

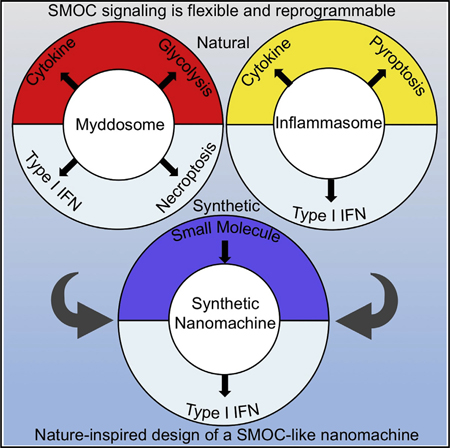
SUMMARY
The signaling organelles of the innate immune system consist of oligomeric protein complexes known as supramolecular organizing centers (SMOCs). Examples of SMOCs include myddosomes and inflammasomes, which respectively induce transcription-dependent and -independent inflammatory responses. The common use of oligomeric structures as signaling platforms suggests multifunctionality, but each SMOC has a singular biochemically defined function. Here, we report that the myddosome is a multifunctional organizing center. In addition to promoting inflammatory transcription factor activation, the myddosome drives the rapid induction of glycolysis. We identify the kinase TBK1 as a myddosome component that promotes glycolysis, but not nuclear factor κB (NF-κB) activation. Synthetic immunology approaches further diversified SMOC activities, as we created interferon- or necroptosis-inducing myddosomes, inflammasomesthat induce interferon responses instead of pyroptosis, and a SMOC-like nanomachine that induces interferon expression in response to a chemical ligand. These discoveries demonstrate the flexibility of immune signaling organelles, which permits the design of user-defined innate immune responses.
In Brief
Innate immune signaling complexes are multifunctional organizing centers that can be rewired to induce user-defined cellular outcomes.
Graphical abstract
INTRODUCTION
The ability to detect and respond to environmental stresses represents one of the key features of living organisms. In the context of host-pathogen interactions, the innate immune system provides a faithful illustration to this principle of life, as failure to rapidly sense or respond to pathogens would cast a fatal stress on the host (Pandey et al., 2014).
Microbial sensing is achieved by a number of structurally unrelated proteins known as pattern recognition receptors (PRRs) (Janeway, 1989). These receptors detect pathogen associated molecular patterns (PAMPs), which are conserved structural components associated uniquely with microorganisms (Pandey et al., 2014). Detection of PAMPs and other microbial activities by PRRs engages numerous cellular processes to eliminate infection and restore homeostasis (Vance et al., 2009). Based on their primary sequence homology, most PRRs are categorized into distinct groups, which include the Toll-like receptors (TLRs), the C-type lectin receptors (CLRs), the nucleotide-binding domain, leucine rich repeat (LRR)-containing proteins (NLRs), and the AIM2-like receptors (ALRs) (Brubaker et al., 2015). Despite this diversity of receptors, unifying themes exist that govern the operation of innate immune signaling pathways. For example, at the level of microbial detection, the concept of pattern recognition explains the activities of structurally distinct families of PRRs (Janeway, 1989). Downstream of microbial detection, however, unifying concepts associated with signal transduction are limited. Indeed, much research has focused on identifying cellular processes and factors that are unique to a specific signaling pathway.
Recent biochemical and structural studies have provided hints that common themes in innate immune signal transduction may exist. For example, PRRs of theTLR, NLR, and RLR families seed the formation of large oligomeric protein complexes that consist of a receptor, an adaptor, and effector enzymes (Kagan et al., 2014). In the TLR pathway, the oligomeric complex is the myddosome and consists of a TLR, the adaptors TIRAP and MyD88, and enzymes of the IRAK family of kinases (Bonham et al., 2014; Lin et al., 2010; Ve et al., 2017). In the NLR pathway, the best-defined oligomeric complex is the inflammasome, which commonly consists of an NLR, the adaptor ASC, and enzymes of the caspase family of proteases (most commonly caspase-1) (Lu et al., 2014). Finally, the oligomeric complex associated with RLR signaling consists of the receptor RIG-I (or MDA5), the MAVS adaptor, and the enzyme tank binding kinase-1 (TBK1) (Jiang et al., 2012). While these complexes share the physiological activity of regulating host defense, they do not share any specific components. Evolutionary pressures may have therefore driven multiple unrelated proteins to organize themselves into common oligomeric structures that ensure host defense. Why would oligomeric protein complexes be commonly utilized by distinct PRRs? One possible explanation is that these complexes provide a scaffold that is modular by design, such that diverse upstream inputs (microbes) can induce their assembly. Once assembled, diverse downstream outputs (defense mechanisms) can be induced via components with distinct effector domains. This idea prompted the classification of these structures as supramolecular organizing centers (SMOCs), which represent the principal subcellular sites of signal transduction and are therefore considered the signaling organelles of the innate immune system (Kagan et al., 2014). However, experimental evidence supporting this speculation has remained sparse. For example, while PRRs are recognized for their ability to induce diverse cellular responses, many of these responses could be explained by the distinct functions of PRR-induced gene products.
The TLR-induced myddosome provides a model to examine the central prediction of the SMOC hypothesis—that these structures represent sites where diverse effector responses emanate. TLRs are transmembrane proteins that reside on the plasma and endosomal membranes (Pandey et al., 2014). They detect a wide range of PAMPs, including bacterial lipopolysaccharides (LPS), lipoproteins, flagellin, and nucleic acids (Pandey et al., 2014). Signal transduction in theTLR pathway is regulated by two SMOCs—the aforementioned myddosome and the poorly defined triffosome (Gay et al., 2014; Lin et al., 2010). The core of the myddosome contains the adaptor MyD88, and the core of the triffosome is thought to contain the adaptor TRIF (Gay et al., 2014). All TLRs induce MyD88-dependent responses, except for TLR3, leading to activation of the inflammatory transcription factors NF-κB and AP-1 (Medzhitov and Horng, 2009). The triffosome is proposed to be assembled by TLR3 or TLR4 to enhance myddosome-dependent NF-κB and AP-1 activation, and to drive type I interferon (IFN) expression (Medzhitov and Horng, 2009). Triffosome-induced IFN expression is linked to its ability to prompt TBK1 to activate the IFN-inducing transcription factor IRF3 (Fitzgerald et al., 2003; Yamamoto et al., 2003). Notably, MyD88-deficient cells display defects in TBK1 activation, but the mechanisms and consequences of this activity are unclear, as MyD88 does not activate IRF3 or induce type I IFN in macrophages (Clark et al., 2011).
In addition to directing pro-inflammatory transcriptional programs, TLRs induce alterations in cellular metabolism (O’Neill et al., 2016). Such metabolic reprogramming is exemplified by the TLR-dependent rapid activation of glycolysis (Everts et al., 2014). While glycolysis induction is increasingly recognized for its importance in inflammation, the means by which TLRs promote this response is unknown (Everts et al., 2014). In particular, the relative roles of the myddosome and triffosome in directing glycolysis are unclear. Also unclear, is whether signals within these SMOCs drive glycolysis directly or indirectly through the upregulation of genes encoding glycolysis-regulatory factors (Tannahill et al., 2013).
Here, we provide direct evidence supporting the proposal that SMOCs are organizing centers, in that the myddosome is the source of diverse effector activities induced upon TLR activation. We identify TBK1 as a novel component of the myddosome and find that this kinase is not necessary for NF-κB or AP-1 activation. Rather, myddosome-associated TBK1 is necessary to induce aerobic glycolysis. Synthetic immunology approaches further diversified SMOC activities, as we engineered myddosomes to induce IFN expression or necroptosis and inflammasomes to induce IFN expression instead of pyroptosis. These findings demonstrate the flexibility of the effector functions of the signaling organelles of our innate immune system.
RESULTS
A MyD88-TBK1 Pathway Commonly Promotes TLR-Dependent Glycolysis
We sought to determine whether all TLRs promote glycolysis in macrophages. To this end, we monitored the extracellular acidification rate (ECAR) resulting from the release of lactate (an end product of glycolysis) into the tissue culture medium (Pelgrom et al., 2016). Stimulation of primary bone-marrow-derived macrophages (BMDMs) with seven ligands that stimulate nine TLRs increased ECAR rapidly (Figure 1A). These ligands stimulate TLR1/TLR2 (P3C), TLR2/TLR6 (P2C), TLR3 (Poly(I:C)), TLR4 (LPS), TLR7/TLR8 (R848), TLR9 (CpG DNA), or TLR13 (ORN Sa19). A detailed analysis was subsequently focused on LPS, P3C, and R848 stimulation, which indicated that these stimuli do not cause discernible changes in mitochondrial oxygen consumption (OCR) during the time frame where glycolysis is induced (Figures S1A and S1B). These findings were made in primary and immortal BMDMs (iBMDMs). The observed increases in ECAR represents glycolysis induction, as pre-treatment of primary BMDMs with 2-DG, a specific inhibitor of the glycolysis regulator hexokinase (Grossbard and Schimke, 1966), blocked TLR-induced ECAR increases (Figure 1B). We found that the glycolytic burst induced by TLR ligands proceeded independent of transcription, as BMDMs treated with actinomycin D were still able to increase ECAR (Figure 1C). As expected, the TLR-induced transcription of Il-1b and Il-6 was prevented by treatment with this drug (Figure S1C). These data demonstrate that the rapid induction of glycolysis in macrophages is a general and non-transcriptional cellular response induced by the TLR family.
Figure 1. A MyD88-TBK1 Pathway Commonly Promotes TLR-inducibie Glycolysis.
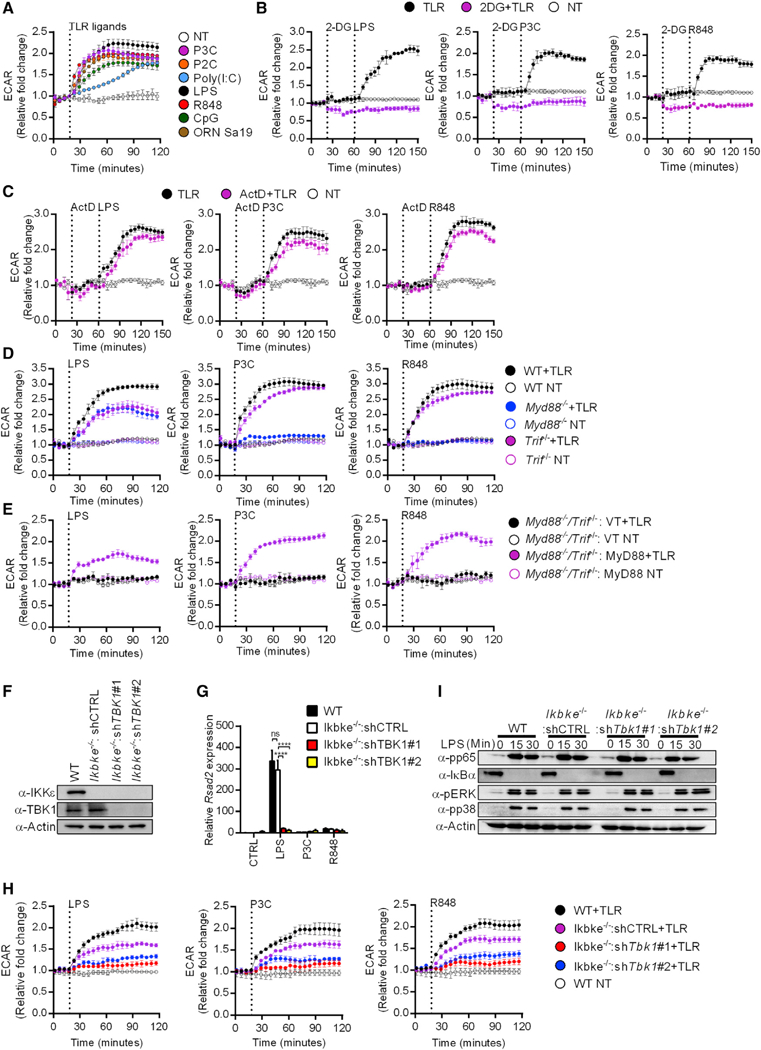
(A) TLR induced changes in the ECAR of primary BMDMs stimulated with the TLR ligands indicated or left untreated (NT) were measured by the Seahorse assay. Data represent mean ± SEM of triplicate wells. Shown is one representative out of 3 independent experiments.
(B) Same as (A) except stimulations were performed with or without 2-DG or left untreated (NT), as indicated.
(C) Same as (A) except stimulations were performed with or without actinomycin D (ActD) or left untreated (NT), as indicated.
(D) Same as (A) except stimulations were performed on primary BMDMs of the genotypes indicated.
(E) Same as (A) except stimulations were performed on Myd88−/−/Trif−/− iBMDMs expressing MyD88 or an empty vector (VT).
(F) Expression levels of TBK1 and IKKε were determined by western analysis in lysates from the indicated iBMDMs.
(G) Indicated iBMDM lines were stimulated as indicated and Rsad2 expression was measured by qPCR.
(H) Same as (A) except stimulations were performed on iBMDMs of the genotypes indicated or left untreated (NT).
(I) Indicated iBMDM lines were pre-treated with cycloheximide and treated with LPS for the times indicated. Western analysis of lysates examined activation of NF-κB (pp65, IκBα) and MAP kinase (pp38, pERK) pathways.
Each panel is a representative experiment of at least 3 independent repeats.
See also Figure S1.
To determine the relative contribution of MyD88 and TRIF to TLR-induced glycolysis, we measured ECAR from wild-type (WT), Myd88−/−, and Trif−/− BMDMs treated with TLR ligands. LPS stimulation induced robust ECAR increases in WT BMDMs, whereas ECAR increases were reduced in Myd88−/− or Trif−/− BMDMs (Figure 1D). MyD88 deficiency abolished the increase in glycolysis upon P3C and R848 stimulation (Figure 1D). Conversely, these ligands triggered comparable ECAR increases in WT and Trif−/− BMDMs (Figure 1D). Therefore, in contrast to TRIF, MyD88 is necessary for optimal glycolytic responses in all TLR pathways examined. To corroborate these observations, we complemented Myd88−/−/Trif−/− iBMDMs, which are deficient for all TLR signaling events, with WT MyD88. This approach allowed us to determine the contribution of MyD88 to TLR-mediated glycolysis from an isogenic background of cells. Cells expressing empty retroviral vectors were completely defective for ECAR induction in response to LPS, P3C, or R848 (Figure 1E). Rescuing MyD88 expression in Myd88−/−/Trif−/− cells restored ECAR increases upon stimulation with all TLR ligands examined (Figure 1E). These findings provide genetic proof for the critical role of MyD88 in promoting TLR-dependent glycolysis.
In DCs, LPS-dependent early glycolysis induction relies on the IKK-related kinases TBK1 and IKKε (Everts et al., 2014). Consistent with these findings, we observed that chemical inhibitors of TBK1-IKKε (BX795 and MRT67307) prevented ECAR increases in TLR-stimulated BMDMs (Figure S1D). To validate our observations, we knocked down TBK1 expression in Ikbke−/− iBMDMs. Ikbke−/− iBMDMs were transduced with two independent short hairpin RNAs (shRNAs) targeting TBK1 (here-after referred to as shTBK1#1 and shTBK1#2). TBK1-targeting shRNAs led to more than 90% depletion of the TBK1 protein from Ikbke−/− iBMDMs (Figure 1F). We functionally verified the loss of TBK1, as TBK1-deficient cells were defective for the LPS-induced expression of the IFN-stimulated gene (ISG) Rsad2 (Figure 1G). In comparison to their WT counterparts, Ikbke−/− iBMDMs expressing a control hairpin (shCTRL) were partially defective for ECAR induction upon TLR ligand stimulation (Figure 1H). Cells lacking TBK1 and IKKε displayed profound impairment of ECAR increase in response to all TLR ligands examined (Figure 1H).
In contrast to the effects of TBK1 and IKKε on glycolysis, the LPS-induced early activation of NF-κB and mitogen-activated protein kinase (MAPK) pathways were not affected by the absence of these kinases (Figure 1I). A MyD88-TBK1 signaling axis is therefore dedicated to promoting glycolysis downstream of multiple TLRs.
TBK1 Is a Myddosome Component
To determine whether TBK1 is locally activated within the myddosome, we examined its presence within MyD88-immunoprecipitates after stimulation of cells with LPS, P3C, or R848. We observed that TBK1 was recruited to MyD88 immunoprecipitates under all conditions of TLR stimulation (Figure 2A). Furthermore, myddosome-associated TBK1 was active, as revealed by western analysis using a phospho-specific antibody raised against TBK1 (Figure 2A). It was possible that TRIF signaling facilitates the recruitment of TBK1 to the myddosome. To address this possibility, we introduced a 3x FLAG-tagged MyD88 allele into Myd88−/−/Trif−/− iBMDMs. FLAG immunoprecipitates from LPS-, P3C-, and R848-stimulated cells contained the myddosome components IRAK2, IRAK4, and TRAF6, as well as total TBK1 and phosphorylated TBK1 (Figure 2B). The recruitment of TBK1 to the myddosome was specific, as no such recruitment could be detected in IgG immunoprecipitates (Figure S2A). These experiments eliminate the possibility that TRIF promotes TBK1 association with the myddosome.
Figure 2. TBK1 Is a Component of the Myddosome.
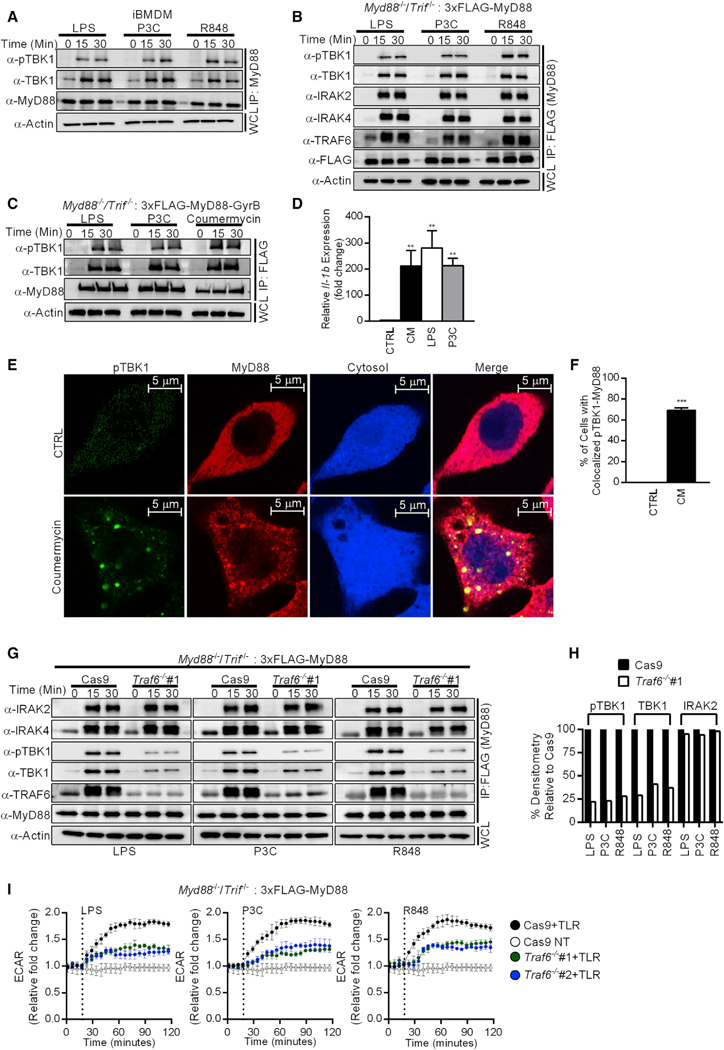
(A) iBMDMs were stimulated with LPS, P3C, and R848 for the times indicated. Components of the myddosome were determined by western analysis after MyD88 immunoprecipitations.
(B) 3xFLAG-MyD88-expressing Myd88−/−/Trif−/− iBMDMs were stimulated with the ligands indicated for the times indicated. Components of the myddosome were determined by western analysis after M2 anti-FLAG immunoprecipitations.
(C) Same as (B) except Myd88−/−/Trif−/− iBMDMs expressing 3xFLAG-MyD88-GyrB were examined.
(D) Myd88−/−/Trif−/− iBMDMs expressing 3xFLAG-MyD88-GyrB were stimulated with coumermycin (CM), LPS, or P3C for 4 h, and Il-1b expression was analyzed by qPCR.
(E and F) Myd88−/−/Trif−/− iBMDMs expressing 3xFLAG-MyD88-GyrB were stimulated with CM for 30 min and fixed. Cells were stained with antibodies detecting FLAG (for MyD88) and pTBK1. Cytosol was visualized by expression of the IRES-GFP from the retroviral vector and was pseudo-colored in blue (E). Quantification of the colocalization between pTBK1 and MyD88 staining (F). Images are representative of at least three independent experiments where more than 100 cells were examined per condition. Scale bar represents 5 μm.
(G and H) Traf6-sufficient (Cas9) and Traf6-deficient (Traf6−/−#1) iBMDMs were stimulated with TLR ligands for the times indicated. Components of the myddosome were determined by western analysis after MyD88 immunoprecipitations (G). Quantification of myddosome-associated pTBK1, TBK1, and IRAK2 was performed by ImageJ (H).
(I) Traf6-sufficient (Cas9) and Traf6-deficient (Traf6−/− #1 and Traf6−/− #2) iBMDMs were stimulated with TLR ligands or not (NT) for the times indicated. Real-time changes in the ECAR were measured by the Seahorse assay. Data represent mean ± SEM of triplicate wells. Shown is one representative experiment out of 3 independent experiments.
For western analysis, each panel is a representative experiment of at least 3 independent repeats.
See also Figure S2.
To corroborate these biochemical analyses, we determined if phosphorylated TBK1 could be detected in myddosomes within intact cells. We used an approach pioneered by Häcker et al. (2006), whereby we fused a GyrB domain (from Escherichia coli DNA gyrase) to the C terminus of a 3x FLAG-MyD88 allele (hereafter referred to as 3x FLAG-MyD88-GyrB). This allele enabled us to chemically induce the entire population of MyD88 to assemble into myddosomes via the compound coumermycin (CM). To validate the functionality of this construct, Myd88−/−/Trif−/− iBMDMs stably transduced with the 3x FLAG-MyD88-GyrB allele were stimulated with CM, LPS, or P3C. CM treatment induced myddosome formation and Il-1b expression to an extent comparable to that induced by LPS and P3C treatment (Figures 2C and 2D), thereby establishing these cells as a suitable model to study myddosome formation. We examined the staining patterns of MyD88 and endogenous phosphorylated TBK1 (pTBK1) in cells stimulated with CM. In untreated cells, MyD88 staining was scattered throughout the cytoplasm (Figure 2E), whereas pTBK1 staining appeared to be dim, as this kinase is inactive at steady state (Figure 2E). Notably, CM treatment induced the formation of distinct “MyD88 specks” resembling the subcellular sites of myddosome formation (Figures 2E and 2F). Notably, MyD88 specks stained positive for pTBK1 (Figures 2E and 2F), indicating that the active kinase was concentrated within these structures. On the contrary, phosphorylated p38 (pp38), which is also activated by MyD88, was not detected within MyD88 specks in CM-stimulated cells (Figure S2B). This kinase was clearly activated by CM, as indicated by its expected nuclear staining pattern (Gong et al., 2010; Figure S2B). These collective observations support the conclusion that TBK1 is recruited to and activated within the myddosome.
To determine the mechanism by which TBK1 is recruited to the myddosome, we first examined the association between TBK1 and individual myddosome components. HA-tagged TBK1 and GFP-tagged myddosome components (MyD88, TIRAP, IRAK2, IRAK4, and TRAF6) were produced in 293T cells in a pairwise manner. Western analysis of HA antibody immunoprecipitations revealed that TRAF6 associated with TBK1 (Figure S2C). Other myddosome components did not form a complex with TBK1 (Figure S2C). TBK1 could also be detected in TRAF6 immuno-precipitates (Figure S2D). These data suggest that TRAF6 forms a complex with TBK1.
To determine the role of TRAF6 in TBK1 recruitment to the myddosome, a pair of RAW264.7 cell lines stably expressing a TRAF6-targeting shRNA (shTRAF6) and a scramble shRNA control (shCTRL) were utilized (West et al., 2011). Western analysis demonstrated the reduction of TRAF6 in shTRAF6 expressing cells (Figure S2E). Functional deficiency of TRAF6 was verified (Deng et al., 2000), as the ability of LPS, P3C, and R848 to induce Il-1b and Il-6 expression was impaired in shTRAF6 cells (Figure S2F). TRAF6 was not required for myddosome assembly, as shTRAF6 expression did not influence IRAK family kinase recruitment to MyD88 immunoprecipitates (Figure S2G). In contrast, shTRAF6 expression diminished the recruitment of total and active TBK1 into myddosomes, as compared to the shCTRL cells (Figure S2G). The defect in TBK1 activation associated with TRAF6 deficiency was also evident in whole cell extracts of TLR-stimulated cells (Figure S2E).
As an alternative approach, TRAF6-deficiencies were generated using CRISPR in Myd88−/−/Trif−/− iBMDMs. This approach allowed us to re-introduce a 3x FLAG-MyD88 allele into these cells to create isogenic lines that permit myddosome assembly in the absence of TRIF. We verified complete loss of TRAF6 in two independent single cell clones of iBMDMs, each containing a distinct gRNA (hereafter referred to as Traf6−/−#1 and Traf6−/−#2) (Figure S2H). These cells displayed the expected defects in TLR-induced gene expression and cytokine secretion (Figures S2I and S2J). TRAF6-deficient cells displayed no defects in myddosome assembly, as IRAK2 and IRAK4 recruitment to MyD88 immuno-precipitates were unaffected by TRAF6 deficiency (Figures 2G and 2H). In contrast, the recruitment of total and active TBK1 into myddosomes was diminished in these cells, as compared to the TRAF6-sufficient cells (Figures 2G and 2H). The requirement of TRAF6 for TLR-induced TBK1 activation was also evident in whole cell extracts of TRAF6-deficient cells (Figure S2H). The requirement of TRAF6 for TBK1 recruitment into myddosomes correlated with a requirement for efficient TLR-induced glycolysis (Figure 2I). TRAF6 is therefore not required for myddosome assembly but rather mediates the recruitment and glycolytic activities of TBK1 within the myddosome.
Myddosomes Can Be Engineered to Induce IFN Responses or Necroptosis
Functionally analogous to MyD88, the MAVS, TRIF, and STING adaptors link PRRs to TBK1 activation. All these adaptors (except MyD88) are able to induce TBK1-mediated IFN expression via IRF3. Embedded within TRIF, MAVS, and STING is a pLxIS motif (p, hydrophilic residue; x, any residue; S, phosphorylation site), which is necessary for TBK1-mediated IRF3 activation (Liu et al., 2015). MyD88does not contain a pLxIS motif. This disparity raised the question of whether the myddosome could be engineered to drive IFN expression and signaling. We reasoned that if the myddosome is truly a modular organizing center, then this organizing center may be flexible in terms of the effector responses that it could induce. To test this hypothesis, synthetic biology-based approaches were used to generate MyD88-pLxIS chimeric alleles that potentially endow the myddosome with the ability to activate IRF3. MyD88-pLxIS alleles were generated by fusing the pLxIS motif from STING to the N terminus or the C terminus of MyD88 (hereafter referred to as MyD88-NpLxIS and MyD88-CpLxIS) (Figure 3A). These constructs were stably expressed in Myd88−/−/Trif−/− iBMDMs, which permitted analysis of their signaling properties at multiple levels. Cells expressing WT MyD88 responded to TLR ligands by inducing Il-1b expression and tumor necrosis factor alpha (TNF-α) secretion (Figures 3B and 3C), as well as TBK1 and NF-κB p65 phosphorylation (Figure S3A). No increases in IRF3 or STAT1 phosphorylation, Rsad2 expression, or IFNβ release were observed in cells expressing WT MyD88 (Figures 3B–3D). These observations are consistent with the role of MyD88 in promoting inflammatory cytokine, rather than type I IFN expression. Cells expressing MyD88-NpLxIS or MyD88-CpLxIS displayed TLR-induced inflammatory activities similar to their WT MyD88-expressing counterparts (Figures 3B, 3C, and S3A). Strikingly, these two synthetic MyD88 variants activated IFN responses upon TLR stimulation, as demonstrated by IRF3 and STAT1 phosphorylation, Rsad2 expression and IFNβ secretion (Figures 3B–3D). Thus, the synthetic MyD88-NpLxIS and MyD88-CpLxIS alleles are capable of activating type I IFN responses, thereby expanding the transcriptional circuits controlled by the myddosome.
Figure 3. Myddosomes Can Be Engineered to Induce Type I IFN Responses.
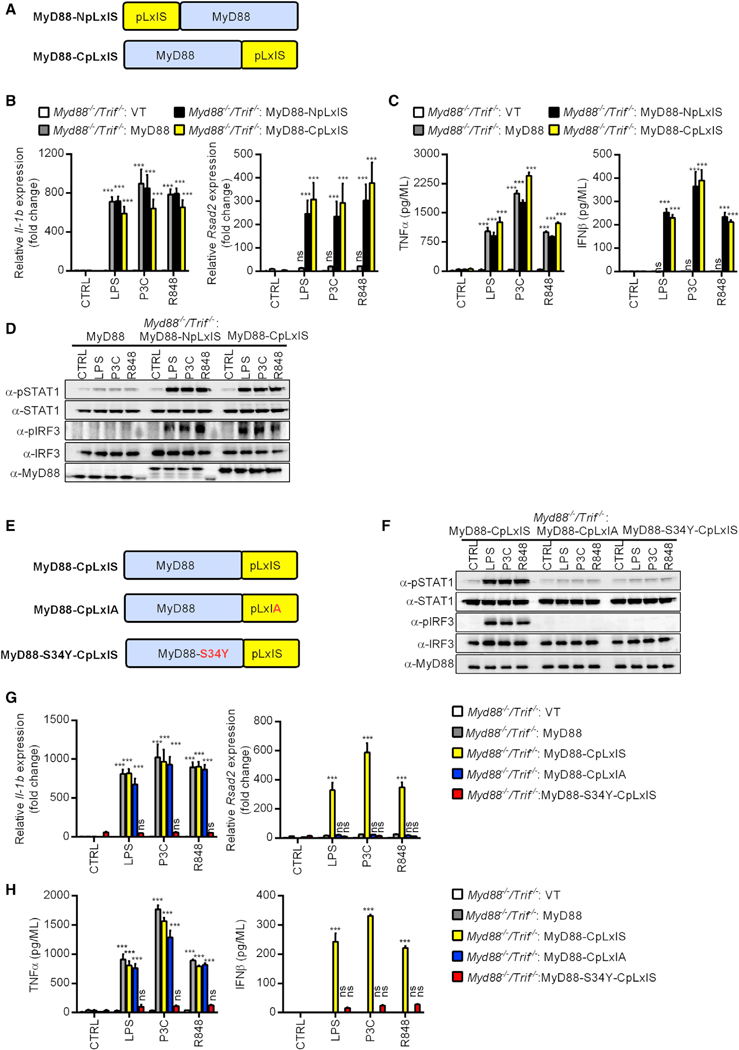
(A) Schematic representation of the MyD88-pLxIS alleles.
(B) Myd88−/−/Trif−/− iBMDMs expressing MyD88, MyD88-NpLxIS, and MyD88-CpLxIS were stimulated with TLR ligands for 4 h. Il-1b and Rsad2 transcripts were determined by qPCR
(C) Myd88−/−/Trif−/− iBMDMs expressing MyD88, MyD88-NpLxIS, and MyD88-CpLxIS were stimulated with TLR ligands for 6 h. Secreted TNF-α and IFNβ were measured by ELISA.
(D) Myd88−/−/Trif−/− iBMDMs expressing MyD88, MyD88-NpLxIS, and MyD88-CpLxIS were stimulated with LPS, P3C or R848 for 90 min and lysed. Abundance of the proteins indicated was determined by western analysis.
(E) Schematic representation of the selected MyD88-CpLxIS mutant alleles.
(F) Myd88−/−/Trif−/− iBMDMs expressing MyD88-CpLxIS and its mutant alleles were treated with TLR ligands for 90 min and lysed. Abundance of the proteins indicated was determined by western analysis.
(G) Myd88−/−/Trif−/− iBMDMs expressing MyD88-CpLxIS and its mutant alleles were treated with TLR ligands for 4 h. Il-1b and Rsad2 transcripts were determined by qPCR.
(H) Myd88−/−/Trif−/− iBMDMs expressing MyD88-CpLxIS and its mutant alleles were treated with TLR ligands for 4 h. Secreted TNFα and IFNβ were measured by ELISA.
For western analysis, each panel is a representative experiment of at least 3 independent repeats.
See also Figure S3.
To determine the mechanism by which these synthetic MyD88 alleles activated IFN expression, we chose the MyD88-CpLxIS variant for these studies and generated two mutant alleles (Figure 3E). The first mutant is MyD88-CpLxIA, which abolishes the Serine residue critical for IRF3 activation upon TBK1 phosphorylation (Liu et al., 2015; Tanaka and Chen, 2012). The second mutant is MyD88-S34Y-CpLxIS, which contains an intact pLxIS motif but impairs MyD88 oligomerization (George et al., 2011; Nagpal et al., 2011).
Whereas cells expressing MyD88-CpLxIS induced the aforementioned IFN-associated responses to TLR stimulation, none of these responses were observed in cells expressing the mutant MyD88-CpLxIA allele (Figures 3F–3H). In contrast, MyD88-CpLxIS and MyD88-CpLxIA displayed a comparable ability to induce Il-1b expression, TNF-α production (Figures 3G and 3H), and TBK1 and p65 phosphorylation (Figure S3B). Cells harboring the oligomerization-deficient allele, MyD88-S34Y-CpLxIS, were poorly responsive to all TLR ligands examined, which was revealed by their defects in inflammatory cytokine and type I IFN expression and signaling (Figures 3F–3H and S3B). These results indicate that upon myddosome oligomerization, the pLxIS motif within MyD88 links TBK1 activation to IFN expression.
To determine if the myddosome could be programmed to induce cellular responses beyond distinct transcriptional programs, we focused on necroptosis. Necroptosis is defined as caspase-independent cell death, which is executed by the kinases RIP1 and RIP3, and mixed lineage kinase domain-like protein (MLKL) (Galluzzi et al., 2017). Importantly, TLR signaling via MyD88 does not directly activate necroptosis (Kaiser et al., 2013).
We reasoned that because oligomerization is a shared mechanism between necroptosis initiation and myddosome signaling, then a MyD88-RIP3 chimeric allele may promote necroptosis. To test this hypothesis, we generated Myd88−/−/Trif−/− iBMDMs stably expressing WT MyD88, MyD88-RIP3 (Figure 4A), or an empty vector. Hallmarks of necroptosis include the loss of plasma-membrane integrity and the release of the cytosolic enzyme lactase dehydrogenase (LDH) (Galluzzi et al., 2017). The loss of membrane integrity allows for the labeling of intracellular nucleic acids by propidium iodide (PI), a membrane impermeable compound. Therefore, we use PI staining and LDH release to measure TLR-induced necroptosis. Myd88−/−/ Trif−/− iBMDMs expressing an empty retroviral vector or WT MyD88 did not stain with PI or release LDH after TLR stimulation (Figures 4B–4D; Video S1). In contrast, within cells harboring MyD88-RIP3, TLR stimulation led to rapid PI staining and LDH release from cells, as both markers became detectable within an hour of ligand stimulation (Figures 4B–4D; Video S2). This process was dependent on RIP3 activity because GSK872, a chemical inhibitor of RIP3 (Kaiser et al., 2013), blocked PI staining and LDH release from MyD88-RIP3 expressing cells in the presence of TLR ligands (Figures 4E and 4F). Moreover, visual examination of TLR-stimulated cells revealed that cells harboring MyD88-RIP3 displayed morphological features indicative of cell lysis (e.g., phase dense shrunken cell corpses) (Figure 4G). Such morphological changes were suppressed by pre-treatment of GSK872 (Figure 4G). The morphological changes observed were distinct from those induced by the apoptosis-inducing agent staurosporine (e.g., membrane blebbing, generation of apoptotic bodies) (Galluzzi et al., 2012; Figure S4A). Furthermore, staurosporine treatment induced PARP cleavage, a marker of apoptosis (Green and Kroemer, 1998; Figure S4B). No cleaved PARP could be detected from the MyD88-RIP3 expressing cells stimulated with TLR ligands (Figure S4B), highlighting that the death program triggered by this allele is distinct from apoptosis. Consistent with the caspase-independent nature of necroptosis, the pan-caspase inhibitor ZVAD failed to prevent membrane rupture induced by MyD88-RIP3 upon TLR activation (Figures S4C and S4D). In WT cells, RIP3 functions downstream of RIP1 to induce necroptosis. Because the myddosome provides a platform to oligomerize RIP3 directly, we predicted that the execution of MyD88-RIP3-mediated cell death would bypass the requirement of RIP1. Indeed, TLR-induced PI staining and LDH release from MyD88-RIP3 expressing cells were unaffected by blockade of RIP1 activity using the inhibitor Nec-1 (Figures S4C and S4D). Importantly, pre-treatment of BMDMs with ZVAD followed by LPS stimulation led to the induction of necroptosis, which could be prevented by Nec-1 (Figure S4E). This experiment demonstrates the efficacy of the chemical inhibitors used in our assays. The myddosome can therefore be programmed to induce necroptosis.
Figure 4. Myddosomes Can Be Engineered to Promote RIP3-Dependent Necroptosis.
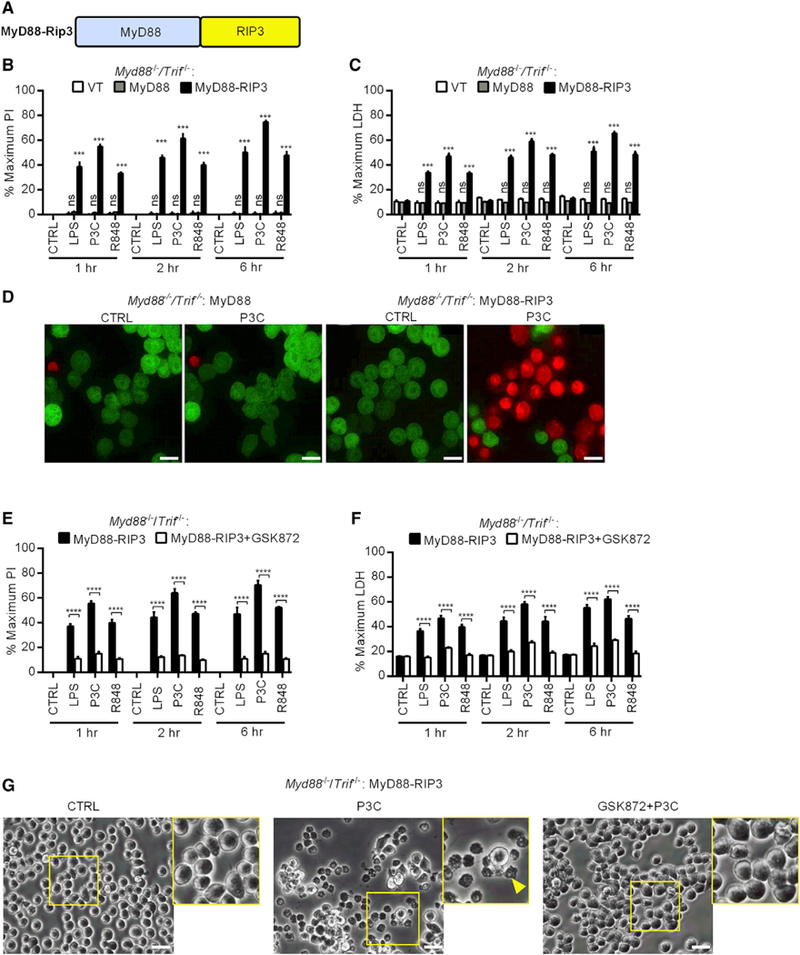
(A) Schematic representation of the MyD88-RIP3 allele.
(B and C) Myd88−/−/Trif−/− iBMDMs expressing MyD88, MyD88-RIP3, or an empty vector (VT) were stimulated with LPS, P3C, or R848 for the times indicated. Membrane rupture was determined by PI staining (B) and extracellular LDH was quantified (C).
(D) Myd88−/−/Trif−/− iBMDMs expressing MyD88 or MyD88-RIP3 were analyzed by confocal microscopy. Cells were stimulated with P3C in Pl-containing medium. One image was captured every 3 min for ~60 min. Shown are representative frames from a capture (see Videos S1 and S2 for full-length movies). Scale bar represents 10 μm.
(E and F) Myd88−/−/Trif−/− iBMDMs expressing MyD88-RIP3 were pre-treated (or not) with GSK872 and stimulated with TLR ligands for the indicated times. Membrane rupture was determined by PI staining (E) and extracellular LDH was quantified (F).
(G) Cells were treated as described in (E) and (F). Images of cell morphology were taken 1 h post-stimulation. The arrow head highlights a dead cell. Scale bar represents 10 μm. Images are representative of at least three independent experiments.
See also Figure S4.
Inflammasomes Can Be Programmed to Prevent Pyroptosis and Induce IFN Responses
Similar to the myddosomes, inflammasomes represent a class of SMOCs that play a central role in inflammation (Rathinam and Fitzgerald, 2016). We reasoned that if inflammasomes operate by similar principles to the myddosomes, then their effector functions could be programmed to induce user-defined responses. We therefore sought to shift the activities of a given in-flammasome from a SMOC that drives pyroptosis into one that induces gene expression. This endeavor required a system that did not involve any other transcription inducing stimulus (also known as priming). The NAIP-NLRC4 inflammasome provides such a system, as this SMOC can be activated by cytosolic bacterial flagellin without the need of any other stimulus (Franchi et al., 2006; Miao et al., 2006). To deliver flagellin to the cytosol, we utilized flatox, an anthrax toxin derivative that consists of a lethal factor-flagellin fusion protein (LFn-Fla) and protective antigen (PA) (von Moltke et al., 2012). When PA and LFn-Fla are combined, flatox is formed, which delivers flagellin into the cytosol where it activates pyroptosis (von Moltke et al., 2012).
To eliminate the pyroptosis-inducing activity of the NAIP-NLRC4 inflammasome, we generated a catalytically inactive caspase-1 allele (C285A), which was stably produced in Caspase1−/−/Caspase11−/− iBMDMs (Figure 5A). Flatox treatment induced the death of WT iBMDMs, as evidenced by LDH release (Figure 5B). LFn-Fla treatments in the absence of PA led to no LDH release (Figure 5B). In contrast, flatox treatment was unable to induce the death of Caspase1−/−/11−/− iBMDMs or Caspase1−/−/11−/− iBMDMs expressing inactive caspase-1 (Figure 5B). Having eliminated the pyroptosis-inducing activity of the NAIP-NLRC4 inflammasome, we determined if this inflammasome could be engineered to induce an IFN response. To this end, the pLxIS motif from STING was appended onto the N or C terminus of catalytically inactive caspase-1 (hereafter referred to as Casp1-NpLxIS and Casp1-CpLxIS) (Figure 5A). These proteins were stably produced in Caspase1−/−/11−/− iBMDMs, and the resultant cells were treated with flatox.
Figure 5. Inflammasomes Can Be Programmed to Prevent Pyroptosis and Induce IFN Responses.
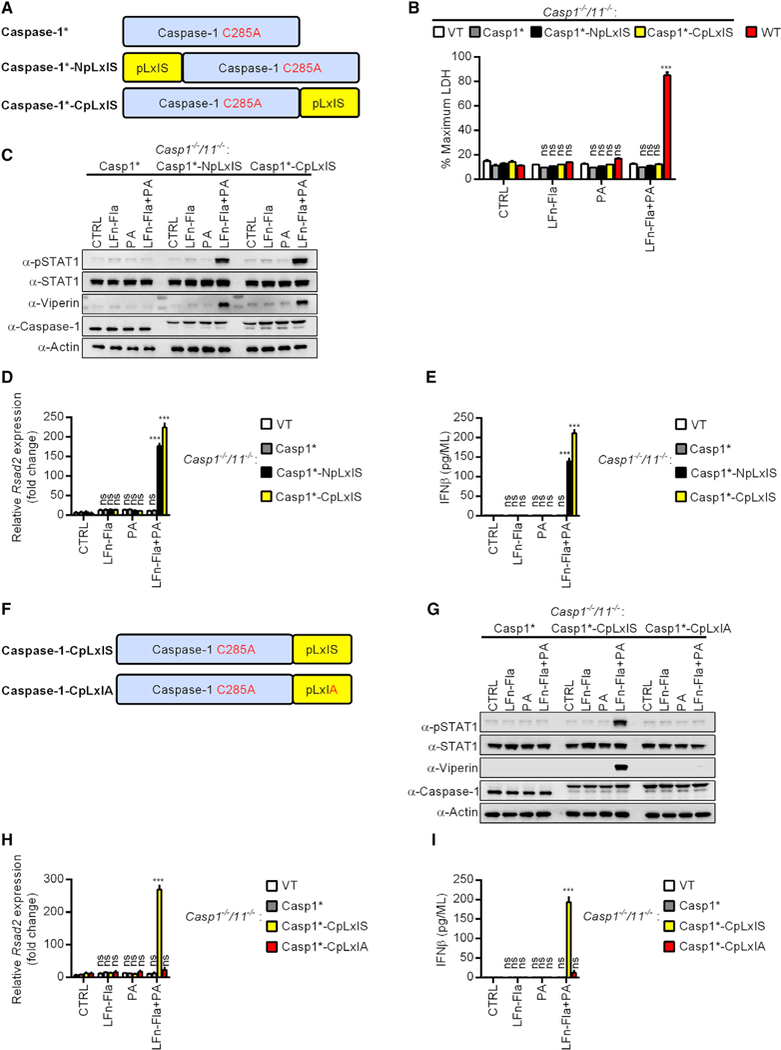
(A) Schematic representation of the caspase alleles.
(B) Casp1−/−/Casp11−/− iBMDMs expressing caspase-1, caspase-1-NpLxIS, caspase-1-CpLxIS, or an empty vector (VT) were stimulated with flatox (LFn-Fla + PA) or its individual components: PA, LFn-Fla or not for 5 h. WT iBMDMs were used as positive control. Pyroptosis was quantified by measuring the extracellular LDH.
(C) Same as (B) except the abundance of the proteins indicated was examined by western blot.
(D) Same as (B) except Rsad2 expression was determined by qPCR.
(E) Same as (B) except secreted IFNß was measured by ELISA.
(F) Schematic representation of the caspase alleles.
(G) Casp1−/−/Casp11−/− iBMDMs expressing caspase-1, caspase-1-CpLxIS, or caspase-1-CpLxIA were treated with flatox (LFn-Fla + PA) or its individual components: PA, LFn-Fla or not for 5 h. Abundance of the proteins indicated was examined by western blot. Actin was probed as loading control.
(H) Casp1−/−/Casp11−/− iBMDMs expressing caspase-1, caspase-1-CpLxIS, caspase-1-CpLxIA, or an empty vector (VT) were treated with flatox (LFn-Fla + PA) or its individual components: PA, LFn-Fla or not for 5 h. Cells were lysed and mRNA was extracted. The level of Rsad2 expression was determined by qPCR.
(I) Casp1−/−/Casp11−/− iBMDMs expressing caspase-1, caspase-1-CpLxIS, caspase-1-CpLxIA, or an empty vector (VT) were treated with flatox (LFn-Fla + PA) or its individual components: PA, LFn-Fla or not for 5 h. Secreted IFNβ was measured by ELISA.
*The C285A mutation. For western analysis, each panel is a representative experiment of at least 3 independent repeats.
Within cells expressing Casp1-NpLxIS or Casp1-CpLxIS, flatox induced STAT1 phosphorylation along with Rsad2 expression and production of its gene product viperin (Figures 5C and 5D). Flatox also induced the secretion of IFNβ from these cells (Figure 5E). Cells producing catalytically inactive caspase-1 did not elicit any IFN expression or activity (Figures 5C–5E). Notably, mutation of the critical serine residue in the pLxIS motif (Casp1-CpLxIA) eliminated all IFN-inducing activities of the synthetic caspase-1 protein (Figures 5F–5I). These data establish that the NAIP-NLRC4 inflammasome can be engineered to abolish its ability to induce pyroptosis and further programmed to induce IFN responses.
Creation of a Synthetic SMOC-like Nanomachine that Drives IFN Expression in Response to a Small Molecule
Our demonstration of the signaling flexibility of myddosomes and inflammasomes suggests that neither SMOC is intrinsically restricted to the types of cellular responses they can naturally induce. Rather, the oligomerizing scaffold within the core of these organelles may serve as a platform upon which any signaling module can be attached. We propose that despite their apparent natural complexity, SMOCs are rather simple organelles that contain two functional units—an oligomerizing scaffold and an effector domain. If this hypothesis is correct, then one should be able to build a wholly synthetic SMOC-like nanomachine that induces user-defined effector functions.
To test this idea, we used as a scaffold the FKBP (F36V) protein (Clackson et al., 1998), which is capable of dimerization, but not oligomerization, in response to the small molecule homodimerizer B/B (Yang et al., 2000; Figure 6A). In cells expressing two copies of FKBP fused to the pLxIS motif from STING (2xFKBP-pLxIS), the dimerizer B/B was unable to induce STAT1 phosphorylation or viperin production (Figure 6B). In contrast, when we further multimerized FKBP, such that scaffolds of dimers can be created (i.e., oligomers), we found that FKBP-pLxIS induced these IFN-associated responses to B/B (Figure 6B). Three copies of FKBP fused to pLxIS (3xFKBP- pLxIS) linked B/B treatment to STAT1 phosphorylation and viperin production and four copies of FKBP (4xFKBP-pLxIS) was even more rapid and robust (Figure 6B). Thus, the speed and strength of the responses induced by B/B correlated with the number of FKBP repeats present in our nanomachines. This observation is consistent with the core principle of the SMOC hypothesis—that supramolecular oligomers serve as scaffolds for signaling. Further analysis revealed that 4xFKBP-pLxIS induced the expression of Rsad2 and the chemokine Cxcl10, as well as IFNβ secretion (Figures 6C and 6D). These findings were made in 293T cells which are defective for STING signaling (Burdette et al., 2011) suggesting that the pLxIS motif we attached to FKBP does not require endogenous STING for activity. Corroborating evidence to support this conclusion was provided by the expression of the 4xFKBP-pLxIS nanomachine in STING-deficient iBMDMs. As was observed in HEK293T cells, WT iBMDMs expressing 4xFKBP-pLxIS induced STAT1 phosphorylation, viperin production, Rsad2 and Cxcl10 expression, and the secretion of IFNβ (Figures 6E–6G). In contrast, cells expressing an empty retroviral vector (VT) were unresponsive to B/B and did not display any evidence of IFN expression or activities (Figures 6E–6G). In STING-deficient iBMDMs, the 4xFKBP-pLxIS nanomachine retained the ability to induce all of the aforementioned IFN responses, although Rsad2 expression and viperin production was more robustly induced in WT cells (Figures 6E–6G). The ability to build a synthetic SMOC-like nanomachine supports that idea that, at their most fundamental level, SMOCs do indeed consist of two functional units—an oligomerizing platform and an effector domain. All other proteins that are present within a natural SMOC probably function to modulate one or both of these core activities.
Figure 6. A Synthetic SMOC-like Nanomachine Drives IFN Expression in Response to a Small Molecule.
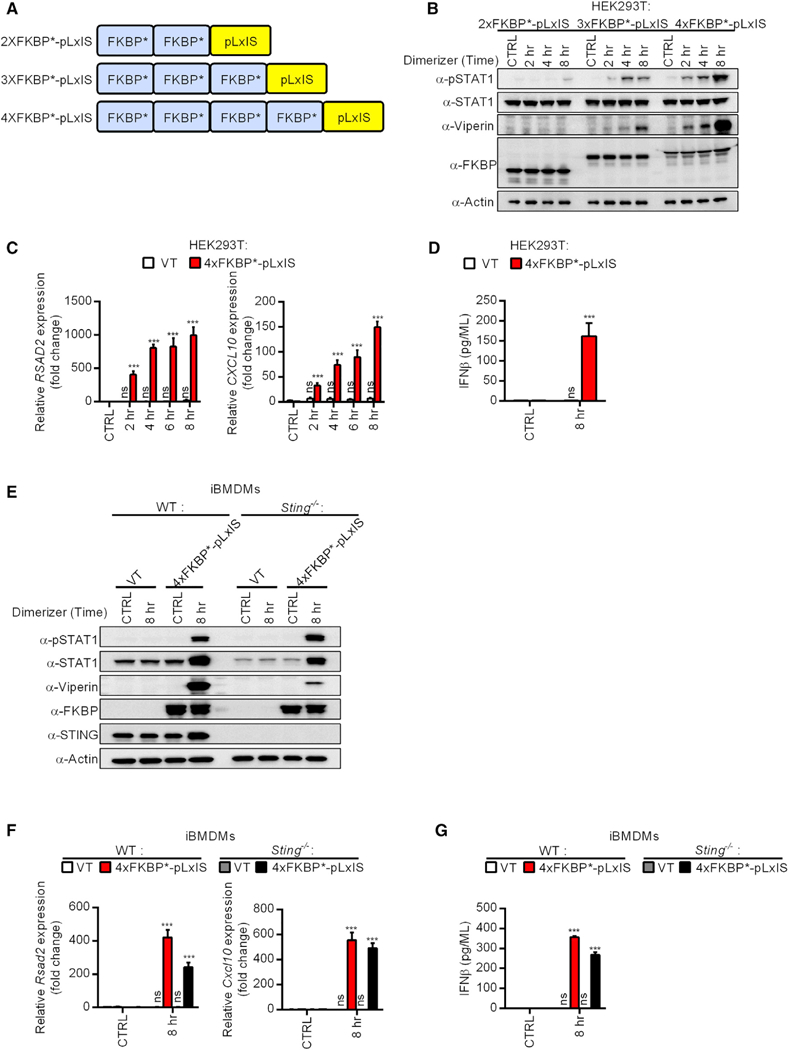
(A) Schematic representation of the FKBP*-pLxIS alleles. *The F36V mutation allows for the recognition of the small molecule B/B homodimerizer, which does not engage endogenous FKBP.
(B) HEK293T cells expressing the indicated FKBP*-pLxIS alleles were treated with B/B (1 μM) for the times indicated. Abundance of the proteins indicated was examined by western analysis.
(C) Same as (B) except RSAD2 and CXCL10 transcripts were measured by qPCR.
(D) Same as (B) except secreted IFNβ was measured by ELISA after 8 h of B/B stimulation.
(E) WT and Sting−/− iBMDMs expressing 4xFKBP-pLxIS or an empty vector (VT) were treated with B/B for 8 h. Abundance of the proteins indicated was determined by western analysis.
(F) Same as (E) except Rsad2 and Cxcl10 transcripts were measured by qPCR.
(G) Same as (E) except secreted IFNβ was measured by ELISA.
For western analysis, each panel is a representative experiment of at least 3 independent repeats.
DISCUSSION
In this study, we provide experimental support to our central hypothesis that the myddosome is a bona fide organizing center. This SMOC must now be considered as the subcellular site of TLR signals that induce NF-κB activation and glycolysis. These findings not only provide insight into TBK1 activation in the TLR pathway, but also expand the physiological functions of the myddosome (from a regulator of transcription to a regulator of glycolysis). We do note, however, that the early glycolytic induction mediated by myddosome-associated TBK1 is just the beginning of profound host metabolic alterations induced by PRR signaling (O’Neill et al., 2016). Indeed, AKT, the master regulator of metabolism, is also regulated by divergent factors which include PI3K, mTOR, and BCAP (Huang et al., 2016; Krawczyk et al., 2010; Troutman et al., 2012). Many of these factors facilitate the long-term commitment of professional phagocytes to glycolysis (O’Neill et al., 2016). These findings, coupled with our data, emphasize the importance of understanding how the myddosome coordinates short- and long-term metabolic needs upon TLR activation. It is possible that other transcription-inducing SMOCs, such as the RLR-MAVS complex and the triffosome, induce glycolytic responses via TBK1-dependent events.
Our identification of TBK1 and TRAF6 as myddosome components is notable, because all of the previously characterized myddosome components share similar domains that allow for homotypic protein-protein interactions (Lin et al., 2010). In particular, all TLRs, TIRAP, and MyD88 possess a Toll-inter- leukin-1 receptor homology domain (TIR) domain (Pandey et al., 2014). MyD88 and IRAK kinases also contain death domains (DD) (Pandey et al., 2014). In cell-free systems, self-association of these domains drives the formation of higher-ordered helical structures (Lin et al., 2010; Ve et al., 2017). However, neither TBK1 norTRAF6 harbor a DD or a TIR domain. These results suggest that within cells, the myddosome is not merely a stack of proteins containing TIR domains and DDs. Additional host proteins (non-TIR-, non-DD-containing) might be recruited to the myddosome upon microbial sensing to promote distinct effector responses.
While our findings highlight the modular nature of myddosome signaling, this modularity could have been restricted to cellular processes shaped by evolution. Alternatively, it was possible that the modularity of SMOCs would enable us to create unique signaling circuits. Our data that synthetic myddosomes promote IFN expression and necroptosis provide affirmative proof to the latter scenario. Also consistent with this idea is our ability to engineer the NAIP-NLRC4 inflammasome to prevent pyroptosis and instead induce IFN expression. Thus, the inflammasome does not have to be a pyroptosis-inducing machine; nature just selected it to be one. The inflammasome has the capacity to operate as a transcription-inducing machine. Likewise, the myddosome has the capacity to operate as a necroptosis- or IFN-inducing machine. This symmetry of observations made with different SMOCs suggests that we have uncovered a common principle associated with innate immune system operation—all SMOCs may have the capacity for user-defined reprogramming.
Based on our findings, we propose that all SMOCs derived from one or more ancestral proto-SMOCs, which were simple two-unit platforms consisting of an oligomerization scaffold and an effector domain. Throughout evolution, proto-SMOCs acquired the ability to engage upstream receptors and downstream effectors, and then diversified into the SMOCs we study today. But at their core, they all still contain the generic two-unit platform that is amenable to synthetic biology-based engineering. Consistent with this idea is our ability to create a SMOC-like synthetic nanomachine that induces IFN expression upon oligomerization stimulated by a small molecule.
While this study was designed to advance our basic knowledge of the innate immune system, our findings raise the possibility that synthetic immunology-based approaches could be used to engineer unique and beneficial cellular responses upon PRR activation. Indeed, these approaches may be considered akin to those taken to rewire signaling pathways in lymphocytes to generate chimeric antigen receptor (CAR) T cell therapies (June et al., 2018).
In summary, our data suggest unifying themes exist to explain the operation of the diverse signaling proteins and pathways in the innate immune system. Just as the concept of pattern recognition transcends the specific mechanisms by which PRRs and PAMPs interact, the two-unit SMOC concept may represent a cornerstone feature that explains the diverse inflammatory activities of PRRs. Our discoveries provide a mandate to explore the natural and potentially programmable features of other organizing centers within (and beyond) the innate immune system.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jonathan C. Kagan (jonathan.kagan@childrens.harvard.edu).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mice
All Mouse strains used in the study were purchased from the Jackson Laboratories. See “Key Resources Table” for details. All mice were maintained in a specific pathogen-free facility and all animal experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of Boston Children’s Hospital.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-HA | Sigma-Aldrich | Cat# 11867423001; RRID:AB_390919 |
| Mouse monoclonal anti-FLAG (M2) | Sigma-Aldrich | Cat# F1804; RRID:AB_262044 |
| Mouse monoclonal anti-β-Actin | Sigma-Aldrich | Cat# A5441; RRID:AB_476744 |
| Mouse monoclonal anti-phospho p38 | BD Biosciences | Cat# 612288; RRID:AB_399605 |
| Rat Monoclonal anti-Caspase 1 (5B10) | Thermo Fisher | Cat# 14-9832-82; RRID:AB_2016691 |
| Rabbit polyclonal anti-FKBP12 | Abcam | Cat# ab24373; RRID:AB_732383 |
| Rabbit monoclonal anti-phospho STAT-1 | BD Biosciences | Cat# 612132; RRID:AB_399503 |
| Rabbit monoclonal anti-phospho IRF3 (4D4G) | Cell Signaling Technology | Cat# 4947s; RRID:AB_823547 |
| Rabbit monoclonal anti-phospho p65 (93H1) | Cell Signaling Technology | Cat# 3033S; RRID:AB_331284 |
| Mouse monoclonal anti-phospho ERK1/2 | Cell Signaling Technology | Cat# 9106s; RRID:AB_331768 |
| Rabbit monoclonal anti-phospho p38 (D3F9) | Cell Signaling Technology | Cat# 4511; RRID:AB_2139682 |
| Rabbit monoclonal anti-IKKε | Cell Signaling Technology | Cat# 3416; RRID:AB_1264180 |
| Rabbit monoclonal anti-TBK1 | Cell Signaling Technology | Cat# 3504; RRID:AB_2255663 |
| Rabbit monoclonal anti-IRF3 (D83B9) | Cell Signaling Technology | Cat# 4302; RRID:AB_1904036 |
| Rabbit monoclonal anti-STAT1(D1K9Y) | Cell Signaling Technology | Cat# 14994; RRID:AB_2737027 |
| Rabbit monoclonal anti-phospho TBK1 (D52C2) | Cell Signaling Technology | Cat# 5483; RRID:AB_10693472 |
| Mouse monoclonal anti-TRAF6 (T2–1SC) | Biolegend | Cat# 654502; RRID:AB_2561868 |
| Rabbit polyclonal anti-TRAF6 | Santa Cruz | Cat# Sc-7221; RRID:AB_793346 |
| Goat polyclonal anti-MyD88 | R&D | Cat# AF3109; RRID:AB_2146703 |
| Pierce High Capacity Streptavidin Agarose | Thermofisher | Cat# 20359 |
| Pierce Anti-HA Agarose | Thermofisher | Cat# 26182; RRID:AB_2532162 |
| ANTI-FLAG® M2 Affinity Gel | Sigma-Aldrich | Cat# A2220; RRID:AB_10063035 |
| Rabbit polyclonal anti-IRAK4 | NA | Gift of Dr. Akira |
| Rabbit polyclonal anti-IRAK2 | Prosci | Cat# 3595; RRID:AB_735436 |
| Mouse monoclonal anti-Viperin | Biolegend | Custom made |
| Normal Goat IgG Control | R&D Systems | Cat# AB-108-C |
| IgG1 Isotype Control | Sigma-Aldrich | Cat# M5284 |
| Chemicals and recombinant proteins | ||
| E. coli LPS Serotype O111:B4 | Enzo Life Sciences | Cat# ALX-581-012-L002 |
| Pam3CSK4 (VacciGrade) | InvivoGen | Cat# vac-pms |
| Pam2CSK4 | InvivoGen | Cat# tlrl-pm2s-1 |
| Poly(I:C) | InvivoGen | Cat# tlrl-pic |
| R848 (VacciGrade) | InvivoGen | Cat# vac-r848 |
| ORN Sa19 | InvivoGen | Cat# tlrl-orn19 |
| CpG (ODN1826) | InvivoGen | Cat# tlrl-1826 |
| BX795 | InvivoGen | Cat# tlrl-bx7 |
| MRT67307 | Cayman | Cat# 19916 |
| Actinomycin D | Sigma-Aldrich | Cat# A9415 |
| 2-Deoxy-D-glucose (2-DG) | Sigma-Aldrich | Cat# D6134 |
| Puromycin | Sigma-Aldrich | Cat# P9620 |
| B/B Homodimerizer | Clontech | Cat# 635059 |
| n-Dodecyl β-D-maltoside | Sigma-Aldrich | Cat# D4641 |
| Cholesteryl hemisuccinate | Sigma-Aldrich | Cat# C6512 |
| Propidium iodide solution | Sigma-Aldrich | Cat# P4864 |
| Coumermycin A1 | Sigma-Aldrich | Cat# C9270 |
| ZVAD | InvivoGen | Cat# tlrl-vad |
| Necrostatin-1 | Cayman Chemical | Cat# 11658 |
| GSK872 | Selleckchem | Cat# S8465 |
| Protein G Sepharose 4 Fast Flow Media | Fisher Scientific | Cat# 45-000-116 |
| Recombinant Protective Antigen (PA) | Russell Vance Laboratory | N/A |
| Lethal factor Flagellin fusion protein (LFn-Fla) | Russell Vance Laboratory | N/A |
| Critical Commercial Assays | ||
| Mouse IFNβ ELISA Kit | InvivoGen | Cat# luex-mifnb |
| Mouse TNFα ELISA Kit | ThermoFisher | Cat# 88–7324-88 |
| Human IFNβ ELISA Kit | InvivoGen | Cat# luex-hifnb |
| LDH Cytotoxicity Assay Kit | Thermo Scientific | Cat# 88953 |
| Experimental Models: Cell lines | ||
| WT iBMDMs | Jonathan Kagan Laboratory | N/A. |
| Myd88−/−/Trif−/− iBMDMs | Jonathan Kagan Laboratory | N/A |
| Myd88−/−/Trif−/−/Traf6−/− iBMDMs | Jonathan Kagan Laboratory | N/A |
| Ikbke−/− iBMDMs | Jonathan Kagan Laboratory | N/A |
| Ikbke−/− iBMDMs expressing shCTRL | Jonathan Kagan Laboratory | N/A |
| Ikbke−/− iBMDMs expressing shTBK1#1 | Jonathan Kagan Laboratory | N/A |
| Ikbke−/− iBMDMs expressing shTBK1#2 | Jonathan Kagan Laboratory | N/A |
| Casp1 −/−/Casp11 −/− iBMDMs | Jonathan Kagan Laboratory | N/A |
| Sting−/− iBMDMs | Jonathan Kagan Laboratory | N/A |
| RAW264.7 macrophage-like cell line | Jonathan Kagan Laboratory | N/A |
| RAW264.7 expressing shCTRL | Jonathan Kagan Laboratory | Gift of Dr. Ghosh PMID: 21525932 |
| RAW264.7 expressing shTraf6 | Jonathan Kagan Laboratory | Gift of Dr. Ghosh PMID: 21525932 |
| Primary bone marrow-derived macrophages (BMDMs) | Jonathan Kagan Laboratory | N/A |
| HEK293T | Jonathan Kagan Laboratory | N/A |
| Oligonucleotides | ||
| Traf6-targeting sgRNA1 | Jonathan Kagan Laboratory | GATGGAACTGAGACATCTCG |
| Traf6-targeting sgRNA2 | Jonathan Kagan Laboratory | GGAGATCCAGGGCTACGATG |
| Recombinant DNA | ||
| pGIPZ TBK1-targeting shRNA | Dharmacon-GE | V3LMM_454840 |
| RMM4532-EG56480 glycerol set | V3LMM_454842 | |
| pLentiCRISPRv2 TRAF6-targeting sgRNA 1 | Jonathan Kagan Laboratory | N/A |
| pLentiCRISPRv2 TRAF6-targeting sgRNA 2 | Jonathan Kagan Laboratory | N/A |
| pCMV-FLAG-TRAF6 | Jonathan Kagan Laboratory | N/A |
| pEGFP-c1 | Jonathan Kagan Laboratory | N/A |
| pEGFP-c1-MyD88 | Jonathan Kagan Laboratory | N/A |
| pEGFP-c1-TRAF6 | Jonathan Kagan Laboratory | N/A |
| pEGFP-c1-IRAK2 | Jonathan Kagan Laboratory | N/A |
| pEGFP-c1-IRAK4 | Jonathan Kagan Laboratory | N/A |
| pEGFP-c1-TIRAP | Jonathan Kagan Laboratory | N/A |
| pcDNA-TBK1-HA | Jonathan Kagan Laboratory | N/A |
| psPAX2 Addgene Plasmid #12260 | psPAX2 Addgene Plasmid #12260 | psPAX2 Addgene Plasmid #12260 |
| pCMV-VSV-G Addgene Plasmid #8454 | pCMV-VSV-G Addgene Plasmid #8454 | pCMV-VSV-G Addgene Plasmid #8454 |
| pMSCV-IRES-GFP | Jonathan Kagan Laboratory | N/A |
| pMSCV-IRES-GFP various MyD88 alleles | Jonathan Kagan Laboratory | N/A |
| pMSCV-IRES-GFP various Caspase-1 (C285A) alleles | Jonathan Kagan Laboratory | N/A |
| pMSCV-IRES-GFP-FKBP-(F36V)-pLxIS alleles | Jonathan Kagan Laboratory | N/A |
| pCL-Eco Addgene Plasmid #12371 | pCL-Eco Addgene Plasmid #12371 | pCL-Eco Addgene Plasmid #12371 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | 000664 |
| Mouse: B6.129P2(SJL)-Myd88tm1.1Defr/J | The Jackson Laboratory | 009088 |
| Mouse: C57BL/6J-Ticam1Lps2/J | The Jackson Laboratory | 005037 |
| Mouse: B6.Cg-Ikbketm1Tman/J | The Jackson Laboratory | 006908 |
| TaqMan probes | ||
| Mouse Rsad2 | Thermo Scientific | Mm00491265_m1 |
| Mouse Cxcl10 | Thermo Scientific | Mm00445235_m1 |
| Mouse Il-6 | Thermo Scientific | Mm00446190_m1 |
| Mouse Il-1b | Thermo Scientific | Mm00434228_m1 |
| Mouse Gapdh | Thermo Scientific | Mm99999915_g1 |
| Human RSAD2 | Thermo Scientific | Hs00369813_m1 |
| Human CXCL10 | Thermo Scientific | Hs01124252_g1 |
| Human GAPDH | Thermo Scientific | Hs02786624_g1 |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/download.html |
| Deposited Data | ||
| Uncropped western analysis from all blots in this study | Mendeley | https://data.mendeley.com/datasets/6bw75vhgg7/1 |
Differentiation of Primary Bone Marrow Derived Macrophages
To generate M-CSF conditioned medium for macrophage differentiation, L929 cells were cultured in complete DMEM. Supernatants from L929 fibroblasts (containing M-CSF) were cleared of cellular debris by spinning at 400 × g for 5 min, and were further passed through a 0.22 mm filter. The filtered supernatants were added to RPMI medium containing 15% FBS at the final concentration of 30%, which is the conditioned medium for primary BMDM culture. Leg bones of mice of various genotypes were surgically removed. Cleaned bones were cut with scissors and flushed with sterile PBS pH 7.4 via syringe. Bone marrow suspension was passed through a 70 mm cell strainer to exclude clumps. Cells were cultured in non-treated 10 cm tissue culture dishes in the conditioned medium, and were fed with 5 mL of additional conditioned medium on day 3 of differentiation. Differentiated BMDMs were used for experiments at day 7 or 8.
Cell lines, Transfection, and Retroviral Transduction
Immortalized bone marrow derived macrophages (iBMDMs) were cultured in DMEM containing 10% FBS, Penicillin and Streptomycin (Pen+Strep), and supplements of L-glutamine and sodium pyruvate. PBS (pH 7.4) containing EDTA (2.5 mM) was used to detach cells for passage or plate for assays. HEK293T cells were cultured in complete DMEM. Cells were washed in PBS and then detached in culture flasks with 0.25% Trypsin. For transient overexpression in HEK293T cells, HA-tagged TBK1 was cloned into the pcDNA vector. MyD88, TIRAP, IRAK2, IRAK4, TRAF6 were cloned into pEGFPc1; TRAF6 was cloned into the pCMV-FLAG vector. For retroviral transduction, all FKBP, Caspase-1, and MyD88 alleles used in this study were cloned into the pMSCV-IRES-GFP vector.
To generate cell lines stably expressing transgenes, retroviral particles were produced by transfecting 293T cells with plasmids pCL-Eco, pCMV-VSV-G, and pMSCV-IRES-GFP containing the gene of interest. For lentiviral-mediated shRNA expression, lentiviral particles were produced by transfecting 293T cells with plasmids psPAX2, pCMV-VSV-G, and lentiviral vector expressing TBK1-targeting shRNAs or a control non-targeting scramble shRNA.
Plasmids were transfected into HEK293T cells in 10 cm dishes at a confluency of 50%–70% with lipofectamine 3000 and media was changed 24 hr post transfection and viral supernatants were collected 24 hr post media change. Viral supernatants were spun at 400 x g to remove cellular debris, then passed through a 0.45 mm PVDF filter via syringe. Polybrene was added to the filtered supernatants (5 μg/ml), and the supernatants were then used to transduce iBMDMs via spin-fection at 1250 × g for 60 min at room temperature. The cell lines expressing MSCV-IRES-GFP constructs were sorted based on GFP expression to ensure comparable levels of transgene expression. For shRNA-mediated gene knock down, cell lines stably expressing shRNA or sgRNA constructs were selected by puromycin (25 μg/ml).
Generation of MyD88−/−/Trif−/−/Traf6−/− iBMDMs
The Traf6’-targeting lentivirus was packaged using lentiCRISPRv2 system. MyD88−/−/Trif−/− iBMDMs were plated into a 6-well tissue culture plate and were transduced the next day with the lentiviral particle expressing Cas9 and two Traf6-specific guide RNAs (GATGGAACTGAGACATCTCG and GGAGATCCAGGGCTACGATG). At 48 hr-post transduction, fresh media containing 25 mg/ml puromycin was added to select for cells transduced with the Traf6-targeting lentivirus. The puromycin resistant cells were further subjected to single-cell cloning by limited dilution. After culturing in puromycin-containing media for an additional 2–3 weeks, single colonies were picked, expanded, and then analyzed for TRAF6 protein expression by western analysis. MyD88−/−/Trif−/− iBMDMs expressing the lentiCRISPRv2 vector with a GFP targeting sequence were used as controls. The resultant Traf6-sufficient and Traf6-deficient lines were transduced with retroviral particles expressing 3xFLAG-MyD88. The expression levels of MyD88 were adjusted to a similar extent by fluorescence activated cell sorting (FACs) based on the IRES-GFP reporter.
Immortalization Protocol for Bone Marrow Derived Macrophages
Primary BMDMs for immortalization were cultured in complete RPMI with 15% FBS, 30% L929 conditioned supernatant and antibiotics. Conditioned supernatant collected from the CREJ2 cell line carrying the J2 retrovirus was used to immortalize primary BMDMs. In brief, differentiated primary BMDMs (day 7) were further incubated with 50% J2 conditioned supernatant and 50% L929 conditioned supernatant for 7 days, with one new batch of mixed J2 supernatant and L929 supernatant added at day 3. Transduced BMDMs were then cultured in complete DMEM plus 30% L929 supernatant until 90% confluent. Cells were then passed into new medium containing 25% L929 supernatant. Following this trend, the L929 supernatant concentration in complete DMEM was decreased by 5% during each passage. The immortalization process was completed when the BMDMs grew normally in complete DMEM in the absence of L929 supernatant.
METHOD DETAILS
Gene expression analysis ELISA
RNA was isolated from cell cultures 4 hours after stimulation using Qiashedder (QIAGEN) and GeneJET RNA Purification Kit (Life Technologies). Purified RNA was analyzed for gene expression on a CFX384 real time cycler (Bio-rad) using TaqMan RNA-to-CT 1-Step Kit (Applied Biosystems) with probes specific for Rsad2, Il-1b, Il-6, Cxcl10 and Gapdh.
ELISA were performed to measure secreted TNFα and IFNβ. Cell culture supernatants were cleared of cell debris by spinning 96 well plates at 400 × g for 5 min. Supernatants were transferred to new 96 well plates. Concentrations of TNFα and IFNβ were measured following the manufacturer’s protocols. The working concentration of the B/B homodimerizer is 1 μM.
PI staining and LDH release quantification
For experiments measuring end-point PI staining, PI (5 μM) was included in the culture media to monitor pore formation at the last 30 min for each incubation period. A Tecan plate reader was used to measure PI staining (excitation 535 nm, emission 617 nm). upernatants were assayed for LDH release at indicated time points using the Pierce LDH cytotoxicity colorimetric assay kit per the manufacturer’s instruction. The Tecan plate reader was used to measure LDH release (absorbance 490 nm and 680 nm). Detergent-treated cells were used as positive control for LDH release quantification.
Western blotting and immunoprecipitation
For western analysis of signaling pathway activation, primary BMDMs (1 × 106) or iBMDMs (0.5 × 106) were seeded in 12 well plates and stimulated with ligands for indicated periods, and subsequently lysed in 300 μL 1×SDS containing 8 M UREA. Lysates were incubated at 65°C for 15 min. Before SDS-PAGE separation, lysates were passed through a BD 1 mL sub-Q syringe attached to a 26G needle to reduce viscosity. 15 μL of individual samples (15–20 μg protein from whole cell extract) were separated by SDS-PAGE followed by western analysis.
For myddosome isolation, iBMDMs (5 × 106) were stimulated with ligands for indicated times, and subsequently lysed in 400 μL of lysis buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5% Glycerol, 1 mM Sodium deoxycholate and 1% NP40. Protease inhibitors and phosphatase inhibitors were added prior to cell lysis. Lysates were spun at top speed for 15 min at a table-top centrifuge in the cold room (at 4°C). The cleared supernatants were collected and 80 μl of the supernatants was saved as sample inputs. 0.5 μg of the anti-MyD88 antibody with 15 μl (bed volume) of protein G Sepharose (for endogenous MyD88) or 15 μl (bed volume) of anti-FLAG (M2) agarose (for 3 × FLAG-MyD88 alleles) was added to the remaining supernatants and the incubation were allowed to proceed for 4–5 hr at 4°C on a nutator. The beads containing the protein complexes were then washed for 3 times with lysis buffer, and 60 μL of SDS loading buffer was added. The protein complexes were further eluted by heating at 65°C for 15 min. A portion of eluted protein complexes (20 μl) were separated by SDS-PAGE and analyzed by western blot. Working concentrations of TLR ligands for the myddosome assay are: LPS (100 μg/ml), P3C (1 μg/ml), and R848 (1 μg/ml). Coumermycin A1 was used at the working concentration of 0.5 μM.
Generation of synthetic biology molecules
To generate MyD88-NpLxIS and MyD88 CpLxIS alleles, the cDNA sequence encoding the mouse STING pLxIS motif (340–378 aa) was fused in tandem then attached to the cDNA sequence encoding the MyD88 protein either at the 5-prime end (for MyD88-NpLxIS) or at the 3-prime end (for MyD88-CpLxIS). The mouse Caspase-1 (C285A)-pLxIS alleles and the FKBP (F36V)-pLxIS alleles were generated in the same manner. The fusion cDNAs were then synthesized as gBLOCKs via IDT. The mutant alleles were also synthesized as gBLOCKs. To generate the MyD88-RIP3 allele, the cDNA sequence encoding the full-length mouse RIP3 was attached the MyD88-encoding cDNA sequence at the 3-prime end. The fusion cDNA was then synthesized as a gBLOCK via IDT. All synthetic DNA sequences were optimized to avoid internal repeats and for optimal expression in murine cells via the IDT online program.
Microscopic imaging of cell morphology and live cell imaging
To determine cell morphology after TLR stimulation, Myd88−/−/Trif−/− iBMDMs expressing MyD88 and MyD88-RIP3 were seeded in 12-well plates (0.5 × 106 per well) and were subjected to indicated treatments (Staurosporine 1 μM; TLR ligands 1μg/ml; GSK872 1 μM) to induce cell death. For static image capture, a Nikon Eclipse TS100 microscope was used with 40 × magnification. Images were processed with the “NIS-Elements F” software. Representative images were chosen from at least three randomly chosen fields from one representative experiment of three independent experiments.
For live cell imaging, stable iBMDM lines (2 × 106 per well) seeded into a 35 mm glass bottom dish (Ibidi) were incubated with TLR ligands in PI (5 μM)-containing media. Images were acquired with the Zeiss Axiovert 200M inverted confocal microscope for 1 hr with images taken in every 3 min.
For confocal imaging of CM (0.5 μM)-induced myddosome formation. Cells were fixed at room temperature for 30 min, permeabilized with 0.2% Triton X-100 for 5 min at room temperature and permeabilized with 2% goat serum in PBS supplemented with 50 mM ammonium chloride. Primary and secondary antibody staining were performed following product instructions. Working concentrations of primary antibodies were used as the following: pTBK1 (1:100), pp38 (1:100), FLAG (M2) (1:100). Samples were imaged by the Zeiss 880 laser scanning confocal microscope.
Seahorse metabolic analysis
Extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) were measured from TLR-stimulated primary BMDMs and iBMDMs with a Seahorse XFe96 Extracellular Flux Analyzer instrument. For real-time measurement of ECAR and OCR, primary BMDMs (5 ×104 per well) and iBMDMs (7.5 ×104 per well) were seed in a seahorse 96 well plate in complete DMEM medium. Cells were allowed to attach to the assay plate for 4–5 h, then cells were washed one time with serum-free seahorse assay medium and incubated in seahorse assay medium containing 5% FBS, 10 mM Glucose and 2 mM Glutamine in 37°C incubator without CO2 for 60 min. ECAR and OCR was measured under basal conditions, after treatment with TLR ligands or inhibitors (or their combination). In the assays that involved in pretreatment of cells with inhibitors, inhibitors were injected into the assay plate seeded with cells by the Seahorse Analyzer. The cells were then incubated with the inhibitors for 45 min, followed by the injection of TLR ligands. Working concentrations of the indicated inhibitors were listed as follows: 2-DG (25 mM), BX795 (5 μM), MRT67307 (2.5 μM), actinomycin D (1.5 μg/ml). Working concentrations of the TLR ligands were listed as follows: P3C (1 μg/ml), P2C (1 μg/ml), Poly (I:C) (20 μg/ml), LPS (100 ng/ml), R848 (1 μg/ml), CpG (10 μM), and ORN Sa19 (1 μg/ml) Data represent mean ± SEM of triplicate wells. Shown is one representative experiment out of three independent experiments.
Flatox-mediated activation of the NAIP-NLRC4 inflammasome
1×106 WT or Casp1−/−/Casp11−/− iBMDMs expressing Caspase-1, Caspase-1-NpLxIS, Caspase-1-CpLxIS were treated with the individual components of flatox, PA (2 μg/ml), LFn-Fla (0.5 μg/ml), the functional flatox [PA (2 μg/ml) + LFn-Fla (0.5 μg/ml)], or not for 5 h. Cells were lysed. The expression of different Caspase-1 alleles and activation the type I-IFN (pSTAT1/STAT1 and viperin) pathway were examined by western blot. Actin was probed as loading control. Cell culture supernatants were collected to measure the release of IFNβ and LDH.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance for experiments with more than two groups was tested with One way Anova and Tukey multiple comparison tests were performed. When comparisons between only two variables were made, unpaired two tailed t test was used to assess statistical significance. Adjusted p values were calculated with Prism7 (Graphpad) or with Excel. Asterisk coding, also indicated in figure legends, is depicted as follows: *: p ≤ 0.05; **: p ≤ 0.01; ***: p ≤ 0.001; ****: p ≤ 0.0001. Data presented are representative of at least 3 independent repeats unless otherwise designated. Data with error bars are represented as mean ± SEM.
DATA AND SOFTWARE AVAILABILITY
Uncropped western analysis of all blots in this study can be found at the following URL: https://data.mendeley.com/datasets/6bw75vhgg7/1
Supplementary Material
Highlights.
Myddosomes represent organizing centers (SMOCs) that induce glycolysis and NF-κB
The kinase TBK1 is a myddosome component that activates glycolysis but not NF-κB
Myddosomes and inflammasomes can be engineered to induce user-defined responses
SMOC design principles allow creation of interferon-inducing synthetic nanomachines
ACKNOWLEDGMENTS
We thank members of the Kagan and Zanoni labs for helpful discussions. We thank R. Vance for sharing flatox. This work was supported by NIH (AI133524, AI093589, AI116550, and P30DK34854 to J.C.K.). J.C.K. holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Well-come Fund. Y.T. is supported by the Jane Coffin Childs Memorial Fund for Medical Research (the Merck Fellow).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures and two videos and can be found with this article online at https://doi.org/10.1016/j.cell.2019.01.039.
DECLARATION OF INTERESTS
The authors and Boston Children’s Hospital have filed a patent application related to the application of synthetic innate immune constructs. The authors have no financial interests to declare.
REFERENCES
- Bonham KS, Orzalli MH, Hayashi K, Wolf AI, Glanemann C, Weninger W, Iwasaki A, Knipe DM, and Kagan JC (2014). A promiscuous lipid-binding protein diversifies the subcellular sites of toll-like receptor signal transduction. Cell 156, 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker SW, Bonham KS, Zanoni I, and Kagan JC (2015). Innate immune pattern recognition: a cell biological perspective. Annu. Rev. Immunol. 33, 257–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, and Vance RE (2011). STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clackson T, Yang W, Rozamus LW, Hatada M, Amara JF, Rollins CT, Stevenson LF, Magari SR, Wood SA, Courage NL, et al. (1998). Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc. Natl. Acad. Sci. USA 95, 10437–10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K, Takeuchi O, Akira S, and Cohen P (2011). The TRAF-associated protein TANK facilitates cross-talk within the IkappaB kinase family during Toll-like receptor signaling. Proc. Natl. Acad. Sci. USA 108, 17093–17098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, and Chen ZJ (2000). Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103, 351–361. [DOI] [PubMed] [Google Scholar]
- Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, Redmann V, Freitas TC, Blagih J, van der Windt GJ, et al. (2014). TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKe supports the anabolic demands of dendritic cell activation. Nat. Immunol. 15, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, and Maniatis T (2003). IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4, 491–496. [DOI] [PubMed] [Google Scholar]
- Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagir-dar R, Inohara N, Vandenabeele P, Bertin J, Coyle A, et al. (2006). Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol. 7, 576–582. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, et al. (2012). Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 19, 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Chan FK, and Kroemer G (2017). Necroptosis: Mechanisms and Relevance to Disease. Annu. Rev. Pathol. 12, 103–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay NJ, Symmons MF, Gangloff M, and Bryant CE (2014). Assembly and localization of Toll-like receptor signalling complexes. Nat. Rev. Immunol. 14, 546–558. [DOI] [PubMed] [Google Scholar]
- George J, Motshwene PG, Wang H, Kubarenko AV, Rautanen A, Mills TC, Hill AV, Gay NJ, and Weber AN (2011). Two human MYD88 variants, S34Y and R98C, interfere with MyD88-IRAK4-myddosome assembly. J. Biol. Chem. 286, 1341–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X, Ming X, Deng P, and Jiang Y (2010). Mechanisms regulating the nuclear translocation of p38 MAP kinase. J. Cell. Biochem. 110, 1420–1429. [DOI] [PubMed] [Google Scholar]
- Green D, and Kroemer G (1998). The central executioners of apoptosis: caspases or mitochondria? Trends Cell Biol. 8, 267–271. [DOI] [PubMed] [Google Scholar]
- Grossbard L, and Schimke RT (1966). Multiple hexokinases of rat tissues. Purification and comparison of soluble forms. J. Biol. Chem. 241, 3546–3560. [PubMed] [Google Scholar]
- Hacker H, Redecke V, Blagoev B, Kratchmarova I, Hsu LC, Wang GG, Kamps MP, Raz E, Wagner H, Hacker G, et al. (2006). Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 439, 204–207. [DOI] [PubMed] [Google Scholar]
- Huang SC, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, and Pearce EJ (2016). Metabolic Reprogramming Mediated by the mTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity 45, 817–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway CA Jr. (1989). Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 54, 1–13. [DOI] [PubMed] [Google Scholar]
- Jiang X, Kinch LN, Brautigam CA, Chen X, Du F, Grishin NV, and Chen ZJ (2012). Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 36, 959–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June CH, O’Connor RS, Kawalekar OU, Ghassemi S, and Milone MC (2018). CART cell immunotherapy for human cancer. Science 359,1361–1365. [DOI] [PubMed] [Google Scholar]
- Kagan JC, Magupalli VG, and Wu H (2014). SMOCs: supramolecular organizing centres that control innate immunity. Nat. Rev. Immunol. 14, 821–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, and Mocarski ES (2013). Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 288, 31268–31279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, and Pearce EJ (2010). Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115, 4742–4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SC, Lo YC, and Wu H (2010). Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 465, 885–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, and Chen ZJ (2015). Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630. [DOI] [PubMed] [Google Scholar]
- Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schroder GF, Fitzgerald KA, Wu H, and Egelman EH (2014). Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 756,1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R, and Horng T (2009). Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 9, 692–703. [DOI] [PubMed] [Google Scholar]
- Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, and Aderem A (2006). Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1betavia Ipaf. Nat. Immunol. 7, 569–575. [DOI] [PubMed] [Google Scholar]
- Nagpal K, Plantinga TS, Sirois CM, Monks BG, Latz E, Netea MG, and Golenbock DT (2011). Natural loss-of-function mutation of myeloid differentiation protein 88 disrupts its ability to form Myddosomes. J. Biol. Chem. 286, 11875–11882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LA, Kishton RJ, and Rathmell J (2016). A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 16, 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey S, Kawai T, and Akira S (2014). Microbial sensing by Toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb. Perspect. Biol. 7, a016246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelgrom LR, van der Ham AJ, and Everts B (2016). Analysis of TLR-Induced Metabolic Changes in Dendritic Cells Using the Seahorse XF(e)96 Extracellular Flux Analyzer. Methods Mol. Biol. 1390, 273–285. [DOI] [PubMed] [Google Scholar]
- Rathinam VA, and Fitzgerald KA (2016). Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 165, 792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, and Chen ZJ (2012). STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal. 5, ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al. (2013). Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troutman TD, Hu W, Fulenchek S, Yamazaki T, Kurosaki T, Bazan JF, and Pasare C (2012). Role for B-cell adapter for PI3K (BCAP) as a signaling adapter linking Toll-like receptors (TLRs) to serine/threonine kinases PI3K/Akt. Proc. Natl. Acad. Sci. USA 109, 273–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance RE, Isberg RR, and Portnoy DA (2009). Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe 6, 10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ve T, Vajjhala PR, Hedger A, Croll T, DiMaio F, Horsefield S, Yu X, Lavrencic P, Hassan Z, Morgan GP, et al. (2017). Structural basis of TIR-domain-assembly formation in MAL- and MyD88-dependent TLR4 signaling. Nat. Struct. Mol. Biol. 24, 743–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, van Rooi-jen N, Brown CR, Krantz BA, Leppla SH, Gronert K, and Vance RE (2012). Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature 490, 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, and Ghosh S (2011). TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472, 476–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Take- uchi O, Sugiyama M, Okabe M, Takeda K, and Akira S (2003). Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301, 640–643. [DOI] [PubMed] [Google Scholar]
- Yang W, Rozamus LW, Narula S, Rollins CT, Yuan R, Andrade LJ, Ram MK, Phillips TB, van Schravendijk MR, Dalgarno D, et al. (2000). Investigating protein-ligand interactions with a mutant FKBP possessing a designed specificity pocket. J. Med. Chem. 43, 1135–1142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Uncropped western analysis of all blots in this study can be found at the following URL: https://data.mendeley.com/datasets/6bw75vhgg7/1