

Summary
Membraneless organelles (MLOs) are liquid-like subcellular compartments providing spatiotemporal control to biological processes. This study reveals that cellular stress leads to the incorporation of the adaptor protein SINTBAD (TBKBP1) into membraneless, cytosolic speckles. Determination of the interactome identified >100 proteins forming constitutive and stress-inducible members of an MLO that we termed SINT-speckles. SINT-speckles partially colocalize with activated TBK1, and deletion of SINTBAD and the SINT-speckle component AZI2 leads to impaired TBK1 phosphorylation. Dynamic formation of SINT-speckles is positively controlled by the acetyltransferase KAT2A (GCN5) and antagonized by heat shock protein-mediated chaperone activity. SINT-speckle formation is also inhibited by the autophagy-initiating kinases ULK1/2, and knockdown of these kinases prevented focal TBK1 phosphorylation in a pathway-specific manner. The phlebovirus-encoded non-structural protein S enhances ULK1-mediated TBK1 phosphorylation and shows a stress-induced translocation to SINT-speckles, raising the possibility that viruses can also target this signaling hub to manipulate host cell functions.
Subject Areas: Biological Sciences, Biochemistry, Molecular Biology, Cell Biology
Graphical Abstract
Highlights
-
•
The SINTBAD interactors constitute a membraneless organelle, the SINT-speckle
-
•
SINT-speckles are composed of constitutive and inducible components
-
•
SINT-speckle formation is controlled by ULK1, KAT2A, and chaperone activity
-
•
Components of SINT-speckles control the threshold of TBK1 activation
Biological Sciences; Biochemistry; Molecular Biology; Cell Biology
Introduction
Spatial and temporal organization of cellular signaling pathways heavily relies on compartmentalization. Structural surfaces for signal transduction can be formed by membranes, components of the cytoskeleton, membrane-surrounded organelles, or membrane-less organelles (MLOs). These MLOs occur in the cytoplasm and nucleoplasm of eukaryotic cells and have recently also been discovered in bacteria (Uversky, 2017, Brangwynne, 2013, Al-Husini et al., 2018). These liquid-like subcellular compartments are believed to be formed by phase separation (Boeynaems et al., 2018), a process that involves the spontaneous separation of a supersaturated solution into a dense and a dilute phase. The liquid-like nature of MLOs allows fusion and fission events and a dynamic exchange of components. MLOs comprise a variety of subcompartments including nucleoli, promyelocytic leukemia (PML) nuclear bodies, P bodies, and stress granules (SGs) (Boeynaems et al., 2018, Darling et al., 2018, Wheeler and Hyman, 2018). The dense phase inside MLOs contains a high concentration of proteins, which may facilitate biochemical reactions and control signaling thresholds from these very crowded environments (Boeynaems et al., 2018, Gomes and Shorter, 2018, Woodruff et al., 2017). In addition, MLOs can act as dynamic buffers for RNAs and proteins. This buffering function also serves as a passive noise filter and thus reduces the inherent randomness of chemical reactions (Saunders et al., 2012). Different MLOs have a variety of functions ranging from the expression of rRNAs and pre-assembly of ribosomes (nucleoli) to the organization of the spindle apparatus (centrosomes) and mRNA splicing (splicing speckles) (Bernardi and Pandolfi, 2007, Boulon et al., 2010, Brangwynne, 2013, Hyman et al., 2014). Aberrant forms of MLOs occur upon failure of the protein quality control system or by mutation of MLO-resident proteins, often causing age-related diseases such as amyotrophic lateral sclerosis, inclusion body myopathy, and frontotemporal dementia (Hock and Polymenidou, 2016, Malinovska et al., 2013, Ramaswami et al., 2013, Taylor et al., 2016). Furthermore, various viruses hijack MLO proteins to aid in their replication (Dhillon and Rao, 2018, Möller and Schmitz, 2003, Nakagawa et al., 2018, Reineke and Lloyd, 2013). MLOs are dynamically formed by an interplay between RNA and intrinsically disordered proteins (IDPs) that typically harbor low-sequence-complexity domains enriched in polar side chains (Arg, Gln, Glu, Ser, Lys) or structure-breaking amino acids (Gly, Pro) (Uversky and Dunker, 2010). The dynamic formation of MLOs can be regulated by post-translational modifications such as acetylation, SUMOylation, or phosphorylation (Bernardi and Pandolfi, 2007, de la Vega et al., 2011, Saito et al., 2019, Wippich et al., 2013) or by environmental cues such as changes in temperature, pH, or osmolarity (Uversky, 2017). Interestingly, a small interfering RNA (siRNA) screen identified several kinases as regulators of MLO formation, including the Ser/Thr kinase TBK1 as a regulator of splicing speckles (Berchtold et al., 2018).
TBK1 has been initially identified as a component of the antiviral response based on its ability to phosphorylate and thus activate IRF3 or IRF7 transcription factors, which in turn leads to inducible expression of type I interferons (IFN) (Fitzgerald et al., 2003, Sharma et al., 2003). TBK1, IRF3, and further components for induced type I IFN signaling are known to translocate to perinuclear punctate structures (Saitoh et al., 2009, Seo et al., 2016). Recent years have witnessed the identification of many additional TBK1 functions, which range from regulation of mitotic microtubule dynamics to the regulation of tumor necrosis factor-induced cell death (Lafont et al., 2018, Pillai et al., 2015, Xu et al., 2018). Furthermore, stress-regulated TBK1 functions comprise its role in autophagy, mitophagy, and xenophagy (Heo et al., 2015, Pilli et al., 2012, Wild et al., 2011). More recent evidence shows that TBK1 directly inhibits the AMP-activated protein kinase to increase energy storage and to repress respiration in adipose tissue, thus mediating a cross talk between immune signaling and metabolism (Zhao et al., 2018). Basal TBK1 signaling is also required for the development of KRAS-driven cancers (Barbie et al., 2009). Given the involvement of TBK1 in so many stress signaling pathways, TBK1 is a frequent target of viral proteins affecting its localization or protein-protein interactions (Liu et al., 2018, Onorati et al., 2016).
How can one single kinase such as TBK1 contribute to so many different signaling pathways? One possible answer to this conundrum might rely on the differential association of TBK1 to various adapter proteins, which co-determine its function (Goncalves et al., 2011). These adapter proteins include SINTBAD (TBKBP1), TANK (I-TRAF), and AZI2 (NAP1) and serve to assist in substrate binding and also affect the subcellular localization of TBK1 (Helgason et al., 2013). Although the adapter proteins lack any intrinsic enzymatic activity, they can affect biological functions, as exemplified by AZI2- and SINTBAD-regulated intracellular xenophagy of Salmonella (Thurston et al., 2009). SINTBAD also facilitates activation of the autophagy-initiating kinase ULK1 to control interleukin (IL)-15-induced autophagy in natural killer T cells (Zhu et al., 2018).
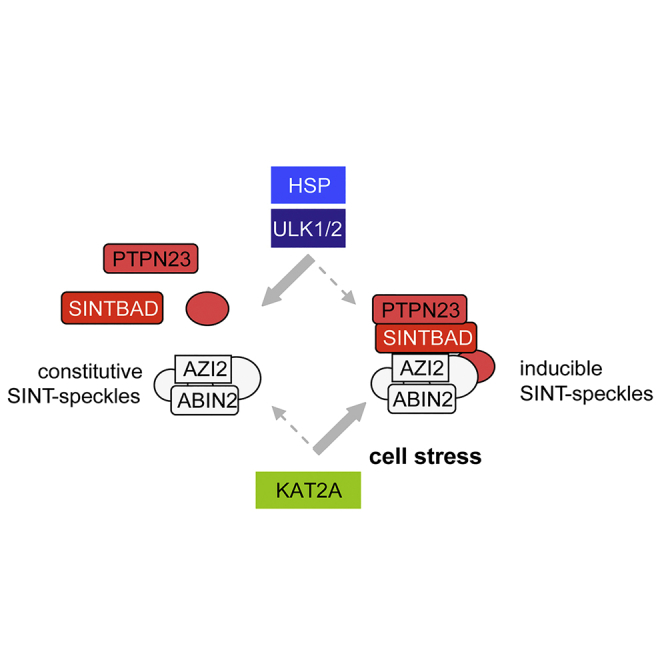
Here we have studied the intracellular distribution of SINTBAD and found its stress-regulated incorporation into cytosolic speckles not corresponding to the known MLO members that were tested. Determination of the SINTBAD interactome allowed the identification of proteins contained in constitutive and inducible SINT-speckles or as regulators of this dynamic process. The formation of inducible SINT-speckles is inhibited by the constitutive chaperone activity of heat shock proteins (HSPs) and ULK1 signaling, whereas KAT2A (also referred to as GCN5) promotes SINT-speckle formation. The SINT-speckle components AZI2 and SINTBAD determine the threshold of TBK1 activation, which partially occurs within SINT-speckles. Knockdown of ULK1/2 was sufficient to trigger SINT-speckle formation, but interfered with focal TBK1 activation in a pathway-specific fashion.
Results
Cell Stress Triggers the Formation of SINTBAD-Containing Speckles
Many proteins participating in the induction of innate immune response signaling have a propensity to form inducible polymers or filaments, which show a reduced solubility in standard lysis buffers (David et al., 2018, Pellegrini et al., 2018, Vajjhala et al., 2017). To test the possible impact of cellular stress on the solubility of SINTBAD, HeLa cells were exposed to heat shock for different periods, followed by fractionation into a cytosolic and a nuclear/insoluble fraction representing not only chromatin proteins but also poorly soluble or aggregated proteins. A western blot analysis revealed that treatment with heat shock triggered the time-dependent transition of SINTBAD from the soluble to the nuclear/insoluble fraction, whereas this dynamic redistribution between the fractions did not occur for the SINTBAD-related proteins AZI2 or TANK (Figure 1A). A similar inducible redistribution of SINTBAD also occurred in arsenite-treated 293T cells (Figure 1B), showing that various stressors can trigger the relocation of SINTBAD in different cell types. It was also interesting to test whether SINTBAD translocation to the insoluble fraction is also seen in response to further different stimuli representing inflammatory conditions or osmotic or proteotoxic stress. These results show that most adverse agents, with the exception of inflammatory stimuli, triggered translocation of SINTBAD to the nuclear/insoluble fraction (Figure 1C). This behavior was seen for the endogenous proteins and also for the adapter proteins when expressed at moderate levels (Figures S1A and S1B).
Figure 1.

Stress-Inducible Formation of SINTBAD-Containing Speckles
(A) HeLa cells were exposed for the indicated periods to heat shock, and cells were harvested and fractionated into cytosolic (cyto.) and nuclear/insoluble (nucl./insol.) extracts. Western blot was performed to detect the dynamic relocalization of the endogenous adapter proteins SINTBAD, AZI2, and TANK. The detection of phosphorylated ERK1/2 (T202/Y204), tubulin, and histone H3 serves as a control for the treatment, cytosolic, and nuclear/insoluble fraction, respectively. The positions of molecular weight markers are indicated.
(B) 293T cells were treated with 0.5 mM arsenite for the indicated times and further analyzed as in (A).
(C) Different cell lines were treated with various stressors; the exact conditions are indicated in the Transparent Methods section. Following these stimulations, cells were fractionated into cytosolic and nuclear/insoluble extracts, followed by the analysis of SINTBAD translocation as described in (A and B). The occurrence of SINTBAD translocation to the nuclear/insoluble fraction is indicated by a +.
(D) Double-deficient (sgAZI2/sgSINTBAD) U2OS cells stably expressing FLAG-SINTBAD or transfected to express moderate amounts of FLAG-AZI2 or FLAG-TANK were treated with arsenite (0.5 mM, 1 h) or heat shock (43°C, 1 h) and analyzed by indirect immunofluorescence using anti-FLAG antibodies. Nuclear DNA was stained with Hoechst; scale bars, 10 μm. The percentage of cells showing a localization as displayed in the figure is indicated.
To test whether the regulated solubility change of SINTBAD is associated with alterations of its intracellular distribution, immunofluorescence studies were performed in U2OS cells that are ideally suited and widely used for the characterization of subcellular compartmentation. In the absence of a commercial antibody faithfully detecting the endogenous SINTBAD protein in immunofluorescence studies, we generated SINTBAD-deficient U2OS cells by CRISPR/Cas9-mediated genome editing (Figures S2A and S2B) that were stably reconstituted to express moderate amounts of SINTBAD fused to an N-terminal FLAG tag. We created cell clones showing SINTBAD expression levels resembling the expression of the endogenous protein and showing dynamic relocalization to the insoluble fraction (Figures S3A and S3B). These cells were used to investigate its distribution in cells exposed to arsenite, heat shock, sorbitol, or Earle's balanced salt solution (EBSS) starvation medium. Untreated control cells showed SINTBAD as a largely cytosolic protein, whereas the various cell stressors triggered the formation of speckles occurring mainly in the cytosol and to a minor extent in the nucleus (Figures 1D and S4), reflecting the transition of SINTBAD from the cytosolic to the nuclear/insoluble fraction. Staining of FLAG-tagged TANK did not reveal any arsenite or heat shock-induced changes of its cytosolic localization, whereas AZI2 already showed a speckled distribution in unstressed cells that was not further influenced by the indicated stressors (Figure 1D).
It was then interesting to directly compare the intracellular colocalization of the three TBK1-binding adapter proteins in unstressed and stressed cells. Cell treatment with arsenite or thermal stress triggered the formation of speckles, whereas TANK remained largely cytosolic and showed only minor colocalization with focal SINTBAD (Figure 2A). The speckled localization of AZI2 in control and stress-exposed cells showed no significant overlap with TANK (Figure 2B). Interestingly, the coexpression of AZI2 and SINTBAD triggered the formation of speckles even in unstressed cells and displayed a very high degree of colocalization within these MLOs (Figure 2C).
Figure 2.

Comparative Analysis of TBK1 Adapter Protein Localization in Control and Stress-Exposed Cells
(A–C) U2OS cells were transfected to express moderate amounts of FLAG-TANK and hemagglutinin (HA)-SINTBAD (A), FLAG-AZI2 and Myc-TANK (B), or FLAG-AZI2 and HA-SINTBAD (C) as shown. Cells remained untreated or were exposed to arsenite or heat shock and subsequently analyzed by immunofluorescence with the indicated antibodies. Representative examples are shown, nuclear DNA was stained with Hoechst. Scale bars, 10 μm; the percentage of cells showing the displayed phenotype is indicated.
Speckle Formation of SINTBAD Is Antagonized by the Chaperone Activity of Heat Shock Proteins
To test the reversibility of inducible speckle formation, cells were exposed to heat shock and then further grown at 37°C to follow the fate of speckles over time. The size and number of speckles decreased in a time-dependent manner, and speckles were not detectable after 3 h of cell recovery, as revealed by fluorescence microscopy (Figure 3A) and its quantitative analysis (Figure 3B). The resolution of speckles occurred also in the absence of de novo protein synthesis (Figure S5A) and in the presence of different lysosome inhibitors (Figure S5B), suggesting that lysosomal degradation and autophagic processes do not significantly contribute to this process. As chaperone function prevents aberrant phase transition of SGs (Mateju et al., 2017) it was interesting to test whether well-characterized inhibitors of HSP70 (VER155008, Pifithrin-μ) or HSP90 (geldanamycin, radicicol) affect SINTBAD localization (Massey et al., 2010, Roe et al., 1999). Inhibition of HSP70 or HSP90 function resulted in the formation of speckles even in the absence of stress signals (Figure 3C), revealing the importance of continuous chaperone function for the cytosolic localization of SINTBAD. Coimmunoprecipitation experiments showed the interaction between SINTBAD and HSP70, which was impaired under thermal stress conditions (Figure 3D). The interaction between SINTBAD and HSP90 was only detectable after coexpression of TBK1 (Figure 3E), suggesting a rather indirect interaction that can be controlled by the relative abundance of the known HSP90 interactor TBK1 (Yang et al., 2006) or that the interaction is phosphorylation dependent.
Figure 3.

HSP-Mediated Chaperone Activity Is Required to Prevent SINTBAD Speckle Formation
(A) Reconstituted U2OS cells stably expressing FLAG-SINTBAD were treated with heat shock to induce speckle formation and then placed back in an incubator at 37°C for different times to allow recovery from heat shock. Speckle formation of SINTBAD was analyzed by immunofluorescence microscopy. Scale bar, 10 μm; the percentage of cells (n = 100) showing the displayed phenotype is indicated.
(B) SINTBAD distribution in (A) was quantified by analyzing 100 cells per condition.
(C) U2OS cells stably expressing FLAG-SINTBAD were treated with the indicated chaperone inhibitors (40 μM Ver155008, 20 μM Pifithrin-μ, 5 μM geldanamycin, and 1 μM radicicol) for 2 h and then analyzed by immunofluorescence. Scale bar, 10 μm; the percentage of cells showing the displayed phenotype is indicated.
(D) 293T cells were transfected to express FLAG-SINTBAD together with HA-HSP70. One day later, one cell dish was treated with heat shock as shown and cells were lysed. Lysates were split into two aliquots, one being used for immunoprecipitation (IP) using an anti-HA antibody. The other aliquot was used as an input control sample to ensure correct and comparable protein expression. The samples were further analyzed by western blot using appropriate antibodies as indicated.
(E) 293T cells were transfected to express FLAG-HSP90 together with HA-SINTBAD or Myc-TBK1 as shown. IP was performed using anti-FLAG antibodies, followed by western blot analysis of the IP and input samples.
SINTBAD-Containing Speckles Show No Colocalization with Cellular Organelles or Known MLOs
TBK1 and several of its interactors including optineurin and mitochondrial antiviral-signaling protein are known to be recruited to membrane-surrounded organelles such as mitochondria (Fang et al., 2017, Moore and Holzbaur, 2016) or upon RNA virus infection to the Golgi apparatus (Pourcelot et al., 2016). Thus, we asked whether SINTBAD would be found in association with these organelles. Costaining experiments with untreated and arsenite- or heat shock-treated cells showed significant colocalization neither with mitochondria (Figure 4A) nor with Golgi, lysosomes, peroxisomes, endosomes, or endoplasmic reticulum (Figure S6). In addition, we costained speckles with marker proteins of several known MLOs. These experiments showed colocalization of speckles neither with the SG marker proteins eIF4G (Figure 4B) or Ras GTPase-activating protein-binding protein 1 (G3BP1) nor with the P body marker DCP1a (Figures S7A and S7B).
Figure 4.

SINTBAD Does Not Significantly Colocalize with Membrane-Surrounded Organelles or Stress Granules
(A) U2OS cells stably expressing FLAG-SINTBAD were left untreated or exposed to arsenite or heat shock and costained with an antibody recognizing the mitochondrial marker AIFM1. A representative picture is shown. Scale bar, 10 μm.
(B) The experiments were done as in (A) with the difference that antibodies detecting the stress granule marker eIF4G were used for costaining with FLAG-SINTBAD.
Identification of SINT-Speckles
As none of these experiments related the SINTBAD-containing speckles to known subcellular structures, we aimed to identify the SINTBAD interactome by mass spectrometry. Cytosolic and nuclear/insoluble fractions were prepared from 293T cells, transfected with moderate levels of FLAG-tagged SINTBAD, followed by immunoprecipitation and identification of coprecipitating proteins by mass spectrometry as schematically shown in Figure 5A. For further bioinformatic analysis we considered proteins identified in two independent biological and technical replicates and defined high-confidence interactors by three different criteria as specified in detail in Figure S8A. This analysis revealed 150 high-confidence SINTBAD interactors of which 27% were found in the cytosol, 58% in the nuclear/insoluble fraction, and 15% in both fractions. A list of the high-confidence interactors and the complete mass spectrometry dataset is given in Table S1. To validate the interaction between SINTBAD and some of the interactors by an independent experimental approach we performed coimmunoprecipitation experiments. These experiments confirmed the results of the mass spectrometry analysis and are exemplified for the interaction of SINTBAD and the tyrosine phosphatase PTPN23 or the autophagy regulator AMBRA1 (Figure 5B). Also, binding of ABIN2 (A20-binding inhibitor of nuclear factor-κB activation 2, also referred to as TNIP2) to SINTBAD was confirmed, and mapping experiments revealed the importance of the N-terminal region of SINTBAD for this interaction (Figure 5C). Interestingly, expression of SINTBAD and all of its mutants led to an increase in ABIN2 protein levels by an unknown mechanism. The other tested interactions are listed in Table S1. The identified interacting proteins are known to serve various biological functions, as displayed in Figure 5D. While the biggest group is represented by enzymes and regulators of metabolic processes, two very prominent groups represent proteins with relevance in membrane trafficking or autophagy and also mitosis and cytoskeleton dynamics. The two smallest groups are represented by regulators of innate immunity and proteins mediating transcription, probably assisting in expression of inflammatory gene expression. To test whether mutual interactions have already been described for some of the SINTBAD-interacting proteins, we analyzed the interactors in the STRING database. This analysis revealed the existence of known protein interaction networks between the 150 proteins, as visualized in Figure 5E. We then compared the intracellular localization of SINTBAD with that of some of its interacting proteins. These experiments revealed that some of the proteins such as PTPN23 exactly mirrored the behavior of SINTBAD, as shown in Figure 5F. PTPN23 localizes in the cytosol of control cells and forms speckles after induction of cell stress. Costaining with SINTBAD revealed a complete colocalization in speckles after treatment with heat shock (Figure 5F) or arsenite (Figure S8B), confirming PTPN23 as a bona fide constituent of speckles. According to the ability of SINTBAD to bind and colocalize with further proteins we term these speckles inducible SINT-speckles.
Figure 5.

Characterization of the SINTBAD Interactome and Identification of SINT-Speckles
(A) Schematic display of the experimental setup used to identify SINTBAD-interacting proteins from cytosolic and nuclear/insoluble fractions.
(B) 293T cells were transfected to express hemagglutinin (HA)-SINTBAD along with GFP-AMBRA1 or GFP-PTPN23. One day later, immunoprecipitation (IP) was performed using GFP-Trap beads. IP and input samples were further analyzed by western blot using appropriate antibodies as indicated.
(C) An expression plasmid encoding HA-tagged ABIN2 was transfected into 293T cells together with various FLAG-SINTBAD truncation mutants (ΔN1: 106–615 amino acid [aa], ΔN2: 165–615 aa, ΔC: 1–520 aa). After IP, samples were analyzed by western blot as shown.
(D) Known functions of the respective SINTBAD interactors were retrieved from databases and Pubmed searches. Proteins were assigned to the functional groups as displayed; proteins with several functions are found in more than one group. Only the five largest functional groups (>10 proteins per group) are shown.
(E) Visualization of interacting protein networks in the SINTBAD interactome using the STRING database (Version 11.0). Line thickness indicates the strength of data support. SINTBAD (TBKBP1) is highlighted in dark green and written in bold. Proteins that were tested for their interaction with SINTBAD by co-IP (data not shown) are marked in green or red, indicating a confirmed or unconfirmed interaction, respectively.
(F) U2OS cells were transfected to express the SINTBAD interactor PTPN23 alone and were left untreated or exposed to heat shock, followed by the analysis of GFP-PTPN23 localization by fluorescence microscopy. In addition, cells cotransfected to express GFP-PTPN23 and FLAG-SINTBAD were treated the same way. A representative experiment is shown. Scale bar, 10 μm; the percentage of cells showing the displayed phenotype is indicated. The right part schematically summarizes the intracellular localization of the proteins.
Regulation of SINT-Speckle Formation by KAT2A and ULK1/2
Further costaining experiments revealed that some interactors can trigger the formation of SINT-speckles even in unstressed cells. An example of such a positive regulator is the lysine acetyltransferase KAT2A. Expression of this predominantly nuclear protein triggered the formation of SINT-speckles even in unstressed cells and also increased the fraction of nuclear speckles (Figure 6A). Another interactor triggering the formation of SINT-speckles was ABIN2, as its expression resulted in SINT-speckle formation even in unstressed cells (Figure 6B). The dominant effect of ABIN2 also occurred for PTPN23 (Figure 6B, lower) and resembles that of AZI2, which was sufficient to trigger incorporation of SINTBAD into speckles (see Figure 2C). Accordingly, AZI2 and ABIN2 always occurred in speckles when expressed either alone (Figures 1D and 6B) or together (Figure S9). Thus, AZI2 and ABIN2 are components of constitutive SINT-speckles already occurring in unstressed cells. These proteins function as positive regulators of SINT-speckle formation, whereas expression of the already known SINTBAD interactor ULK1 (Zhao et al., 2018) revealed its inhibitory function for the incorporation of SINTBAD into SINT-speckles in control and heat shock- (Figure 6C) as well as in arsenite-treated cells (Figure S10). A kinase-inactive ULK1 K46I point mutant (Chan et al., 2009), in contrast to the ULK1 wild-type, formed speckles upon cellular stress when expressed alone (Figure 6D). Coexpression of the kinase-inactive mutant with SINTBAD resulted in the formation of SINT-speckles even in unstressed cells, emphasizing the importance of ULK1 kinase activity for controlling SINT-speckle formation. To determine the consequences of ULK knockdown on SINT-speckle formation we interfered with the expression of ULK1 and also ULK2, as both kinases can have redundant functions (Lee and Tournier, 2011, Li et al., 2016). Loss of both kinases already triggered the formation of SINT-speckles even in unstressed cells (Figure 6E), supporting the finding that constitutive ULK signaling is important for restriction of SINT-speckle formation. These kinase-dependent regulatory processes could enable regulation of SINT-speckle formation, as MLOs are highly dynamic and their formation is typically also regulated during the cell cycle (Rai et al., 2018). This also applies to SINT-speckles, which do not occur in unstressed or stressed mitotic cells (Figure 6F).
Figure 6.

Characterization and Regulation of SINT-Speckles
(A) U2OS cells were transfected to express the SINTBAD interactor KAT2A alone and were left untreated or exposed to heat shock, followed by the analysis of FLAG-KAT2A localization by fluorescence microscopy. In addition, cells cotransfected to express FLAG-KAT2A and hemagglutinin (HA)-SINTBAD were treated the same way. Scale bar, 10 μm. The right part schematically summarizes the intracellular localization of the proteins.
(B) Upper: The experiment was done as in (A) with the difference that HA-ABIN2 was expressed either alone or together with FLAG-SINTBAD as shown. Lower: Cells were cotransfected to express GFP-PTPN23 and HA-ABIN2, followed by exposure to heat shock and the analysis of colocalization by immunofluorescence as shown. Scale bars, 10 μm.
(C) U2OS cells were transfected to express HA-ULK1 alone or together with FLAG-SINTBAD. Cells were left untreated or exposed to heat shock, stained, and analyzed by fluorescence microscopy. Scale bars, 10 μm. The right part schematically summarizes the results.
(D) The experiment was performed as in (A), except that a kinase-inactive HA-ULK1-K46I mutant was transfected into U2OS cells.
(E) Reconstituted SINTBAD-deficient U2OS cells were treated for three days with siRNAs specifically targeting ULK1 and ULK2 or alternatively with a control siRNA. Cells were then analyzed by fluorescence microscopy for the intracellular distribution of SINTBAD as shown. Scale bars, 10 μm.
(F) Reconstituted SINTBAD-deficient U2OS cells were exposed to arsenite or heat shock, and FLAG-SINTBAD was costained with antibodies against phosphorylated TBK1 (S172) as a centrosome marker (Pillai et al., 2015). The DNA was stained with Hoechst, and only mitotic cells were analyzed. Scale bars, 5 μm.
SINT-Speckle Components Control the Amplitude and Localization of Activated TBK1
As expression of ULK1 can trigger TBK1 phosphorylation (Zhao et al., 2018), we tested the effect of ULK1 expression on the localization of phosphorylated TBK1. ULK1-expressing cells lacked any areas with focal TBK1 phosphorylation, as the activated kinase was found in the cytosol (Figure 7A), revealing that ULK1 can control the distribution of phosphorylated TBK1. To test whether loss of ULK1 and ULK2 affects TBK1 phosphorylation in response to thermal stress, the expression of both kinases was downregulated with specific siRNAs (Figure S11). Immunofluorescence analysis of control cells revealed the heat shock-activated TBK1 in focal structures mainly in the nucleus with a partial overlap with cytosolic SINT-speckles. Downregulation of ULK1/2 largely inhibited the heat shock-induced phosphorylation of TBK1 (Figure 7B), suggesting an important contribution of these kinases for this activation pathway. In contrast, arsenite-induced TBK1 phosphorylation was not changed by ULK1/2 knockdown and occurred to a significant part in SINT-speckles (Figure 7B), showing that the ULK kinases control phosphorylation of TBK1 in a pathway-specific manner.
Figure 7.

Regulation of TBK1 Phosphorylation by Inducible SINT-Speckles
(A) U2OS cells were transfected to express hemagglutinin (HA)-ULK1 and stained for the localization of HA-ULK1 and phosphorylated TBK1 (S172) as shown.
(B) U2OS cells stably expressing FLAG-SINTBAD were treated for 3 days with siRNAs specifically targeting ULK1 and ULK2 or alternatively with a scrambled siRNA (siSCR). Cells were left untreated or exposed to arsenite or heat shock and analyzed by immunofluorescence microscopy for the intracellular distribution of SINTBAD and phosphorylated TBK1 with specific antibodies. Areas of colocalization are shown by arrows. Scale bar, 10 μm; the percentage of cells showing the displayed phenotype is given.
(C) U2OS cells stably expressing FLAG-SINTBAD were treated with arsenite or heat shock and stained for the intracellular localization of SINTBAD and TBK1 with specific antibodies. Scale bar, 10 μm; the percentage of cells showing the displayed phenotype is given.
(D) U2OS wild-type (WT) cells and two U2OS cell clones (DKO #1 and #2) lacking SINTBAD and AZI2 expression due to CRISPR/Cas9-mediated gene editing were treated for the indicated periods with 0.5 mM arsenite. Phosphorylation of TBK1 and MAP kinases (p38, ERK1/2) was determined by immunoblotting with phospho-specific antibodies as shown, to ensure successful cell stimulation. The position of a non-specific band is indicated by an asterisk.
(E) 293T cells were transfected to express HA-tagged ULK1 WT along with FLAG-tagged SINTBAD or AZI2 as shown. After 1 day cell lysates were prepared and analyzed by immunoblotting for the phosphorylation of TBK1.
It was then interesting to test whether cell stress also leads to changes in the intracellular distribution of TBK1. Treatment with arsenite or heat shock resulted in a partial recruitment of TBK1 to SINT-speckles (Figure 7C), suggesting that this previously identified interaction (Ryzhakov and Randow, 2007) also occurs in MLOs. To investigate a possible contribution of SINTBAD for the activation of TBK1, SINTBAD and AZI2 double-deficient U2OS cells were treated for various periods with arsenite and TBK1 activation was assessed with a phospho-specific antibody by immunoblotting. Knockout of SINTBAD alone had no effect (data not shown), whereas cells lacking SINTBAD and AZI2 (Figure S2) showed reduced TBK1 phosphorylation (Figure 7D). To investigate the contribution of SINTBAD and AZI2 for ULK1-induced TBK1 phosphorylation by an independent experimental approach, cells were transfected to express ULK1 together with SINTBAD and/or AZI2. Immunoblotting revealed that ULK1-triggered TBK1 phosphorylation was further enhanced by SINTBAD and AZI2 (Figure 7E), corroborating the finding that both adaptor proteins contribute to control of the TBK1 activation threshold.
A Fraction of Phlebovirus Non-structural Protein S Associates with SINT-Speckles
The severe fever thrombocytopenia syndrome phlebovirus non-structural protein S (NSs) targets the ABIN2/p105 complex to activate proviral signaling cascades (Choi et al., 2019). As ABIN2 is a core component of SINT-speckles it was interesting to investigate whether the NSs protein also can be recruited to these MLOs. Immunofluorescence analysis not only confirmed the described cytosolic localization of NSs (Choi et al., 2019) but also revealed a fraction of NSs in colocalization with ABIN2 (Figure 8A). Induction of cell stress by arsenite or heat shock resulted in an increased recruitment of NSs to SINT-speckles (Figure 8A). The expression of NSs also increased UKL1-induced TBK1 phosphorylation (Figure 8B). In summary, these data show that a virus-encoded protein affecting cellular signaling pathways such as NSs can inducibly associate with SINT-speckles.
Figure 8.

The Phlebovirus NSs Protein Inducibly Translocates to SINT-Speckles and Enhances ULK1-Triggered TBK1 Phosphorylation
(A) U2OS cells were transfected to express hemagglutinin (HA)-ABIN2 together with FLAG-NSs, treated with arsenite or heat shock as shown, and analyzed by indirect immunofluorescence. A representative experiment is shown; nuclear DNA was stained with Hoechst. Scale bars, 10 μm; the percentage of cells showing the displayed phenotype is indicated.
(B) 293T cells were transfected to express HA-ULK1 and FLAG-NSs. Total cell lysates were analyzed by immunoblotting for the phosphorylation of TBK1 (S172) as shown.
Discussion
SINT-Speckles
Here we identified the stress-induced translocation of SINTBAD to SINT-speckles, as revealed by immunofluorescence and cell fractionation experiments. These SINT-speckles form a subcellular compartment with no significant overlap to other characterized MLOs (Darling et al., 2018). SINTBAD contains two low-complexity domains containing 12.9% Gln (residues 106–346) and 27.7% Pro (residues 340–535). Core components of constitutive SINT-speckles such as ABIN2 and AZI2, and also the inducible interactor SINTBAD, are predicted to harbor long intrinsically disordered regions (Figure 9A, Table S1), which are typical for MLO-resident proteins (Elbaum-Garfinkle et al., 2015, Nott et al., 2015, Uversky and Dunker, 2010). Accordingly, about one-third of the SINT-speckle proteins (47 from 150) are predicted to have >40% disordered regions (Table S1). The mechanisms controlling the formation of constitutive SINT-speckles are not known and might involve post-translational modifications such as ULK1/2-mediated phosphorylation. Another possible mechanism could involve the bridging of two ABIN2 dimers by binding to M1-linked tri-ubiquitin chains, which might facilitate ABIN2 assembly to higher-order signaling complexes (Lin et al., 2017). Also, changes in the relative expression levels of ABIN2 speckle components affect the formation of these MLOs, explaining the abundant finding that expression of a given protein such as ABIN2 or AZI2 can affect the intracellular localization of its interactors. This behavior is characteristic for MLO formation, and accordingly, also overexpression of SG components such as TIA1 or G3BP1 is sufficient to trigger formation of SGs (Kedersha and Anderson, 2007). This implies that physiological variations in the amounts of SINT-speckle proteins can already affect speckle formation. Regulation of ABIN2 protein levels occurs in the presence of increased glucose levels or by the kinases TPL2 or IKKα/β (Chen et al., 2013, Leotoing et al., 2011, Nanda et al., 2018), and it will thus be interesting to investigate whether these situations will affect the formation of SINT-speckles. The formation of inducible SINT-speckles is regulated by several mechanisms, as schematically shown in Figure 9B. SINT-speckle formation is triggered by the acetyltransferase KAT2A. A recent study showed the relevance of acetylation of low-complexity domains for the formation of SGs (Saito et al., 2019), and it will be interesting to investigate whether the enzymatic activity of KAT2A contributes to its ability to promote inducible SINT-speckle formation. Formation of inducible SINT-speckles is antagonized by the kinase activity of its component ULK1, raising the possibility of an autoregulatory control of speckle homeostasis. Interestingly, a recent study showed that, vice versa, SINTBAD is required for ULK1 phosphorylation occurring during the cell type-specific induction of autophagy (Zhu et al., 2018), suggesting a mutual cross talk between these SINT-speckle components. Depletion of ULK1/2 leads to the formation of SINT-speckles in unstimulated cells. As expression of a kinase-inactive ULK1 mutant results in the spontaneous formation of SINT-speckles it is reasonable to assume an important contribution of the kinase function for this process. Also, other protein kinases function to restrict the formation of MLOs. The related kinases HIPK1 and HIPK2 decrease the size and number of PML-NBs during the cell cycle (Berchtold et al., 2018), and active DYRK3 facilitates the dissolution of several types of MLOs during mitosis (Rai et al., 2018). Kinase-dependent mechanisms might also contribute to the elimination of SINT-speckles during mitosis, a process that is accompanied by massive SINTBAD phosphorylation (Figures S12A and S12B). MLO composition can be modulated in response to stress (Boulon et al., 2010, Dellaire and Bazett-Jones, 2004), and accordingly this study reveals the recruitment of SINTBAD to SINT-speckles in response to a variety of cell stresses or after inhibition of the chaperone function of HSPs. Heat shock-induced speckle incorporation of SINTBAD can probably also be explained by the temperature-regulated SINTBAD-HSP70 interaction. The relevance of HSP70 was also shown for the prevention of aberrant SG formation (Mateju et al., 2017), and it will be interesting to reveal whether HSPs play a general role in the control of MLO integrity, as recently implicated by the analysis of the cellular chaperone network (Rizzolo et al., 2017). This study revealed the existence of a large functional chaperone supercomplex and its preferential interaction with proteins forming foci or condensates under stress conditions.
Figure 9.

Potential Mechanisms Allowing Formation of SINT-Speckles
(A) The amino acid sequences of the indicated proteins were analyzed by the PONDR VSL2 and VL3 prediction tools for the identification of unstructured regions, which are highlighted by bold bars.
(B) Schematic model summarizing the regulatory events allowing assembly and disassembly of inducible SINT-speckles.
Possible Functions of Inducible SINT-Speckles
SINTBAD together with AZI2 controls the threshold of TBK1 phosphorylation, as revealed by loss-of-function and gain-of-function experiments. Active and phosphorylated TBK1 in arsenite-treated cells was largely occurring in the cytosol and showed considerable colocalization with SINT-speckles. In contrast, heat shock-induced TBK1 phosphorylation was mainly nuclear and showed only a partial overlap with SINT-speckles. These differential intracellular localizations together with their distinct dependency on upstream UKL1/2 signals suggest that several pathways lead to TBK1 phosphorylation. Thus it is conceivable that SINT-speckles serve as sites of TBK1 phosphorylation in a stimulus- and context-specific manner. The occurrence of phosphorylated TBK1 in SINT-speckles and also outside from these MLOs can be explained by the fact that only a fraction of TBK1 is found in SINT-speckles at a given time point. A further possible explanation is derived from the mechanism of TBK1 activation, where the initial activation of the kinase leads to rapid interdimer trans-autophosphorylation of its activation loop (Ma et al., 2012). This implies that after primary activation of the kinase (probably facilitated by the high local protein density in SINT-speckles) active TBK1 can rapidly spread to create high local concentrations at substrate sides. This local enrichment of phosphorylated TBK1 is frequently seen by immunofluorescence and can occur in diverse subcellular localizations (Moharir et al., 2018, Pourcelot et al., 2016). The localization of TBK1 is also controlled by differential interaction with adaptor proteins including SINTBAD, TANK, and AZI2, which compete for binding to a C-terminal interaction domain in TBK1 (Goncalves et al., 2011). Contrary to the initial assumption that the TBK1 adaptor proteins control the antiviral function of the kinase, recent publications have shown their dispensability for IRF3 activation (Fang et al., 2017).
Further possible functions of inducible SINT-speckles might be derived from a set of SINTBAD interactors, which fall into several categories (see Figure 5D). The smallest group of SINTBAD interactors comprises the expected group of innate immune regulators, but interestingly the largest group is formed by enzymes and regulators of cell metabolic pathways controlling glucose phosphorylation, pyruvate decarboxylation, fatty acid synthesis, and amino acid metabolism. This might indicate a role of SINT-speckles in metabolic regulation as a mediator of the cross talk between innate immunity and metabolism (Hotamisligil, 2017, Joseph et al., 2018, Jung et al., 2019). Interestingly, the ULK kinases have also been implicated in the regulation of glucose metabolic fluxes (Li et al., 2016) and lipid metabolism (Ro et al., 2013). In addition, metabolic processes co-determine effector functions and cell fate decisions of cells from the innate and adaptive immune systems (Ganeshan and Chawla, 2014, Odegaard and Chawla, 2013). Vice versa, immunomodulatory signals such as cytokines directly regulate metabolic hormones or pathways (Könner and Brüning, 2011, Matsuki et al., 2003). The second largest group is formed by proteins involved in vesicle trafficking and autophagy, which is consistent with a previous study identifying ABIN2 as a hub protein binding to components of the endosomal sorting complex (Banks et al., 2016). This set of interactors might be also relevant for the recently uncovered role of SINTBAD as a regulator of IL-15-induced autophagy (Zhu et al., 2018), adding to the emerging role of innate immune signaling proteins such as TBK1 and TRAF6 for the formation of autophagosomes (Nazio et al., 2013, Pilli et al., 2012, Shi and Kehrl, 2010, Thurston et al., 2009). The third largest group of the SINTBAD interactome comprises regulators of mitosis and components of the cytoskeleton, in line with previous reports, documenting a function of TBK1 as a centrosome-associated regulator of mitotic microtubule dynamics (Pillai et al., 2015, Kim et al., 2013). Although this study focuses on the identification and cell biological characterization of SINT-speckles, future studies must comprehensively characterize the function(s) of constitutive and inducible SINT-speckles.
SINT-speckles might also be of pathophysiological relevance in virus infections or protein aggregation diseases. This study shows that a significant fraction of the phlebovirus NSs protein colocalize with ABIN2. It will be interesting to investigate the functional consequences of this association as well as the molecular mechanisms leading to this interaction, as the NSs protein is a structured protein (Barski et al., 2017). ABIN2 is also bound by the rabies virus-encoded M protein, which in turn controls the expression of inflammatory target genes (Besson et al., 2017), and also other components of SINT-speckles assigned to the functional group “inflammation and infection” (see Figure 5D) is targeted by viruses or involved in the antiviral response (Schmitz et al., 2014). Another possible pathophysiological scenario is due to the function of MLOs as signaling hubs in a crowded microenvironment, which can come at the cost of unwanted protein aggregation. Interestingly, partial loss of TBK1 causes protein misfolding diseases, namely, familial amyotrophic lateral sclerosis and frontotemporal dementia (Freischmidt et al., 2015, Gijselinck et al., 2015). Also, mutations in TBK1 and its substrate protein optineurin contribute to frontotemporal lobar degeneration (Le Ber et al., 2015, Pottier et al., 2015). This disease is also associated with an elevated SINTBAD expression (Broce et al., 2018), and it will be very interesting to reveal in future studies whether the formation and function of SINT-speckles is affected in neurodegenerative diseases.
Limitations of the Study
In the present study we define components of constitutive and inducible SINT-speckles and reveal their regulation by ULK1/2, HSPs, and KAT2A. However, we have not identified all biological functions exerted by this dynamically regulated protein assembly.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Yvonne Horn, Markus Schwinn, and Georgette Stovall for excellent technical assistance. We thank the following colleagues for providing plasmids: Dr. A. Chariot (University of Liège, Belgium), Dr. B. Song (Emory University School of Medicine, Atlanta, USA), Dr. C. A. Tanase (University of Bucharest, Romania), Dr. F. Cecconi (University of Rome, Italy), Dr. R. Beyaert (VIB-Ghent University, Belgium), Dr. S. H. Tooze (The Francis Crick Institute, London, UK), Dr. Ezra Burstein (UT Southwestern, Dallas, Texas, USA), Dr. J. U. Jung (University of Southern California, Los Angeles, USA), and Dr. F. Zhang (Harvard University, Boston, USA). This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - Projektnummer 109546710 - TRR81, Projektnummer 197785619 - SFB1021, and the Excellence Cluster Cardio-Pulmonary System (ECCPS, EXC 147/2).
Author Contributions
V.V.S. and M.L.S. conceived the study, V.V.S., M.S., M. Krüger, and S.J. performed and evaluated experiments, M.L.S. wrote the manuscript, and M. Kracht finalized the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: September 27, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.08.001.
Supplemental Information
References
- Al-Husini N., Tomares D.T., Bitar O., Childers W.S., Schrader J.M. alpha-proteobacterial RNA degradosomes assemble liquid-liquid phase-separated RNP bodies. Mol. Cell. 2018;71:1027–1039. doi: 10.1016/j.molcel.2018.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks C.A., Boanca G., Lee Z.T., Eubanks C.G., Hattem G.L., Peak A., Weems L.E., Conkright J.J., Florens L., Washburn M.P. TNIP2 is a hub protein in the NF-kappaB network with both protein and RNA mediated interactions. Mol. Cell. Proteomics. 2016;15:3435–3449. doi: 10.1074/mcp.M116.060509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbie D.A., Tamayo P., Boehm J.S., Kim S.Y., Moody S.E., Dunn I.F., Schinzel A.C., Sandy P., Meylan E., Scholl C. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski M., Brennan B., Miller O.K., Potter J.A., Vijayakrishnan S., Bhella D., Naismith J.H., Elliott R.M., Schwarz-Linek U. Rift Valley fever phlebovirus NSs protein core domain structure suggests molecular basis for nuclear filaments. Elife. 2017;6 doi: 10.7554/eLife.29236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berchtold D., Battich N., Pelkmans L. A systems-level study reveals regulators of membrane-less organelles in human cells. Mol. Cell. 2018;72:1035–1049. doi: 10.1016/j.molcel.2018.10.036. [DOI] [PubMed] [Google Scholar]
- Bernardi R., Pandolfi P.P. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 2007;8:1006–1016. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- Besson B., Sonthonnax F., Duchateau M., Ben K.Y., Larrous F., Eun H., Hourdel V., Matondo M., Chamot-Rooke J., Grailhe R., Bourhy H. Regulation of NF-kappaB by the p105-ABIN2-TPL2 complex and RelAp43 during rabies virus infection. PLoS. Pathog. 2017;13:e1006697. doi: 10.1371/journal.ppat.1006697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeynaems S., Alberti S., Fawzi N.L., Mittag T., Polymenidou M., Rousseau F., Schymkowitz J., Shorter J., Wolozin B., Van Den Bosch L. Protein phase separation: a new phase in cell biology. Trends Cell Biol. 2018;28:420–435. doi: 10.1016/j.tcb.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulon S., Westman B.J., Hutten S., Boisvert F.M., Lamond A.I. The nucleolus under stress. Mol. Cell. 2010;40:216–227. doi: 10.1016/j.molcel.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne C.P. Phase transitions and size scaling of membrane-less organelles. J. Cell Biol. 2013;203:875–881. doi: 10.1083/jcb.201308087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broce I., Karch C.M., Wen N., Fan C.C., Wang Y., Tan C.H., Kouri N., Ross O.A., Hoglinger G.U., Muller U. Immune-related genetic enrichment in frontotemporal dementia: an analysis of genome-wide association studies. PLoS. Med. 2018;15:e1002487. doi: 10.1371/journal.pmed.1002487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan E.Y., Longatti A., McKnight N.C., Tooze S.A. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol. Cell. Biol. 2009;29:157–171. doi: 10.1128/MCB.01082-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.Y., Chou H.C., Chen Y.H., Chan H.L. High glucose-induced proteome alterations in hepatocytes and its possible relevance to diabetic liver disease. J. Nutr. Biochem. 2013;24:1889–1910. doi: 10.1016/j.jnutbio.2013.05.006. [DOI] [PubMed] [Google Scholar]
- Choi Y., Park S.J., Sun Y., Yoo J.S., Pudupakam R.S., Foo S.S., Shin W.J., Chen S.B., Tsichlis P.N., Lee W.J. Severe fever with thrombocytopenia syndrome phlebovirus non-structural protein activates TPL2 signalling pathway for viral immunopathogenesis. Nat. Microbiol. 2019;4:429–437. doi: 10.1038/s41564-018-0329-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling A.L., Liu Y., Oldfield C.J., Uversky V.N. Intrinsically disordered proteome of human membrane-less organelles. Proteomics. 2018;18:e1700193. doi: 10.1002/pmic.201700193. [DOI] [PubMed] [Google Scholar]
- David L., Li Y., Ma J., Garner E., Zhang X., Wu H. Assembly mechanism of the CARMA1-BCL10-MALT1-TRAF6 signalosome. Proc. Natl. Acad. Sci. U S A. 2018;115:1499–1504. doi: 10.1073/pnas.1721967115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Vega L., Fröbius K., Moreno R., Calzado M.A., Geng H., Schmitz M.L. Control of nuclear HIPK2 localization and function by a SUMO interaction motif. Biochim. Biophys. Acta. 2011;1813:283–297. doi: 10.1016/j.bbamcr.2010.11.022. [DOI] [PubMed] [Google Scholar]
- Dellaire G., Bazett-Jones D.P. PML nuclear bodies: dynamic sensors of DNA damage and cellular stress. Bioessays. 2004;26:963–977. doi: 10.1002/bies.20089. [DOI] [PubMed] [Google Scholar]
- Dhillon P., Rao C.D. Rotavirus induces formation of remodeled stress granules and P bodies and their sequestration in viroplasms to promote progeny virus production. J. Virol. 2018;92 doi: 10.1128/JVI.01363-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaum-Garfinkle S., Kim Y., Szczepaniak K., Chen C.C., Eckmann C.R., Myong S., Brangwynne C.P. The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc. Natl. Acad. Sci. U S A. 2015;112:7189–7194. doi: 10.1073/pnas.1504822112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R., Jiang Q., Zhou X., Wang C., Guan Y., Tao J., Xi J., Feng J.M., Jiang Z. MAVS activates TBK1 and IKKepsilon through TRAFs in NEMO dependent and independent manner. PLoS. Pathog. 2017;13:e1006720. doi: 10.1371/journal.ppat.1006720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald K.A., McWhirter S.M., Faia K.L., Rowe D.C., Latz E., Golenbock D.T., Coyle A.J., Liao S.M., Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- Freischmidt A., Wieland T., Richter B., Ruf W., Schaeffer V., Muller K., Marroquin N., Nordin F., Hubers A., Weydt P. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015;18:631–636. doi: 10.1038/nn.4000. [DOI] [PubMed] [Google Scholar]
- Ganeshan K., Chawla A. Metabolic regulation of immune responses. Annu. Rev. Immunol. 2014;32:609–634. doi: 10.1146/annurev-immunol-032713-120236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijselinck I., Van M.S., van der Zee J., Sieben A., Philtjens S., Heeman B., Engelborghs S., Vandenbulcke M., De B.G., Baumer V. Loss of TBK1 is a frequent cause of frontotemporal dementia in a Belgian cohort. Neurology. 2015;85:2116–2125. doi: 10.1212/WNL.0000000000002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes E., Shorter J. The molecular language of membraneless organelles. J. Biol. Chem. 2018;294:7115–7127. doi: 10.1074/jbc.TM118.001192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves A., Burckstummer T., Dixit E., Scheicher R., Gorna M.W., Karayel E., Sugar C., Stukalov A., Berg T., Kralovics R. Functional dissection of the TBK1 molecular network. PLoS One. 2011;6:e23971. doi: 10.1371/journal.pone.0023971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helgason E., Phung Q.T., Dueber E.C. Recent insights into the complexity of Tank-binding kinase 1 signaling networks: the emerging role of cellular localization in the activation and substrate specificity of TBK1. FEBS Lett. 2013;587:1230–1237. doi: 10.1016/j.febslet.2013.01.059. [DOI] [PubMed] [Google Scholar]
- Heo J.M., Ordureau A., Paulo J.A., Rinehart J., Harper J.W. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol. Cell. 2015;60:7–20. doi: 10.1016/j.molcel.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock E.M., Polymenidou M. Prion-like propagation as a pathogenic principle in frontotemporal dementia. J. Neurochem. 2016;138(Suppl 1):163–183. doi: 10.1111/jnc.13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil G.S. Foundations of immunometabolism and implications for metabolic health and disease. Immunity. 2017;47:406–420. doi: 10.1016/j.immuni.2017.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman A.A., Weber C.A., Julicher F. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014;30:39–58. doi: 10.1146/annurev-cellbio-100913-013325. [DOI] [PubMed] [Google Scholar]
- Joseph A.M., Monticelli L.A., Sonnenberg G.F. Metabolic regulation of innate and adaptive lymphocyte effector responses. Immunol. Rev. 2018;286:137–147. doi: 10.1111/imr.12703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J., Zeng H., Horng T. Metabolism as a guiding force for immunity. Nat. Cell Biol. 2019;21:85–93. doi: 10.1038/s41556-018-0217-x. [DOI] [PubMed] [Google Scholar]
- Kedersha N., Anderson P. Mammalian stress granules and processing bodies. Methods Enzymol. 2007;431:61–81. doi: 10.1016/S0076-6879(07)31005-7. [DOI] [PubMed] [Google Scholar]
- Kim J.Y., Welsh E.A., Oguz U., Fang B., Bai Y., Kinose F., Bronk C., Remsing Rix L.L., Beg A.A., Rix U. Dissection of TBK1 signaling via phosphoproteomics in lung cancer cells. Proc. Natl. Acad. Sci. U S A. 2013;110:12414–12419. doi: 10.1073/pnas.1220674110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Könner A.C., Brüning J.C. Toll-like receptors: linking inflammation to metabolism. Trends Endocrinol. Metab. 2011;22:16–23. doi: 10.1016/j.tem.2010.08.007. [DOI] [PubMed] [Google Scholar]
- Lafont E., Draber P., Rieser E., Reichert M., Kupka S., de M.D., Draberova H., von M.A., Bhamra A., Henderson S. TBK1 and IKKepsilon prevent TNF-induced cell death by RIPK1 phosphorylation. Nat. Cell Biol. 2018;20:1389–1399. doi: 10.1038/s41556-018-0229-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Ber I., De Septenville A., Millecamps S., Camuzat A., Caroppo P., Couratier P., Blanc F., Lacomblez L., Sellal F., Fleury M.C. TBK1 mutation frequencies in French frontotemporal dementia and amyotrophic lateral sclerosis cohorts. Neurobiol. Aging. 2015;36:3116. doi: 10.1016/j.neurobiolaging.2015.08.009. [DOI] [PubMed] [Google Scholar]
- Lee E.J., Tournier C. The requirement of uncoordinated 51-like kinase 1 (ULK1) and ULK2 in the regulation of autophagy. Autophagy. 2011;7:689–695. doi: 10.4161/auto.7.7.15450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leotoing L., Chereau F., Baron S., Hube F., Valencia H.J., Bordereaux D., Demmers J.A., Strouboulis J., Baud V. A20-binding inhibitor of nuclear factor-kappaB (NF-kappaB)-2 (ABIN-2) is an activator of inhibitor of NF-kappaB (IkappaB) kinase alpha (IKKalpha)-mediated NF-kappaB transcriptional activity. J. Biol. Chem. 2011;286:32277–32288. doi: 10.1074/jbc.M111.236448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T.Y., Sun Y., Liang Y., Liu Q., Shi Y., Zhang C.S., Zhang C., Song L., Zhang P., Zhang X. ULK1/2 constitute a bifurcate node controlling glucose metabolic fluxes in addition to autophagy. Mol. Cell. 2016;62:359–370. doi: 10.1016/j.molcel.2016.04.009. [DOI] [PubMed] [Google Scholar]
- Lin S.M., Lin S.C., Hong J.Y., Su T.W., Kuo B.J., Chang W.H., Tu Y.F., Lo Y.C. Structural insights into linear tri-ubiquitin recognition by A20-binding inhibitor of NF-kappaB, ABIN-2. Structure. 2017;25:66–78. doi: 10.1016/j.str.2016.11.005. [DOI] [PubMed] [Google Scholar]
- Liu X., Main D., Ma Y., He B. Herpes simplex virus 1 inhibits TANK-binding kinase 1 through formation of the Us11-Hsp90 complex. J. Virol. 2018;92 doi: 10.1128/JVI.00402-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X., Helgason E., Phung Q.T., Quan C.L., Iyer R.S., Lee M.W., Bowman K.K., Starovasnik M.A., Dueber E.C. Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc. Natl. Acad. Sci. U S A. 2012;109:9378–9383. doi: 10.1073/pnas.1121552109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinovska L., Kroschwald S., Alberti S. Protein disorder, prion propensities, and self-organizing macromolecular collectives. Biochim. Biophys. Acta. 2013;1834:918–931. doi: 10.1016/j.bbapap.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Massey A.J., Williamson D.S., Browne H., Murray J.B., Dokurno P., Shaw T., Macias A.T., Daniels Z., Geoffroy S., Dopson M. A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother. Pharmacol. 2010;66:535–545. doi: 10.1007/s00280-009-1194-3. [DOI] [PubMed] [Google Scholar]
- Mateju D., Franzmann T.M., Patel A., Kopach A., Boczek E.E., Maharana S., Lee H.O., Carra S., Hyman A.A., Alberti S. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 2017;36:1669–1687. doi: 10.15252/embj.201695957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuki T., Horai R., Sudo K., Iwakura Y. IL-1 plays an important role in lipid metabolism by regulating insulin levels under physiological conditions. J. Exp. Med. 2003;198:877–888. doi: 10.1084/jem.20030299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moharir S.C., Bansal M., Ramachandran G., Ramaswamy R., Rawat S., Raychaudhuri S., Swarup G. Identification of a splice variant of optineurin which is defective in autophagy and phosphorylation. Biochim. Biophys. Acta Mol. Cell Res. 2018;1865:1526–1538. doi: 10.1016/j.bbamcr.2018.08.009. [DOI] [PubMed] [Google Scholar]
- Möller A., Schmitz M.L. Viruses as hijackers of PML nuclear bodies. Arch. Immunol. Ther. Exp. (Warsz.) 2003;51:295–300. [PubMed] [Google Scholar]
- Moore A.S., Holzbaur E.L. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. U S A. 2016;113:E3349–E3358. doi: 10.1073/pnas.1523810113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa K., Narayanan K., Wada M., Makino S. Inhibition of stress granule formation by middle east respiratory syndrome coronavirus 4a accessory protein facilitates viral translation, leading to efficient virus replication. J. Virol. 2018;92 doi: 10.1128/JVI.00902-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda S.K., Nagamori T., Windheim M., Amu S., Aviello G., Patterson-Kane J., Arthur J.S.C., Ley S.C., Fallon P., Cohen P. ABIN2 function is required to suppress DSS-induced colitis by a Tpl2-independent mechanism. J. Immunol. 2018;201:3373–3382. doi: 10.4049/jimmunol.1700614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazio F., Strappazzon F., Antonioli M., Bielli P., Cianfanelli V., Bordi M., Gretzmeier C., Dengjel J., Piacentini M., Fimia G.M., Cecconi F. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 2013;15:406–416. doi: 10.1038/ncb2708. [DOI] [PubMed] [Google Scholar]
- Nott T.J., Petsalaki E., Farber P., Jervis D., Fussner E., Plochowietz A., Craggs T.D., Bazett-Jones D.P., Pawson T., Forman-Kay J.D., Baldwin A.J. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell. 2015;57:936–947. doi: 10.1016/j.molcel.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard J.I., Chawla A. The immune system as a sensor of the metabolic state. Immunity. 2013;38:644–654. doi: 10.1016/j.immuni.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onorati M., Li Z., Liu F., Sousa A.M.M., Nakagawa N., Li M., Dell′Anno M.T., Gulden F.O., Pochareddy S., Tebbenkamp A.T.N. Zika virus disrupts phospho-TBK1 localization and mitosis in human neuroepithelial stem cells and radial glia. Cell Rep. 2016;16:2576–2592. doi: 10.1016/j.celrep.2016.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini E., Desfosses A., Wallmann A., Schulze W.M., Rehbein K., Mas P., Signor L., Gaudon S., Zenkeviciute G., Hons M. RIP2 filament formation is required for NOD2 dependent NF-kappaB signalling. Nat. Commun. 2018;9:4043. doi: 10.1038/s41467-018-06451-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai S., Nguyen J., Johnson J., Haura E., Coppola D., Chellappan S. Tank binding kinase 1 is a centrosome-associated kinase necessary for microtubule dynamics and mitosis. Nat. Commun. 2015;6:10072. doi: 10.1038/ncomms10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilli M., Arko-Mensah J., Ponpuak M., Roberts E., Master S., Mandell M.A., Dupont N., Ornatowski W., Jiang S., Bradfute S.B. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. 2012;37:223–234. doi: 10.1016/j.immuni.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottier C., Bieniek K.F., Finch N., van de Vorst M., Baker M., Perkersen R., Brown P., Ravenscroft T., van B.M., Nicholson A.M. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 2015;130:77–92. doi: 10.1007/s00401-015-1436-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourcelot M., Zemirli N., Silva Da C.L., Loyant R., Garcin D., Vitour D., Munitic I., Vazquez A., Arnoult D. The Golgi apparatus acts as a platform for TBK1 activation after viral RNA sensing. BMC. Biol. 2016;14:69. doi: 10.1186/s12915-016-0292-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai A.K., Chen J.X., Selbach M., Pelkmans L. Kinase-controlled phase transition of membraneless organelles in mitosis. Nature. 2018;559:211–216. doi: 10.1038/s41586-018-0279-8. [DOI] [PubMed] [Google Scholar]
- Ramaswami M., Taylor J.P., Parker R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell. 2013;154:727–736. doi: 10.1016/j.cell.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reineke L.C., Lloyd R.E. Diversion of stress granules and P-bodies during viral infection. Virology. 2013;436:255–267. doi: 10.1016/j.virol.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzolo K., Huen J., Kumar A., Phanse S., Vlasblom J., Kakihara Y., Zeineddine H.A., Minic Z., Snider J., Wang W. Features of the chaperone cellular network revealed through systematic interaction mapping. Cell Rep. 2017;20:2735–2748. doi: 10.1016/j.celrep.2017.08.074. [DOI] [PubMed] [Google Scholar]
- Ro S.H., Jung C.H., Hahn W.S., Xu X., Kim Y.M., Yun Y.S., Park J.M., Kim K.H., Seo M., Ha T.Y. Distinct functions of Ulk1 and Ulk2 in the regulation of lipid metabolism in adipocytes. Autophagy. 2013;9:2103–2114. doi: 10.4161/auto.26563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe S.M., Prodromou C., O′Brien R., Ladbury J.E., Piper P.W., Pearl L.H. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem. 1999;42:260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- Ryzhakov G., Randow F. SINTBAD, a novel component of innate antiviral immunity, shares a TBK1-binding domain with NAP1 and TANK. EMBO J. 2007;26:3180–3190. doi: 10.1038/sj.emboj.7601743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M., Hess D., Eglinger J., Fritsch A.W., Kreysing M., Weinert B.T., Choudhary C., Matthias P. Acetylation of intrinsically disordered regions regulates phase separation. Nat. Chem. Biol. 2019;15:51–61. doi: 10.1038/s41589-018-0180-7. [DOI] [PubMed] [Google Scholar]
- Saitoh T., Fujita N., Hayashi T., Takahara K., Satoh T., Lee H., Matsunaga K., Kageyama S., Omori H., Noda T. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. U S A. 2009;106:20842–20846. doi: 10.1073/pnas.0911267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders T.E., Pan K.Z., Angel A., Guan Y., Shah J.V., Howard M., Chang F. Noise reduction in the intracellular pom1p gradient by a dynamic clustering mechanism. Dev. Cell. 2012;22:558–572. doi: 10.1016/j.devcel.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz M.L., Kracht M., Saul V.V. The intricate interplay between RNA viruses and NF-kappaB. Biochim. Biophys. Acta. 2014;1843:2754–2764. doi: 10.1016/j.bbamcr.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo M., Lee S.O., Kim J.H., Hong Y., Kim S., Kim Y., Min D.H., Kong Y.Y., Shin J., Ahn K. MAP4-regulated dynein-dependent trafficking of BTN3A1 controls the TBK1-IRF3 signaling axis. Proc. Natl. Acad. Sci. U S A. 2016;113:14390–14395. doi: 10.1073/pnas.1615287113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S., tenOever B.R., Grandvaux N., Zhou G.P., Lin R., Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- Shi C.S., Kehrl J.H. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci. Signal. 2010;3:ra42. doi: 10.1126/scisignal.2000751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J.P., Brown R.H., Jr., Cleveland D.W. Decoding ALS: from genes to mechanism. Nature. 2016;539:197–206. doi: 10.1038/nature20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston T.L., Ryzhakov G., Bloor S., von M.N., Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 2009;10:1215–1221. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- Uversky V.N. Intrinsically disordered proteins in overcrowded milieu: membrane-less organelles, phase separation, and intrinsic disorder. Curr. Opin. Struct. Biol. 2017;44:18–30. doi: 10.1016/j.sbi.2016.10.015. [DOI] [PubMed] [Google Scholar]
- Uversky V.N., Dunker A.K. Understanding protein non-folding. Biochim. Biophys. Acta. 2010;1804:1231–1264. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vajjhala P.R., Ve T., Bentham A., Stacey K.J., Kobe B. The molecular mechanisms of signaling by cooperative assembly formation in innate immunity pathways. Mol. Immunol. 2017;86:23–37. doi: 10.1016/j.molimm.2017.02.012. [DOI] [PubMed] [Google Scholar]
- Wheeler R.J., Hyman A.A. Controlling compartmentalization by non-membrane-bound organelles. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018;373 doi: 10.1098/rstb.2017.0193. pii: 20170193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild P., Farhan H., McEwan D.G., Wagner S., Rogov V.V., Brady N.R., Richter B., Korac J., Waidmann O., Choudhary C. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–233. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wippich F., Bodenmiller B., Trajkovska M.G., Wanka S., Aebersold R., Pelkmans L. Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell. 2013;152:791–805. doi: 10.1016/j.cell.2013.01.033. [DOI] [PubMed] [Google Scholar]
- Woodruff J.B., Ferreira G.B., Widlund P.O., Mahamid J., Honigmann A., Hyman A.A. The centrosome is a selective condensate that nucleates microtubules by concentrating tubulin. Cell. 2017;169:1066–1077. doi: 10.1016/j.cell.2017.05.028. [DOI] [PubMed] [Google Scholar]
- Xu D., Jin T., Zhu H., Chen H., Ofengeim D., Zou C., Mifflin L., Pan L., Amin P., Li W. TBK1 suppresses RIPK1-driven apoptosis and inflammation during development and in aging. Cell. 2018;174:1477–1491. doi: 10.1016/j.cell.2018.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K., Shi H., Qi R., Sun S., Tang Y., Zhang B., Wang C. Hsp90 regulates activation of interferon regulatory factor 3 and TBK-1 stabilization in Sendai virus-infected cells. Mol. Biol. Cell. 2006;17:1461–1471. doi: 10.1091/mbc.E05-09-0853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P., Wong K.I., Sun X., Reilly S.M., Uhm M., Liao Z., Skorobogatko Y., Saltiel A.R. TBK1 at the crossroads of inflammation and energy homeostasis in adipose tissue. Cell. 2018;172:731–743. doi: 10.1016/j.cell.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L., Xie X., Zhang L., Wang H., Jie Z., Zhou X., Shi J., Zhao S., Zhang B., Cheng X., Sun S.C. TBK-binding protein 1 regulates IL-15-induced autophagy and NKT cell survival. Nat. Commun. 2018;9:2812. doi: 10.1038/s41467-018-05097-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.