3806–12. 10.1021/jacs.5b13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nothaft H, & Szymanski CM (2010). Protein glycosylation in bacteria: Sweeter than ever, Nature Reviews Microbiology, 8(11), 765–78. 10.1038/nrmicro2383. [DOI] [PubMed] [Google Scholar]
- Olivier NB, Chen MM, Behr JR, & Imperiali B (2006). In vitro biosynthesis of UDP-N,N’-diacetylbacillosamine by enzymes of the Campylobacter jejuni general protein glycosylation system, Biochemistry, 45, 13659–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid CW, Stupak J, Szymanski CM, & Li J (2009). Analysis of bacterial lipid-linked oligosaccharide intermediates using porous graphitic carbon liquid chromatography-electrospray ionization mass spectrometry: Heterogeneity in the polyisoprenyl carrier revealed, Analytical Chemistry, 81(20), 8472–8. 10.1021/ac9013622. [DOI] [PubMed] [Google Scholar]
- Rejzek M, Mukhopadhyay B, Wenzel CQ, Lam JS, & Field RA (2007). Direct oxidation of sugar nucleotides to the correspondinguronic acids: Tempo and platinum-based procedures, Carbohydrate Research, 342, 460–66. [DOI] [PubMed] [Google Scholar]
- Rejzek M, Sri Kannathasan V, Wing C, Preston A, Westman EL, Lam JS, … Field RA (2009). Chemical synthesis of UDP-Glc-2,3-diNAcA, a key intermediate in cell surface polysaccharide biosynthesis in the human respiratory pathogens B. pertussis and P. aeruginosa, Organic and Biological Chemistry, 7(6), 1203–10. 10.1039/b819607a. [DOI] [PubMed] [Google Scholar]
- Schirm M, Soo EC, Aubry AJ, Austin J, Thibault P, & Logan SM (2003). Structural, genetic and functional characterization of the flagellin glycosylation process in helicobacter pylori, Molecular Microbiology, 48(6), 1579–92. doi: <Go to ISI>://000183369500012. [DOI] [PubMed] [Google Scholar]
- Schmidt MA, Riley LW, & Benz I (2003). Sweet new world: Glycoproteins in bacterial pathogens, Trends in Microbiology, 11(12), 554–61. doi: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=14659687. [DOI] [PubMed] [Google Scholar]
- Schoenhofen IC, McNally DJ, Brisson JR, & Logan SM (2006). Elucidation of the cmp-pseudaminic acid pathway in helicobacter pylori: Synthesis from UDP-N-acetylglucosamine by a single enzymatic reaction, Glycobiology, 16(9), 8C–14C. 10.1093/glycob/cwl010. [DOI] [PubMed] [Google Scholar]
- Schoenhofen IC, McNally DJ, Vinogradov E, Whitfield D, Young NM, Dick S, … Logan SM (2006). Functional characterization of dehydratase/aminotransferase pairs from helicobacter and campylobacter: Enzymes distinguishing the pseudaminic and bacillosamine biosynthetic pathways Journal of Biological Chemistry, 281(2), 723–32. doi: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16286454. [DOI] [PubMed] [Google Scholar]
- Schoenhofen IC, Vinogradov E, Whitfield DM, Brisson JR, & Logan SM (2009). The CMP-legionaminic acid pathway in Campylobacter: Biosynthesis involving novel GDP-linked precursors, Glycobiology, 19(7), 715–25. 10.1093/glycob/cwp039. [DOI] [PubMed] [Google Scholar]
- Scott NE, & Cordwell SJ (2015). Enrichment and identification of bacterial glycopeptides by mass spectrometry, Methods in Molecular Biology, 1295, 355–68. 10.1007/978-1-4939-2550-6_25. [DOI] [PubMed] [Google Scholar]
- Sharon N (2007). Celebrating the golden anniversary of the discovery of bacillosamine, the diamino sugar of a bacillus, Glycobiology, 17, 1150–55. [DOI] [PubMed] [Google Scholar]
- Sharon N, & Jeanloz RW (1960). The diaminohexose component of a polysaccharide isolated from Bacillus subtilis, Journal of Biological Chemistry, 235, 1–5. doi: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=14445588. [PubMed] [Google Scholar]
- Singh S, Singh U, Hogan SE, & Feingold DS (1990). Formation of UDP-2-acetamido-2-deoxy-l-galactose and UDP-2-acetamido-2-deoxy-L-galacturonic acid by Pseudomonas aeruginosa, Journal of Bacteriology, 172, 299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stimson E, Virji M, Makepeace K, Dell A, & Morris HR (1995). Meningococcal pilin: A glycoproteinsubstituted with digalactosyl 2,4-diacetamido-2,4,6-trideoxyhexose, Molecular Microbiology, 17, 1201–04. [DOI] [PubMed] [Google Scholar]
- Swiezewska E, Sasak W, Mankowski T, Jankowski W, Vogtman T, Krajewska I, … Chojnacki T (1994). The search for plant polyprenols, Acta Biochimica Polonica, 41(3), 221–60. doi: https://www.ncbi.nlm.nih.gov/pubmed/7856395. [PubMed] [Google Scholar]
- Thibault P, Logan SM, Kelly JF, Brisson JR, Ewing CP, Trust TJ, & Guerry P (2001). Identification of the carbohydrate moieties and glycosylation motifs in Campylobacter jejuni flagellin, Journal of Biological Chemistry, 276(37), 34862–70. doi: <Go to ISI>://000171024600070. [DOI] [PubMed] [Google Scholar]
- VanDyke DJ, Wu J, Logan SM, Kelly JF, Mizuno S, Aizawa S, & Jarrell KF (2009). Identification of genes involved in the assembly and attachment of a novel flagellin N-linked tetrasaccharide important for motility in the archaeon Methanococcus maripaludis, Molecular Microbiology, 72(3), 633–44. 10.1111/j.1365-2958.2009.06671.x. [DOI] [PubMed] [Google Scholar]
- Voisin S, Houliston RS, Kelly J, Brisson J-R, Watson D, Bardy SL, … M. LS (2005). Identification and characterization of the unique N-linked glycan common to the flagellins and S-layer glycoprotein of Methanococcus voltae, J Biol Chem, 280, 16586–93. [DOI] [PubMed] [Google Scholar]
- Williams JT, Corcilius L, Kiefel MJ, & Payne RJ (2016). Total synthesis of native 5,7-diacetylpseudaminic acid from N-acetylneuraminic acid, Journal of Organic Chemistry, 81(6), 2607–11. 10.1021/acs.joc.5b02754. [DOI] [PubMed] [Google Scholar]
- Wu R, Beauchamps MG, Laquidara JM, & Sowa JR Jr. (2012). Ruthenium-catalyzed asymmetric transfer hydrogenation of allylic alcohols by an enantioselective isomerization/transfer hydrogenation mechanism, Angewandte Chemie International Edition, 51(9), 2106–10. 10.1002/anie.201107910. [DOI] [PubMed] [Google Scholar]
- Young NM, Brisson JR, Kelly J, Watson DC, Tessier L, Lanthier PH, … Szymanski CM (2002). Structure of the N-linked glycan present on multiple glycoproteins in the gram-negative bacterium, campylobacter jejuni, Journal of Biological Chemistry, 277(45), 42530–39. doi: <Go to ISI>://000179081200015. [DOI] [PubMed] [Google Scholar]
- Zunk M, Williams J, Carter J, & Kiefel MJ (2014). A new approach towards the synthesis of pseudaminic acid analogues, Org Biomol Chem, 12(18), 2918–25. 10.1039/c3ob42491j. [DOI] [PubMed] [Google Scholar]