

SUMMARY
Recent findings are consistent with a slow but constant shift towards reduced sensitivity of Mycosphaerella graminicola to azole fungicides, which target the CYP51 gene. The goal of this study was to elucidate the evolutionary mechanisms through which CYP51‐based mutations associated with altered sensitivity have evolved in M. graminicola over space and time. To accomplish this, we sequenced and compared a portion of the CYP51 gene encompassing the main mutations associated with altered sensitivity towards demethylation inhibitor fungicides. The CYP51 gene showed an extraordinary dynamic shift consistent with a selective haplotype replacement both in space and in time. No mutations associated with increased resistance to azoles were found in non‐European populations. These mutations were also absent in the oldest collections from Europe, whereas they dominated in the recent European populations. Intragenic recombination was identified as an important evolutionary process in populations affected by high fungicide selection, suggesting the creation of novel alleles among existing mutations as a potential source of novel resistance alleles. We propose that CYP51 mutations giving resistance in M. graminicola arose only locally (perhaps in Denmark or the UK) and were then spread eastward across Europe through wind‐dispersed ascospores. We conclude that recurring cycles of recombination coupled with selection due to the widespread use of azole fungicides will increase the frequency of novel mutants or recombinants with higher resistance. Long‐distance gene flow due to wind dispersal of ascospores will move the resulting new alleles to new areas following the prevailing wind directions. A selective replacement favouring haplotypes with various coding mutations at the target site for azole fungicides during the last 5–10 years is the most likely cause of the decrease in sensitivity reported for many azole fungicides in the same period.
INTRODUCTION
The development of fungicide resistance represents the acquisition of a novel pathogen phenotype that can severely disrupt disease management in agricultural ecosystems. While there is a wealth of information on the molecular mechanisms giving rise to fungicide resistance (Ma and Michailides, 2005), considerably less is known about the underlying evolutionary processes that generate and spread novel resistance alleles. The unpredictable temporal and spatial scales over which fungicide resistance develops motivates research that seeks to understand the underlying evolutionary processes as a first step toward development of improved models for predicting the emergence and spread of fungicide resistance. In particular, we need to understand how diversity for fungicide resistance is generated, distributed and maintained in populations of fungal plant pathogens. Two common scenarios to explain the rapid development and widespread occurrence of fungicide resistance are (1) simultaneous selection in many populations of a rare pre‐existing variant that was already widely distributed or (2) origin of a new allele in one population followed by spread of the new mutation to other populations through gene or genotype flow. Conclusive evidence in favour of one or the other evolutionary process is rare because the necessary data must include samples that have not been exposed to fungicide selection pressure either in space or in time. The creation of novel alleles through intragenic recombination among existing mutations at a locus is a third potential source of novel resistance alleles.
Mycosphaerella graminicola (anamorph Septoria tritici) is a widespread fungal pathogen of wheat that is especially damaging in areas with a humid, temperate climate, such as north‐western Europe, resulting in up to 40% economic loss (Eyal, 1981). Control in Europe relies heavily on the use of fungicides, in particular on sterol demethylation inhibitors (DMIs). The reliance on this class of fungicides increased following the recent appearance and very fast spread of M. graminicola isolates with a high degree of resistance to strobilurin or QoI fungicides (Fraaije et al., 2005).
Previous studies detected a wide variation in the baseline sensitivity of isolates to DMI fungicides (Gisi et al., 1997; Stergiopoulos et al., 2003), with differences of up to 40‐fold between isolates from the same country. Recent findings are consistent with a slow but constant shift towards reduced sensitivity of M. graminicola to DMI fungicides, although initially this shift was not considered strong enough to compromise disease control (Gisi et al., 2005; Leroux et al., 2005; Mavroeidi and Shaw, 2005). Leroux et al. (2007) reported various amino acid substitutions in field isolates of M. graminicola that expressed different levels of sensitivity to DMI fungicides. Isolates with a combination of amino acid substitutions including I381V showed a reduced sensitivity to DMI fungicide, as compared with isolates lacking the I381V substitution. Interestingly, the highest levels of fungicide resistance where observed among the most recently collected isolates. We recently showed that there was a unimodal and continuous variation in the level of resistance to the DMI fungicide cyproconazole among over 140 isolates sampled from untreated wheat fields in Switzerland, Israel, Oregon and Australia (Zhan et al., 2006). This finding was consistent with the polygenic nature of the resistance to azoles that was described earlier (De Waard, 1994; Stergiopoulos et al., 2003). We also found that the population differentiation among the five field populations for cyproconazole tolerance (Q ST) was significantly higher than the corresponding population differentiation for neutral restriction fragment length polymorphism (RFLP) markers (F ST), indicating that the Swiss population had been subjected to selection that led to local adaptation, even though all of the populations originated from unsprayed fields (Zhan et al., 2005). The Swiss population had a lower than expected variation in cyproconazole resistance with a skewed distribution. Sequence analysis of the CYP51 gene revealed that the Swiss isolates possessed various point mutations and a 6‐bp deletion, called ΔY459/G460, that had been associated with a decrease in sensitivity. However, all these isolates lacked the amino acid substitutions Y137F and I381V, both associated with reduced sensitivity (Cools et al., 2005a). The other populations did not possess any of the mentioned mutations; however, we found the amino acid substitution S188N in all populations. Interestingly, the frequency of S188N varied dramatically among populations, ranging from as low as 2% in Oregon to 30% in Israel, 50% in Australia and 65% in Switzerland, though the Israeli and Australian populations did not show any sign of increased resistance. Leroux et al. (2007) found that this mutation was always associated with the double deletion ΔY459/G460 and with another substitution, N513K, in a collection of French isolates.
The goal of this study was to elucidate the evolutionary mechanisms through which CYP51‐based mutations associated with altered sensitivity have evolved in M. graminicola over space and time. To accomplish this, we sequenced and compared a portion of the CYP51 gene encompassing the main mutations associated with sensitivity towards DMI fungicides. We compared geographically distinct populations of M. graminicola subjected to either high DMI pressure (Europe) or low/no DMI pressure (non‐European populations). To address the temporal dimension of evolution, we compared isolates from European populations sampled from the same regions over a 13‐year period.
RESULTS
A 1371‐bp‐long fragment of the CYP51 gene (total size 1897 bp) was sequenced for 341 isolates of M. graminicola. Nucleotide positions throughout the paper were numbered according to the whole gene sequence deposited in GenBank (accession no. AY730587). A total of 90 sites (6.5%) were polymorphic, mostly due to base substitutions, and these defined 108 distinct haplotypes (Appendix 1; sequences deposited in GenBank with accession numbers EU418016–EU418123).
Population genetic parameters are listed in Table 1 for the three major population groupings, non‐European, old European and recent European, respectively. As expected, differences were detected among individual populations that probably reflect different population histories. For example, isolates from Oregon R (i.e. collected from a wheat cultivar resistant to M. graminicola) showed a significantly reduced number of polymorphic sites (S = 4; P = 0.002) compared with isolates collected from the susceptible cultivar in the same field (Oregon S, S = 17). An interpretation of this observation is the selection of only a few haplotypes adapted to the resistant wheat cultivar and subsequent selection of closely related haplotypes carrying the corresponding CYP51 alleles (Zhan et al., 2006). Recent European populations exhibited the highest number of CYP51 alleles, but many more isolates from this group were analysed and haplotype diversity was similar to the other groups.
Table 1.
Origin and summary of genetic diversity for the Mycosphaerella graminicola populations used in this study.
| Designation | Population | Year of collection | Reference | n | S | K | Hd | Pi |
|---|---|---|---|---|---|---|---|---|
| Non‐European | Oregon R. | 1995 | Zhan et al., 2005 | 11 | 4 | 5 | 0.78 | 0.001 |
| Oregon S. | 1995 | Zhan et al., 2005 | 13 | 17 | 6 | 0.72 | 0.002 | |
| Israel | 1992 | Zhan et al., 2005 | 15 | 22 | 11 | 0.95 | 0.004 | |
| Australia | 2002 | Zhan et al., 2005 | 14 | 29 | 8 | 0.86 | 0.009 | |
| combined | 53 | 46 | 27 | 0.92 | 0.005 | |||
| ‘Old’‐European | Germany | 1992 | Banke et al., 2004 | 11 | 19 | 4 | 0.60 | 0.003 |
| Ukraine | 1992 | Banke et al., 2004 | 19 | 9 | 3 | 0.61 | 0.002 | |
| Denmark | 1992 | Banke et al., 2004 | 12 | 23 | 5 | 0.74 | 0.003 | |
| United Kingdom | 1994 | Banke et al., 2004 | 22 | 26 | 12 | 0.76 | 0.003 | |
| combined | 64 | 38 | 22 | 0.86 | 0.003 | |||
| ‘Recent’‐European | Switzerland | 1999 | Zhan et al., 2005 | 25 | 32 | 10 | 0.76 | 0.007 |
| Denmark 1 | 2005 | This paper | 10 | 20 | 6 | 0.91 | 0.004 | |
| Denmark 2 | 2005 | This paper | 21 | 20 | 5 | 0.55 | 0.004 | |
| Germany Q | 2004 | This paper | 86 | 36 | 26 | 0.83 | 0.007 | |
| Germany D | 2004 | This paper | 82 | 33 | 25 | 0.80 | 0.005 | |
| combined | 224 | 43 | 65 | 0.84 | 0.005 |
n, number of isolates; S, number of polymorphic sites; K, number of haplotypes; Hd, haplotype diversity; Pi, nucleotide diversity.
The majority of haplotypes were unique for each population grouping with few shared haplotypes among populations (Table 2). For example, non‐European and old European populations shared only 8 and 9% of their haplotypes, indicating significant spatial differentiation that was confirmed with an amova. The proportion of total genetic variation attributed to among‐group differentiation was 6.88%, resulting in a highly significant F CT = 0.133, P < 0.001. Old European and recent European populations sampled from the same region but at different points in time also differed significantly in haplotype composition, suggesting significant temporal differentiation. The recent European populations shared only 3% of their haplotypes with the old European populations, suggesting that the European CYP51 gene pool experienced a nearly complete replacement of old haplotypes by new haplotypes.
Table 2.
Proportion of shared haplotypes between populations grouped according to Table 1.
| Non‐European | Old European | Recent European | |
|---|---|---|---|
| Non‐European | 0.92 | 0.08 | 0.04 |
| Old European | 0.09 | 0.86 | 0.09 |
| Recent European | 0.02 | 0.03 | 0.97 |
Proportions of unique/not‐shared haplotypes are on the diagonal.
Neutral coalescent simulations showed that for both the non‐European and the old European populations, the observed number of haplotypes did not deviate significantly from expectations under the K test and assuming no intragenic recombination (Table 3). In contrast, the number of observed haplotypes (46) among the recent European populations was much higher than the expected number of 28 haplotypes, indicating a highly significant departure from neutral expectations. The four‐gamete test indicated different levels of intragenic recombination among the populations. The identified pairs of polymorphic loci along the CYP51 alignments translated into a minimum of three and two recombination events in the history of non‐European and old European populations, respectively. A minimum of eight recombination events were detected in the recent European populations. Figure 1 summarizes the results of the TOPALI analyses. This analysis found significant recombination only in the recent European populations with a breakpoint located around nucleotide position 1647. This suggests that some recent haplotypes acquired a combination of mutations located on both sides of this breakpoint by intragenic recombination rather than through a series of independent mutations in the same ancestral allele.
Table 3.
Expected number of haplotypes (K‐test).
| Parameter | Non‐European populations | Old European Populations | Recent European populations |
|---|---|---|---|
| Sample size, n | 53 | 64 | 224 |
| Segregating sites, S | 46 | 38 | 43 |
| Observed haplotypes, K O | 23 | 21 | 46 |
| Probability, P (K = K O) | 0.150 | 0.216 | < 0.001 |
| Expected haplotypes, K E | 20 | 19 | 28 |
| 95% C.I. for K E | 13–27 | 12–25 | 19–36 |
Expectations (means) of parameters, P values, and 95% confidence intervals (CI) are based on 100 000 coalescent simulations under an infinite‐sites model and assuming no intragenic recombination. P (K = K O) refers to the proportion of simulations yielding a value as low as or lower than the observed K O.
Figure 1.
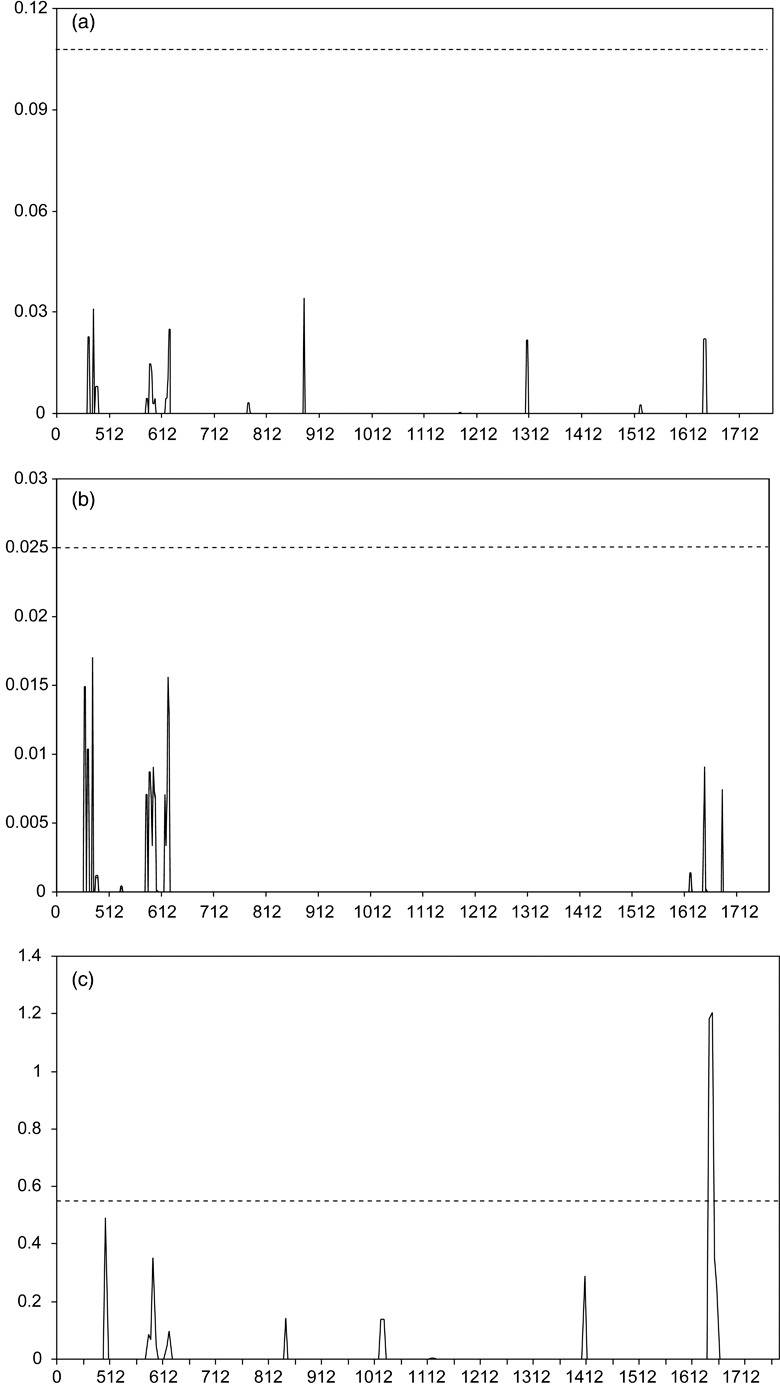
Identification of intragenic recombination breakpoints in the CYP51 gene of M. graminicola using the DSS method implemented in TOPALI. Horizontal axes represent nucleotide positions and dashed lines are significance thresholds for the DSS values indicated on the vertical axes: (a) non‐European populations, (b) old European populations, (c) recent European populations.
Occurrence of specific mutations previously associated with altered sensitivity
As reported earlier (Zhan et al., 2006), analysis of the CYP51 sequences in the Swiss population showed several mutations in the amino acids from positions 459 to 461 [numbering of amino acids follows the convention used in Cools et al. (2005b) and represent nucleotide positions 1647–1654; see Appendix 1]. Most of these mutations were present in the recent isolates from Germany and Denmark, together with some additional variants (Table 4). In particular the 6‐bp deletion ΔY459/G460 was present at a high level in all of the recent European populations. No mutations associated with increased resistance to azoles were found in the populations from Oregon 1992, Israel 1992 and Australia 2002. These mutations were also absent in the oldest collections from Europe, such as the isolates from Germany 1992, Ukraine 1992 and United Kingdom 1994. These sensitive alleles are considered to represent the ancestral situation and are henceforth referred to as ‘wild types’.
Table 4.
Shift of mutation frequencies over time across European populations of M. graminicola for amino acid positions affecting the azole sensitivity of the CYP51 gene. All other old European and non‐European populations show the wild type.
| Amino acid substitution | Frequency (%) of specific mutations | |||||
|---|---|---|---|---|---|---|
| Germany 1992 | Denmark 1992 | Switzerland 1999 | Denmark 2005 | Germany Quarnbek | Germany Dedelow | |
| Wild type | 100 | 33 | 1 | 1 | ||
| ΔY459/G460 | 63 | 37 | 57 | 66 | ||
| G460D | 58 | 10 | 2 | |||
| Y461H | 3 | 52 | 28 | 15 | ||
| Y461S | 16 | 2.5 | 1 | 6 | ||
| Y459P | 3 | |||||
| Y461D | 3 | |||||
| Y459D | 7.5 | 10 | 8 | |||
| Y459S | 2 | |||||
| ΔY459 | 2 | |||||
| Y459C | 8 | |||||
| ΔG460 | 1 | 2 | ||||
| I381V | 95 | 83 | 37 | |||
The I381V amino acid substitution (nucleotide position 1418; Fraaije et al., 2007) was completely absent in all non‐European and all old European populations. While I381V was also absent in the recent Swiss population, it was found at high frequencies in the recent German (±85%) and Danish (95%) populations. Amino acid substitution I381V was frequently found in combination with other mutations located on the opposite side of the identified recombination breakpoint. For example ±90% of the recent German isolates with this mutation also carried the 6‐bp deletion ΔY459/G460. This finding supports the hypothesis that some combinations of CYP51 mutations evolved through intragenic recombination and subsequently reached high frequencies due to reduced azole sensitivity compared with the parental haplotypes.
Gene flow and phylogenetic relationships of CYP51 haplotypes
The highest migration rates were estimated from Danish into German populations, ranging from 3 to 20 migrants per generation (Table 5). Gene flow in the opposite direction from German to Danish populations was clearly lower (range 0.07–0.40), indicating that the majority of gene flow occurred in a west‐to‐east direction.
Table 5.
Pairwise likelihood estimates of directional migration rates expressed as the number of migrants per generation (2Nm) between recent European populations (95% confidence intervals in parentheses).
| Switzerland | Denmark 1 | Denmark 2 | Germany Q | Germany D | |
|---|---|---|---|---|---|
| Switzerland | — | 0.00 (0.00–0.25) | 0.09 (0.05–0.43) | 1.11 (0.24–2.63) | 2.52 (1.08–3.87) |
| Denmark 1 | 0.89 (0.32–1.68) | — | 1.49 (0.74–2.63) | 11.88 (8.63–14.95) | 3.52 (1.81–5.63) |
| Denmark 2 | 0.17 (0.00–0.40) | 0.13 (0.03–0.44) | — | 20.77 (15.44–24.98) | 6.27 (4.63–8.36) |
| Germany Q | 0.26 (0.11–0.61) | 0.09 (0.05–0.32) | 0.40 (0.22–0.68) | — | 5.54 (3.40–7.21) |
| Germany D | 0.29 (0.10–0.85) | 0.09 (0.01–0.24) | 0.07 (0.04–0.12) | 1.217 (0.467–2.102) | — |
Donor populations are shown on the left and receiving populations are given along the top. Note that populations Denmark 1, 2, and Germany Q, D represent a geographical west‐to‐east transect.
The genealogies of haplotypes were reconstructed with median‐joining networks (Fig. 2). The networks obtained for the non‐European populations (Fig. 2a) and for the old European populations (Fig. 2b) were quite similar. Both networks identified one or two main haplotypes occurring at high frequencies with ‘star‐like’ structures, consistent with recent population expansion. Both networks were well resolved and only few missing haplotypes were found. Both networks also showed little reticulation with only two loops identified in each network, suggesting the rare occurrence of homoplasy due to recurrent mutations or intragenic recombination. Two recombinant haplotypes were detected in each group, representing 7 and 18% of the haplotypes in the respective populations (Table 6). The network for the recent European populations also identified two major haplotypes and showed evidence of population expansion. However, in sharp contrast to the previous networks, extensive homoplasy was evidenced by numerous network reticulations, suggesting a significantly increased number of recurrent mutations or intragenic recombination events. Although there was no difference with regard to non‐sensitive sites compared with non‐European populations, recent European populations contained a large proportion (32%) of recombinant haplotypes combining different mutations correlated with azole sensitivity. These recombinant haplotypes made up 16% of all isolates in the recent populations and many of these carried mutations associated with the highest levels of azole resistance. Overall, the total number of recombinant haplotypes in recent European populations was six times higher compared with non‐European populations experiencing less azole selection.
Figure 2.
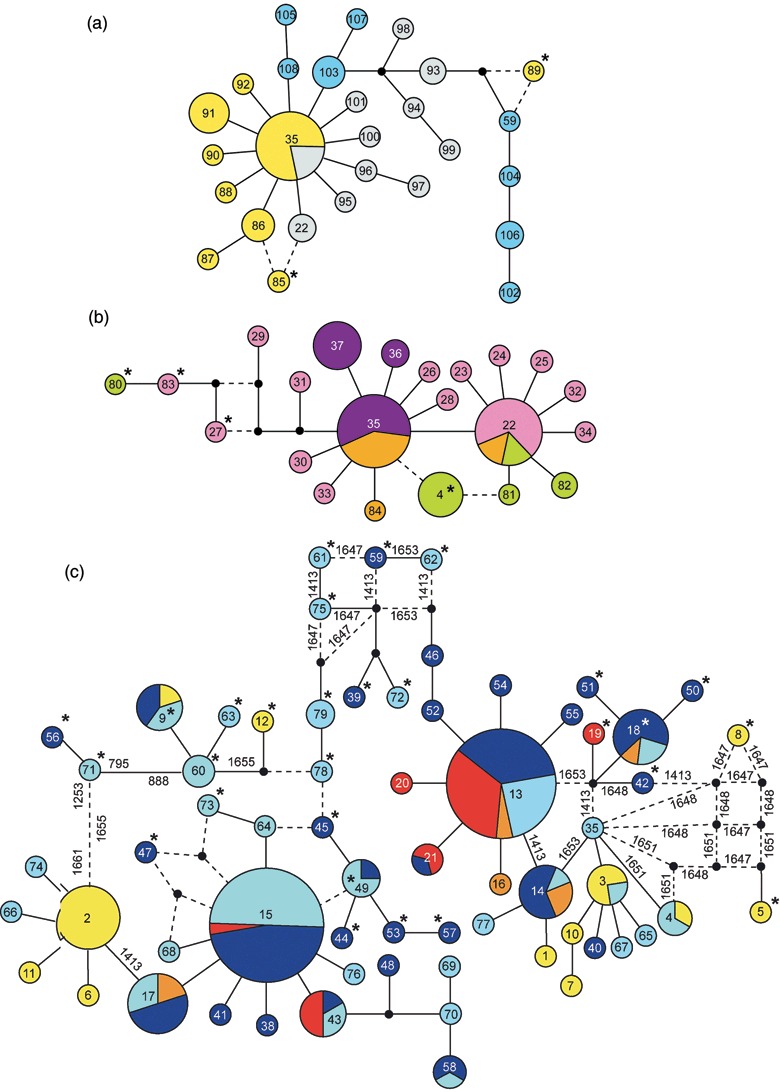
Median‐joining networks depicting the genealogy of CYP51 haplotypes indicated by circles. Circle size is proportional to haplotype frequency and colours denote the sampled populations. Small black circles represent median vectors/hypothetical haplotypes. For the sake of visual clarity, mutated positions are only indicated on the reticulations if nucleotide sites associated with reduced azole sensitivity were involved. Branch lengths connecting haplotypes are not proportional to distance. (a) Network including the non‐European populations of M. graminicola. Yellow, Oregon; grey, Israel; blue, Australia. (b) Old European populations. Purple, Ukraine; pink, UK; green, Denmark; orange, Germany. (c) Recent European populations. Light blue, Germany D; dark blue, Germany Q; yellow, Switzerland; red, Denmark 2; orange, Denmark 1. Haplotype numbers are indicated within circles and recombinant haplotypes are marked with asterisks (see also Appendix 1).
Table 6.
Proportions of recombinant haplotypes and recombinant isolates (in parentheses) detected with NETWORK (Fig. 2).
| Non‐sensitive sites | Sensitive sites | Total | |
|---|---|---|---|
| Non‐European | 0.07 (0.04) | 0 | 0.07 (0.04) |
| Old European | 0 | 0.18 (0.14) | 0.18 (0.14) |
| Recent European | 0.10 (0.05) | 0.32 (0.16) | 0.43 (0.21) |
Haplotypes are categorized according to the intragenic recombination of nucleotide sites associated with reduced azole sensitivity.
Because recombination violates the assumption in traditional molecular phylogenetics that there is only one evolutionary history in a sequence data set, the potentially recombined haplotypes identified in Fig. 2 were excluded from the maximum‐parsimony (MP) and maximum‐likelihood (ML) analyses. Both analyses resulted in identical topologies. The likelihood phylogeny of the reduced data set is reconstructed in Fig. 3. Two main phylogenetic clades emerged from this analysis. Whereas one clade (top in Fig. 3) contained almost all haplotypes observed in non‐European and old European populations as well as some related haplotypes from the recent Europe populations carrying azole‐associated mutations, the other clade was composed almost exclusively of recent European haplotypes carrying mutations significantly lowering azole sensitivity. The various resistance‐associated mutations were not dispersed across the phylogeny but were clustered in distinct haplotype groups, suggesting localized evolution of mutated haplotypes rather than independent and recurrent mutations in unrelated genetic backgrounds.
Figure 3.

Phylogenetic relationship of M. graminicola haplotypes based on a likelihood analysis of CYP51 sequence data, excluding recombinant haplotypes identified in Fig. 2. Arrows indicate basal haplotypes for particular clusters of related azole‐associated mutations (indicated in different colours). The central haplotype H36 was also identified as the potential root in a statistical parsimony‐based analysis (data not shown).
DISCUSSION
To understand better the role of the CYP51 gene in the observed shift in azole sensitivity, we analysed the CYP51 sequence variability in a set of European populations that were sampled along a west‐to‐east transect at different points in time. The three main results were (1) the CYP51 gene showed an extraordinary dynamic shift consistent with a selective replacement both in space and in time, (2) the CYP51 gene showed a significantly higher number of intragenic recombination events in populations affected by high fungicide selection and (3) most mutations associated with resistance against azole fungicides probably originated only once (or twice in the case of amino acid substitution I381V) and were subsequently dispersed.
Evidence for a west‐to‐east selective replacement in time
In the earlier work by Zhan et al. (2006) we analysed cyproconazole sensitivity among global populations of M. graminicola and found that only the European population possessed a decreased level of sensitivity to cyproconazole. The difference in sensitivity was correlated with the presence of various mutations at CYP51. The mutations conferring lower sensitivity (see Table 4) were absent in geographically distant populations from Israel, the USA and Australia.
Triggered by these first observations of locally restricted haplotype groups, we extended this research with a temporal component by comparing CYP51 sequences of older European populations with their more recent counterparts. We found a dramatic increase in the frequency of mutations associated with azole resistance in the recent populations (Table 4). For example, whereas the 1992 German population was composed only of wild‐type isolates, the 2004 German populations contained only 1% wild‐type CYP51 alleles. Similarly, the UK population sampled in 1992 did not show any CYP51 mutations related to resistance. We do not have data for a more recent population from the UK, but Cools et al. (2005a) followed the evolution in time of mutations in field isolates from Rothamsted, UK, and reported that 69% of isolates from a triazole‐treated field carried CYP51 mutations associated with resistance in 2001, with a prevalence of Y461H, while the ΔY459/G460 deletion was still relatively rare. In their untreated plots there was still a substantial percentage of wild‐type isolates, with only 29% of isolates carrying mutations. The situation changed significantly in the following 2 years. In 2003, 88% of the isolates from Kent carried the ΔY459/G460 deletion.
We conclude from these observations and from the results presented herein that there has been a significant change in haplotype frequencies at the CYP51 gene in European populations of M. graminicola over the past 10–15 years. Since this change in frequency involved the almost complete extinction of haplotypes lacking mutations associated with azole resistance (i.e. ‘wild‐type’) and replacement by haplotypes carrying mutations associated with increased azole resistance, we consider this evolutionary process to represent a selective replacement.
The German isolates from 2004 were sampled from two distant locations, Quarnbek (west) and Dedelow (east). We did not find differences in the frequency of the various mutations at the 459–461 amino acids between these populations, but we found a significant difference in the frequency of the I381V amino acid substitution with 83% of isolates in Quarnbek having I381V compared with 37% of the isolates in Dedelow. This amino acid substitution was present in 95% of the recent Danish isolates we sequenced and is reported to be common in the UK as well (Fraaije et al., 2007). Our interpretation of these results is that there currently exists a gradient in the distribution of CYP51 alleles across Europe, with the mutations most strongly associated with high levels of azole resistance at the highest frequency in the northern‐ and westernmost populations, and at a lower frequency in the southern‐ and easternmost populations sampled.
There was substantial regional variation in triazole use (and hence potentially different selection pressure) among the populations examined. Thus, high levels of use in western populations (e.g. UK) and low levels of use in eastern Europe (e.g. Ukraine) could at least partially account for the observed gradient of CYP51 alleles. Nevertheless, we believe this distribution mainly reflects the air‐dispersal of ascospores from north‐west to south‐east following the prevailing wind directions across Europe. This hypothesis is corroborated by our analysis of directional gene flow indicating high numbers of migrants moving in a west‐to‐east direction, and a virtual lack of migrants moving in the opposite direction. Furthermore, recent European populations shared only two haplotypes (H04 and H35) with non‐European and old European populations. Both haplotypes were found at high frequencies in old populations but were rare in recent populations (four isolates in total). For example, among recent populations haplotype H35 was detected in only one isolate in the easternmost population of Dedelow (Germany D). In contrast, H35 is central in both the networks of non‐European and old‐European populations (Fig. 2a,b), as well as in the phylogenetic trees including all non‐recombined haplotypes (Fig. 3). Haplotype H35 was identified as the most ancestral haplotype in a statistical parsimony‐based network analysis using the program TCS (Clement et al., 2000; data not shown), additionally supporting a west‐to‐east selective replacement not only geographically but also over time.
Origin of CYP51 resistant haplotypes through mutation and intragenic recombination
A central question is whether resistance‐associated mutations arose once and then spread to other populations or whether they arose through independent mutation events (homoplasy) in different genetic backgrounds. A related question is how often new alleles arise through intragenic recombination between pre‐existing mutations. The number of origins of pesticide resistance mutations is important not only for our understanding of the evolution of resistance but also for modelling its spread and for developing appropriate management strategies. Two scenarios to explain the rapid development and widespread occurrence of fungicide resistance are (1) selection of a rare pre‐existing variant distributed among many populations or (2) origin of the mutation in one population and rapid spread of the new mutation to surrounding populations by gene flow.
Although we cannot rule out the possibility of undetected rare haplotypes in old European populations, the data collected thus far suggest that important azole‐associated amino acid substitutions such as I381V or ΔY459/G460 (as well as combinations of these) arose locally de novo and relatively recently. The phylogenetic analysis showed that haplotypes carrying the same resistance mutation were related by descent. For example, all haplotypes carrying the ΔY459/G460 deletion were connected with each other (Fig. 3). Furthermore, from this group of haplotypes another group arose carrying in addition the I381V amino acid substitution, strongly suggesting that each group of haplotypes shared a common ancestor. The I381V amino acid substitution is arguably contributing the most to azole resistance in M. graminicola. According to our phylogenetic analysis, this is the only mutation arising twice in different ancestral haplotypes, namely H13 and H17 (Fig. 3). In a recent study, Leroux et al. (2007) reported evidence that both I138V and amino acid substitution V136A evolved in different ancestral lineages. Unfortunately, these findings are not readily comparable with our study because of fundamental methodological differences. For example, Leroux et al. (2007) compared the distribution of specific ‘haplotypes’ among fungicide sensitivity‐based phenotypes of M. graminicola. These haplotype definitions, however, were based on the presence of amino acid substitutions (such as I381V), whereas our conclusions were based on the entire nucleotide sequence information and included specific phylogenetic analyses.
Considering both the phylogeny and the migration analysis, we propose that CYP51 mutations giving resistance in M. graminicola arose only locally and were then spread across Europe by wind‐dispersed ascospores. An alternative hypothesis is that the resistance mutations and combinations of these mutations arose at different locations independently from each other. We consider the latter hypothesis less plausible because the evolution of identical haplotypes across 1371 bp of nucleotides, including exons and introns, shared among distant populations as illustrated in the network is very unlikely given the known genetic diversity of M. graminicola at other loci (Zhan et al., 2005).
We also found strong evidence that intragenic recombination played an important role in shaping the diversity observed in CYP51 haplotypes. All recombination analyses suggested that intragenic recombination occurred in all populations, but a larger number of recombinants were found in recent European populations. No intragenic recombination events were identified between nucleotide sites associated with altered azole sensitivity in the non‐European populations. In contrast, the frequency of these recombination events was high (32%) in recent European populations compared with old European populations (18%; Table 6, Fig. 2). We believe that these recombinants have very recently increased to a high frequency because they exhibit higher levels of azole resistance.
In general, the CYP51 gene exhibits a very high level of genetic variation at both the nucleotide and the amino acid level. This appears counterintuitive at first because strong directional selection imposed by fungicide treatments might be expected to reduce genetic diversity at selected loci such as CYP51. Furthermore, it is generally assumed that resistance mutations carry a fitness cost for the pathogen, thus giving advantage only in the environment where selection is applied. The lack of resistance mutations in the absence of strong fungicide selection in non‐European populations supports these assumptions. Two non‐exclusive hypotheses can explain this observation. First, if the presence of resistance mutations at the CYP51 gene carries a fitness cost, this could be ameliorated by additional compensatory mutations in other regions of the gene. These compensatory mutations could generate significant diversity in the overall gene sequence as found in HIV1 (Nijhuis et al., 1999) and Escherichia coli (Levin et al., 2000). Second, many related DMI fungicides have been used across Europe over the last 30 years, and these are known to differ in their efficacy against different haplotypes (as shown in Fraaije et al., 2007), probably reflecting differing abilities to interfere with the CYP51 proteins produced by these haplotypes. In practice, different farmers use different azole fungicides and thus the wheat fields of Europe present a mosaic of different selective environments that can support a high diversity of differentially adapted CYP51 haplotypes that are selected by different azole compounds.
In conclusion, we hypothesize that mutations conferring azole resistance appeared first in north‐western Europe (e.g. Denmark or the UK). Mutations affecting amino acid positions 459–461 were already present in Denmark in 1992. By 1999, isolates with CYP51 mutations encoding azole resistance were already prevalent in central Europe, represented by the Swiss population. New amino acid substitutions, such as I381V, as well as combinations of mutations conferring increased resistance appeared later either by de novo mutations or through intragenic recombination. The highly resistant CYP51 alleles spread eastward through wind‐dispersed ascospores and are still moving eastwards, as indicated by the lower frequency of the I381V amino acid substitution in the easternmost sample from Germany. We hypothesize that the eastern regions of Europe represent the current leading edge of an ongoing selective replacement eliminating all old European haplotypes that had higher levels of azole sensitivity. Our hypothesis of a rapid and widespread selective replacement is consistent with the known population biology of M. graminicola. Analysis of field populations of M. graminicola with RFLP and sequence markers indicated that populations recombined regularly (Chen and McDonald, 1996) and that there was little genetic differentiation among populations from the same region (Linde et al., 2002; McDonald et al., 1999). The earlier findings that sequence haplotypes and RFLP alleles occurred at similar frequencies among field populations from different regions on the same continent suggested that we could consider the European population as panmictic (Banke et al., 2004; Zhan et al., 2003). The selection caused by widespread use of azole fungicides for several decades led to a steady decrease in the DMI sensitivity of European M. graminicola populations (Gisi et al., 2005; Leroux et al., 2005). Recurring cycles of selection and recombination are expected to increase the frequency of novel mutants or recombinants with higher resistance to a class of fungicides with a similar mode of action. Long‐distance gene flow due to wind dispersal of ascospores will move the resulting new alleles to new areas following the prevailing wind directions. The resulting selective replacement of haplotypes with various coding amino acid substitutions at the target site for azole fungicides across the continent, in particular during the last 5–10 years, is the most likely cause of the decrease in sensitivity reported for many azole fungicides in the same period.
EXPERIMENTAL PROCEDURES
Origins of M. graminicola isolates
Origins and collection dates for the isolates included in this study are summarized in Table 1. To address the temporal component of this study, samples included isolates from Germany and Denmark collected in 1992 (henceforth called old populations) and 2004–2005 (recent populations). Additional old samples were collected in the UK and Ukraine, and a more recent population was collected in Switzerland. The spatial component was addressed by (1) sampling European populations on a west–east transect reflecting the main direction for movement of wind‐dispersed ascospores, and by (2) adding non‐European populations that were both geographically distant and under less fungicide selection pressure.
Culture conditions and DNA isolation
Isolations from infected leaves were made as described previously (McDonald et al., 1994). Briefly, leaves were surface sterilized and single cirri were cultured on YMA plates (4 g/L yeast extract, 4 g/L sucrose, 4 g/L malt extract, 50 µg/mL kanamycin sulphate) then transferred to liquid shake cultures (YSB medium (10 g/L yeast extract, 10 g/L sucrose, 50 µg/mL kanamycin sulphate) and propagated for further analysis. DNA isolation was performed as described earlier (Linde et al., 2002). Briefly, 2‐mL aliquots of liquid cultures were pelleted in a table‐top centrifuge, and the pellets were freeze‐dried, ground in a bead‐beater mill and extracted by using a DNeasy Qiagen plant mini kit (Qiagen, Germany) according to the manufacturer's instructions.
PCR fingerprinting and CYP51 sequencing
New German samples and Danish populations were evaluated for clonality using rep‐PCR (R. Sommerhalder, personal communication) using the following primers: BOX 1 AR (5′‐CTACGGCAAGGCGACGCTGACG‐3′) and ERIC 2 (5′‐AAGTAAGTGACTGGGGTGAGCG‐3′) (Versalovic et al., 1991, 1994). PCR conditions were as follows: 2 min at 94 °C followed by 30 s denaturation at 94 °C, 1 min annealing at 52 °C and 5 min extension at 65 °C. These three steps were repeated for 32 cycles and followed by a last incubation of 8 min at 65 °C. PCR products were loaded onto 1% agarose gels stained with ethidium bromide and photographed. Isolates sharing the same rep‐PCR pattern were treated as clones. Only one representative of each clone was sequenced.
CYP51 was sequenced as previously described (Zhan et al., 2006). Sequence St1 (GenBank accession no. AY730587) was used to design primers for gene amplification and sequencing. The primers used to amplify the CYP51 gene were EBI112 (5′‐TTCAGCACGCTCGCCATCCTCC‐3′, F) and CypFra2r (5′‐TCCTTCTCCTCCCTCCTCTC‐3′, R) (Microsynth, Switzerland), generating a 1794‐bp fragment. Purified PCR products were sequenced with the BigDye Terminator v3.0 Cycle Sequencing kit (Applied Biosystems, Foster City, CA) according to the manufacturer's instructions. The primers used for sequencing were CypFra1f (5′‐CATATGATGATTGCGCTGCT‐3′, F), CypFra1r (5′‐CGGCTGAACAAACTGCTGTA‐3′, R), Cyp380 (5′‐GAGATATACAGCCCGCTGAC‐3′, F) and Cyp1337 (5′‐CGAGAGGTTGGCGTATGTGAG‐3′, R). The sequencing products were purified on Sephadex G50 superfine DNA‐grade resin (Amersham, Switzerland) and loaded into the ABI PRISM R 3100 Genetic analyser under standard conditions. Sequences were aligned and edited manually using Sequencher 4.5 (Gene Code Corp., Ann Arbor, MI).
Genetic analyses
Population‐genetic analyses of the sequence data were performed using the program DNASP v. 4.10 (Rozas et al., 2003). Evidence for intragenic recombination within CYP51 was tested in three ways. (1) Neutral coalescent simulations (excluding recombinant haplotypes) were used to test for selective neutrality and demographic equilibrium using DNASP. These simulations embody the ‘haplotype number test’ (K test; Depaulis and Veuille, 1998) and the coalescent‐based expectations of K (Ke) were estimated under the assumption that recombination had not occurred in the history of the sample. Simulations were based on segregating sites and 100 000 replicates. (2) A phylogeny‐based differences in the sum of squares (DSS) approach implemented in TOPALI v2 (Milne et al., 2004) was used to detect intragenic recombination breakpoints in multiple sequence alignments. Briefly, the program uses a sliding window and compares the phylogeny on both sides of the window using the least squares method. DSS values are then plotted against the centre of the corresponding windows with larger peaks marking the intragenic recombination breakpoints. (3) Recombination within a segment flanked by a pair of polymorphic nucleotide sites is suggested when all four possible combinations are observed (‘four‐gamete test’; Hudson and Kaplan, 1985). This presence of four‐gamete pairs entails a considerable level of homoplasy (i.e. genetic variability not originated by descent) that can be depicted as ‘reticulations’ in a median network (Bandelt et al., 1999; http://www.fluxus‐engineering.com/sharenet.htm).
We applied ML and MP analyses using the computer program PAUP* 4.0b10 (Swofford, 2002) to assess phylogenetic relationships among haplotypes. The hierarchical likelihood‐ratio test (LRT) implemented in MODELTEST 3.06 (Posada and Crandall, 1998) was used to determine the substitution model that best fits the data set for the ML analysis. The MP analysis using all equal weights was performed under the heuristic search option (50 replicate searches with random addition of taxa). A bootstrap analysis was performed to test for statistical significance of the trees generated with 500 pseudoreplicates and under the fast stepwise addition option for MP. Finally, migration among recent European populations (excluding recombinant haplotypes) was estimated with MIGRATE version 1.7.3 (Beerli and Felsenstein, 2001). MIGRATE is based on the coalescent theory and uses an ML approach to estimate asymmetrical gene flow among populations. Following the authors’ recommendations, we made a first MIGRATE run with the default values using F ST to find the start parameters. Parameters obtained from this initial run were used to re‐run the program with the following Markov chain settings: short chains ten, long chains five, averaging over five independent replicates with different random number seeds and a four‐chain heating scheme.
Supporting information
Supporting info item
ACKNOWLEDGEMENTS
We are most grateful to Andreas von Tiedemann and Christine Schäfer (Georg‐August‐University of Göttingen) for generously providing isolates from recent German populations. This study was funded by the ETH Zurich.
REFERENCES
- Bandelt, H.‐J. , Forster, P. and Röhl, A. (1999) Median‐joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48. [DOI] [PubMed] [Google Scholar]
- Banke, S. , Peschon, A. and McDonald, B.A. (2004) Phylogenetic analysis of globally distributed Mycosphaerella graminicola populations based on three DNA sequence loci. Fungal Genet. Biol. 41, 226–238. [DOI] [PubMed] [Google Scholar]
- Beerli, P. and Felsenstein, J. (2001) Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc. Natl Acad. Sci. USA 98, 4563–4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, R.‐S. and McDonald, B.A. (1996) Sexual reproduction plays a major role in the genetic structure of populations of the fungus Mycosphaerella graminicola . Genetics, 142, 1119–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement, M. , Posada, D. and Crandall, K.A. (2000) TCS: a computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1659. [DOI] [PubMed] [Google Scholar]
- Cools, H.J. , Fraaije, B.A. and Lucas, J.A. (2005a) Molecular examination of Septoria tritici isolates with reduced sensitivities to triazoles In: Modern Fungicides and Antifungal Compounds IV (Dehne H.W., Gisi U., Kuck K.H., Russell P.E. and Lyr H., eds), pp. 103–114. Alton, UK: BCPC. [Google Scholar]
- Cools, H.J. , Fraaije, B.A. and Lucas, J.A. (2005b) Molecular mechanisms correlated with changes in triazole sensitivity in isolates of Mycosphaerella graminicola . Proceedings of the BCPC Congress, Crop Science and Technology 2005, 1, 267–274. [Google Scholar]
- De Waard, M.A. (1994) Resistance to fungicides which inhibit sterol 14‐(‐demethilation, an historical perspective In: Fungicide Resistance (Heaney S., Slawson D., Hollomon D.W., Smith M., Russell P.E. and Parry D.W., eds), pp. 3–10. Alton, UK: BCPC. [Google Scholar]
- Depaulis, F. and Veuille, M. (1998) Neutrality tests based on the distribution of haplotypes under an infinite‐site model. Mol. Biol. Evol. 15, 1788–1790. [DOI] [PubMed] [Google Scholar]
- Eyal, Z. (1981) Integrated control of Septoria diseases of wheat. Plant Dis. 65, 763–768. [Google Scholar]
- Fraaije, B.A. , Cools, H.J. , Fountaine, J. , Lovell, D.J. , Motteram, J. , West, J.S. and Lucas, J.A. (2005) Role of ascospores in further spread of QoI‐resistant cytochrome b alleles (G143A) in field populations of Mycosphaerella graminicola . Phytopathology, 95, 933–941. [DOI] [PubMed] [Google Scholar]
- Fraaije, B.A. , Cools, H.J. , Kim, S.‐H. , Motteram, J. , Clark, W.S. and Lucas, J.A. (2007) A novel substitution I381V in the sterol 14α‐demethylase (CYP51) of Mycosphaerella graminicola is differentially selected by azole fungicides. Mol. Plant Pathol. 8, 245–254. [DOI] [PubMed] [Google Scholar]
- Gisi, U. , Hermann, D. , Ohl, L. and Steden, C. (1997) Sensitivity profiles of Mycosphaerella graminicola and Phytophthora infestans populations to different classes of fungicides. Pestic. Sci. 51, 290–298. [Google Scholar]
- Gisi, U. , Pavic, L. , Stanger, C. , Hugelshofer, U. and Sierotzki, H. (2005) Dynamics of Mycosphaerella graminicola populations in response to selection by different fungicides In: Modern Fungicides and Antifungal Compounds IV (Dehne H.W., Gisi U., Kuck K.H., Russell P.E. and Lyr H., eds), pp. 89–101. Alton, UK: BCPC. [Google Scholar]
- Hudson, R.R. and Kaplan, N.L. (1985) Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics, 111, 147–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroux, P. , Gredt, M. , Walker, A.S. , Moinard, J.M. and Caron, D. (2005) Resistance of the wheat leaf blotch pathogen Septoria tritici to fungicides in France In: Modern Fungicides and Antifungal Compounds IV (Dehne H.W., Gisi U., Kuck K.H., Russell P.E. and Lyr H., eds), pp. 115–124. Alton, UK: BCPC. [Google Scholar]
- Leroux, P. , Albertini, C. , Gautier, A. , Gredt, M. and Walker, A.‐S. (2007) Mutations in the CYP51 gene correlated with changes in sensitivity to sterol 14(‐demethylation inhibitors in field isolates of Mycosphaerella graminicola . Pest Manag. Sci. 63, 688–698. [DOI] [PubMed] [Google Scholar]
- Levin, B.R. , Perrot, V. and Walker, N. (2000) Compensatory mutations, antibiotic resistance and the population genetics of adaptive evolution in bacteria. Genetics, 154, 985–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linde, C.C. , Zhan, J. and McDonald, B.A. (2002) Population structure of Mycosphaerella graminicola: from lesions to continents. Phytopathology, 92, 946–955. [DOI] [PubMed] [Google Scholar]
- Ma, Z.H. and Michailides, T.J. (2005) Advances in understanding molecular mechanisms of fungicide resistance and molecular detection of resistant genotypes in phytopathogenic fungi. Crop Prot. 24, 853–863. [Google Scholar]
- Mavroeidi, V.I. and Shaw, M.W. (2005) Sensitivity distributions and cross‐resistance patterns of Mycosphaerella graminicola to fluquinconazole, prochloraz and azoxystrobin over a period of 9 years. Crop Prot. 24, 259–266. [Google Scholar]
- McDonald, B.A. , Miles, J. , Nelson, L.R. and Pettway, R.E. (1994) Genetic variability in nuclear‐DNA in field populations of Stagonospora nodorum . Phytopathology, 84, 250–255. [Google Scholar]
- McDonald, B.A. , Zhan, J. , Yarden, O. , Hogan, K. , Garton, J. and Pettway, R.E. (1999) The population genetics of Mycosphaerella graminicola and Stagonospora nodorum In: Septoria on Cereals: a Study of Pathosystems (Lucas J.A., Bowyer P. and Anderson H.M., eds), pp. 44–69. Wallingford, UK: CABI Publishing. [Google Scholar]
- Milne, I. , Wright, F. , Rowe, G. , Marshal, D.F. , Husmeier, D. and McGuire, G. (2004) TOPALI: software for automatic identification for recombinant sequences within DNA multiple alignments. Bioinformatics, 20, 1806–1807. [DOI] [PubMed] [Google Scholar]
- Nijhuis, M. , Schuurman, R. , De Jong, D. Erickson, J. , Gustchina, E. , Albert, J. , Schipper, P. , Gulnik, S. and Boucher, C.A.B. (1999) Increased fitness of drug resistant HIV‐1 protease as a result of acquisition of compensatory mutations during suboptimal therapy. AIDS, 13, 2349–2359. [DOI] [PubMed] [Google Scholar]
- Posada, D. and Crandall, K.A. (1998) Modeltest: testing the model of DNA substitution. Bioinformatics, 14, 817–818. [DOI] [PubMed] [Google Scholar]
- Rozas, J. , Sanches‐DelBarrio, J.C. , Messeguer, X. and Rozas, R. (2003) DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics, 19, 2496–2497. [DOI] [PubMed] [Google Scholar]
- Stergiopoulos, I. , Van Nistelrooy, J.G.M. , Kema, G.H.J. and De Waard, M.A. (2003) Multiple mechanisms account for variation in base‐line sensitivity to azole fungicides in field isolates of Mycosphaerella graminicola . Pest Manag. Sci. 59, 1333–1343. [DOI] [PubMed] [Google Scholar]
- Swofford, D.L. (2002) PAUP*. Phylogenetic Analysis Using Parsimony (and other methods), Version 4. Sunderland, MA: Sinauer Associates. [Google Scholar]
- Versalovic, J. , Koeuth, T. and Lupski, J.R. (1991) Distribution of repetitive DNA‐sequences in Eubacteria and application to fingerprinting of bacterial genomes. Nucleic Acids Res. 19, 6823–6831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versalovic, J. , Schneider, M. , De Bruijn, F.J. and Lupski, J.R. (1994) Genomic fingerprinting of bacteria using repetitive sequence‐based polymerase chain reaction. Method. Mol. Cell. Biol. 5, 25–40. [Google Scholar]
- Zhan, J. , Pettway, R.E. and McDonald, B.A. (2003) The global genetic structure of the wheat pathogen Mycosphaerella graminicola is characterized by high nuclear diversity, low mitochondrial diversity, regular recombination, and gene flow. Fungal Genet. Biol. 38, 286–297. [DOI] [PubMed] [Google Scholar]
- Zhan, J. , Linde, C.C. , Jürgens, T. , Merz, U. , Steinebrunner, F. and McDonald, B.A. (2005) Variation for neutral markers is correlated with variation for quantitative traits in the plant pathogenic fungus Mycosphaerella graminicola . Mol. Ecol. 14, 2683–2693. [DOI] [PubMed] [Google Scholar]
- Zhan, J. , Stefanato, F.L. and McDonald, B.A. (2006) Selection for increased cyproconazole tolerance in Mycosphaerella graminicola through local adaptation and in response to host resistance. Mol. Plant Pathol. 7, 259–268. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item