

Abstract
Traumatic brain injury (TBI) is a neurodegenerative disorder for which no effective pharmacological treatment is available. Glucagon-like peptide 1 (GLP-1) analogues such as Exenatide have previously demonstrated neuro-trophic and neuroprotective effects in cellular and animal models of TBI. However, chronic or repeated administration was needed for efficacy. In this study, the pharmacokinetics and efficacy of PT302, a clinically available sustained-release Exenatide formulation (SR-Exenatide) were evaluated in a concussive mild (m)TBI mouse model. A single subcutaneous (s.c.) injection of PT302 (0.6, 0.12, and 0.024mg/kg) was administered and plasma Exenatide concentrations were time-dependently measured over 3 weeks. An initial rapid regulated release of Exenatide in plasma was followed by a secondary phase of sustained-release in a dose-dependent manner. Short-and longer-term (7 and 30 day) cognitive impairments (visual and spatial deficits) induced by weight drop mTBI were mitigated by a single post-injury treatment with Exenatide delivered by s.c. injection of PT302 in clinically translatable doses. Immunohistochemical evaluation of neuronal cell death and inflammatory markers, likewise, cross-validated the neurotrophic and neuroprotective effects of SR-Exenatide in this mouse mTBI model. Exenatide central nervous system concentrations were 1.5% to 2.0% of concomitant plasma levels under steady-state conditions. These data demonstrate a positive beneficial action of PT302 in mTBI. This convenient single, sustained-release dosing regimen also has application for other neurological disorders, such as Alzheimer’s disease, Parkinson’s disease, multiple system atrophy and multiple sclerosis where prior preclinical studies, likewise, have demonstrated positive Exenatide actions.
Keywords: Mild traumatic brain injury, PT302, Exenatide, Exendin-4, Glucagon-like peptide-1, Incretin mimetic, Neurodegeneration, Neuroinflammation
1. Introduction
The incretin glucagon-like peptide 1 (GLP-1), a proteolytic product of proglucagon produced by intestinal cells, is a gut-derived circulating peptide hormone that potentiates glucose-dependent insulin secretion by pancreatic β-cells following food ingestion (Campbell and Drucker, 2013a; Tolhurst et al., 2009). In addition to its best known in-sulinotropic actions, GLP-1 confers glucose sensitivity to glucose-resistant β-cells and acts as a trophic agent by inducing pancreatic β-cell proliferation and neogenesis, and by inhibiting β-cell apoptosis (Chon and Gautier, 2016; Koehler et al., 2015). As a consequence of the glucose homeostatic actions and enhancement of insulin signaling afforded by GLP-1, agents that activate the GLP-1 receptor (GLP-1R), such as long acting GLP-1 analogues, have been developed for the treatment of type 2 diabetes mellitus (T2DM), and appear to be well tolerated, efficacious and widely used (Drucker, 2018; Nauck, 2016).
Increasing evidence indicates that GLP-1 and analogues also exert multiple extra-pancreatic actions that are independent of glucose homeostasis and are mediated by GLP-1R activation at other sites (Campbell and Drucker, 2013b; Gallwitz, 2014). GLP-1R have been found in multiple organs, including the central nervous system, where GLP-1Rs are widely expressed on neurons throughout the brain, localized to dendrites (Hamilton and Hölscher, 2009; Heppner et al., 2015), as well as on glia during an inflammatory response (Kim et al., 2009; Yun et al., 2018). GLP-1 and analogues appear to freely enter the brain (Kastin et al., 2002; Kastin and Akerstrom, 2003) and elicit multiple cellular events within it, including effects on neuroinflammation, mitochondrial function, and cellular proliferation (Athauda and Foltynie, 2018; Athauda and Foltynie, 2016; Kim et al., 2017; Li et al., 2016; Salcedo et al., 2012). Furthermore, multiple studies have demonstrated neurotrophic and neuroprotective actions of GLP-1R stimulation across animal models of neurodegeneration (Athauda and Foltynie, 2016, 2018; Kim et al., 2017; Li et al., 2016; Salcedo et al., 2012). Consequently, long acting GLP-1 analogues are being evaluated as a new treatment strategy for neurological disorders and, in particular, for neurodegenerative conditions. As the insulinotropic actions of GLP-1 and longer-acting agonists are glucose-dependent, advantageously and unlike other current agents for diabetes treatment, their pharmacological actions are not associated with a risk of inducing hypoglycemia (Campbell and Drucker, 2013a; Drucker, 2018; Nauck, 2016).
Several proof-of-concept clinical studies have shown promise in Parkinson’s disease (PD), and others remain ongoing in Alzheimer’s disease (AD) (Antonsen et al., 2018; Athauda et al., 2017, 2018; Gejl et al., 2016). Exenatide (Exendin-4), a stable non-hydrolyzable GLP-1 analogue, in particular, has shown efficacy in recent PD clinical studies (Aviles-Olmos et al., 2014, 2013; Athauda et al., 2017), and can hence be considered a prime candidate among other GLP-1 analogues to reposition for neurological disorders consequent to its brain penetration and proven safety in T2DM. In prior preclinical research that supported clinical translation for neurological disorders, Exenatide proved particularly efficacious when delivered continuously by a micro-osmotic pump in order to maintain brain target concentrations within a therapeutic range (Bassil et al., 2017; Eakin et al., 2013; Li et al., 2010a, 2012; Rachmany et al., 2013, 2017; Tweedie et al., 2013, 2016b). The implantation of drug-containing pumps is less practical in human studies; however, the application of sustained-release (SR) technology to Exenatide provides the ability to potentially maintain steady-state concentrations over weeks to months after a single acute subcutaneous (s.c.) administration. Such technology provides the opportunity to optimize the beneficial potential of drug treatment for a chronic disorder, particularly where disease associated cognitive impairments may impact compliance.
PT302 is a SR-Exenatide formulation with a long duration dosing profile and is under clinical development by Peptron Inc. (Daejeon, Republic of Korea) for the treatment of T2DM (Gu et al., 2014). PT302 is manufactured utilizing an ultrasonic spray drying process to generate microspheres containing 2% Exenatide. These microspheres are comprised of large polymers, such as poly(lactic-co-glycolic acid) (PLGA), and are coated with L-lysine to minimize the initial burst-related adverse effects of Exenatide that can occur following its administration. In the current study, PT302 was evaluated time-dependently in a concussive mild traumatic brain injury (mTBI) mouse model by utilizing behavioral tests and immunohistochemistry to evaluate the neuroprotective, anti-inflammatory and cognitive benefits of PT302 across a range of doses of potential clinical relevance. Pharmacokinetic studies in parallel evaluated Exenatide release in plasma and central nervous system entry.
2. Materials and methods
2.1. Experimental animals
For mouse mTBI and pharmacokinetic studies, male ICR mice, 30 to 40g body weight, were used, and housed at five per home cage under a constant 12-h light/dark cycle, at room temperature (22 ± 2°C). Animals had unlimited access to food (Purina) and water. For each assessment time point and treatment regimen in behavioral and immunohistochemistry studies, an animal was utilized only once. For pharmacokinetic studies to evaluate plasma levels of Exenatide released following PT302 administration, one or more time-dependent blood samples (approximately 0.25 mL) were obtained from the submandibular vein (Golde et al., 2005). For rat pharmacokinetic studies, adult male Sprague-Dawley rats (2 months old) were evaluated and housed two per cage under a constant 12-h light/dark cycle, at room temperature (22 ± 2°C) with unlimited access to food (Purina) and water.
All studies were performed under experimental protocols approved by either the Animal Care and Use Committee of the Intramural Research Program, National Institute on Aging (Baltimore, MD, USA) (Protocol No. 331-TGB-2018) or the Sackler Faculty of Medicine Ethics Committee (Tel Aviv, Israel) (Protocol No. M-11–086). All animal study methods were carried out in accordance with the National Institutes of Health (DHEW publication 85–23, revised, 1995). The animal numbers evaluated in each assessment group, and the experimental measures and times at which they were performed were selected based upon our prior studies. An analysis of the variance of data from these prior studies was evaluated in order to minimize the numbers of animals used based on power calculations.
2.2. Mouse closed head mild traumatic brain injury
A weight drop-induced head injury was employed in our studies using a head trauma device that has previously been described (Milman et al., 2005; Tweedie et al., 2007; Zohar et al., 2003). The device consisted of a metal tube (80 cm long, 13 cm inner diameter) together with a sponge placed under the tube to support the head of each mouse during the procedure. Mice were anesthetized by inhalation of isoflurane and placed under the device. A metal weight (30 g) was then dropped from the top of the tube to strike the head of the mouse on the right temporal side between the corner of the eye and the ear. Immediately following the injury, mice were returned to their original cages for recovery and observation. Sham animals (control mice not subjected to mTBI) were handled similarly. Specifically, they were anesthetized with gaseous isofluorane, aligned under the weight drop device, but without weight drop, and were then returned to their original cage for recovery and observation. Notably, and in accord with the mild nature of our TBI challenge and our prior studies (Milman et al., 2005; Tweedie et al., 2007; Zohar et al., 2003), sham and mTBI challenged animals were indistinguishable following recovery from anesthesia, and at 1 and 24 h observation times thereafter. No animals demonstrated distress of any kind. Body (rectal) temperature was monitored (pediatric digital thermometer) in a subset of animals prior to and post mTBI, both with and without PT302 administration, to assess for changes in animal core body temperature between sham and peptide treated animals. No difference was found between these groups of mice. These mTBI conditions (a 30 g weight and 30 g mouse) were created to mirror human falling on their head from a three foot fall, or a clash of heads between two humans of approximately equal weight. These are considered mild concussive injuries (Deselms et al., 2016).
2.3. PT302 preparation and administration
PT302 is a sustained-release formulation of Exenatide. The powdered PT302 formulation used in our study (Lot PT3025014) was clinical grade material, similar to that used in prior human studies (Gu et al., 2014), and contained a mixture of polymers (98%) and Exenatide (2%). The composition of the diluent used to prepare the PT302 suspension was 0.5% carboxymethylcellulose sodium, 5.0% D-mannitol and 0.1% Tween 80 (pH 6.66) in sterile, double distilled water. PT302 was prepared in diluent one hour prior to administration, maintained on wet ice (4 °C), and thoroughly mixed (by vortex) immediately before each animal injection to ensure a homogeneous distribution of PT302 within the diluent. A single s.c. injection was made into the back of the neck region in dosed rodents. A similar volume of vehicle was administered to other animals. No difference in blood glucose levels was measured between animals administered PT302 and vehicle.
2.3.1. Mouse pharmacokinetic studies
All pharmacokinetic studies were performed in mice with the sole exception of those evaluating the central nervous system uptake of Exenatide from plasma into CSF, which were undertaken in rats (as detailed below). Initial dose range studies were performed in mice without mTBI (PT302 dose evaluation range 0.1 to 2.0 mg/kg) with blood sampling 7 days later to ascertain the dose-dependent linearity of plasma Exenatide levels. To evaluate whether Exenatide release from PT302 and plasma Exenatide levels were not altered by mTBI challenge, a single dose of PT302 (0.6 mg/kg) was administered to mTBI challenged mice 1 h after injury, as well as to sham mice. Blood samples were then withdrawn at 12, 24 and 72 h. Finally, three doses of PT302 were selected from the original dose ranging 7 day study (low: 0.024 mg/kg, medium: 0.12 mg/kg and high: 0.6 mg/kg) and time-dependent samples were collected before dosing and at 1, 24, 168, 336, and 504 h after dosing to define the release of Exenatide and maintenance of plasma levels after a single PT302 dose.
2.3.2. Rat pharmacokinetic studies
Sprague-Dawley rats were used to evaluate PT302 entry into the central nervous system as the use of rats, rather than mice, allowed the sampling of a sufficient volume of CSF (approximately 100 ul) for quantification of Exenatide levels. Our pilot studies of quantifying Exenatide levels in rodent brain samples demonstrated that unspecific background signal was too high to accurately allow determination of levels below 100pg/mL. As it has been established that the permeability of the blood-brain barrier is similar between mice and rats within the experimentally determined values of the drug transport measure, the permeability coefficient-surface area product (PS) showing a correlation of 1:1 between the two species across a broad range of drugs (Murakami et al., 2000), we therefore evaluated Exenatide uptake into CSF as a measure comparable with human studies using the rat as an animal model. As prior studies have demonstrated that there is no change in the integrity of the blood-brain barrier following a 30 g mTBI challenge in mice (Pan et al., 2003), our studies to compare Exenatide CSF and plasma levels did not involve a TBI challenge.
To separate groups of animals (N = 4 to 5 per group), Exenatide was administered s.c. in the form of either (i) PT302 containing 0.46mg or 0.92mg Exenatide, (ii) via mini pump (Alzet model 2ML2, Cupertino, CA) delivering 7.0 pM/kg/min or 15.0 pM/kg/min as an alternative means to maintain steady-state plasma levels, or (iii) twice daily immediate release Exenatide (4.6 or 10 μg/kg daily). Pumps were implanted aseptically via a small incision in the back of the neck under isoflurane anesthesia. Plasma and CSF (from cisterna magna) samples were obtained from all animals on day 14 after the initiation of Exenatide administration, which for immediate release Exenatide was measured at 90min following the final s.c. dose.
Across our pharmacokinetic studies, blood samples were collected into heparinized tubes that contained aprotinin; samples were centrifuged (10,000g × 4min at 4°C), and plasma collected and immediately stored at −80 °C. Thereafter, plasma Exenatide concentrations were quantified with an Exenatide fluorescent immunoassay kit (Phoenix Pharmaceuticals Inc., Burlingame, CA). An organic solvent extract was obtained by adding methanol to the plasma that contained either the Exenatide standards or the in vivo obtained test samples. Our calibration curves ranged from 20 to 5000 pg/mL, and quality control samples were prepared at the concentrations of 40, 150, and 300 pg/ mL. CSF samples were withdrawn from animals and immediately frozen at −80°. Calibration curves were prepared from 1 to 1000 pg/mL. All samples, including the Exenatide standards, the quality control samples, and the test samples, were assayed in duplicate. Fluorescence was detected with a Tecan GENios (Grödig, Austria) microplate reader at an excitation wavelength of 320 nm and an emission wavelength of 420 nm. The bioanalysis method was validated and satisfied the applicable US Food and Drug Administration guidelines (guidance for industry: bioanalytical method validation, 2001) with regard to the criteria of specificity, recovery (extractability), linearity, reproducibility, accuracy, and stability.
2.4. Behavioral tests
Fig. 1 provides a diagrammatic scheme of the study design of mTBI induction, drug (PT302) and vehicle administration, and immunohistochemical and behavioral evaluations. Behavioral assessments were conducted 7 or 30 days following mTBI challenge. Each mouse was used at one time point only. At 7 days, all three doses of PT302 (low, medium and high) were evaluated, while at 30 days only the low and high doses were assessed. All equipment used in the behavioral tests was thoroughly cleaned with 70% ethanol between each session to minimize any olfactory influences on mouse behavior. Mouse cognitive tests were performed using an unbiased operator basis. The assessments employed were as follows: elevated plus maze, novel object recognition and the Y-maze paradigm. Our laboratories utilize these behavioral tests routinely (Baratz et al., 2015; Deselms et al., 2016; Li et al., 2015; Rachmany et al., 2013, 2017; Tamargo et al., 2017; Tweedie et al., 2013, 2016a). All behavioral studies were performed during the light phase of the light/dark cycle. The animal numbers utilized within each of the behavioral studies are provided within the Figures.
Fig. 1.
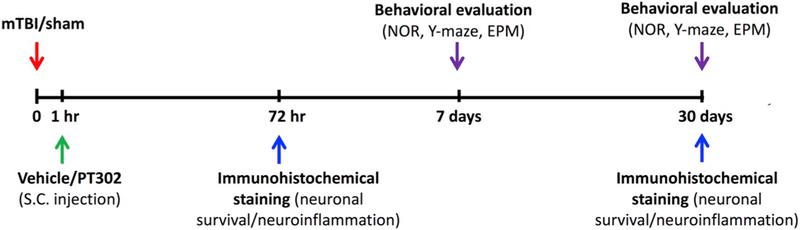
Scheme of study design time line: Animals were exposed to sham/mTBI and 1 h later, were administered PT302 or vehicle as a single s.c injection. Immunohistochemical staining to evaluate neuronal survival and neuroinflammation were performed 72 h/30 days following mTBI challenge. Behavioral tests to assess cognitive abilities were carried out 7 days/30 days following mTBI. Each experimental time point was performed on a different group of mice. (NOR: novel object recognition paradigm; EPM: elevated plus maze paradigm).
2.5. Elevated plus maze paradigm
The elevated plus maze was chosen to evaluate anxiety-like behavior, as previously described (Alcalay et al., 2004). This test relies on the preference of rodents to explore enclosed darkened environments rather than open, light and elevated environments. The apparatus consisted of a four-armed black Plexiglass maze (each at 90° to one another), in which two of the arms had low walls (open-30 × 5 × 1 cm) and the other two arms possessed high walls (closed-30 × 5 × 15 cm). The alike arms faced each other, and all had no roof. For evaluation, each mouse was gently placed in the middle of the apparatus facing one of the open arms, and was allowed to explore the arena for 5 min. The time that mice spent within the open arms and the number of entries to the open/closed arms were recorded. A longer total time spent within the open arms is associated with lower anxiety levels.
2.6. Novel object recognition paradigm
The novel object recognition task was selected to quantitatively evaluate the recognition and visual memory of the mice, as previously described (Edut et al., 2011). This paradigm relies on the inclination of rodents to investigate novel objects within their environment, as compared to known ones. The behavioral arena entailed the use of an open field black Plexiglass box (59 × 59 × 20 cm in dimension). At 48 h prior to the test, mice were individually placed within the empty arena to allow habituation for a 5 min duration. At 24 h prior to the test, each mouse was exposed to two identical objects within the same arena for 5 min. On the test day, one of these objects was replaced with a novel one, and mice were permitted to explore the arena for 5 min. During each trial, the amount of time spent near the novel and the old objects was measured. A preference index was adapted from Dix and Aggleton (1999) and calculated as follows: (time near novel object - time near old object) / (time near novel object + time near old object).
2.7. Y-maze paradigm
The Y-maze paradigm was selected to quantitatively probe the spatial memory of mice, as previously described (Baratz et al., 2010). This test relies on the preference of rodents to explore new environments in lieu of already familiar ones. The Y-maze apparatus was comprised of a three-armed black Plexiglass maze with each arm separated by 120°. These arms measured 8 × 30 × 15 cm and were distinguishable from one another only by the presence of different spatial cues placed within each (explicitly, a triangle, a square, and a circle). The start arm was chosen randomly. In the first session of the test, each mouse was placed within the start arm and permitted to explore one arm while the other one was blocked for 5 min. Thereafter, the mouse was returned to its home cage for a two min duration. Meanwhile, the arena was cleaned with ethanol 70%. During the second test session, the previously closed arms were opened, and the mouse was permitted to explore all three arms for 2 min. The time each mouse spent within the old and new arms was measured and documented. Again, a preference index was adapted from Dix and Aggleton (1999) and calculated as follows: (time in new arm − time in familiar arm) / (time in new arm + time in familiar arm).
2.8. Immunohistochemical staining
At either 72 h or 30 days following either injury or the sham procedure, mice were anesthetized with a combination of ketamine (100 mg/kg) and xylazine (10 mg/kg), and perfused transcardially with 10 mL phosphate buffered saline (PBS) followed by 20 mL of 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (pH 7.4). Thereafter, the brain was removed, fixed overnight in 4% PFA, and then placed in 1% PFA. Prior to sectioning, brains were transferred to 30% sucrose for 48 h. Frozen coronal sections (30 μm) were cut on a cryostat and collected serially from bregma at ~ −1.28 mm to ~ −2.4 mm. The sections were placed in a cryoprotectant solution containing phosphate buffer, ethylene glycol and glycerin, and stored at −20 °C. The freefloating sections were first blocked by 0.1% Triton X-100 in phosphate-buffered saline (PBST) and 10% normal horse serum for 1 h at 25 °C, and then incubated for 24 h at 4 °C with appropriate antibodies specific for staining key features of interest in our studies.
NeuN antibodies are widely used to label neurons, as the vast majority are strongly NeuN positive, and NeuN immunoreactivity has been widely used to differentiate neurons from glia and other cell types in tissue culture and in immunohistological sections in vivo (Herculano-Houzel and Lent, 2005). We hence probed sections with a mouse primary antibody raised against NeuN (Millipore; MAB377, 1:50 in incubation buffer). By contrast, glial cells were visualized by mouse anti-GFAP (glial fibrillary acidic protein) (1:500, Dako, Cat#Z0334, Agilent, Santa Clara, CA). Subsequently, sections were incubated for 1 h at 25 °C with donkey anti-mouse IgG H&L secondary antibody (Abcam, Alexa Flour® 488 ab150109, 1:500 in incubation buffer). Control sections were incubated without primary antibody. In addition, select sections were counterstained, as required, to aid visualization of brain structures. Finally, sections were mounted on dry gelatin-coated slides for fluorescence visualization using a Leica SP5 confocal microscope (Leica, Germany). For each section, images were taken from up to four fields: the temporal cortex (lateral to the hippocampus), the CA1 and CA3 region, and the dentate gyrus within the hippocampus. The images were then analyzed using imageJ 1.50i software. The number of NeuN stained cells from a series of sections (4–5 sections per animal) were averaged to provide a mean value per animal. The average measurements from each animal were used to generate an overall treatment group mean that was used for statistical analysis.
Fluoro-Jade (FJ) and, in particular FJC stain is a fluorochrome widely used to label degenerating neurons in ex vivo studies of the central nervous system (Schmued, 2016). It is a highly specific stain for degenerating neurons due to its high affinity for neuronal cells, and has been widely used in quantifying neuronal loss in TBI (Chen et al., 2014; Baratz et al., 2015). For FJC staining, brain sections were first immersed in a solution containing 1% NaOH in 80% ethanol for 5 min. They were then rinsed for 2 min in 70% ethanol, washed in distilled water, and incubated in 0.06% potassium permanganate solution for 10 min. Following a water wash, slides were incubated in the FJC staining solution (obtained by adding 4 mL 0.01% FJC stock solution in distilled water to 96 mL of 0.1% acetic acid) for 10 min. After 3 washes with distilled water, slides were fully air-dried on a slide warmer, cleared in xylene and coverslipped with DPX mountant. FJC positive (FJC +) cells were counted for each region in both hemispheres, by a FV 1000MPE Olympus multiphoton laser scanning microscope (Olympus Corp., Tokyo, Japan).
Increased expression of the inflammatory markers, such as the ionized calcium-binding adapter molecule 1(Iba1) and tumor necrosis factor-α (TNF-α), is associated with activated microglial cells within the brain (Benakis et al., 2014; Ito et al., 1998). For Iba1/TNF-α double labelling, sections were incubated for 48 h with Iba1 antibody (polyclonal goat anti-Iba1 1:200, Abcam, USA) and TNF-α antibody (TNF-α polyclonal rabbit anti-TNF-α 1:800, Abbiotec, USA) at 4°C. After PBS washing, sections were incubated for 2 h at 25 °C with a donkey anti-goat secondary antibody (donkey anti-goat IgG (H + L) cross adsorbed secondary antibody Alexa Fluor 555 1:400, ThermoFisher, USA). A three-step detection was used to increase the signal of TNF-α using a biotin-conjugated IgG (IgG (H + L) Biotin-Goat anti rabbit 1:500, Invitrogen, USA) incubated for 3 h at 25 °C and streptavidin-fluorescein (1:200, Vector, UK) for 2h at 25 °C. Washing between these steps was done with a 0.2% TritonX100 solution and PBS respectively. Control sections were incubated without primary antibody, and select sections were counterstained, as required, to aid visualization of brain structures. For Iba1, immunofluorescence analysis was undertaken by ImageJ 1.47v software, and a stack of three images was obtained for each acquired field in order to identify Iba1 + microglial cells. For each microglial cell, the body and primary processes were outlined, and the area occupied by each microglial cell was measured, to show any morphological changes related to activation processes.
Qualitative and quantitative analyses for Iba1 and TNF-α were performed using a FV 1000MPE, Olympus confocal laser scanning microscope. Z-series images were processed by ImageJ 1.47v and the volume of co-localized elements was measured by Imaris 7.4.2 as follows: for each dataset, a co-localization channel was automatically generated by the software. In the final stacks, a region of interest for each brain region (CA1, CA3, DG and temporal cortex) per hemisphere of each animal was chosen, and the volume of the elements of interest was calculated, summed and expressed as volume/μm3.
2.9. Data analysis
All results are presented as mean ± SEM and were analyzed by either Prism or SPSS V 20 software. For behavioral studies, one-way ANOVA tests were performed for comparisons between multiple data sets, followed by either Dunnett’s or Bonferroni post hoc analysis. t-tests were also used for direct comparisons in some studies. For significance *p < 0.05, **p < 0.01, ***p < 0.001. The results from the analysis of NeuN, FJC, GFAP, Iba1, and TNFα positively stained cells were statistically analyzed with a one-way analysis of variance (ANOVA) followed by a Tukey’s post-hoc test. The level of significance was set at p ≤ 0.05. For all histochemical studies, measurements were made by observers blinded to the treatment groups, and for immunohistochemistry, additional controls were carried out by omission of the primary antibody, as noted above.
3. Results
All pharmacokinetic, behavioral and immunohistochemical studies were undertaken in mice, except for studies evaluating the central nervous system uptake of Exenatide by measuring drug in CSF and plasma which involved the use of rats. Across all studies, Exenatide administration as well as mTBI (whether implemented separately or together) were well tolerated and not associated with observed changes in ‘basic well-being,’ a concept that underlies the combined health and wellness of an animal (Edut et al., 2011). This parameter combined subjective evaluations, such as grooming and appearance, righting skills, ambulation, and blinking reflex, with objective measurements that included weight, body temperature, anxiety-like behavior, and motor skills.
3.1. Pharmacokinetic properties
Initial pharmacokinetic studies measured plasma Exenatide levels achieved at 7 days following increasing single s.c. doses of PT302 to evaluate dose-dependence. As shown in Fig. 2A, single PT302 doses of 0.1 to 2.0 mg/kg generated approximately linear elevations in plasma levels of Exenatide in mice that ranged from 370 to 4320 pg/mL. On the basis of this study, and on our recent pharmacokinetic analyses in humans administered a routine clinical dose of Exenatide demonstrating median peak serum Exenatide concentrations of 543 3 pg/mL (Athauda et al., 2017), three PT302 doses were selected (0.024, 0.12 and 0.6 mg/ kg) to cover the range of plasma Exenatide levels potentially achievable in humans given PT302 (Gu et al., 2014) for time-dependent evaluation of Exenatide plasma levels in mice. Illustrated in Fig. 2B, these single s.c. PT302 administrations generated a dose-dependent biphasic plasma Exenatide pharmacokinetic profile with an initial peak occurring at 1 h and a maximal concentration at 14 days. Steady-state levels of plasma Exenatide were maintained for 21 days (the last monitored sample time) and descriptive statistics for the pharmacokinetic parameters of PT302 are summarized in Table 1.
Fig. 2.
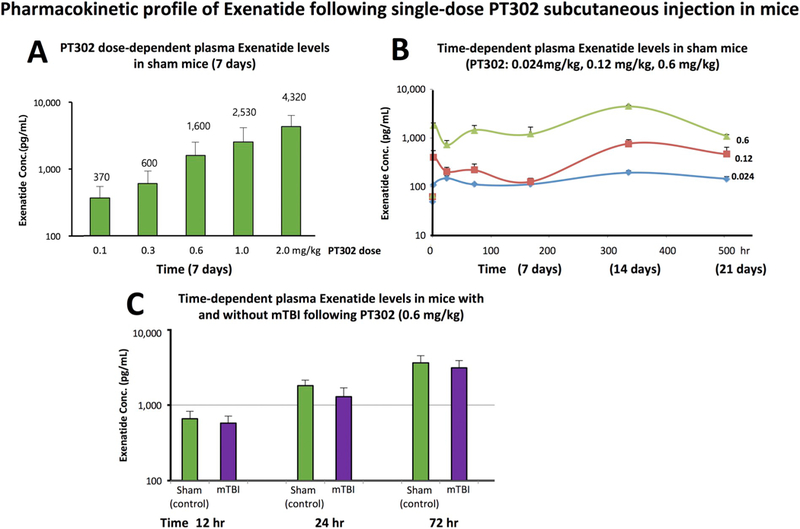
PT302 generates rapid, steady-state plasma levels of Exenatide following a single subcutaneous injection to mice. (A) Dose-dependent levels of Exenatide achieved in plasma at 7 days following PT302 administration (0.1 to 2.0 mg/kg) to separate cohorts of mice (N = 4 to 5 per group). The mean Exenatide concentration (pg/mL) is noted above each PT302 dose. (B) Time-dependent plasma concentrations of Exenatide (pg/mL) achieved following a single administration of PT302 (0.024, or 0.12 or 0.6 mg/kg) to separate groups of mice (N = 4 to 5 per group, sampling times: 1, 24,168, 336, and 504 h). For pharmacokinetic parameters see Table 1. (C) Time-dependent plasma Exenatide levels achieved following a single dose of PT302 (0.6 mg/kg) in mice with and without a mTBI challenge (N = 4 to 5 per group, sampling times: 12, 24, and 72 h). Plasma Exenatide concentrations were no different between groups with and without mTBI at each evaluated time (p > 0.05, paired t-test). Data are presented as a Mean ± S.E.M. of measurements.
Importantly, our recent pharmacokinetic analyses in humans administered a routine clinical dose of Exenatide (Bydureon 2 mg) demonstrated a median peak serum Exenatide concentration of 543–3 pg/mL utilizing the same assay and procedures (Athauda et al., 2017). Our PT302 doses evaluated in Fig. 2B provide plasma Exenatide levels that bracket clinically achievable levels.
Table 1.
Pharmacokinetic parameters of PT302 in mice after single subcutaneous administration.
| Parameter | PT302 dose |
||
|---|---|---|---|
| 0.024 mg/kg (n = 6) |
0.12 mg/kg (n = 6) | 0.6 mg/kg (n = 6) | |
| Immediate peak (1 h) (pg/mL) | 105 ± 5.3 | 389.7 ± 143 | 1756.2 ± 291.8 |
| CMax (pg/mL) | 190.5 ± 19.1 | 754.4 ± 158.7 | 4380 ± 328.4 |
| Tmax (d) | 14 | 14 | 14 |
| CAve (pg/mL) | 144.8 | 414.4 | 2246 |
| AUClast (ngh/mL) | 72,960 | 208,863 | 1,132,000 |
Cmax: maximal plasma concentration of Exenatide; Tmax: time of maximal concentration; CAve: average plasma concentration achieved across the 21 day study; AUClast: area under curve (i.e., the time-dependent concentration) for plasma Exenatide to the last recorded time point (21 days).
Notable, our recent pharmacokinetic analyses in humans administered a routine clinical dose of Exenatide (Bydureon 2 mg) demonstrated a median peak serum Exenatide concentration of 543–3 pg/mL utilizing the same assay procedures (Athauda et al., 2017).
Importantly for these pharmacokinetic measures across all doses evaluated was that the initial plasma peak concentration of Exenatide, associated with the initial release burst property of PT302, was well controlled and provided a value that was less than the ultimate Cmax. This value matched the CAve plasma concentration of Exenatide over the 21 day period of the study, with the initial burst peak reaching 72.5%, 94% and 78.2% of their respective CAve concentrations for the 0.024, 0.12 and 0.6 mg/kg PT302 doses.
To evaluate whether or not the plasma pharmacokinetic profile of Exenatide release from PT302 was affected by TBI, a single s.c. PT302 dose (0.6 mg/kg) was time-dependently evaluated in animals with and without a mTBI insult. As shown in Fig. 2C, time-dependent plasma Exenatide levels were no different between animals challenged with mTBI versus a sham procedure.
3.2. Elevated plus maze paradigm
Anxiety-like behavior was evaluated using the elevated plus maze test and was measured by the time spent in the open arm. A shorter period spent in the open maze arm suggests raised anxiety levels. mTBI challenge had no effect on the duration mice spent within the open arm when measured 7 days post injury. Likewise, all PT302 doses (0.024, 0.12 and 0.6 mg/kg) proved not significantly different from sham animals (Supplemental Fig. 1(left)). Evaluation of separate cohorts of mice at 30 days post mTBI provided similar results across groups, in which the highest and lowest doses of PT302 (0.024 and 0.6 mg/kg) were appraised (Supplemental Fig. 1(right)). Together, these results suggest that anxiety is not a limiting factor in relation to the cognitive evaluations undertaken by the novel object recognition and Y-maze paradigms, below. This is important to know in light of reports of elevated anxiety in other mild, moderate and severe TBI mouse models (Washington et al., 2012).
3.3. Novel Object recognition paradigm
The novel object recognition paradigm was performed 7 days after mTBI to evaluate visual memory. The time that mice spent investigating the novel/familiar object was recorded, and a preference index was then calculated (Fig. 3A). mTBI challenge resulted in impaired performance with mice showing no differentiation between the novel and the familiar objects. PT302 treatment for mTBI demonstrated a dose-dependent mitigation of this impairment, with 0.024 mg/kg exhibiting an improvement trend, 0.12 mg/kg a partial recovery (*p < 0.05 vs. mTBI vehicle and sham mice), and 0.6 mg/kg a full recovery of visual memory (**p < 0.01 vs. mTBI vehicle, and not significantly different from sham mice: Fig. 3A). Visual-memory was determined to be similarly impaired when evaluated 30 days post injury in the mTBI vehicle group (***p < 0.001 vs. sham mice). Treatment with PT302 0.6 mg/kg fully ameliorated this impairment (**p < 0.01 vs. mTBI vehicle, with no significant difference vs. sham mice). PT302 0.024 mg/kg provided partial mitigation (*p < 0.05 vs. mTBI vehicle and sham mice: Fig. 3B).
Fig. 3.
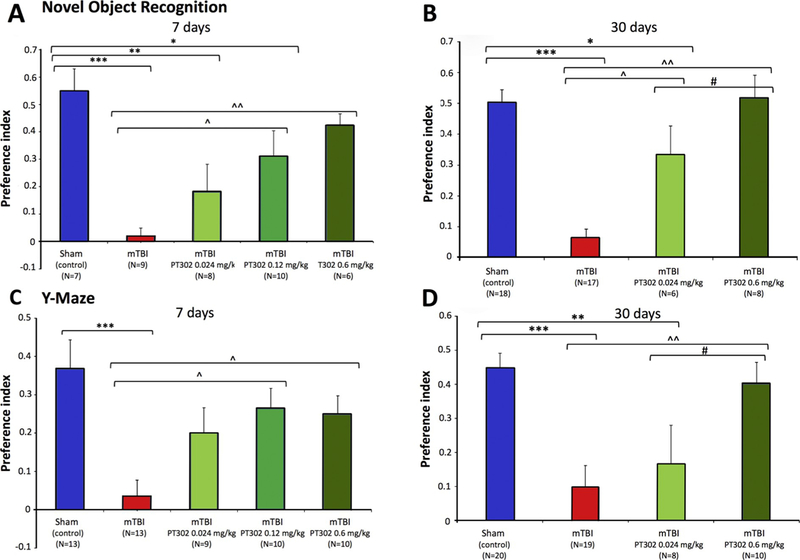
PT302 released Exenatide dose-dependently ameliorates concussive mTBI cognitive impairments following a single subcutaneous dose post injury. (A and B) mTBI mice demonstrate a deficit in visual memory compared with sham (control) animals (***p ≤ 0.001) at 7 and 30 days, respectively. PT302 administration dose-dependently mitigated this behavioral deficit at 0.12 and 0.6 mg/kg (^p ≤ 0.05 and ^^p ≤ 0.01, respectively, vs. mTBI vehicle) at 7 days, and at 0.024 and 0.6 mg/kg (^p ≤ 0,05 and ^^p ≤ .01, respectively, vs. mTBI vehicle) at 30 days. (C and D) mTBI mice demonstrate a significant deficit in spatial memory compared with sham (control) animals at 7 and 30 days, respectively (***p ≤ .001). PT302 administration significantly ameliorated this damage at both 7 days (^p ≤ 0.05 for both 0.12 and 0.6 mg/kg) and 30 days (^^p ≤ 0.01 for 0.6 mg/kg).
Data are presented as a Mean ± S.E.M. of measurements, with animal number (N) in parenthesis.
Statistical significance: * p ≤ .05, ** p ≤ .01, *** p ≤ .001 vs. Sham (control) mice (i.e., no mTBI). ^p ≤ .05, ^^p ≤ .01 vs. mTBI, and # p ≤ .05 vs. PT302 + mTBI.
3.3.1. Y-maze paradigm
The Y-maze test was undertaken 7 days following mTBI challenge to assess spatial-memory. The periods spent exploring the new/old arms of the maze were recorded, and a preference index was determined (Fig. 3C). mTBI challenge resulted in a spatial memory deficit, with mice spending similar times within the maze arms. This deficit was ameliorated by PT302 administered at 0.12 mg/kg and 0.6 mg/kg, with the lower 0.024 mg/kg dose showing a trend toward improvement (*p < 0.05: PT302 0.12 and 0.6 mg/kg vs. mTBI vehicle mice; Fig. 3C). Spatial memory was similarly assessed in separate cohorts of mice at 30 days, with mTBI vehicle challenged mice, likewise, showing a reduced preference index, indicative of spatial memory dysfunction. Similarly, 0.6 mg/kg PT302 fully ameliorated this impairment (**p < 0.01: PT302 0.6 mg/kg vs. mTBI vehicle group), and a dose of 0.024 mg/kg provided a trend toward mitigation that failed to reach significance (Fig. 3D).
3.4. Immunohistochemical staining
Effect on neuronal survival: FJC is widely used to label degenerating neuronal cells ex vivo and is specific and sensitive for evaluating cellular loss immunohistochemically. Evaluated at 72 h following mTBI challenge (Fig. 4A), vehicle exposed TBI challenged animals displayed a consistent and statistically significant elevation in FJC-expressing cells throughout the hippocampus (dentate gyrus and CA3 and CA1 regions) as well as in temporal cortex adjacent to the hippocampus. This varied from a 50% increase in degenerating cell number in the CA1, to over a 2-fold rise in CA3 and cerebral cortex. As illustrated by representative images (Fig. 4B), this cellular loss was diffuse and occurred throughout each of these brain regions. Notably and in a concentration-dependent manner, PT302 substantially mitigated the elevation in FJC positive cells across each evaluated brain area. Specifically, PT302 at a dose of 0.6 mg/ kg counteracted mTBI-induced neurodegeneration across all areas (p < 0.01 in CA3 and DG, p < 0.05 in CTX and CA1); the lower dose of 0.12 mg/kg demonstrated significant effects in the CA3 and temporal cortex regions (p < 0.05, p < 0.01, respectively) with strong trends evident in the other areas evaluated.
Fig. 4.
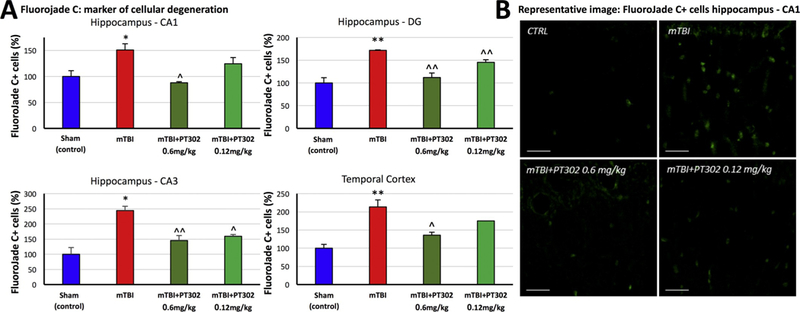
PT302 subcutaneous injection post mTBI mitigates cellular loss in brain areas impacted by TBI (evaluated at 72 h post TBI). (A) mTBI induces an increase in FluoroJade C-positive (FJC + ) cells, compared to sham (control) mice, across the hippocampal CA1 and CA3 regions, dentate gyrus and temporal cortex adjacent to the hippocampus. This rise was dose-dependently mitigated by PT302 (0.12 and 0.6 mg/kg). (B) Representative images of FJC staining within the CA1 region of hippocampus (bar =30 μm) (CTRL: sham control group).
Data are presented as a Mean ± S.E.M. of measurements (N =5 per treatment group). Statistical significance: * p ≤ .05, ** p ≤ .01 significantly different from sham (control) animals without TBI. ^ p ≤ .05, ^^ p ≤ .01 significantly different from mTBI group.
To extend this observation across time and technique, anti-NeuN immunohistochemical staining, widely used to label neurons, was performed to assess neuronal survival 30 days post mTBI (Fig. 5A). Three areas were evaluated within the brain: the temporal cortex, the CA3 region of the hippocampus and the dentate gyrus. mTBI challenge led to a decline in neuronal counts within all three regions at 30 days, compared to all other groups (**p < 0.01), indicating significant neuronal loss at 30 days following the injury (Fig. 5A and D). Treatment with PT302, as a single dose of 0.6 mg/kg following mTBI, preserved neuronal counts in these regions, thus mitigating mTBI-induced neuronal loss (**p < 0.01, Fig. 5A). Additionally, shown in Fig. 5 (B and C) is cellular expression of GFAP, an intermediate filament protein predominantly expressed by astrocytes and widely used as an astroglial marker within brain. mTBI caused a mild elevation in GFAP expressing cells at 30 days across brain regions that failed to reach statistical significance. This elevation was not evident in PT302 treated animals.
Fig. 5.
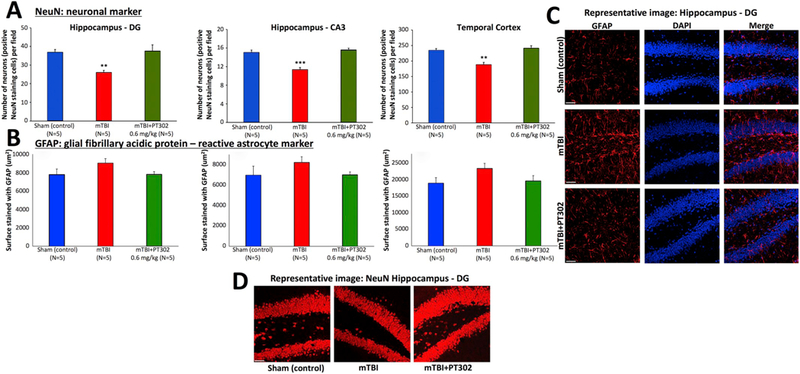
PT302 subcutaneous injection post mTBI mitigates neuronal loss in brain areas impacted by TBI (evaluated at 30 days post TBI). (A) Anti-NeuN immunohistochemical staining, to label neurons, allowed quantification of neuronal survival 30 days post mTBI in hippocampal dentate gyrus and CA3 regions, and adjacent temporal cortex. mTBI challenge induced a decline in NeuN positive cells across all three regions (**p ≤ .01, ***p ≤ .001 vs. sham (control) mice), which was fully mitigated by a single s.c. dose of PT302 (0.6 mg/kg) (with no significant difference in counts between PT302 mTBI and sham (control) groups). (B) GFAP immunohistochemistry staining was used to visualize reactive astrocytes. mTBI induced a mild elevation in GFAP expressing cells across brain regions that failed to reach statistical significance (p > .05 vs. sham (control) mice). This elevation was not found in PT302 treated animals. (C) Representative images of GFAP and DAPI, and merged staining, and (D) of NeuN staining within the dentate gyrus region of hippocampus across groups of mice (bar = 50 μm). Data are presented as a Mean ± S.E.M. of measurements (N = 5 per group).
Effect on mTBI-induced inflammation: Iba1 is a microglia/macrophage-specific calcium-binding protein that participates in membrane ruffling and phagocytosis in activated microglia, in which its levels increase. mTBI challenge induced an elevation in Iba1 immunoreactivity at 72 h (Fig. 6A and B), as evaluated in vehicle treated mTBI mice vs. sham controls across all analyzed brain regions (p < 0.05 for CA1; p < 0.001 for CA3, dentate gyrus and temporal cortex). In sham control mice, microglial cells displayed a resting morphology with a small soma, long and thin processes. After mTBI microglia showed an activated morphology, characterized by a larger cell body with shorter and thicker processes. Notably as illustrated in Fig. 7, Iba1 expressing cells in mTBI vehicle mice demonstrated a greater co-localization with antibodies to the proinflammatory cytokine TNF-α, thereby providing further support for their activated state. PT302 substantially inhibited mTBI-induced microglial activation (Figs. 6 and 7), with PT302 0.6 mg/kg proving effective across all the brain regions (p < 0.001 in temporal cortex; p < 0.01 in CA1, CA3 and dentate gyrus vs. mTBI vehicle) and 0.12 mg/kg effective across all areas (p < 0.001 temporal cortex, p < 0.01 CA1, p < 0.05 dentate gyrus vs. mTBI vehicle) except for the CA1 region. mTBI-induced colocalization between TNF-α and Iba1 was significantly and substantially inhibited by both PT302 doses (Fig. 7).
Fig. 6.
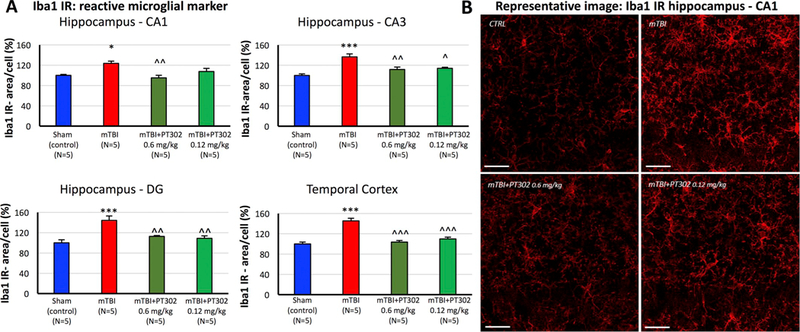
PT302 subcutaneous post-treatment of mTBI mice inhibited microglial activation in hippocampus and adjacent temporal cortex at 72h. (A) Microglial activation was evaluated by quantifying Ibal immunoreactivity (IR) (area/cell) across hippocampal CA1, CA3 and dentate gyrus regions and adjacent temporal cortex. mTBI induced a significant elevation in Ibal IR across all analyzed brain areas, which was dose-dependently mitigated by PT302. (B) Representative images of Ibal IR within the hippocampal CA1 region across animal groups (bar = 30μm) (CTRL: sham (control) group). Data are presented as a Mean ± S.E.M. of measurements (N = 5 per treatment group). Statistical significance: *p < 0.05, ***p < 0.001 vs. sham (control) animals without TBI.
^p < 0.05, ^^p < 0.01 ^^^p <0.001 vs. mTBI.
Fig. 7.
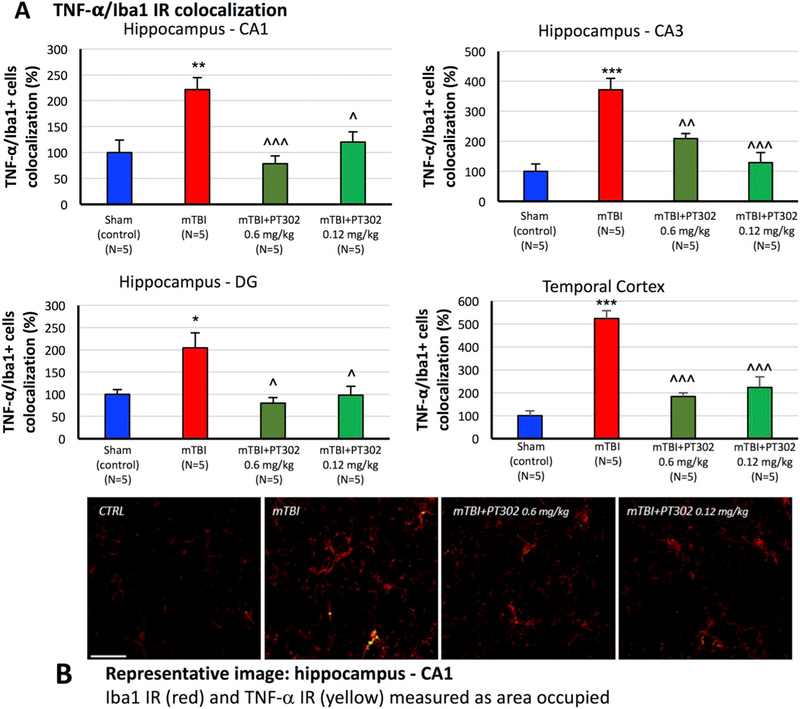
PT302 subcutaneous injection post mTBI inhibits TNF-α generation within activated microglial (as evaluated by TNF-α/Iba1 co-localization at 72 h post TBI). (A) Iba1 IR (red) and TNF-α IR co-localization (yellow) was measured as the area occupied in microglia across hippocampal CA1, CA3 and dentate gyrus regions and adjacent temporal cortex. mTBI induced a significant increase in TNF-α/Iba1 IR co-localization across all analyzed areas, which was significantly mitigated by PT302 (0.6 and 0.12 mg/kg). Data are presented as a Mean ± S.E.M. of measurements (N = 5 per treatment group). *p < 0.05, **p < 0.01, ***p < 0.001 vs. sham (control) group without TBI.
^p < 0.05, ^^p < 0.01, ^^^p < 0.001 vs. mTBI.
(B) Representative images of Iba1/TNF-α IR co-localization within the CA1 region of hippocampus across groups of mice (bar = 30 μm) (CTRL: sham (control) group). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To evaluate the time-dependence of mTBI-induced neuroinflammation and PT302 amelioration, Iba1 expression was also quantified immunohistochemically in mice at 30 days post injury. As illustrated in Fig. 8, the elevation in Iba1 levels evident at 72 h post injury had largely abated by day 30 and there was no significant difference between sham control and mTBI vehicle mice. Notably, however, animals treated with a single dose of 0.6 mg/kg PT302 demonstrated reduced levels of Iba1 expression as compared to sham control mice (p < 0.001 temporal cortex and CA3; p,0.01 dentate gyrus), indicative of sustained anti-inflammatory action.
Fig. 8.
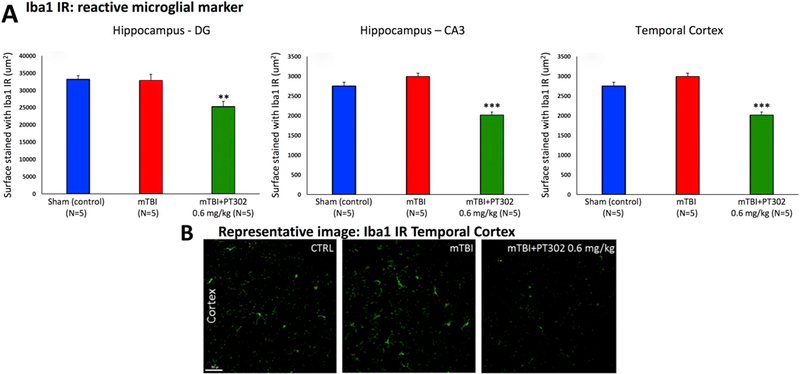
PT302 subcutaneous injection post mTBI reduces microglial cell activation in brain areas impacted by mTBI to lower than sham control levels (evaluated at 30 days post mTBI). (A) Whereas elevations in Iba1 immunoreactivity (IR) found at 72 h after mTBI (see Fig. 6) had subsided by 30 days (mTBI no different from sham (control) group, p > .05) across hippocampal CA3 and dentate gyrus regions and adjacent temporal cortex, PT302 (0.6 mg/kg) induced a decline in Iba1 IR to levels lower than sham (control) values. (B) Representative images of Iba1 IR within the temporal cerebral cortex across groups of mice (bar = 50 μm) (CTRL: sham (control) group). Data are presented as a Mean ± S.E.M. of measurements (N = 5 per treatment group), ** p ≤ .01, *** p ≤ .01 significantly different from sham (control) animals without TBI.
3.5. Plasma and CSF exenatide levels
Exenatide was administered in the form of (i) sustained-release PT302, (ii) via mini pump and (iii) as a twice daily injection – all by the s.c. route – and plasma and CSF samples were obtained 14 days later from rats. As shown in Fig. 9, Exenatide was detected in the plasma and CSF in both s.c. PT302 administered and pump implanted groups. In contrast, it was detected in plasma but not CSF in animals injected twice daily with Exenatide over the 14 day period, with a lower limit of assay detection for Exenatide of 6.9 pg/mL. The determined CSF/ plasma ratio for two doses of Exenatide appraised by each route of s.c. administration is for PT302: 0.015 ± 0.004, pump: 0.02 ± 0.007, and twice daily Exenatide injection: 0.001. This was determined by dividing the lower limit of detection for the assay by the plasma Exenatide concentration. This data demonstrates that the sustained release of Exenatide provides CNS levels in the order of 1.5% to 2.0% of concomitant plasma levels under steady-state conditions (with no significant difference (p = 0.54) between Exenatide administration as PT302 or via pump).
Fig. 9.
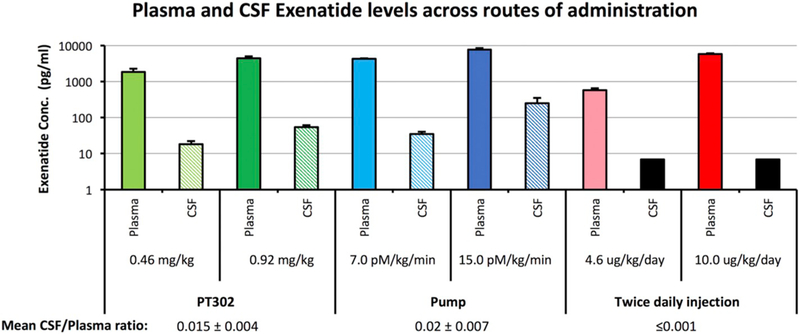
Plasma and CSF Exenatide levels across routes of Exenatide administration. Exenatide was administered in the form of (i) PT302 (0.46 and 0.92 mg/kg), (ii) Alzet mini-pump filled with Exenatide (7.0 and 15.0 pM/kg/min) and (iii) twice daily Exenatide injection (4.6 and 10.0 μg/kg/day) - all by the s.c. route. Plasma and CSF levels of Exenatide were measured 14 days later by fluorescent immunoassay kit (upper panel), to provide a CSF/plasma concentration ratio across the doses evaluated (lower panel). The black box shown for the CSF concentration from twice daily Exenatide injection represents the lower limit of detection value of the assay. Data are presented as a Mean ± S.E.M. of measurements (N = 4 to 5 per treatment group). The CSF/plasma concentration ratio for Exenatide generated by PT302 and pump are not significantly different (p = 0.54).
Importantly, our recent pharmacokinetic analyses in humans with PD administered a routine clinical dose of Exenatide (Bydureon 2 mg) demonstrated CSF Exenatide concentrations of 11.4–11.7 pg/mL utilizing the same assay and procedures (Athauda et al., 2017).
4. Discussion
TBI is a leading cause of death and long-term disability in the developed world with >10 million people suffering a TBI event worldwide annually (Hyder et al., 2007; Ruff et al., 2012); of these, some 1.7 million occur within the US. Indeed, by 2020, TBI is predicted to comprise the third largest portion of the global disease burden (Hyder et al., 2007). By far the majority of these TBIs (80 to 95%) are mild to moderate in nature (Tagliaferri et al., 2006). With improvements in survival rate following initial injury, TBI can result in substantial and lifelong cognitive, physical, and behavioral impairments that require long-term access to health care and disability services (Shi et al., 2013; Tagliaferri et al., 2006), and some 5.3 million Americans live with such debilities (Langlois et al., 2006; Prins and Giza, 2012). TBI symptoms can occasionally resolve within the first year after injury, but up to 70–90% of patients continue to manifest prolonged and often permanent neurocognitive dysfunction. In the light of the lack of currently available therapeutic options (Moppett, 2007), rapidly moving new promising treatment strategies to clinical evaluation is clearly important, particularly when centered on a drug class such as GLP-1R agonists (also termed incretin mimetics), which has proven efficacious and well tolerated in the treatment of another disorder (T2DM [Drucker, 2018; Nauck, 2016]). This approach allows rapid repurposing as a potential TBI treatment. In the present study, we demonstrate that a single s.c. administration of sustained-release Exenatide, in the form of PT302 clinical material, mitigates TBI-induced cognitive impairments, neuronal loss and neuroinflammation in a dose-dependent manner and, importantly, at a clinically translatable plasma concentration.
The endogenous incretin peptide GLP-1 together with glucose-dependent insulinotropic polypeptide (GIP) are released from the L and K cells of the small intestine, respectively, during food digestion (Campbell and Drucker, 2013b; Tolhurst et al., 2009), and bind to their corresponding receptors (GLP-1R and GIP-R) on pancreatic β-cells. Receptor stimulation induces the intracellular activation of adenylyl cyclase, cyclic adenosine monophosphate (cAMP) accumulation, protein kinase A (PKA) activation, and liberation of insulin to regulate blood glucose levels (Campbell and Drucker, 2013a; Drucker, 2018; Russell, 2013). Such insulin release is “glucose-dependent”, only occurring when blood levels are elevated (Campbell and Drucker, 2013b; Drucker, 2018; Gallwitz, 2014), and thus the use of incretin mimetics is not associated with the classical hypoglycemic liability of most other T2DM medications. Notably, these incretins and incretin mimetics additionally provide trophic and protective actions on pancreatic β-cells (Campbell and Drucker, 2013a; Chon and Gautier, 2016; Drucker, 2018; Koehler et al., 2015; Russell, 2013), which our previous studies and those of others demonstrate translate to neurons (Athauda et al., 2017; Athauda and Foltynie, 2016; Hölscher, 2018; Kim et al., 2017; Li et al., 2016; Li et al., 2012; Li et al., 2010a; Li et al., 2009; Perry et al., 2007; Perry et al., 2002a; Perry et al., 2002b; Salcedo et al., 2012) that similarly express GLP-1R and GIPR (Athauda and Foltynie, 2016, 2018; Hölscher, 2018; Kim et al., 2017; Li et al., 2016; Salcedo et al., 2012; Verma et al., 2018).
The activation of the GLP-1R on neurons generates potent neuro-trophic and neuroprotective actions across cellular and animal models of brain injury and neurodegeneration (Athauda and Foltynie, 2016, 2018; Hölscher, 2018; Kim et al., 2017; Li et al., 2016; Salcedo et al., 2012; Verma et al., 2018), which include multiple models of TBI (Eakin et al., 2013; Greig et al., 2014; Hakon et al., 2015; Rachmany et al., 2017; Rachmany et al., 2013; Tweedie et al., 2016a; Tweedie et al., (2013), spinal cord injury (Sun et al., 2018), stroke and subarachnoid hemorrhage (Li et al., 2009; Xie et al., 2018) as well as degenerative disorders such as AD, PD, multiple system atrophy, ALS, Huntington’s disease and peripheral neuropathy (Athauda and Foltynie, 2018; Athauda and Foltynie, 2016; Bassil et al., 2017; Bertilsson et al., 2008; Harkavyi and Whitton, 2010; Hölscher, 2018; Kim et al., 2017; Li et al., 2012; Li et al., 2010a; Li et al., 2009; Li et al., 2016; Salcedo et al., 2012). Because of numerous key measures of improved outcome cross-validated between such animal models as well as across independent laboratories at different institutions, interest in the evaluation of incretin mimetics in humans with neurodegenerative disorders has grown, and has spawned clinical trials in PD and AD [(Athauda et al., 2017; Athauda and Foltynie, 2018; Aviles-Olmos et al., 2013, 2014; Gejl et al., 2016); and ongoing clinical trials: NCT03456687, NCT02953665, NCT01843075, NCT01469351], as well as a growing interest in other neurological disorders including TBI (Greig et al., 2014). Key issues in considering clinical translation are which clinically available GLP-1R agonist to evaluate, and how it should be dosed both to optimize its pharmacokinetics for potential efficacy and to limit possible adverse actions.
Our prior studies on TBI largely focused on Exenatide, administered at a clinically translatable dose via a s.c. 7 day pump in order to (i) maintain steady-state plasma and target drug concentrations and (ii) emulate the use of Exenatide when administered in the form of Bydureon to humans (Eakin et al., 2013; Rachmany et al., 2013, 2017; Tweedie et al., 2013, 2016b). Bydureon is clinical grade Exenatide encapsulated within biodegradable 0.06 mm diameter PLGA microspheres to provide once weekly s.c. dosing. It contains the identical active material used in twice daily injections of Exenatide as immediate release Byetta (Deyoung et al., 2011). The degradation of the PLGA polymer to release Exenatide occurs via natural (e.g., non-catalyzed) hydrolysis of the ester linkages to produce lactic acid and glycolic acid, which are naturally occurring substances that are ultimately cleared as carbon dioxide and water. Bydureon was not available for preclinical studies. When imitating its use by Exenatide administration via a 7 day s.c. pump, it mitigated short (7 day) and longer term (30 day) mTBI-induced memory impairments in mice challenged with both concussive (30 g weight drop) and blast (7 m from 500 g of TNT detonation) injury (Rachmany et al., 2013, 2017; Tweedie et al., 2013, 2016b).
A caveat of our past studies of Exenatide in mouse TBI models is that placing pumps filled with Exenatide into humans is not a particularly viable option, and peptide release from a s.c. 7 day pump implanted in mice may not precisely imitate the time-dependence achieved with clinical material in a clinically approved sustained release injectable form. The recent development of PT302 similarly encapsulates Exenatide in biodegradable PLGA microspheres (0.02 mm diameter). However, the development of a proprietary ultrasonic spray drying process and use of L-lysine coating makes a formulation available that represses the initial release burst of peptide, provides a once every two-week treatment in humans, and can be administered via a smaller needle to minimize potential injection pain (Gu et al., 2014). With this clinical material, we show here the long-term, controlled, sustained release of Exenatide from PT302, demonstrating dose-dependence (Fig. 2A) and early maintenance of steady-state plasma levels for up to 21 days after a single s.c. dose to mice (Fig. 2B). This pharmacokinetic profile is in accord with that determined in prior human and rat studies (Chen et al., 2018; Gu et al., 2014). Similar to these studies, Exenatide exposure during the initial burst release phase following PT302 administration proved low and consistent, reaching a plasma concentration of less than the CMax and in line with the average time-dependent Exenatide concentrations (CAve) achieved across doses. This initial burst is associated with the rapid release of drug at the microsphere surface that, if poorly controlled, may result in a spike in drug levels and induction of adverse actions such as nausea and vomiting (Lewis and Kumar, 2012). Also evident from our time-dependent analysis of plasma Exenatide concentrations generated by PT302 (Fig. 2B), is that there was a negligible ‘lag phase’ (i.e., a decline in released drug levels) following the initial burst peak before achieving relatively stable steady-state (CAve) Exenatide levels; the difference between the CAve and CMax values was < 2-fold (Table 1). A smaller CAve and CMax differential for a drug delivery system is associated with improved extended release while maintaining the peptide in a stable form in vivo (Deyoung et al., 2011; Lewis and Kumar, 2012). Our prior pharmacokinetic studies of Exenatide in humans demonstrated that a routine clinical dose of Exenatide (Bydureon 2 mg s.c. once weekly) provided a median peak serum Exenatide concentration of 543 3 pg/mL (Athauda et al., 2017), as assessed using the same assay and procedures as in the present study. Evident in Fig. 2B and Table 1, the plasma levels of Exenatide associated with the PT302 doses evaluated in the current study compare favorably with those achievable and well tolerated in humans.
Notably, PT302 administration dose-dependently mitigated mTBI-induced short-and long-term cognitive impairments in our mouse model, as evaluated in both the Y-maze and novel object recognition paradigms at 7 and 30 days in separate cohorts of animals. The lack of any effect in the elevated maze paradigm suggests that anxiety is not a confound in these measures of spatial and recognition memory, which is important in light of recent reports that some TBI models may induce anxiety-like behaviors (Washington et al., 2012), and that GLP-1 has potential anxiogenic/antidepressant actions (Anderberg et al., 2016; Isacson et al., 2011). Enduring spatial and visual/recognition learning deficits have similarly been detected after both mild and moderate TBI in humans (Beers, 1992; Chuah et al., 2004; Konrad et al., 2011; Skelton et al., 2000). Hence our results not only replicate the efficacy of a clinically translatable dose of Exenatide delivered by s.c. pump seen in our former preclinical mTBI studies (Rachmany et al., 2013, 2017; Tweedie et al., 2013, 2016b), but also have relevance for the human disorder. Additionally, our current PT302 study extends our prior work by demonstrating mitigation of mTBI-induced cellular loss in brain regions associated with cognition, specifically within the hippocampus and cerebral cortex both at 72 h and 30 days post injury. Furthermore, Exenatide release from PT302 ameliorated mTBI-induced neuroinflammation, as evaluated by Iba1 labelled microglia and TNF-α co-localization in activated microglia at 72 h. Iba1 is a calcium-binding protein whose expression in brain is restricted to microglial cells (Ito et al., 1998). Under physiological (healthy, basal) conditions, microglia predominantly exhibit a quiescent phenotype typified by a ramified morphology, the downregulation of activation antigens like major histocompatibility complex, and low constitutive expression of Iba1 (Frank et al., 2007; Ladeby et al., 2005). In contrast, under a broad range of neuropathological conditions associated with neuroinflammation and including TBI, microglia transform into an activated state characterized by a de-ramified morphology, an upregulation of Iba1 and antigen presentation molecules, the generation of pro-inflammatory cytokines typified by TNF-α, the production of reactive oxygen and nitrogen species, and display of phagocytic activity (Frank et al., 2007; Ladeby et al., 2005; Streit et al., 1999). Interestingly, whereas our evaluation of Iba1 labelled microglia at 30 days following mTBI indicated that injury-induced neuroinflammation had largely subsided, mTBI animals administered PT302 demonstrated yet lower levels of activated microglia than the sham control group (without mTBI), thereby confirming the anti-inflammatory actions of Exenatide described across a broad number of preclinical animal models of neural injury.
Many of the key molecular events underpinning the neurotrophic, neuroprotective and anti-inflammatory actions of GLP-1 and analogs have been characterized in neuronal cultures and brain. In large part, they parallel the cascades activated in pancreatic cells associated with their trophic/protective properties and are independent of the glycemic effects. Neuronal GLP-1 activation generates an almost immediate increase in intracellular cAMP, and the activation of PKA and phosphoinositide 3-kinase (PI3K), as well as their downstream pathways (Athauda and Foltynie, 2018, 2016; Kim et al., 2017; Li et al., 2010b; Salcedo et al., 2012). Together, these pathways augment cell survival and foster neuroprotection by activating calcium channels, increasing protein synthesis, mitochondrial biogenesis and inhibiting apoptosis and inflammation (Athauda and Foltynie, 2016, 2018; Kim et al., 2017; Li et al., 2010b; Salcedo et al., 2012).
Activated microglia are a major source of a broad range of toxic compounds, including superoxide, nitric oxide, eicosanoids and cytokines (Kim et al., 2009; Matsuo et al., 1995) that, if elevated, can initiate and drive neuroinflammation (Frankola et al., 2011). A growing literature indicates that GLP-1R activation provides anti-inflammation, either directly or indirectly, which our study confirms. In support of this view, GLP1R protein is highly expressed in microglia, in comparison to its expression in astrocytes and neurons (Iwai et al., 2006; Yun et al., 2018). Microglia have additionally been shown to express the proglucagon gene and to both generate and secrete GLP-1 (Kappe et al., 2012; Kim et al., 2009). Multiple previous studies have demonstrated that Exenatide prevents the activation of microglia both in cell culture in response to LPS stimulation and in vivo following many injury paradigms (Gonsalves et al., 2016; Gullo et al., 2017; Kim et al., 2009; Lee et al., 2018; Li et al., 2012; Teramoto et al., 2011; Wu et al., 2018; Yun et al., 2018) that result in reduced M1 phenotype expression as well as a decline in the generation and secretion of deleterious moities such as TNF-α and IL-10. These changes result in a mitigation of resulting neuronal toxicity, whether mediated directly by these proinflammatory products or via their ability to activate astrocytes into toxic A1 astrocytes (Kim et al., 2009; Yun et al., 2018). From a translational perspective, prospective biomarkers associated with TBI-induced neuronal cell death as well as astrocyte inflammation, and their mitigation by drugs, can potentially be followed in plasma derived exosomes to aid development from preclinical into clinical studies and to gain a signal of target engagement (Karnati et al., 2018). Proteins associated with neuroinflammation and preprogrammed neuronal cell death can potentially be followed in plasma sampled exosomes enriched for astrocytic and neuronal origin, respectively (Mustapic et al., 2017).
Our studies demonstrate a clear dose-response relationship for PT302 across both behavioral (visual/spatial cognition) and immunohistochemical (cellular loss/neuroinflammation) measures related to the mitigation of mTBI induced impairments. This suggests that such measures lie on the linear portion of the sigmoidal dose-response curve, and that brain uptake of released Exenatide is a particularly important factor among those governing efficacy. Our evaluation of CSF/plasma concentrations of Exenatide administration over a 14 day period to achieve a steady-state by the use of s.c. PT302 or a pump demonstrated CSF levels were 1.5% to 2.0% of concurrent plasma levels across doses, which is in line with the 2% uptake of Exenatide from plasma to CSF measured in a recent human clinical trial of Exenatide as Bydureon in PD (Athauda et al., 2017). This contrasts with administration of Exenatide by twice daily administration (mimicking Byetta), in which the relatively short half-life of the peptide, 2.4 h in humans and shorter still in rodents (Molina Vega et al., 2018), precludes the compound from reaching steady-state concentrations in plasma. Our prior studies in humans with Parkinson’s disease that responded to Exenatide treatment demonstrated median CSF Exenatide levels of 11 4 to 11.7 pg/mL following a routine clinical dose (Bydureon 2 mg, s.c., once weekly). Our current results in rodents indicate that the preservation of constant Exenatide levels in plasma, whether by a sustained release injectable formulation or by pump, provides a driving force to reach greater levels within the central nervous system (providing levels that exceed twice daily immediate release Exenatide formulation, and that are in the realm for predicted human efficacy). In this regard, sustained release Exenatide formulations such as s.c. PT302 may be the optimal way for evaluating the utility of Exenatide as a treatment strategy for mTBI and other neurological disorders.
Our studies to evaluate CSF/plasma Exenatide levels following systemic dosing were undertaken in rat, rather than mouse, to allow collection of a sufficient sample volume for analysis. This decision is supported by a study from Murakami and colleagues (Murakami et al., 2000) establishing that the permeability of the blood-brain barrier is similar between mice and rats, by demonstrating a correlation of 1:1 between the two species across a broad range of drugs. A prior study of the same mouse mTBI model used in our study (30 g weight drop in a 30 g mouse) demonstrated integrity of the blood-brain barrier following mTBI challenge (Pan et al., 2003) by evaluating the brain uptake of i.v. administered radiolabeled albumin and IL-1β, which were unchanged between sham and mTBI challenged mice at 1 h, 24 h and 1 week after mTBI. In supplemental studies, we evaluated plasma and brain levels of the i.v. administered vascular marker Evans Blue dye, that binds to serum albumin, following a more substantial 50 g mTBI challenge in 30 g mice, and found no significant difference between sham and mTBI groups (Supplemental Fig. 2). Our measured CSF/plasma ratio values of Exenatide are hence reflective of central nervous system uptake under conditions of normal blood-brain barrier integrity, but could be increased during conditions associated with barrier breakdown, as can occur in other forms of brain injury.
It is now understood that TBI represents a process, rather than an event, and recent epidemiological studies have revealed that TBI is a powerful environmental risk factor for the early development of dementia, PD and AD (Fann et al., 2018; Gardner and Yaffe, 2015). This view is supported by gene expression studies demonstrating an upregulation of pathways leading to PD and AD in mTBI preclinical models (Goldstein et al., 2012; Greig et al., 2014; Tweedie et al., 2016a; Tweedie et al., 2013). Our current studies evaluating clinical grade PT302 demonstrate a dose-dependent amelioration of neuronal loss, neuroinflammation and cognitive impairments induced by mTBI in a well characterized close head preclinical model of the disorder. They importantly cross-validate and extend prior studies of Exenatide administered by s.c. pump in the same animal model (Tweedie et al., 2013) as well as Exenatide efficacy across other TBI animal models (Eakin et al., 2013; Rachmany et al., 2017; Tweedie et al., 2016b). Whereas our prior studies used a s.c. implanted mini pump to generate steady-state Exenatide levels and imitate a s.c. sustained release Exenatide formulation, PT302 provides this reality and allows the evaluation of clinical grade material, as in the present study. Collectively our current studies, together with previous literature demonstrating the mitigation of multiple mTBI impairments in cognition and brain pathology by Exenatide, as well as the reversal of mTBI-provoked gene pathways leading to PD and AD (Tweedie et al., 2016a, 2013), support the view that sustained release Exenatide warrants evaluation in humans as a new mTBI treatment strategy.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
This research was supported in part by (i) the Technological Innovation R&D Program (S2174574) funded by the Small and Medium Business Administration (Republic of Korea); (ii) Peptron Inc., Daejeon, Republic of Korea; (iii) the Bio and Medical Technology Development Program(NRF-2014M3A9B5073868) of the National Research Foundation (NRF) fundedby the Ministry of Science, ICT and Future Planning (MSIP) of Korea, (iv) NINDS, NIH grant NS 094152 to BJH, (v) the Intramural Research Program of the National Institute on Aging, National Institutes of Health, USA, (vi) the Ari and Regine Aprijaskis Fund, and by a grant from the Dr. Miriam and Sheldon G. Adelson Chair for the Biology of Addictive Diseases at Tel-Aviv University, Tel Aviv, Israel. This work was performed in partial fulfillment of the requirements for a Ph.D. degree of MB, Sackler Faculty of Medicine, Tel Aviv University, Israel.
Footnotes
Competing financial interests
H—K Kim, DS Kim and H—I Choi are employees of Peptron Inc. The Intramural Research Program of the National Institute on Aging, NIH, and Peptron Inc. have a Cooperative Research and Development Agreement to develop Exenatide as a treatment strategy for neurodegenerative disorders for which NIA and Peptron Inc. hold patent rights via the work of H—K Kim, DS Kim, HI-Choi and NH Greig.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2018.11.023.
References
- Alcalay RN, Giladi E, Pick CG, Gozes I, 2004. Intranasal administration of NAP, a neuroprotective peptide, decreases anxiety-like behavior in aging mice in the elevated plus maze. Neurosci. Lett. 361, 128–131. [DOI] [PubMed] [Google Scholar]
- Anderberg RH, Richard JE, Hansson C, Nissbrandt H, Bergquist F, Skibicka KP, 2016. GLP-1 is both anxiogenic and antidepressant; divergent effects of acute and chronic GLP-1 on emotionality. Psychoneuroendocrinology 65, 54–66. 10.1016/j.psyneuen.2015.11.021. [DOI] [PubMed] [Google Scholar]
- Antonsen KK, Klausen MK, Brunchmann AS, le Dous N, Jensen ME, Miskowiak KW, Fisher PM, Thomsen GK, Rindom H, Fahmy TP, Vollstaedt-Klein S, Benveniste H, Volkow ND, Becker U, Ekstrøm C, Knudsen GM, Vilsbøll T, Fink-Jensen A, 2018. Does glucagon-like peptide-1 (GLP-1) receptor agonist stimulation reduce alcohol intake in patients with alcohol dependence: study protocol of a randomised, double-blinded, placebo-controlled clinical trial. BMJ Open 8, e019562. 10.1136/bmjopen-2017-019562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athauda D, Foltynie T, 2016. The glucagon-like peptide 1 (GLP) receptor as a therapeutic target in Parkinson’s disease: mechanisms of action. Drug Discov. Today 21, 802–818. 10.1016/j.drudis.2016.01.013. [DOI] [PubMed] [Google Scholar]
- Athauda D, Foltynie T, 2018. Protective effects of the GLP-1 mimetic exendin-4 in Parkinson’s disease. Neuropharmacology 136, 260–270. 10.1016/].neuropharm.2017.09.023. [DOI] [PubMed] [Google Scholar]
- Athauda D, Maclagan K, Skene SS, Bajwa-Joseph M, Letchford D, Chowdhury K, Hibbert S, Budnik N, Zampedri L, Dickson J, Li Y, Aviles-Olmos I, Warner TT, Limousin P, Lees AJ, Greig NH, Tebbs S, Foltynie T, 2017. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet 390, 1664–1675. 10.1016/S0140-6736(17)31585-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athauda D, Maclagan K, Budnik N, Zampedri L, Hibbert S, Skene SS, Chowdhury K, Aviles-Olmos I, Limousin P, Foltynie T, 2018. What Effects Might Exenatide have on Non-Motor Symptoms in Parkinson’s Disease: a Post Hoc Analysis. J. Parkinsons. Dis. 8, 247–258. 10.3233/JPD-181329. [DOI] [PubMed] [Google Scholar]
- Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Ell P, Soderlund T, Whitton P, Wyse R, Isaacs T, Lees A, Limousin P, Foltynie T, 2013. Exenatide and the treatment of patients with Parkinson’s disease. J. Clin. Invest. 123, 2730–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Kahan J, Ell P, Whitton P, Wyse R, Isaacs T, Lees A, Limousin P, Foltynie T, 2014. Motor and Cognitive Advantages Persist 12 months after Exenatide Exposure in Parkinson’s. Disease. J. Parkinsons. Dis. 4, 337–344. [DOI] [PubMed] [Google Scholar]
- Baratz R, Rubovitch V, Frenk H, Pick CG, 2010. The Influence of Alcohol on Behavioral Recovery after mTBI in mice. J. Neurotrauma 27, 555–563. [DOI] [PubMed] [Google Scholar]
- Baratz R, Tweedie D, Wang J-Y, Rubovitch V, Luo W, Hoffer BJ, Greig NH, Pick CG, 2015. Transiently lowering tumor necrosis factor-a synthesis ameliorates neuronal cell loss and cognitive impairments induced by minimal traumatic brain injury in mice. J. Neuroinflammation 12, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassil F, Canron M-H, Vital A, Bezard E, Li Y, Greig NH, Gulyani S, Kapogiannis D, Fernagut P-O, Meissner WG, 2017. Insulin resistance and exendin-4 treatment for multiple system atrophy. Brain 140, 1420–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers SR, 1992. Cognitive effects of mild head injury in children and adolescents. Neuropsychol. Rev. 3, 281–320. [DOI] [PubMed] [Google Scholar]
- Benakis C, Garcia-Bonilla L, Iadecola C, Anrather J, 2014. The role of microglia and myeloid immune cells in acute cerebral ischemia. Front. Cell. Neurosci. 8, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertilsson G, Patrone C, Zachrisson O, Andersson A, Dannaeus K, Heidrich J, Kortesmaa J, Mercer A, Nielsen E, Rönnholm H, Wikström L, 2008. Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of parkinson’s disease. J. Neurosci. Res. 86, 326–338. [DOI] [PubMed] [Google Scholar]
- Campbell JE, Drucker DJ, 2013a. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 17, 819–837. [DOI] [PubMed] [Google Scholar]
- Campbell JE, Drucker DJJ, 2013b. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 17, 819–837. [DOI] [PubMed] [Google Scholar]
- Chen Y, Mao H, Yang KH, Abe l.T., Meaney DF, 2014. A modified controlled cortical impact technique to model mild traumatic brain injury mechanics in mice. Front. Neurol. 5, 100 10.3389/fneur.2014.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Yu S-J, Li Y, Lecca D, Glotfelty E, Kim HK, Choi H-I, Hoffer BJ, Greig NH, Kim D-S, Wang Y, 2018. Post-treatment with PT302, a long-acting Exendin-4 sustained release formulation, reduces dopaminergic neurodegeneration in a 6-Hydroxydopamine rat model of Parkinson’s disease. Sci. Rep. 8 (10722). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chon S, Gautier JF, 2016. An Update on the effect of Incretin-based Therapies on β-Cell Function and Mass. Diabetes Metab. J. 40, 99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuah YML, Maybery MT, Fox AM, 2004. The long-term effects of mild head injury on short-term memory for visual form, spatial location, and their conjunction in well-functioning university students. Brain Cogn. 56, 304–312. [DOI] [PubMed] [Google Scholar]
- Deselms H, Maggio N, Rubovitch V, Chapman J, Schreiber S, Tweedie D, Kim DS, Greig NH, Pick CG, 2016. Novel pharmaceutical treatments for minimal traumatic brain injury and evaluation of animal models and methodologies supporting their development. J. Neurosci. Methods 272, 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deyoung MB, MacConell L, Sarin V, Trautmann M, Herbert P, 2011. Encapsulation of exenatide in poly-(D,L-lactide-co-glycolide) microspheres produced an investigational long-acting once-weekly formulation for type 2 diabetes. Diabetes Technol. Ther. 13, 1145–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dix SL, Aggleton JP, 1999. Extending the spontaneous preference test of recognition: evidence of object-location and object-context recognition. Behav. Brain Res. 99, 191–200. [DOI] [PubMed] [Google Scholar]
- Drucker DJ, 2018. Mechanisms of Action and Therapeutic Application of Glucagon-like Peptide-1. Cell Metab. 27, 740–756. [DOI] [PubMed] [Google Scholar]
- Eakin K, Li Y, Chiang Y-H, Hoffer BJ, Rosenheim H, Greig NH, Miller JP, 2013. Exendin-4 ameliorates traumatic brain injury-induced cognitive impairment in rats. PLoS One 8, e82016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edut S, Rubovitch V, Schreiber S, Pick CG, 2011. The intriguing effects of ecstasy (MDMA) on cognitive function in mice subjected to a minimal traumatic brain injury (mTBI). Psychopharmacology 214, 877–889. [DOI] [PubMed] [Google Scholar]
- Fann JR, Ribe AR, Pedersen HS, Fenger-Grøn M, Christensen J, Benros ME, Vestergaard M, 2018. Long-term risk of dementia among people with traumatic brain injury in Denmark: a population-based observational cohort study. Lancet Psychiatry 5, 424–431. [DOI] [PubMed] [Google Scholar]
- Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF, 2007. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav. Immun. 21, 47–59. [DOI] [PubMed] [Google Scholar]
- Frankola KA, Greig NH, Luo W, Tweedie D, 2011. Targeting TNF-α to elucidate and ameliorate neuroinflammation in neurodegenerative diseases. CNS Neurol. Disord. Drug Targets 10, 391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallwitz B, 2014. Extra-pancreatic effects of incretin-based therapies. Endocrine 47, 360–371. [DOI] [PubMed] [Google Scholar]
- Gardner RC, Yaffe K, 2015. Epidemiology of mild traumatic brain injury and neuro-degenerative disease. Mol. Cell. Neurosci. 66, 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gejl M, Gjedde A, Egefjord L, Møller A, Hansen SB, Vang K, Rodell A, Brandgaard H, Gottrup H, Schacht A, Møller N, Brock B, Rungby J, 2016. Alzheimer’s disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: randomized, placebo-controlled, double-blind clinical trial. Front. Aging Neurosci. 8 (108). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golde WT, Gollobin P, Rodriguez LL, 2005. A rapid, simple, and humane method for submandibular bleeding of mice using a lancet. Lab Anim. (NY). 34, 39–43. [DOI] [PubMed] [Google Scholar]
- Goldstein LE, Fisher AM, Tagge CA, Zhang X-L, Velisek L, Sullivan JA, Upreti C, Kracht JM, Ericsson M, Wojnarowicz MW, Goletiani CJ, Maglakelidze GM, Casey N, Moncaster JA, Minaeva O, Moir RD, Nowinski CJ, Stern RA, Cantu RC, Geiling J, Blusztajn JK, Wolozin BL, Ikezu T, Stein TD, Budson AE, Kowall NW, Chargin D, Sharon A, Saman S, Hall GF, Moss WC, Cleveland RO, Tanzi RE, Stanton PK, McKee AC, 2012. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med 4, 134ra60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonsalves A, Lin C-M, Muthusamy A, Fontes-Ribeiro C, Ambròsio AF, Abcouwer SF, Fernandes R, Antonetti DA, 2016. Protective effect of a GLP-1 analog on ischemia-reperfusion induced blood-retinal barrier breakdown and inflammation. Invest. Ophthalmol. Vis. Sci. 57, 2584–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig NH, Tweedie D, Rachmany L, Li Y, Rubovitch V, Schreiber S, Chiang Y-H, Hoffer BJ, Miller J, Lahiri DK, Sambamurti K, Becker RE, Pick CG, 2014. Incretin mimetics as pharmacologic tools to elucidate and as a new drug strategy to treat traumatic brain injury. Alzheimers Dement. 10, S62–S75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Cho S-H, Kim J, Shin D, Seol E, Lee H, Lim KS, Shin S-G, Jang I-J, Yu K-S, 2014. Pharmacokinetic properties and effects of PT302 after repeated oral glucose loading tests in a dose-escalating study. Clin. Ther. 36, 101–114. [DOI] [PubMed] [Google Scholar]
- Gullo F, Ceriani M, D’Aloia A, Wanke E, Constanti A, Costa B, Lecchi M, 2017. Plant polyphenols and exendin-4 prevent hyperactivity and TNF-α release in LPS-treated in vitro neuron/astrocyte/microglial networks. Front. Neurosci. 11 (500). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakon J, Ruscher K, Romner B, Tomasevic G, 2015. Preservation of the blood brain barrier and cortical neuronal tissue by liraglutide, a long acting glucagon-like-1 analogue, after experimental traumatic brain injury. PLoS One 10, e0120074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton A, Hölscher C, 2009. Receptors for the incretin glucagon-like peptide-1 are expressed on neurons in the central nervous system. Neuroreport 20, 1161–1166. [DOI] [PubMed] [Google Scholar]
- Harkavyi A, Whitton PS, 2010. Glucagon-like peptide 1 receptor stimulation as a means of neuroprotection. Br. J. Pharmacol. 159, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner KM, Kirigiti M, Secher A, Paulsen SJ, Buckingham R, Pyke C, Knudsen LB, Vrang N, Grove KL, 2015. Expression and distribution of glucagon-like peptide-1 receptor mRNA, protein and binding in the male nonhuman primate (Macaca mulatta) brain. Endocrinology 156, 255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herculano-Houzel S, Lent R, 2005. Isotropic fractionator: a simple, rapid method for the quantification of total cell and neuron numbers in the brain. J. Neurosci. 25, 2518–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hölscher C, 2018. Novel dual GLP-1/GIP receptor agonists show neuroprotective effects in Alzheimer’s and Parkinson’s disease models. Neuropharmacology 136, 251–259. [DOI] [PubMed] [Google Scholar]
- Hyder AA, Wunderlich CA, Puvanachandra P, Gururaj G, Kobusingye OC, 2007. The impact of traumatic brain injuries: a global perspective. NeuroRehabilitation 22, 341–353. [PubMed] [Google Scholar]
- Isacson R, Nielsen E, Dannaeus K, Bertilsson G, Patrone C, Zachrisson O, Wikström L, 2011. The glucagon-like peptide 1 receptor agonist exendin-4 improves reference memory performance and decreases immobility in the forced swim test. Eur. J. Pharmacol. 650, 249–255. [DOI] [PubMed] [Google Scholar]
- Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S, 1998. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res. Mol. Brain Res. 57, 1–9. [DOI] [PubMed] [Google Scholar]
- Iwai T, Ito S, Tanimitsu K, Udagawa S, Oka J-I, 2006. Glucagon-like peptide-1 inhibits LPS-induced IL-1β production in cultured rat astrocytes. Neurosci. Res. 55, 352–360. [DOI] [PubMed] [Google Scholar]
- Kappe C, Tracy LM, Patrone C, Iverfeldt K, Sjöholm Ä, 2012. GLP-1 secretion by microglial cells and decreased CNS expression in obesity. J. Neuroinflammation 9 (766). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnati HK, Garcia JH, Tweedie D, Becker RE, Kapogiannis D, Greig NH, 2018. Neuronal enriched eExtracellular vesicle proteins as biomarkers for traumatic brain injury. J. Neurotrauma. 10.1089/neu.2018.5898. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastin AJ, Akerstrom V, 2003. Entry of exendin-4 into brain is rapid but may be limited at high doses. Int. J. Obes. 27, 313–318. [DOI] [PubMed] [Google Scholar]
- Kastin AJ, Akerstrom V, Pan W, 2002. Interactions of Glucagon-like Peptide-1 (GLP-1) with the Blood-Brain Barrier. J. Mol. Neurosci. 18, 07–13. [DOI] [PubMed] [Google Scholar]
- Kim S, Moon M, Park S, 2009. Exendin-4 protects dopaminergic neurons by inhibition of microglial activation and matrix metalloproteinase-3 expression in an animal model of Parkinson’s disease. J. Endocrinol. 202, 431–439. [DOI] [PubMed] [Google Scholar]
- Kim DS, Choi H-I, Wang Y, Luo Y, Hoffer BJ, Greig NH, 2017. A New Treatment Strategy for Parkinson’s Disease through the Gut-Brain Axis: the Glucagon-like Peptide-1 Receptor Pathway. Cell Transplant. 26, 1560–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler JA, Baggio LL, Cao X, Abdulla T, Campbell JE, Secher T, Jelsing J, Larsen B, Drucker DJ, 2015. Glucagon-like Peptide-1 Receptor Agonists increase Pancreatic Mass by Induction of Protein Synthesis. Diabetes 64, 1046–1056. [DOI] [PubMed] [Google Scholar]
- Konrad C, Geburek AJ, Rist F, Blumenroth H, Fischer B, Husstedt I, Arolt V, Schiffbauer H, Lohmann H, 2011. Long-term cognitive and emotional consequences of mild traumatic brain injury. Psychol. Med. 41, 1197–1211. [DOI] [PubMed] [Google Scholar]
- Ladeby R, Wirenfeldt M, Garcia-Ovejero D, Fenger C, Dissing-Olesen L, Dalmau I, Finsen B, 2005. Microglial cell population dynamics in the injured adult central nervous system. Brain Res. Rev. 48, 196–206. [DOI] [PubMed] [Google Scholar]
- Langlois JA, Rutland-Brown W, Wald MM, 2006. The Epidemiology and Impact of Traumatic Brain Injury: A Brief Overview. J. Head Trauma Rehabil. 21, 375–378. [DOI] [PubMed] [Google Scholar]
- Lee C-H, Jeon SJ, Cho KS, Moon E, Sapkota A, Jun HS, Ryu JH, Choi JW, 2018. Activation of Glucagon-like Peptide-1 Receptor Promotes Neuroprotection in Experimental Autoimmune Encephalomyelitis by reducing Neuroinflammatory responses. Mol. Neurobiol. 55, 3007–3020. [DOI] [PubMed] [Google Scholar]
- Lewis A, Kumar R, 2012. Bydureon Development at Alkermes: Interview with Rajesh Kumar [WWW Document]. URL. https://www.controlledreleasesociety.org/publications/intrack/Pages/it0002.aspx, Accessed date: 11 July 2018.
- Li Y, Perry T, Kindy MS, Harvey BK, Tweedie D, Holloway HW, Powers K, Shen H, Egan JM, Sambamurti K, Brossi A, Lahiri DK, Mattson MP, Hoffer BJ, Wang Y, Greig NH, 2009. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc. Natl. Acad. Sci. U. S. A. 106, 1285–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Duffy KB, Ottinger MA, Ray B, Bailey JA, Holloway HW, Tweedie D, Perry T, Mattson MP, Kapogiannis D, Sambamurti K, Lahiri DK, Greig NH, 2010a. GLP-1 Receptor Stimulation Reduces Amyloid-ß Peptide Accumulation and Cytotoxicity in Cellular and Animal Models of Alzheimer’s Disease. J. Alzheimers Dis. 19, 1205–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Tweedie D, Mattson MP, Holloway HW, Greig NH, 2010b. Enhancing the GLP-1 receptor signaling pathway leads to proliferation and neuroprotection in human neuroblastoma cells. J. Neurochem. 113, 1621–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chigurupati S, Holloway HW, Mughal M, Tweedie D, Bruestle DA, Mattson MP, Wang Y, Harvey BK, Ray B, Lahiri DK, Greig NH, 2012. Exendin-4 Ameliorates Motor Neuron Degeneration in Cellular and Animal Models of Amyotrophic Lateral Sclerosis. PLoS One 7, e32008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Bader M, Tamargo I, Rubovitch V, Tweedie D, Pick CG, Greig NH, 2015. Liraglutide is neurotrophic and neuroprotective in neuronal cultures and mitigates mild traumatic brain injury in mice. J. Neurochem. 135, 1203–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Li L, Hölscher C, 2016. Incretin-based therapy for type 2 diabetes mellitus is promising for treating neurodegenerative diseases. Rev. Neurosci. 27, 689–711. [DOI] [PubMed] [Google Scholar]
- Matsuo M, Hamasaki Y, Fujiyama F, Miyazaki S, 1995. Eicosanoids are produced by microglia, not by astrocytes, in rat glial cell cultures. Brain Res. 685, 201–204. [DOI] [PubMed] [Google Scholar]
- Milman A, Rosenberg A, Weizman R, Pick CG, 2005. Mild Traumatic Brain Injury Induces Persistent Cognitive Deficits and Behavioral Disturbances in mice. J. Neurotrauma 22, 1003–1010. [DOI] [PubMed] [Google Scholar]
- Molina Vega M, Munoz-Garach A, Tinahones FJ, 2018. Pharmacokinetic drug evaluation of exenatide for the treatment of type 2 diabetes. Expert Opin. Drug Metab. Toxicol. 14, 207–217. [DOI] [PubMed] [Google Scholar]
- Moppett IK, 2007. Traumatic brain injury: assessment, resuscitation and early management. Br. J. Anaesth. 99, 18–31. [DOI] [PubMed] [Google Scholar]
- Murakami H, Takanaga H, Matsuo H, Ohtani H, Sawada Y, 2000. Comparison of blood-brain barrier permeability in mice and rats using in situ brain perfusion technique. Am. J. Physiol. Circ. Physiol. 279, H1022–H1028. [DOI] [PubMed] [Google Scholar]
- Mustapic M, Eitan E, Werner JK Jr., Berkowitz ST, Lazaropoulos MP, Tran J, Goetzl EJ, Kapogiannis D, 2017. Plasma extracellular vesicles enriched for neuronal origin: a potential window into brain pathologic processes. Front. Neurosci. 11 (278). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauck M, 2016. Incretin therapies: highlighting common features and differences in the modes of action of glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors. Diabetes Obes. Metab. 18, 203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan W, Kastin AJ, Rigai T, McLay R, Pick CG, 2003. Increased hippocampal uptake of tumor necrosis factor alpha and behavioral changes in mice. Exp. Brain Res. 149, 195–199. [DOI] [PubMed] [Google Scholar]
- Perry T, Haughey NJ, Mattson MP, Egan JM, Greig NH, 2002a. Protection and Reversal of Excitotoxic Neuronal damage by Glucagon-like Peptide-1 and Exendin-4. J. Pharmacol. Exp. Ther. 302, 881–888. [DOI] [PubMed] [Google Scholar]
- Perry T, Lahiri DK, Chen D, Zhou J, Shaw KTY, Egan JM, Greig NH, 2002b. A novel neurotrophic property of glucagon-like peptide 1: a promoter of nerve growth factor-mediated differentiation in PC12 cells. J. Pharmacol. Exp. Ther. 300, 958–966. [DOI] [PubMed] [Google Scholar]
- Perry T, Holloway HW, Weerasuriya A, Mouton PR, Duffy K, Mattison JA, Greig NH, 2007. Evidence of GLP-1-mediated neuroprotection in an animal model of pyridoxine-induced peripheral sensory neuropathy. Exp. Neurol. 203, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins ML, Giza CC, 2012. Repeat traumatic brain injury in the developing brain. Int. J. Dev. Neurosci. 30, 185–190. [DOI] [PubMed] [Google Scholar]
- Rachmany L, Tweedie D, Li Y, Rubovitch V, Holloway HW, Miller J, Hoffer BJ, Greig NH, Pick CG, 2013. Exendin-4 induced glucagon-like peptide-1 receptor activation reverses behavioral impairments of mild traumatic brain injury in mice. Age (Omaha). 35, 1621–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachmany L, Tweedie D, Rubovitch V, Li Y, Holloway HW, Kim DS, Ratliff WA, Saykally JN, Citron BA, Hoffer BJ, Greig NH, Pick CG, 2017. Exendin-4 attenuates blast traumatic brain injury induced cognitive impairments, losses of synaptophysin and in vitro TBI-induced hippocampal cellular degeneration. Sci. Rep. 7 (3735). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff RL, Riechers RG, Wang X-F, Piero T, Ruff SS, Ruff SS, 2012. A case-control study examining whether neurological deficits and PTSD in combat veterans are related to episodes of mild TBI. BMJ Open 2, e000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell S, 2013. Incretin-based therapies for type 2 diabetes mellitus: a review of direct comparisons of efficacy, safety and patient satisfaction. Int. J. Clin. Pharm. 35, 159–172. [DOI] [PubMed] [Google Scholar]
- Salcedo I, Tweedie D, Li Y, Greig NH, 2012. Neuroprotective and neurotrophic actions of glucagon-like peptide-1: an emerging opportunity to treat neurodegenerative and cerebrovascular disorders. Br. J. Pharmacol. 166, 1586–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, 2016. Development and application of novel histochemical tracers for localizing brain connectivity and pathology. Brain Res. 1645, 31–35. [DOI] [PubMed] [Google Scholar]
- Shi H-Y, Hwang S-L, Lee K-T, Lin C-L, 2013. In-hospital mortality after traumatic brain injury surgery: a nationwide population-based comparison of mortality predictors used in artificial neural network and logistic regression models. J. Neurosurg. 118, 746–752. [DOI] [PubMed] [Google Scholar]
- Skelton RW, Bukach CM, Laurance HE, Thomas KGF, Jacobs JW, 2000. Humans with Traumatic Brain Injuries Show Place-Learning Deficits in Computer-Generated Virtual Space. J. Clin. Exp. Neuropsychol. Neuropsychology, Dev. Cogn. Sect. A 22, 157–175. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Walter SA, Pennell NA, 1999. Reactive microgliosis. Prog. Neurobiol. 57, 563–581. [DOI] [PubMed] [Google Scholar]
- Sun Z, Liu Y, Kong X, Wang R, Xu Y, Shang C, Huo J, Huang M, Zhao F, Bian K, Zhang S, Tu Y, Chen X, 2018. Exendin-4 Plays a protective Role in a Rat Model of Spinal Cord Injury through SERCA2. Cell. Physiol. Biochem. 47, 617–629. [DOI] [PubMed] [Google Scholar]
- Tagliaferri F, Compagnone C, Korsic M, Servadei F, Kraus J, 2006. A systematic review of brain injury epidemiology in Europe. Acta Neurochir. 148, 255–268. [DOI] [PubMed] [Google Scholar]
- Tamargo IA, Bader M, Li Y, Yu S-J, Wang Y, Talbot K, Dimarchi RD, Pick CG, Greig NH, 2017. Novel GLP-1R/GIPR co-agonist “twincretin” is neuroprotective in cell and rodent models of mild traumatic brain injury. Exp. Neurol. 288, 176–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teramoto S, Miyamoto N, Yatomi K, Tanaka Y, Oishi H, Arai H, Hattori N, Urabe T, 2011. Exendin-4, a glucagon-like peptide-1 receptor agonist, provides neuroprotection in mice transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 31, 1696–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolhurst G, Reimann F, Gribble FM, 2009. Nutritional regulation of glucagon-like peptide-1 secretion. J. Physiol. 587, 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweedie D, Milman A, Holloway HW, Li Y, Harvey BK, Shen H, Pistell PJ, Lahiri DK, Hoffer BJ, Wang Y, Pick CG, Greig NH, 2007. Apoptotic and behavioral sequelae of mild brain trauma in mice. J. Neurosci. Res. 85, 805–815. [DOI] [PubMed] [Google Scholar]
- Tweedie D, Rachmany L, Rubovitch V, Lehrmann E, Zhang Y, Becker KG, Perez E, Miller J, Hoffer BJ, Greig NH, Pick CG, 2013. Exendin-4, a glucagon-like peptide-1 receptor agonist prevents mTBI-induced changes in hippocampus gene expression and memory deficits in mice. Exp. Neurol. 239, 170–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweedie D, Fukui K, Li Y, Yu Q-S, Barak S, Tamargo IA, Rubovitch V, Holloway HW, Lehrmann E, Wood WH, Zhang Y, Becker KG, Perez E, Van Praag H, Luo Y, Hoffer BJ, Becker RE, Pick CG, Greig NH, Greig NH, 2016a. Cognitive Impairments Induced by Concussive Mild Traumatic Brain Injury in Mouse are Ameliorated by Treatment with Phenserine via Multiple Non-Cholinergic and Cholinergic Mechanisms. PLoS One 11, e0156493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweedie D, Rachmany L, Rubovitch V, Li Y, Holloway HW, Lehrmann E, Zhang Y, Becker KG, Perez E, Hoffer BJ, Pick CG, Greig NH, 2016b. Blasttraumatic brain injury-induced cognitive deficits are attenuated by preinjury or postinjury treatment with the glucagon-like peptide-1 receptor agonist, exendin-4. Alzheimers Dement. 12, 34–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma MK, Goel R, Krishnadas N, Nemmani KVS, 2018. Targeting glucose-dependent insulinotropic polypeptide receptor for neurodegenerative disorders. Expert Opin. Ther. Targets 22, 615–628. [DOI] [PubMed] [Google Scholar]
- Washington PM, Forcelli PA, Wilkins T, Zapple DN, Parsadanian M, Burns MP, 2012. The effect of injury severity on behavior: a phenotypic study of cognitive and emotional deficits after mild, moderate, and severe controlled cortical impact injury in mice. J. Neurotrauma 29, 2283–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H-Y, Tang X-Q, Liu H, Mao X-F, Wang Y-X, 2018. Both classic Gs-cAMP/ PKA/CREB and alternative Gs-cAMP/PKA/p38β/CREB signal pathways mediate exenatide-stimulated expression of M2 microglial markers. J. Neuroimmunol. 316, 17–22. [DOI] [PubMed] [Google Scholar]
- Xie Z, Enkhjargal B, Wu L, Zhou K, Sun C, Hu X, Gospodarev V, Tang J, You C, Zhang JH, 2018. Exendin-4 attenuates neuronal death via GLP-1R/PI3K/Akt pathway in early brain injury after subarachnoid hemorrhage in rats. Neuropharmacology 128, 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun SP, Kam T-I, Panicker N, Kim S, Oh Y, Park J-S, Kwon S-H, Park YJ, Karuppagounder SS, Park H, Kim S, Oh N, Kim NA, Lee S, Brahmachari S, Mao X, Lee JH, Kumar M, An D, Kang S-U, Lee Y, Lee KC, Na DH, Kim D, Lee SH, Roschke VV, Liddelow SA, Mari Z, Barres BA, Dawson VL, Lee S, Dawson TM, Ko HS, 2018. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zohar O, Schreiber S, Getslev V, Schwartz JP, Mullins PG, Pick CG, 2003. Closed-head minimal traumatic brain injury produces long-term cognitive deficits in mice. Neuroscience 118, 949–955. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.