

ABSTRACT
Background
Hypertrophic cardiomyopathy (HCM) is a common genetic heart disease characterized by ventricular hypertrophy, myocardial fibrosis, and impaired ventricular relaxation. The exact mechanisms by which fibrosis is caused remain unknown.
Hypothesis
Circulating TGF‐β is related to poor prognosis in HCM.
Methods
We compared TGF‐β levels of 49 HCM patients with those of 40 non‐HCM patients. We followed the patients with HCM for 18 months and divided them into 2 groups: low TGF‐β (≤4877 pg/mL) and high TGF‐β (>4877 pg/mL). We compared the 2 groups in terms of brain natriuretic peptide (BNP), echocardiographic parameters, and clinical outcomes including myocardial infarction, arrhythmias, implantable cardioverter‐defibrillator implantation, hospitalization, New York Heart Association (NYHA) class, acute heart failure, and mortality.
Results
The HCM patients had higher TGF‐β levels than those in the control group (P = 0.005). In the follow‐up, those in the high TGF‐β group had higher BNP levels, larger left‐atrial size, thicker interventricular septum, NYHA class, more hospitalizations, and a greater number of clinical adverse events (P < 0.001, P = 0.01, P < 0.001, P = 0.002, P < 0.001 and P = 0.003, respectively). TGF‐β level of >4877 pg/mL can predict adverse events with a specificity of 75% and a sensitivity of 72% (P = 0.014). In multivariate regression analysis, TGF‐β, BNP, and interventricular septum thickness were significantly associated with adverse events (P = 0.028, P = 0.030, and P = 0.034, respectively).
Conclusions
The TGF‐β level is higher in HCM patients and associated with a poor prognosis in HCM.
Introduction
Hypertrophic cardiomyopathy (HCM) is a genetic disorder of the myocardium due to genetic mutations in sarcomere protein genes and is associated with hypertrophy, heart failure, arrhythmias, and sudden cardiac death.1, 2, 3, 4, 5 Myocardial fibrosis occurs as a maladaptive reaction against impaired cardiac relaxation.6, 7, 8 The exact mechanisms by which fibrosis is caused remain unknown. Transforming growth factor‐β (TGF‐β) signaling may be a key mechanism for cardiac fibrosis.9 TGF‐β is one of the major profibrotic cytokines directing fibrosis. In some diseases, such as myocardial infarction (MI), idiopathic pulmonary fibrosis, hepatitis, and chronic kidney disease, TGF‐β plays a pivotal role in uncontrolled fibrosis.10, 11, 12
Myocardial TGF‐β expression is upregulated in experimental models of MI and cardiac hypertrophy and in patients with dilated and hypertrophic cardiomyopathy.9, 13, 14, 15, 16 TGF‐β effects on myocytes and mesenchymal and immune cells in myocardium lead to hypertrophic remodeling and cardiac fibrosis and regulate the matrix metabolism in the overloaded heart via Smad3‐dependent pathways.17 Because of its crucial role in cardiac remodeling, the TGF‐β signaling system may be a therapeutic target for patients with HCM.
There are much data from various experimental and animal studies about the relationship between HCM and TGF‐β, but clinical studies exploring this relationship are scant in the literature.17, 18 Although there are no data to show that high TGF‐β levels can predict clinical outcomes in patients with HCM, the arguments about treatment of HCM focus on reducing increased TGF‐β signaling.
In our study, we compared the levels of TGF‐β between HCM and non‐HCM patients and investigated whether a higher TGF‐β level was a risk factor for adverse cardiac events in HCM patients.
Methods
Between January 2012 and March 2013, this prospective study included patients admitted or referred to our cardiology outpatient clinic and diagnosed as HCM and non‐HCM patients, alongside an age‐ and sex‐matched control group of individuals with no previous cardiac disease. Hypertrophic cardiomyopathy was diagnosed in patients in whom the thickness of the interventricular septum (IVS) was >1.5 cm in the echocardiographic examination and who did not have any other illnesses or causes of IVS thickening, such as aortic stenosis and insufficiency, uncontrolled hypertension, or hypertension grade ≥2. Including the patients with both HCM and hypertension was critical; however, an IVS >1.5 cm is uncommon, even in the patients with uncontrolled hypertension.19 The European Society of Cardiology defines HCM by a wall thickness of ≥15 mm in ≥1 myocardial segments, as measured by any imaging technique (echocardiography, magnetic resonance imaging, or computed tomography), that is not explained solely by loading conditions in the HCM guideline.20
Exclusion criteria were hypertension with grade ≥2 or causing end‐organ damage, or uncontrolled or controlled with multiple drugs; acute inflammation; presence of chronic liver or kidney failure; hematological diseases; and use of angiotensin‐converting enzyme inhibitors. Patients taking angiotensin‐converting enzyme inhibitors, angiotensin II type 1 receptor blockers, or aldosterone inhibitors before being diagnosed with HCM also were not included.
Peripheral blood samples were obtained in the early morning from all subjects by venipuncture of an upper limb. Serum TGF‐β1 levels were measured by a quantitative enzyme‐linked immunosorbent assay (ELISA) technique using a specific TGF‐β1 kit (Human TGF‐β CytoSet; BioSource, Camarillo, CA) according to the manufacturer's instructions. The calibrator consisted of recombinant human TGF‐β1. All samples were measured in duplicate, and respective mean values were calculated. The limit detection of the assay was 30 pg/mL, and the intra‐assay and interassay coefficients of variability were 2.8% and 12.5%, respectively.
We followed the patients with HCM for 18 months. Resting and Valsalva echocardiographic examination and stress testing were done at initial application for all patients; physical examination and electrocardiogram were done every 3 months for asymptomatic patients. Patients with symptoms such as chest pain, dyspnea, syncope, or palpitation were examined with echocardiography and a 24‐hour ambulatory electrocardiogram. In addition, if patients with symptoms had a left ventricular outflow track (LVOT) peak gradient of <50 mm Hg in resting, stress echocardiographic examination was done to evaluate provoked LVOT gradient. Then, patient medications were regulated or the patients were hospitalized. The patients with acute heart failure, with arrhythmias impairing hemodynamic status, and who needed implantation of an implantable cardioverter‐defibrillator (ICD) were hospitalized. Implantable cardioverter‐defibrillators were implanted in the patients that had survived a cardiac arrest due to ventricular fibrillation or ventricular tachycardia and sustained ventricular tachycardia causing syncope or hemodynamic compromise.
In the follow‐up, we recorded mortality and those patients presenting with acute heart failure rates, symptomatic status in New York Heart Association (NYHA) class, MI, stroke, hospitalization rate, and arrhythmic events including paroxysmal and persistent atrial fibrillation (AF), and sustained and nonsustained ventricular tachycardia. We accepted MI, acute heart failure, and ventricular arrhythmic events requiring ICD implantation and cardiac mortality as cardiac adverse events.
The study was performed according to the recommendations of the Declaration of Helsinki on Biomedical Research Involving Human Subjects. Approval of the institutional ethics committee was received, and a signed document from each patient was taken for the study.
Statistical Analysis
All analyses were performed using SPSS version 20.0 (IBM Corp., Armonk, NY) and GPower (Erdfelder, Faul, & Buchner, 1996). Comparison of parametric values between the 2 groups was performed by means of an independent samples t test. Comparisons of nonparametric values between the 2 groups were performed by Mann–Whitney U test. Categorical variables were compared by the χ2 test. In addition, we used an analysis of covariance (ANCOVA) model putting the TGF‐β level as a dependent variable, the presence of HCM as a fixed factor, and age, sex, body mass index (BMI), presence of hypertension, diabetes mellitus (DM), smoking, brain natriuretic peptide (BNP) level, and creatinine (Cr) level as covariates. Logistic regression analysis was used to assess predictors of HCM. Those variables with P < 0.1 by univariate analysis were included in the backward stepwise multivariate logistic regression analysis model, and the respective odds ratios with 95% confidence intervals were calculated. When a significant cutoff value was observed, the sensitivity and specificity values were presented. A Kaplan‐Meier curve was generated to examine the difference in the cardiac adverse event rates in high and low TGF‐β subgroups. The capacity of TGF‐β in predicting adverse events in HCM patients was analyzed using receiver operating characteristic (ROC) curve analyses. A post hoc power analysis was conducted with the sample size of 89, using mean and SDs of the study groups and an α level set to P < 0.05. The effect size of the study population was 0.61 (moderate to large size effect) and the power of the study was 0.81. A 2‐tailed P value of <0.05 was considered statistically significant.
Results
Our study included 49 patients (mean age, 38 ± 22 years; male 57%) as an HCM group and 40 patients (mean age, 36 ± 16 years; male 60%) as the control group. Baseline clinical characteristics including age, sex, smoking, hypertension, DM, and BMI of each group were similar except for hyperlipidemia (all P < 0.05; Table 1). The TGF‐β and BNP levels of HCM patients were significantly higher than those of patients in the control group (P = 0.005 and P = 0.02, respectively; Table 1). There was no difference in other biochemical and hematological parameters between the 2 groups (all P > 0.05; Table 1).
Table 1.
Comparison of Patients With HCM and Control Groups in Terms of TFG‐β and BNP Levels and Baseline Clinical, Biochemical, and Hematological Characteristics
| HCM, n = 49 | Control, n = 40 | P Value | |
|---|---|---|---|
| Age, y | 38 ± 22 | 36 ± 16 | 0.07 |
| Male sex | 28 (57) | 24 (60) | 0.6 |
| Hypertension | 12 (24) | 10 (25) | 0.8 |
| DM | 13 (26) | 11 (27) | 0.7 |
| Hyperlipidemia | 15 (30) | 9 (22) | 0.04 |
| Family history | 12 (24) | 0 (0) | — |
| Smoking | 10 (20) | 11 (27) | 0.06 |
| BMI, kg/m2 | 30 ± 4 | 29 ± 3 | 0.5 |
| Fasting glucose, mg/dL | 99 ± 20 | 101 ± 18 | 0.5 |
| LDL‐C, mg/dL | 122 ± 22 | 131 ± 13 | 0.07 |
| HDL‐C, mg/dL | 47 ± 8 | 49 ± 6 | 0.2 |
| TG, mg/dL | 140 ± 42 | 130 ± 53 | 0.09 |
| TGF‐β, pg/mL | 4240 ± 3413 | 2504 ± 2113 | 0.005 |
| BNP, pg/mL | 119 ± 44.3 | 5.8 ± 3.1 | <0.001 |
| Urea, mg/dL | 35.4 ± 12 | 33.2 ± 13 | 0.4 |
| Cr, mg/dL | 0.9 ± 0.2 | 0.8 ± 0.2 | 0.8 |
| HbA1c, % | 5.7 ± 1.5 | 5.6 ± 1.2 | 0.6 |
| AST, U/L | 22 ± 14 | 24 ± 11 | 0.1 |
| ALT, U/L | 25 ± 12 | 23 ± 8 | 0.5 |
| Hg, g/dL | 13 ± 2 | 13 ± 2 | 0.9 |
| WBC, ×103/mm3 | 5.3 ± 1.3 | 5.5 ± 0.9 | 0.3 |
| Platelet count, ×103/mm3 | 132 ± 24 | 142 ± 28 | 0.2 |
Abbreviations: ALT, alanine transaminase; AST, aspartate transaminase; BMI, body mass index; BNP, brain natriuretic peptide; Cr, creatinine; DM, diabetes mellitus; HbA1c, glycated hemoglobin; HCM, hypertrophic cardiomyopathy; HDL‐C, high‐density lipoprotein cholesterol; Hg, hemoglobin; LDL‐C, low‐density lipoprotein cholesterol; SD, standard deviation; TG, triglycerides; TGF‐β, transforming growth factor‐β; WBC, white blood cell count.
Data are expressed as mean ± SD for normally distributed data and n (%) for categorical variables.
In ANCOVA analysis, TGF‐β levels were significantly higher in the HCM group compared with the control group, even when including age, sex, BMI, presence of hypertension, DM, smoking, BNP level, and Cr level as covariates (P = 0.01). We used the ROC curve analysis to find a cutoff value for predicting adverse events. We found that TGF‐β levels of >4877 pg/mL predicted adverse events in HCM patients with a specificity of 75% and a sensitivity of 72% (area under curve: 0.727, 95% confidence interval: 0.567‐0.887, P = 0.014; Figure 1).
Figure 1.
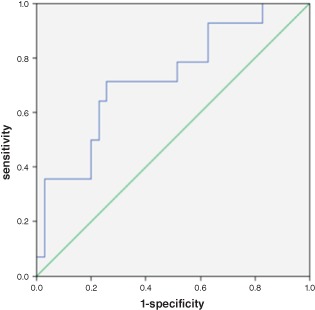
In the ROC curve analysis, TGF‐β levels >4877 pg/mL had a sensitivity of 71% and specificity of 75% in predicting adverse events in HCM patients (area under curve: 0.727, 95% CI: 0.567–0.887, P = 0.014). Abbreviations: CI, confidence interval; HCM, hypertrophic cardiomyopathy; ROC, receiver operating characteristic; TGF‐β, transforming growth factor‐β.
We divided the patients according to the cutoff value into 2 groups, high TGF‐β (>4877 pg/mL; n = 19) and low TGF‐β (≤4877 pg/mL; n = 30). In baseline clinical characteristics, patients with high TGF‐β were younger and had higher hypertension than did patients with low TGF‐β (P = 0.04 and P = 0.01, respectively; Table 2). The 2 groups were similarly matched according to sex, smoking, hyperlipidemia, DM, BMI, and family history (all P < 0.05; Table 2).
Table 2.
Comparison of HCM Patients With High and Low TGF‐β Levels in Terms of Clinical, Echocardiographic, Biochemical, and Hematological Characteristics
| High TGF‐β, n = 19 | Low TGF‐β, n = 30 | P Value | |
|---|---|---|---|
| Age, y | 34 ± 11 | 41 ± 13 | 0.04 |
| Male sex | 11 (57) | 17 (56) | 0.3 |
| Hypertension | 7 (36) | 5 (16) | 0.01 |
| DM | 10 (52) | 16 (53) | 0.2 |
| Hyperlipidemia | 11 (57) | 19 (63) | 0.6 |
| Family history | 5 (26) | 7 (23) | 0.1 |
| Smoking | 4 (21) | 6 (20) | 0.5 |
| BMI, kg/m2 | 31 ± 4 | 28 ± 3 | 0.07 |
| LAD, cm | 4.3 ± 1.2 | 3.9 ± 2.1 | 0.01 |
| LVDD, cm | 5.0 ± 1.2 | 5.1 ± 1.1 | 0.1 |
| LVSD, cm | 3.7 ± 0.8 | 3.8 ± 0.6 | 0.1 |
| IVS, cm | 2.9 ± 0.2 | 2.3 ± 0.4 | <0.001 |
| PW, cm | 2.1 ± 0.1 | 2.0 ± 0.3 | 0.9 |
| LVEF, % | 62 ± 5 | 60 ± 6 | 0.8 |
| Obstructive type | 12 (63) | 13 (43) | 0.3 |
| LVOT peak gradient, mm Hg | 39 ± 19 | 35 ± 33 | 0.4 |
| AF | 6 (31) | 8 (26) | 0.7 |
| Nonsustained VT | 6 (31) | 7 (23) | 0.5 |
| Sustained VT | 2 (10) | 1 (3) | 0.4 |
| ICD implantation | 5 (26) | 3 (10) | 0.2 |
| Acute MI | 1 (5) | 1 (3) | 0.4 |
| NYHA class >1 | 15 (78) | 12 (40) | 0.003 |
| NYHA class | 1.9 ± 0.6 | 1.4 ± 0.9 | 0.002 |
| Acute HF | 5 (26) | 4 (13) | 0.2 |
| Hospitalization | 14 (73) | 12 (40) | <0.001 |
| Mortality | 4 (21) | 2 (6) | 0.1 |
| Adverse eventsa | 10 (52) | 4 (13) | 0.003 |
| Fasting glucose, mg/dL | 99 ± 21 | 98 ± 18 | 0.8 |
| HbA1c, % | 5.5 ± 0.5 | 5.8 ± 0.7 | 0.1 |
| HDL‐C, mg/dL | 44 ± 9 | 49 ± 11 | 0.4 |
| LDL‐C, mg/dL | 129 ± 18 | 125 ± 21 | 0.8 |
| TG, mg/dL | 138 ± 32 | 144 ± 48 | 0.3 |
| TGF‐β, pg/mL | 1869 ± 1392 | 7150 ± 2850 | <0.001 |
| BNP, pg/mL | 149 ± 59 | 28 ± 20 | <0.001 |
| Urea, mg/dL | 34 ± 15 | 36 ± 8 | 0.5 |
| Cr, mg/dL | 0.9 ± 0.2 | 0.95 ± 0.2 | 0.3 |
| AST, U/L | 24 ± 16 | 20 ± 12 | 0.7 |
| ALT, U/L | 28 ± 14 | 22 ± 16 | 0.6 |
| Hg, g/dL | 13 ± 1 | 13 ± 2 | 0.8 |
| WBC, ×103/mm3 | 5.5 ± 1.6 | 4.9 ± 1.8 | 0.1 |
| Platelet count, ×103/mm3 | 138 ± 34 | 130 ± 29 | 0.3 |
| Medications | |||
| Warfarin | 5 (26) | 8 (26) | 0.6 |
| Cordarone | 3 (15) | 2 (6) | 0.07 |
| ACEI | 13 (68) | 18 (60) | 0.2 |
| ARB | 5 (26) | 10 (33) | 0.4 |
| β‐Blocker | 15 (78) | 25 (83) | 0.5 |
| CCB | 4 (21) | 5 (16) | 0.3 |
Abbreviations: ACEI, angiotensin‐converting enzyme inhibitor; AF, atrial fibrillation; ALT, alanine transaminase; ARB, angiotensin receptor blocker; AST, aspartate transaminase; BMI, body mass index; BNP, brain natriuretic peptide; CCB, calcium channel blocker; Cr, creatinine; DM, diabetes mellitus; HbA1c, glycated hemoglobin; HCM, hypertrophic cardiomyopathy; HDL‐C, high‐density lipoprotein cholesterol; HF, heart failure; Hg, hemoglobin; ICD, implantable cardioverter‐defibrillator; IVS, interventricular septum; LAD, left atrial diameter, LDL‐C, low‐density lipoprotein cholesterol; LVDD, left ventricular diastolic diameter; LVEF, left ventricular ejection fraction; LVOT, left ventricular outflow tract; LVSD, left ventricular systolic diameter; MI, myocardial infarction; NYHA, New York Heart Association; PW, posterior wall; SD, standard deviation; TG, triglycerides; TGF‐β, transforming growth factor‐β; WBC, white blood cell count.
Data are expressed as mean ± SD for normally distributed data and n (%) for categorical variables.
Acute HF, acute MI, ventricular arrhythmic events requiring ICD implantation, and cardiac mortality.
Table 3.
Multivariate Regression Analysis of TGF‐β, BNP, LVOT Gradient, Cr, and Baseline Clinical Characteristics of HCM Patients in Predicting Adverse Events
| Variable | OR (95% CI) | P Value |
|---|---|---|
| TGF‐β | 1.312 (1.001‐1.333) | 0.028 |
| BNP | 1.223 (1.010‐1.055) | 0.030 |
| IVS diameter | 1.202 (1.111‐1.302) | 0.032 |
| LVOT gradient | 2.786 (0.459‐17.33) | 0.089 |
| Age | 0.988 (0.786‐1.550) | 0.733 |
| Male sex | 0.320 (0.009‐1.454) | 0.934 |
| Hypertension | 0.344 (0.013‐9.789) | 0.354 |
| DM | 1.222 (0.218‐1.665) | 0.089 |
| Hyperlipidemia | 0.431 (0.021‐5.782) | 0.590 |
| Family history | 3.450 (0.136‐13.30) | 0.345 |
| Smoking | 1.597 (0.391‐2.001) | 0.132 |
| BMI | 0.323 (0.300‐1.889) | 0.388 |
Abbreviations: BMI, body mass index; BNP, brain natriuretic peptide; CI, confidence interval; Cr, creatinine; DM, diabetes mellitus; HCM, hypertrophic cardiomyopathy; IVS, interventricular septum; LVOT, left ventricular outflow tract; OR, odds ratio; TGF‐β, transforming growth factor‐β.
Regarding echocardiographic parameters, there was no difference in the left ventricular systolic and diastolic diameters, posterior wall thickness, LVOT peak gradient, and left ventricular ejection fraction (all P < 0.05; Table 2). Patients with high TGF‐β had a larger left atrium and thicker IVS (P = 0.01 and P < 0.001, respectively; Table 2). In addition, the patients with high TGF‐β had a more obstructive type of HCM, but there was no statistical difference (Table 2). The total number of patients with obstructive HCM was 25, and they had a mean TGF‐β level of 4597 pg/mL, but patients with nonobstructive HCM had a mean TGF‐β level of 3869 pg/mL (P = 0.04).
Patients with high TGF‐β had higher BNP (P < 0.001), and there was no difference in other biochemical or hematological parameters between the 2 groups (all P > 0.05; Table 2).
Considering clinical events during 18 months of follow‐up, patients with high TGF‐β were in a higher NYHA class, and hospitalization and adverse‐event rates were also higher (P = 0.002, P < 0.001, and P = 0.003, respectively; Table 2). There was no difference in MI, AF, ventricular arrhythmias, acute heart failure, ICD implantation, and mortality between the 2 groups (Table 2). In addition, there were no patients with stroke in the follow‐up. In the Kaplan‐Meier curve, the rate of cardiac adverse events was significantly higher in the high TGF‐β subgroup compared with the low TGF‐β subgroup (log‐rank P < 0.01; Figure 2). In multivariate regression analysis, TGF‐β, BNP, and IVS thickness were significantly associated with adverse events (P = 0.028, P = 0.030, and P = 0.034, respectively).
Figure 2.
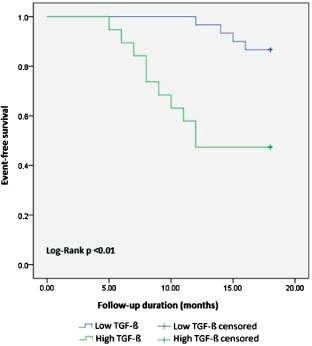
In the Kaplan‐Meier curve, the rate of cardiac adverse events was significantly higher in the high TGF‐β subgroup compared with the low TGF‐β subgroup (log‐rank P < 0.01). Abbreviations: TGF‐β, transforming growth factor‐β.
Discussion
Our study showed that HCM patients have significantly increased TGF‐β levels. Increased TGF‐β levels are correlated with worse echocardiographic parameters, including the thickness of the septum and left‐atrial size. In addition, increased TGF‐β levels were related to the obstructive type of HCM. The patients with obstructive HCM had significantly higher TGF‐β levels. Increased TGF‐β levels correlated with cardiac adverse events including mortality, acute heart failure, and life‐threatening ventricular arrhythmias over the follow‐up of 18 months. In addition, the patients with high TGF‐β levels had worse clinical status as an NYHA class, higher BNP levels, and higher hospitalization rates. To our knowledge, there is no clinical study presenting the association between TGF‐β and poor clinical events in HCM in the literature. In addition, the TGF‐β levels of >4877 pg/mL predicted a poor prognosis in HCM with a specificity of 75% and a sensitivity of 72%. In multivariate regression analysis, TGF‐β was an independent risk factor for adverse clinic events as much as BNP and IVS thickness. There are many experimental animal studies examining the relationship between cardiac hypertrophy and fibrosis and TGF‐β signaling9, 13, 15 and some human studies showing only increased TGF‐β signaling in HCM.15, 21 Li et al found that TGF‐β increased gene expression with the use of multiplex reverse transcription–polymerase chain reaction in 8 HCM patients.15 In another study, Li et al found that TGF‐β messenger RNA and protein levels were higher in the hypertrophic septum than in the nonhypertrophic region in biopsy specimens of 8 HCM patients.21 The association between TGF‐β and ventricular hypertrophy was shown in patients with hypertension, and TGF‐β levels also correlated with the amount of ventricular mass.22 In aortic stenosis, TGF‐β levels were found to be related to increased aortic gradient and hypertrophy.23 However, in another study, it was demonstrated that TGF‐β protein and receptor levels were higher in HCM patients than in patients with aortic stenosis, stable angina, and transplanted heart.16
Mutations in the myosin heavy chain gene impair the structure of sarcomere, altering calcium signaling in myocytes, and thus resulting in deterioration of cardiac relaxation.24, 25 The hypertrophy develops against the impairment of relaxation as a maladaptive mechanism. Owing to hypertrophy, restricted coronary flow, and microvascular dysfunction, increased metabolic demand and reactive oxygen species due to abnormal physiology of mutant sarcomeres contribute to the myocardial fibrosis.26, 27 The degree of hypertrophy, the systolic and diastolic ventricular functions, and arrhythmic events are significantly associated with amounts of myocardial fibrosis.28, 29, 30, 31, 32
Moreover, premature death of mutant myocytes, expansion of interstitial matrix, and pathologic changes in myocytes affecting nonmyocyte cells such as mesenchymal cells and fibroblasts cause proliferation of nonmyocyte cells and expression of profibrotic molecules that substantially contribute to fibrosis.9 The exact mechanism expanding the extracellular matrix is still unknown. TGF‐β is a critical molecule for nonmyocyte activation and expansion of the extracellular matrix because it stimulates the cardiac microvascular endothelial cells of myocardium to transform into mesenchymal cells and mesenchymal cells to migrate into the myocardium.7, 17 Whether myocytes or nonmyocyte cells are the origin of increased TGF‐β in HCM is not clearly known. Teekakirikul et al demonstrated a 3‐fold to 4‐fold increase in nonmyocyte proliferation in the hypertrophic regions of mice myocardium, so nonmyocyte proliferation and TGF‐β signaling associated with it constituted together a pivotal mechanism for the fibrotic process.9 In their study, they suggested that early inhibition of TGF‐β signaling should be a therapeutic strategy to prevent fibrosis in HCM.
In previous studies, losartan, an angiotensin 2 type 1 receptor inhibitor, could limit TGF‐β activation and reduce levels of TGF‐β in Marfan syndrome and hypertensive patients.33, 34 Teekakirikul et al observed that losartan reduced cardiac fibrosis and attenuated nonmyocyte proliferation in pre‐HCM mice. In histopathologic sections, there was minimal fibrosis in losartan‐treated mice in comparison with nontreated mice. Losartan failed to reverse hypertrophy and fibrosis but diminished nonmyocyte proliferation in established HCM mice.9
Moreover, TGF‐β signaling is currently under investigation in a wide range of cardiac diseases, including bicuspid aorta, thoracic aorta aneurysm and dissection, MI, heart failure, AF, and dilated cardiomyopathy.35, 36, 37, 38 Based on extensive evidence supporting a substantial role for TGF‐β in cardiac remodeling, TGF‐β signaling inhibition in the fibrotic pathway is a promising treatment in the future in many cardiac diseases, not only HCM.17, 39
Our study clinically supported the in vitro and animal studies demonstrating the correlation between a high level of TGF‐β and an increased amount of cardiac fibrosis. In addition, increased BNP levels were demonstrated to be an independent predictor of morbidity and mortality in HCM.40 Increased BNP levels in the patients with high TGF‐β in our study showed that TGF‐β could be a prognostic marker in HCM.
Study Limitations
The relatively low number of patients and short follow‐up are limitations of our study. Our patients had a higher mortality rate than the expected mortality for HCM. This was because those patients who died were mostly referred from another hospital and in the final stages. In addition, the patients in the study had a higher incidence of DM than the general population. We could not undertake any genetic test or magnetic resonance imaging. However, our study has adequately supported the hypothesis that increased TGF‐β level is associated with a poor prognosis in HCM.
Conclusion
TGF‐β levels are higher in HCM patients and may be a prognostic marker for cardiac adverse events.
The authors have no funding, financial relationships, or conflicts of interest to disclose.
References
- 1. Maskatia SA. Hypertrophic cardiomyopathy: infants, children, and adolescents. Congenit Heart Dis. 2012;7:84–92. [DOI] [PubMed] [Google Scholar]
- 2. Amano Y, Takeda M, Tachi M, et al. Myocardial fibrosis evaluated by Look‐Locker and late gadolinium enhancement magnetic resonance imaging in apical hypertrophic cardiomyopathy: association with ventricular tachyarrhythmia and risk factors. J Magn Reson Imaging. 2014;40:407–412. [DOI] [PubMed] [Google Scholar]
- 3. Subasic K. Living with hypertrophic cardiomyopathy. J Nurs Scholarsh. 2013;45:371–379. [DOI] [PubMed] [Google Scholar]
- 4. Hernández‐Romero D, Orenes‐Piñero E, García‐Honrubia A, et al. Involvement of the –420C > G RETN polymorphism in myocardial fibrosis in patients with hypertrophic cardiomyopathy. J Intern Med. 2014. doi: 10.1111/joim.12334. [DOI] [PubMed] [Google Scholar]
- 5. Brouwer WP, van Dijk SJ, Stienen GJ, et al. The development of familial hypertrophic cardiomyopathy: from mutation to bedside. Eur J Clin Invest. 2011;41:568–578. [DOI] [PubMed] [Google Scholar]
- 6. Song BG, Yang HS, Hwang HK, et al. Correlation of electrocardiographic changes and myocardial fibrosis in patients with hypertrophic cardiomyopathy detected by cardiac magnetic resonance imaging [retraction appears in Clin Cardiol. 2013]. Clin Cardiol. 2013;36:31–35. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7. Zeisberg EM, Tarnavski O, Zeisberg M, et al. Endothelial‐to‐mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. [DOI] [PubMed] [Google Scholar]
- 8. Hina K, Iwasaki K, Nogami K, et al. Progression of left ventricular enlargement in patients with hypertrophic cardiomyopathy: incidence and prognostic value. Clin Cardiol. 1993;16:403–407. [DOI] [PubMed] [Google Scholar]
- 9. Teekakirikul P, Eminaga S, Toka O, et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non‐myocyte proliferation and requires TGF‐β. J Clin Invest. 2010;120:3520–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Medina C, Santos‐Martinez MJ, Santana A, et al. Transforming growth factor‐β type 1 receptor (ALK5) and Smad proteins mediate TIMP‐1 and collagen synthesis in experimental intestinal fibrosis. J Pathol. 2011;224:461–472. [DOI] [PubMed] [Google Scholar]
- 11. Porte J, Jenkins G. Assessment of the effect of potential antifibrotic compounds on total and αVβ6 integrin‐mediated TGF‐β activation. Pharmacol Res Perspect. 2014. doi: 10.1002/prp2.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cai Y, Zhou CH, Fu D, et al. Overexpression of Smad ubiquitin regulatory factor 2 suppresses transforming growth factor‐β–mediated liver fibrosis. J Dig Dis. 2012;13:327–334. [DOI] [PubMed] [Google Scholar]
- 13. Rosenkranz S, Flesch M, Amann K, et al. Alterations of β adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF‐β(1). Am J Physiol Heart Circ Physiol. 2002;283:1253–1262. [DOI] [PubMed] [Google Scholar]
- 14. Meyer A, Wang W, Qu J, et al. Platelet TGF‐β1 contributions to plasma TGF‐β1, cardiac fibrosis, and systolic dysfunction in a mouse model of pressure overload. Blood. 2012;119:1064–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li RK, Li G, Mickle DA, et al. Overexpression of transforming growth factor‐β1 and insulin‐like growth factor‐I in patients with idiopathic hypertrophic cardiomyopathy. Circulation. 1997;96:874–881. [DOI] [PubMed] [Google Scholar]
- 16. Li G, Li RK, Mickle DA, et al. Elevated insulin‐like growth factor‐I and transforming growth factor‐β1 and their receptors in patients with idiopathic hypertrophic obstructive cardiomyopathy: a possible mechanism. Circulation. 1998;II:144–149. [PubMed] [Google Scholar]
- 17. Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)‐β signaling in cardiac remodeling. J Mol Cell Cardiol. 2011;51:600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bujak M, Frangogiannis NG. The role of TGF‐β signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74:184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cuspidi C, Negri F, Muiesan ML, et al. Prevalence and severity of echocardiographic left ventricular hypertrophy in hypertensive patients in clinical practice. Blood Press. 2011;20:3–9. [DOI] [PubMed] [Google Scholar]
- 20. Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35:2733–2779. [DOI] [PubMed] [Google Scholar]
- 21. Li G, Borger MA, Williams WG, et al. Regional overexpression of insulin‐like growth factor‐I and transforming growth factor‐β1 in the myocardium of patients with hypertrophic obstructive cardiomyopathy. J Thorac Cardiovasc Surg. 2002;123:89–95. [DOI] [PubMed] [Google Scholar]
- 22. Almendral JL, Shick V, Rosendorff C, et al. Association between transforming growth factor‐β(1) and left ventricular mass and diameter in hypertensive patients. J Am Soc Hypertens. 2010;4:135–141. [DOI] [PubMed] [Google Scholar]
- 23. Villar AV, Cobo M, Llano M, et al. Plasma levels of transforming growth factor‐β1 reflect left ventricular remodeling in aortic stenosis. PLoS One. 2009;4:e8476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Timmer SA, Germans T, Brouwer WP, et al. Carriers of the hypertrophic cardiomyopathy MYBPC3 mutation are characterized by reduced myocardial efficiency in the absence of hypertrophy and microvascular dysfunction. Eur J Heart Fail. 2011;13:1283–1289. [DOI] [PubMed] [Google Scholar]
- 25. Fatkin D, McConnell BK, Mudd JO, et al. An abnormal Ca(2+) response in mutant sarcomere protein‐mediated familial hypertrophic cardiomyopathy. J Clin Invest. 2000;106:1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ahn HS, Kim HK, Park EA, et al. Coronary flow reserve impairment in apical vs asymmetrical septal hypertrophic cardiomyopathy. Clin Cardiol. 2013;36:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gavallér H, Sepp R, Csanády M, et al. Hypertrophic cardiomyopathy is associated with abnormal echocardiographic aortic elastic properties and arteriograph‐derived pulse‐wave velocity. Echocardiography. 2011;28:848–852. [DOI] [PubMed] [Google Scholar]
- 28. O'Hanlon R, Pennell DJ. Cardiovascular magnetic resonance in the evaluation of hypertrophic and infiltrative cardiomyopathies. Heart Fail Clin. 2009;5:369–387. [DOI] [PubMed] [Google Scholar]
- 29. Menon SC, Eidem BW, Dearani JA, et al. Diastolic dysfunction and its histopathological correlation in obstructive hypertrophic cardiomyopathy in children and adolescents [published correction appears in J Am Soc Echocardiogr. 2010;23:222]. J Am Soc Echocardiogr. 2009;22:1327–1334. [DOI] [PubMed] [Google Scholar]
- 30. Moreo A, Ambrosio G, De Chiara B, et al. Influence of myocardial fibrosis on left ventricular diastolic function: noninvasive assessment by cardiac magnetic resonance and echo. Circ Cardiovasc Imaging. 2009;2:437–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rubinshtein R, Glockner JF, Ommen SR, et al. Characteristics and clinical significance of late gadolinium enhancement by contrast‐enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail. 2010;3:51–58. [DOI] [PubMed] [Google Scholar]
- 32. Inada K, Seiler J, Roberts‐Thomson KC, et al. Substrate characterization and catheter ablation for monomorphic ventricular tachycardia in patients with apical hypertrophic cardiomyopathy. J Cardiovasc Electrophysiol. 2011;22:41–48. [DOI] [PubMed] [Google Scholar]
- 33. Abe M, Okada K, Matsumoto K. Clinical experience in treating hypertension with fixed‐dose combination therapy: angiotensin II receptor blocker losartan plus hydrochlorothiazide. Expert Opin Drug Metab Toxicol. 2009;5:1285–1303. [DOI] [PubMed] [Google Scholar]
- 34. Matt P, Schoenhoff F, Habashi J, et al; GenTAC Consortium . Circulating transforming growth factor‐β in Marfan syndrome. Circulation. 2009;120:526–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xiao H, Lei H, Qin S, et al. TGF‐β1 expression and atrial myocardium fibrosis increase in atrial fibrillation secondary to rheumatic heart disease. Clin Cardiol. 2010;33:149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Felkin LE, Lara‐Pezzi E, George R, et al. Expression of extracellular matrix genes during myocardial recovery from heart failure after left ventricular assist device support. J Heart Lung Transplant. 2009;28:117–122. [DOI] [PubMed] [Google Scholar]
- 37. Frantz S, Hu K, Adamek A, et al. Transforming growth factor‐β inhibition increases mortality and left ventricular dilatation after myocardial infarction. Basic Res Cardiol. 2008;103:485–492. [DOI] [PubMed] [Google Scholar]
- 38. Hillebrand M, Millot N, Sheikhzadeh S, et al. Total serum transforming growth factor‐β1 is elevated in the entire spectrum of genetic aortic syndromes. Clin Cardiol. 2014;37:672–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Edgley AJ, Krum H, Kelly DJ. Targeting fibrosis for the treatment of heart failure: a role for transforming growth factor‐β. Cardiovasc Ther. 2012;30:e30–e40. [DOI] [PubMed] [Google Scholar]
- 40. Geske JB, McKie PM, Ommen SR, et al. B‐type natriuretic peptide and survival in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2013;61:2456–2460. [DOI] [PubMed] [Google Scholar]