

Abstract
Molecularly targeted therapies aim to obstruct cell autonomous programs required for tumor growth. We show that mitogen-activated protein kinase (MAPK) and cyclin-dependent kinase 4/6 inhibitors act in combination to suppress the proliferation of KRAS-mutant lung cancer cells while simultaneously provoking a natural killer (NK) cell surveillance program leading to tumor cell death. The drug combination, but neither agent alone, promotes retinoblastoma (RB) protein-mediated cellular senescence and activation of the immunomodulatory senescence-associated secretory phenotype (SASP). SASP components tumor necrosis factor-α and intercellular adhesion molecule−1 are required for NK cell surveillance of drug-treated tumor cells, which contributes to tumor regressions and prolonged survival in a KRAS-mutant lung cancer mouse model. Therefore, molecularly targeted agents capable of inducing senescence can produce tumor control through non−cell autonomous mechanisms involving NK cell surveillance.
The KRAS oncogene is frequently mutated in several human cancers. It drives tumor-igenesis by constitutively activating growth factor signaling pathways that promote uncontrolled proliferation, namely the mitogen-activated protein kinase (MAPK) or phosphoinositide 3-kinase pathways. Although much effort has been placed on targeting KRAS or its downstream effectors, to date, most therapeutic agents have failed, owing to an inability to sustain inhibition of RAS-driven signaling (1, 2). Combinatorial strategies are being developed to circumvent these effects, for example, by combining MAPK kinase (MEK) inhibitors with upstream receptor tyrosine kinase inhibitors to thwart adaptive resistance mechanisms (3, 4). Another approach involves combining MEK inhibitors with downstream cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitors that, in principle, could more potently block the proliferation of KRAS-mutant cells by simultaneously reducing MAPK-regulated cyclin D levels and directly targeting CDK4 kinase activity (5). In addition to the intrinsic effects on tumor cell proliferation, both MEK and CDK4/6 inhibitors can modulate T cell function as single agents or in combination with T cell checkpoint blockade (6–8).
We explored the cell autonomous and non–cell autonomous effects of combining MEK and CDK4/6 inhibitors using KRAS-mutant tumor models. We first tested a number of highly selective CDK4/6 inhibitors (palbociclib, abemaciclib, ribociclib) in combination with the U.S. Food and Drug Administration–approved MEK inhibitor trametinib in human KRAS-mutant lung and pancreatic cancer cell lines. Compared with treatment with either single agent, the two-drug combination was substantially more effective at inhibiting proliferation as well as phosphorylation of the retinoblastoma (RB) protein, a direct CDK4 and 6 target (Fig. 1A and fig. S1). Accordingly, the combination of trametinib and palbociclib was more effective at impairing tumor growth and inducing tumor stasis in mice harboring a KRAS-mutant lung cancer patient-derived xenograft (PDX), when treated at the maximally tolerated dose for each agent (Fig. 1B) (9, 10). Similar results were also observed in other KRAS-mutant PDX models treated at lower doses (Fig. 1C and fig. S2, A and B), confirming that the combination produces biological effects that neither drug can achieve alone.
Fig. 1. NK cell immunity is required for the efficacy of combination MEK and CDK4/6 inhibitor therapy.
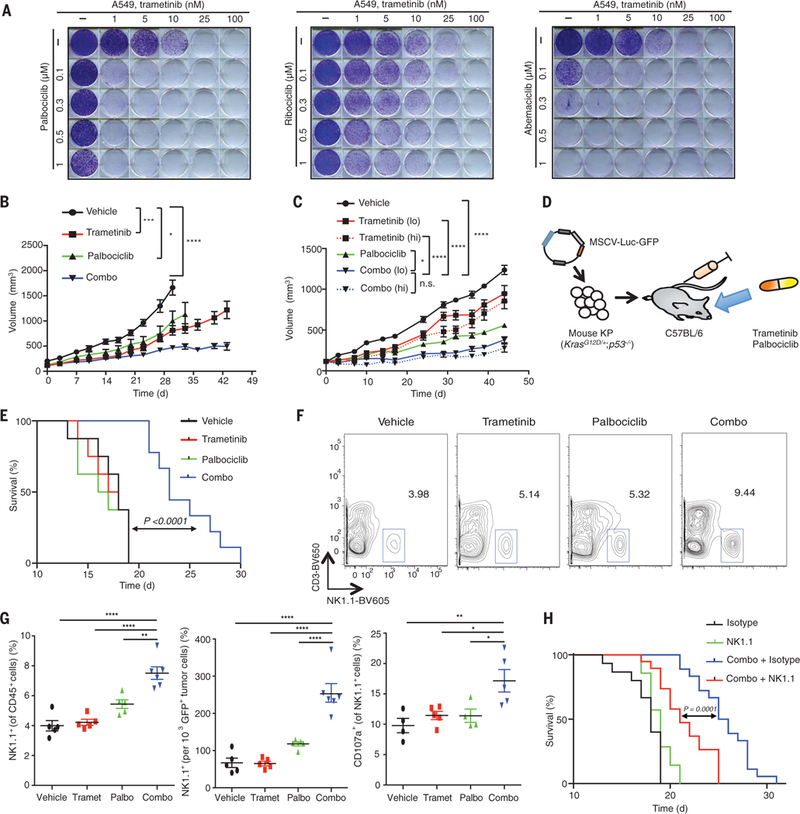
(A) Clonogenic assay of A549 lung cancer cells treated with MEK (trametinib) and/or various CDK4/6 inhibitors (palbociclib, ribociclib, abemaciclib); representative of three biological replicates. (B) Tumor volumes of mice bearing KRAS-mutant MSK-LX27 PDX lung tumors treated with vehicle, trametinib (3 mg/kg body weight), palbociclib (150 mg/kg body weight), or both in combination (Combo) for indicated times (n = 5 mice per group). (C) Tumor volumes of mice bearing KRAS-mutant MSK-LX68 PDX lung tumors treated with vehicle, trametinib [1 mg/kg (lo) or 3 mg/kg (hi) body weight], palbociclib (150 mg/kg body weight), or both in combination for indicated times (n = 8 mice per group). n.s., not significant. (D) Syngeneic KP transplant lung cancer model. (E) Kaplan-Meier survival curve of KP transplant mice treated with vehicle, trametinib (1 mg/kg body weight), palbociclib (100 mg/kg body weight), or both in combination (n ≥ 8 per group) (log-rank test). (F) Representative flow cytometry plots of NK cell populations in lung tumors from KP transplant mice treated for 1 week as in (E). (G) Percentage of NK cells within the CD45+ population (left), total NK cells relative to tumor cell number (middle), and percentage of CD107a+ degranulating NK cells (right) (n ≥ 4 mice per group). Palbo, palbociclib; Tramet, trametinib. (H) Kaplan-Meier survival curve of KP transplant mice treated with vehicle or combined trametinib (1 mg/kg body weight) and palbociclib (100 mg/kg body weight) and either an isotype control antibody (C1.18.4) or NK1.1-depleting antibody (PK136) (n ≥ 8 per group) (log-rank test). (B and C) Two-way ANOVA. (G) One-way ANOVA. Error bars, mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
These human xenograft studies require the use of immunodeficient NOD-scid IL2Rgnull (NSG) mice. To assess whether and to what extent tumor cell responses are altered by the immune system, we made use of an established syngeneic transplant mouse model of lung cancer. Mouse tumor cells derived from a KrasG12D/+;Trp53−/− (KP) lung tumor (11) were engineered to express luciferase and green fluorescent protein (GFP) and tested for drug sensitivity in vitro or after intravenous injection into C57BL/6 immuno-competent mice (Fig. 1D), where they produced aggressive lung adenocarcinomas by 1-week post transplantation. As in the human models, the trametinib plus palbociclib combination synergistically suppressed the growth of cultured KP tumor cells (fig. S2C) and significantly increased the survival of lung tumor–bearing animals (Fig. 1E). This effect was substantially impaired when the same cells were transplanted into immunodeficient NSG mice (fig. S2D and E), suggesting that the immune system might contribute to treatment efficacy.
Immune profiling of lung tumors after 1 week of combination treatment revealed a general influx of CD45+ immune cells compared with single-agent treatment (fig. S3A). Whereas there was no change in B cell or macrophage numbers in tumors after treatment, there was an increased infiltration of CD4+ and CD8+ T cells in the lungs and spleens of both tumor-bearing and naïve (tumor-free) mice after either single-agent or combination therapy. However, changes in CD4+ or CD8+ T cell activation were not observed, as assessed by staining for CD69, KLRG1, and CD107a, a marker of lymphocyte degranulation (figs. S3, B to D; S4, A and B; and S5, A to D). Thus, the drug combination does not appear to produce selective activation of T cells in this lung cancer model.
By contrast, combined treatment with trametinib and palbociclib triggered the selective accumulation of natural killer (NK) cells and a decrease in Gr-1hiCD11b+ myeloid-derived suppressor cells (MDSCs) in the lungs of tumor-bearing mice compared with treatment with either agent alone (Fig. 1, F and G; and figs. S3, E and F; S4, C and D; and S5F). NK cells derived from combination-treated tumors appeared activated and more mature, as indicated by cell surface expression of CD107a and other activating receptors and altered transcriptional profiles showing a reduction in proliferation-associated genes and an increase in expression of genes linked to NK cell maturation and cytotoxicity (Fig. 1G and figs. S4C, S5E, and S6). Combined treatment also led to NK cell accumulation (but not activation) and a decrease in MDSCs in the spleens of tumor-bearing mice but not in the lungs or spleens of naïve mice (figs. S3, F to H, and S5, E to G), suggesting that this immune effect is tumor dependent.
To determine whether specific immune cells contribute to the outcome of combination therapy, we assessed the impact of perturbing immune cell function on the survival of tumor-bearing (immunocompetent) mice after vehicle or trametinib and palbociclib therapy. Neither depletion of macrophages, Gr-1+ granulocytes/MDSCs, and CD4+ and CD8+ T cells using blocking antibodies nor anti–PD-1 immunotherapy (to stimulate exhausted T cells) had any impact on the survival of vehicle or combination-treated mice (figs. S7, A to D, and S8). By contrast, depletion of NK cells using an anti-NK1.1 anti-body (PK136) significantly reduced the survival advantage produced by combination therapy while having no effect on cohorts treated with vehicle or single agents (Fig. 1H and figs. S7E and F, and S8). This effect was independent of the MDSC reduction after treatment (fig. S3F), as Gr-1 depletion did not influence the survival of combination-treated mice in the presence or absence of NK cells (fig. S7B). Hence, in this model, the trametinib and palbociclib combination triggers a potent and selective NK cell–mediated response that contributes to treatment efficacy.
We found the contribution of immune surveillance to the action of an apparently cytostatic drug combination intriguing and set out to study the underlying mechanisms in more detail. CDK4 and 6-mediated phosphorylation of RB (the intended target of trametinib and palbociclib treatment) cancels its growth-suppressive action to facilitate E2F-mediated G1-S progression (5). We confirmed in all our models that combination therapy produced a more potent reduction in RB phosphorylation and proliferation without inducing apoptosis (fig. S9). Besides its role in modulating cell cycle progression, RB plays a crucial role in mediating cellular senescence, a tumor-suppressive program that involves a stable (if not permanent) cell cycle arrest program coupled to an immune modulatory component (12–14). Specifically, senescent cells display an RB-dependent down-regulation of proliferation genes (15, 16) and a concomitant up-regulation of genes encoding a wide range of secretory proteins and other factors that modulate the microenvironment (17). Of note, this secretory program, often referred to as the senescence-associated secretory phenotype (SASP), can have either tumor-suppressive or tumor-promoting functions, depending on context (18–23). In its tumor-suppressive role, RB often collaborates with the p53 tumor suppressor to limit the proliferation of premalignant cells (24), and disruption of both genes is linked to senescence escape during tumorigenesis (25).
To test whether trametinib and/or palbociclib treatment induced senescence in human or mouse KRAS-mutant lung cancer cells, we evaluated a series of markers and functional properties associated with the senescent state (14, 26). Only combination treatment resulted in significant senescence-associated beta-galactosidase (SA-β-gal) activity, the accumulation of senescence-associated heterochromatin foci, loss of the nuclear envelope protein Lamin B1, and increased expression of DEC1, DCR2, and CDKN2B (Fig. 2A and fig. S10).
Fig. 2. Senescence and SASP induction after combination MEK and CDK4/6 inhibitor therapy induces NK cell immune surveillance.
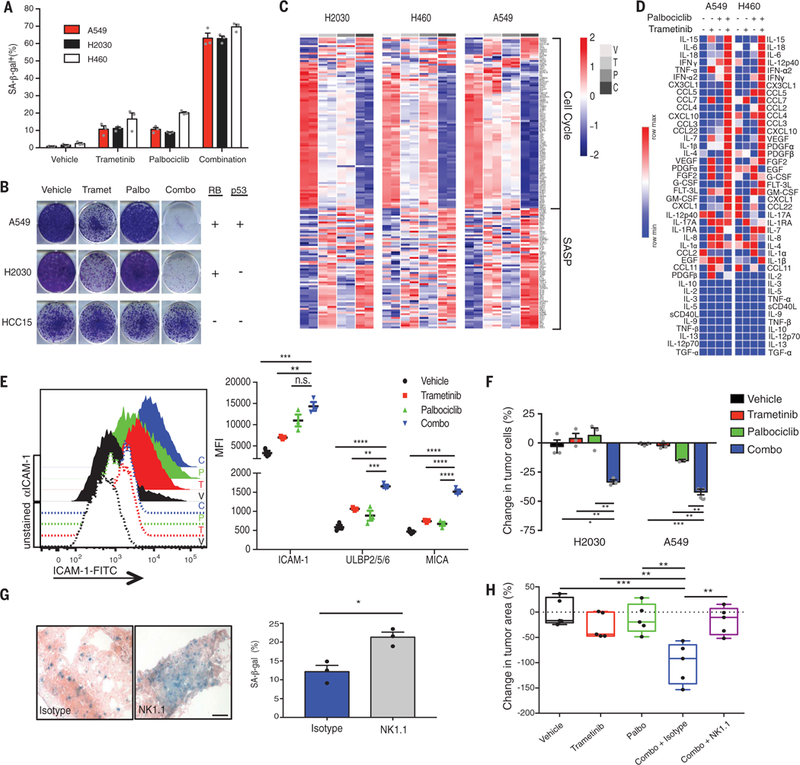
(A) Quantification of SA-β-gal+ cells in human KRAS-mutant lung tumor cell lines after 8-day treatment with trametinib (25 nM) and/or palbociclib (500 nM). Mean of three biological replicates is plotted. (B) Clonogenic assay of human KRAS-mutant lung cancer cells replated in the absence of drugs after 8-day pretreatment as in (A); representative of three biological replicates. RB and p53 genomic status is indicated on right. (C) Heat map of senescence-associated cell cycle and SASP gene expression in human KRAS-mutant lung cancer cell lines after treatment as in (A), as assessed by RNA-seq.Two biological replicates per cell line are shown. C, combination; P, palbociclib; T, trametinib; V, vehicle. (D) Heat map of cytokine array results from human KRAS-mutant lung tumor cells treated with trametinib (25 nM) and/or palbociclib (500 nM) for 8 days. Data are presented as mean of three biological replicates. (E) Flow cytometry analysis of ICAM-1 expression and levels of other NK cell–activating ligands after 8-day treatment of A549 lung tumor cells as described in (A). Quantification of mean fluorescence intensity (MFI) from three biological replicates is shown on the right. (F) Quantification of NK cell cytotoxicity (by live cell imaging) after pretreatment of A549 and H2030 lung tumor cell lines with indicated drugs for 8 days and coculturing with the YT NK cell line at a 10:1 effector to target cell (E:T) ratio for 20 hours in the presence or absence of indicated drugs. Change in tumor cells is normalized to control wells lacking NK cells. Mean of three biological replicates is plotted. (G) Representative SA-β-gal staining of KP transplant lung tumors after 1-week treatment with combined trametinib (1 mg/kg body weight) and palbociclib (100 mg/kg body weight) and either an isotype control antibody (C1.18.4) or NK1.1 depleting antibody (PK136) (scale bar, 50 mm). Quantification shown on right (n = 3 mice per group). (H) Effect of 1-week treatment as in (G) on lung tumor burden (relative to vehicle) (n = 5 mice per group). (E, F, and H) One-way ANOVA. (G) Unpaired two-tailed t test. Error bars, mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Colony-forming assays revealed that many KRAS-mutant cell lines treated with either palbociclib or trametinib alone resumed proliferation upon drug washout, whereas combination-treated cells did not (Fig. 2B and fig. S11, A to B). This durable, senescent-like arrest was RB dependent and did not occur in tumor cells harboring RB genomic loss (Fig. 2B and figs. S11 and S12). Both human and murine KRAS-mutant tumor cells lacking p53 also displayed senescence markers in response to the trametinib and palbociclib combination (Fig. 2B and figs. S11 and S12). Therefore, this treatment can restore senescence to tumor cells that have escaped p53 tumor suppressive programs during tumor evolution provided that RB function is retained.
The senescence program provoked by combined trametinib and palbociclib treatment was also associated with a potent SASP induction. RNA sequencing (RNA-seq) analysis of human KRAS-mutant tumor cell lines after drug treatment revealed that the drug combination produced a greater reduction in proliferation genes and increase in SASP factor expression compared with either treatment alone (Fig. 2C, fig. S13B, and tables S1 and S2). Among the immune modulatory genes that were induced and secreted were chemokines involved in NK cell recruitment (CCL2, CCL4, CCL5, CXCL10, CX3CL1), as well as cytokines that promote NK cell proliferation and activation [interleukin-15 (IL-15), IL-18, tumor necrosis factor–α (TNF-α)] (Fig. 2D and fig. S13, A and C). Many of these same SASP factors were also induced in murine KP lung cancer cells and a PDX lung cancer model in vivo (fig. S13, D and E). Gene Set Enrichment Analysis revealed that signatures linked to oncogene-induced and replicative senescence (22, 27), nuclear factor κB (NF-κB) and TNF-α signaling, and NK cell–mediated cytotoxicity were also selectively enriched in the transcriptional profiles of combination treated cells (fig. S13, F to H). Although not secretory per se, NK cell ligands (which are required for activation of NK cell cytotoxicity and tumor cell targeting) are part of the transcriptional module linked to the SASP (28, 29). We found that intercellular adhesion molecule–1 (ICAM-1) and the NKG2D ligands ULBP2 and MICA were induced after combination treatment in human KRAS-mutant tumor cells, PDXs, and murine KP lung tumor cells (Fig. 2E and fig. S14). Overall, these data suggest that, in addition to a more stable cell cycle arrest conferred by RB-mediated senescence, combined MEK and CDK4/6 inhibition may promote tumor cell immune surveillance through induction of the SASP program.
To explore this possibility, we tested whether NK cells could functionally target tumor cells after combined trametinib and palbociclib treatment. Using an in vitro NK cell coculture assay that quantitatively measures both NK cell–tumor cell interactions and cytotoxicity, we observed that treatment with the drug combination, but not with single agents, triggered the rapid association and eventual killing of KRAS-mutant tumor cells by the human YT NK cell line even in the presence of the drugs (Fig. 2F; fig. S15, A and B; and movies S1 to S4). These results were corroborated using freshly isolated primary human NK cells (fig. S15, C and D) and using the murine system, in which senescent KP cells treated with the drug combination were sufficient to induce degranulation and cytotoxicity in drug-treated splenic NK cells upon coculture (fig. S15, E and F). In vivo, NK cells also appeared to preferentially target senescent cells, as tumors in NK cell–depleted mice that received the trametinib and palbociclib combination were significantly larger and retained more SA-β-gal+ cells compared with those in control mice with NK cells intact (Fig. 2, G and H, and fig. S15G). NK cells are therefore capable of eliminating senescent tumor cells after combined trametinib and palbociclib treatment.
We performed a genetic experiment to disable the SASP program in KP tumor cells. The transcription factor NF-κB is a master regulator of the SASP program but plays only a limited role in senescence-induced cell cycle arrest (30–32). KP tumor cells expressing a well-characterized short hairpin RNA (shRNA) targeting the p65 subunit of NF-κB underwent growth arrest in response to the drug combination but displayed a reduction in many SASP factors and were not targeted by spleen-derived murine NK cells in vitro (Fig. 3, A and B, and fig. S16, A to D). In vivo, tumors derived from p65-suppressed KP cells showed similar levels of NK cell accumulation as tumors expressing a control shRNA targeting the nonexpressed gene Renilla luciferase after combination therapy; however, these infiltrating NK cells were not activated, and the treatment was not as effective (Fig. 3C and fig. 16E). Thus, the SASP appears necessary for therapy-induced NK cell surveillance and the efficacy of the drug combination in vivo.
Fig. 3. NF-κB-mediated SASP and TNF-α secretion is required for treatment-induced NK cell activity.
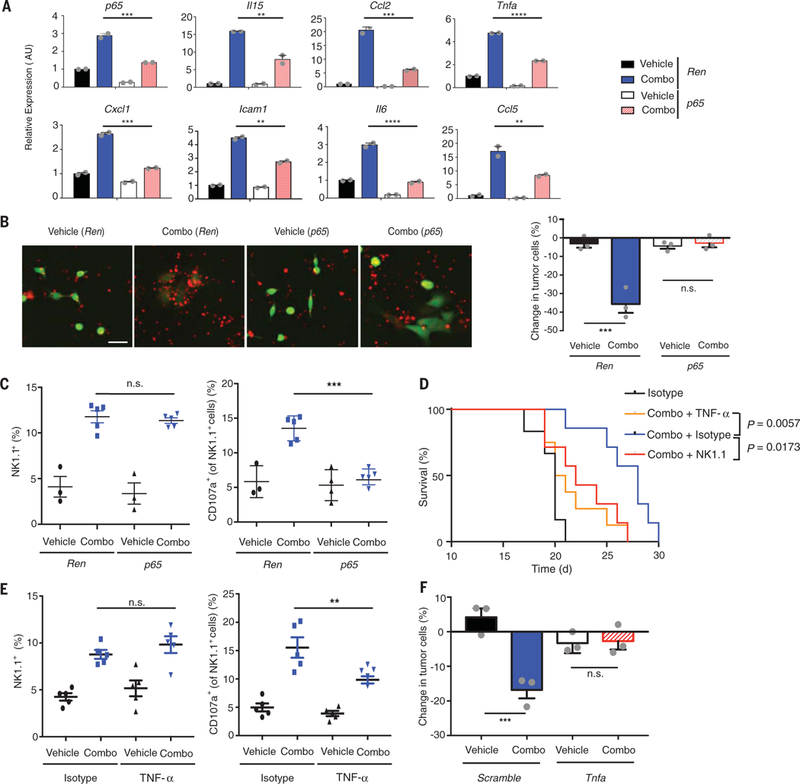
(A) Quantitative reverse transcription polymerase chain reaction analysis of SASP gene expression in KP tumor cells transduced with indicated shRNAs after treatment with vehicle or trametinib (25 nM) and palbociclib (500 nM) for 8 days. Mean of two biological replicates associated with three technical replicates is plotted. AU, arbitrary units. (B) Representative images of KP tumor cells with indicated shRNAs (green) pretreated as in (A) and cocultured with primary murine splenic NK cells (red) at a 20:1 E:T ratio for 20 hours in the presence or absence of indicated drugs (scale bar, 25 μm). Quantification of NK cell cytotoxicity (by live cell imaging) is shown on the right. Change in tumor cells is normalized to control wells lacking NK cells. Mean of three biological replicates is plotted. (C) Flow cytometry analysis of total and CD107a+ degranulating NK cells within the CD45+ population in the lungs after 1-week treatment of mice transplanted with KP tumor cells containing control Renilla (Ren) or p65 shRNAs with vehicle or combined trametinib (1 mg/kg body weight) and palbociclib (100 mg/kg body weight) (n ≥ 3 mice per group). (D) Kaplan-Meier survival curve of KP transplant mice treated with vehicle or combined trametinib (1 mg/kg body weight) and palbociclib (100 mg/kg body weight) and either an isotype control (HRPN), NK1.1 (PK136), or TNF-α (XT3.11) targeting antibody (n ≥ 6 per group) (log-rank test). (E) Flow cytometry analysis as in (C) after 1-week treatment of KP transplant mice with vehicle or combined trametinib (1 mg/kg body weight) and palbociclib (100 mg/kg body weight) and either an isotype control (HRPN) or TNF-α (XT3.11) targeting antibody (n = 5 per group). (F) Quantification of NK cell cytotoxicity (by live cell imaging) toward KP tumor cells with indicated shRNAs pretreated as in (B) and cocultured with primary murine splenic NK cells at a 20:1 E:T ratio for 20 hours in the presence or absence of indicated drugs. (A to C and E and F) One-way ANOVA. Error bars, mean ± SEM. **P < 0.01, ***P < 0.001, ****P < 0.0001.
To pinpoint SASP factors needed for NK cell attack, we tested a range of neutralizing anti-bodies against NF-κB-regulated SASP factors (IL-15, IL-18, TNF-α, CCL2, CCL5) for their ability to alter survival in KP transplant mice treated with the trametinib and palbociclib combination. Only TNF-α depletion reduced animal survival, which was similar in magnitude to that produced by NK cell depletion (Fig. 3D and fig. S16, F and G) and was associated with a reduction in activated NK cells present in the treated tumors (Fig. 3E). This effect was in part due to tumor cell–derived TNF-α, as shRNAs capable of suppressing Tnfa in combination-treated KP cells markedly inhibited NK cell cytotoxicity in vitro (Fig. 3F and fig. S17, A to D). Still, Tnfa suppression (fig. S17E) was not as effective as p65 depletion (fig. S16E) at impairing the survival of tumor-bearing mice after combination treatment, indicating that an interplay between multiple SASP factors is required for NK cell surveillance in vivo.
We also explored potential mechanisms by which NK cells target tumor cells undergoing therapy-induced senescence. Cell surface NK cell ligands and adhesion molecules are up-regulated along with the SASP during many forms of senescence (29). We found that both ICAM-1 and members of the MICA/B, ULBP, Rae-1, and H60 family of NKG2D ligands were increased after combination therapy in an NF-κB-dependent or independent manner, respectively (Figs. 2E and 3A and figs. S14 and 16D). Furthermore, blocking ICAM-1 and its receptor LFA-1 (and to a lesser extent NKG2D) blunted NK cell cytotoxicity in combination-treated tumor cells in live cell imaging and chromium release assays and significantly reduced the survival of combination-treated KP transplant mice in vivo (figs. S15, C and D, and S17, F to H). Therefore, the SASP transcriptional module contributes both secreted (i.e., TNF-α) and cell surface (i.e., ICAM-1) factors to facilitate NK cell surveillance.
The orthotopic KP model represents a flexible syngeneic experimental system but is extraordinarily aggressive with tumors disseminated throughout the lungs, making assessment of tumor response to therapy challenging. To examine the impact of this senescence-inducing therapy on NK cell immune surveillance and tumor progression in a more physiologically relevant setting, we utilized an autochthonous model of KRAS-mutant lung cancer. Genetically engineered KrasLSL-G12D/wt;Trp53flox/flox (KP GEMM) mice were infected intratracheally with an adenovirus expressing Cre recombinase to induce endogenous lung tumor formation (33), leading to the development of focal KRAS-driven tumors that could be monitored using microcomputed tomography (μCT). Consistent with our findings in the syngeneic transplant model, combined trametinib and palbociclib treatment after endogenous tumor formation led to a significant reduction in RB phosphorylation and tumor cell proliferation, accumulation of SA-β-gal+ senescent tumor cells, induction of SASP factors including TNF-α and ICAM-1, and subsequent recruitment of NK cells within lung adenocarcinomas of KP GEMM mice (Fig. 4A and fig. S18, A to D).
Fig. 4. Combination trametinib and palbociclib treatment drives NK cell–mediated lung tumor regressions in genetically engineered mice.
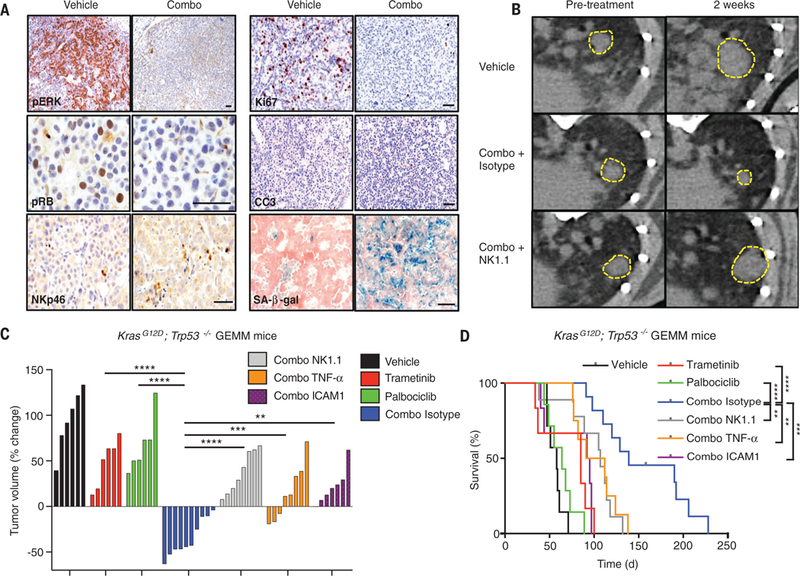
(A) Immunohistochemical staining of KP GEMM tumors treated with vehicle or combined trametinib (1 mg/kg body weight) and palbociclib (100 mg/kg body weight) for 2 weeks (scale bar, 50 μm). CC3, cleaved caspase-3; pERK, phosphorylated extracellular signal–regulated kinase. (B) Representative μCT images of KP GEMM lung tumors prior to treatment and after 2 weeks of treatment with vehicle or combined trametinib (1 mg/kg body weight) and palbociclib (100 mg/kg body weight) and either an isotype control antibody (C1.18.4) or NK1.1 depleting antibody (PK136). Yellow boxes indicate lung tumors. (C) A waterfall representation of the response of each tumor after 2 weeks of treatment with vehicle, trametinib (1 mg/kg body weight), palbociclib (100 mg/kg body weight), or both, and either an isotype control (C1.18.4), NK1.1 (PK136), TNF-α (XT3.11), or ICAM-1 (YN1/1.7.4) blocking antibody (n ≥ 6 per group). (D) Kaplan-Meier survival curve of KP GEMM mice treated as in (C) (n ≥ 6 per group) (log-rank test). (C) One-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
The contribution of NK cells to the activity of the combination therapy was marked. Two weeks post treatment, we observed tumor regressions in mice treated with the combination therapy and an isotype control antibody but not in those mice receiving an NK1.1-depleting anti-body or single-agent trametinib or palbociclib whose tumors continued to progress (Fig. 4, B and C). Moreover, the overall survival of mice treated with trametinib plus palbociclib, which included a notable number of long-term survivors, was significantly longer than mice receiving single-agent treatment or the combination treatment in the context of NK cell depletion (Fig. 4D). Antibody-mediated blockade of SASP factors TNF-α or ICAM-1 also prevented tumor regressions and blunted the survival benefit of the drug combination (Fig. 4, C and D). Therefore, senescence-inducing targeted therapies can lead to tumor control in KRAS-mutant lung tumors, with SASP-provoked and NK cell–mediated immune surveillance facilitating tumor regression and prolonged survival.
Leveraging immunocompetent mouse models of KRAS-mutant lung cancer, we explored the non–cell autonomous effects of combined MEK and CDK4/6 inhibitor therapy, revealing an immune modulatory component to the antitumor response. Complementary to previous work in other systems demonstrating activation of T cell responses using similar monotherapies (6, 7), our studies highlight a distinct mechanism of innate immune attack by NK cells, which act as a natural senolytic to eliminate tumor cells in combination drug–treated mice harboring autochthonous tumors. The ability of NK cells to target drug-treated tumor cells requires the induction of RB-mediated cellular senescence and acquisition of an NF-κB-dependent SASP program that culminates in the secretion of proinflammatory cytokines (e.g., TNF-α) and surface expression of NK cell–activating molecules (e.g., ICAM-1) (fig. S18E). Thus, whereas developing tumors evade both senescence and immune surveillance to become fully malignant, these processes can be reestablished in tumor cells by certain targeted cancer therapies. Although chronic SASP induction can have deleterious consequences in some settings (17, 34), the timely clearance of senescent cells by NK cells in this model establishes senescence induction as a beneficial outcome of targeted therapy, and explains how some cytostatic agents can be cytotoxic in vivo. Because NK cells do not require neoantigen recognition to target tumor cells (28), strategies to exploit and enhance this form of immune surveillance may complement existing efforts to harness adaptive immune surveillance by means of T cell checkpoint blockade immunotherapies.
Supplementary Material
ACKNOWLEDGMENTS
We thank H. Varmus, T. Jacks, and J. P. Morris IV for sharing cell lines; A. Banito, X. Li, D. Alonso-Curbelo, V. Low, H. C. Chen, L. Zamechek, W. Luan, F. Luna, and M. Rowicki for technical assistance and advice; N. Adams and J. Sun for graciously providing murine NKG2D-Fc antibodies and helpful comments on the manuscript; J. Boudreau and K. Hsu for insights concerning in vitro NK cell assays; and other members of the Lowe laboratory for insightful discussions.
Funding: We thank W. H. Goodwin and A. Goodwin and the Commonwealth Foundation for Cancer Research for research support. This work was supported by three grants from the National Cancer Institute (PO1 CA129243–06, P30 CA008748-S5, and U54 0D020355–01), a grant from the Center of Experimental Therapeutics of Memorial Sloan Kettering Cancer Center (MSKCC), and an MSKCC-Parker Institute for Cancer Immunotherapy pilot grant. M.R. was supported by an MSKCC Translational Research Oncology Training Fellowship (NIH T32-CA160001) and an American Cancer Society Postdoctoral Fellowship (129040-PF-16–115-01-TBG). J.L. was supported by a fellowship of the German Research Foundation (DFG). M.J.B. was supported by an American Association for Thoracic Surgery-Andrew Morrow Research Scholarship. T.B. was supported by the William C. and Joyce C. O’Neil Charitable Trust and Memorial Sloan Kettering Single Cell Sequencing Initiative. F.J.S.-R. is an HHMI Hanna Gray Fellow and was partially supported by an MSKCC Translational Research Oncology Training Fellowship (NIH T32-CA160001). P.B.R. was supported in part by a K12 Paul Calebresi Career Development Award for Clinical Oncology (K12 CA184746). E.d.S. received support through a U54 OD020355–01 NIH grant and the Geoffrey Beene Cancer Research Center. C.M.R. was supported by a Stand Up To Cancer grant from the American Association for Cancer Research. S.W.L. is the Geoffrey Beene Chair of Cancer Biology. S.W.L. and C.J.S. are Howard Hughes Medical Institute investigators.
Competing interests: P.B.R. is a shareholder of Pfizer and a consultant for EMD Serono and AstraZeneca and has received an honorarium from Corning. E.M. is an employee and share holder of Novartis. S.W.L. is a founder and scientific advisory board member of Blueprint Medicines and ORIC Pharmaceuticals and received an award and honorarium from Eli Lilly and Company.
Data and materials availability: RNA-seq data presented in this study are deposited in the Gene Expression Omnibus database under accession number GSE110397.
REFERENCES AND NOTES
- 1.Samatar AA, Poulikakos PI, Nat. Rev. Drug Discov. 13, 928–942 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Lito P et al. , Cancer Cell 25, 697–710 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manchado E et al. , Nature 534, 647–651 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prahallad A et al. , Nature 483, 100–103 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Sherr CJ, Beach D, Shapiro GI, Cancer Discov. 6, 353–367 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng J et al. , Cancer Discov. 8, 216–233 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goel S et al. , Nature 548, 471–475 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ebert PJR et al. , Immunity 44, 609–621 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Fry DW et al. , Mol. Cancer Ther. 3, 1427–1438 (2004). [PubMed] [Google Scholar]
- 10.Gilmartin AG et al. , Clin. Cancer Res. 17, 989–1000 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Dimitrova N et al. , Cancer Discov. 6, 188–201 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shay JW, Pereira-Smith OM, Wright WE, Exp. Cell Res. 196, 33–39 (1991). [DOI] [PubMed] [Google Scholar]
- 13.Kuilman T, Michaloglou C, Mooi WJ, Peeper DS, Genes Dev. 24, 2463–2479 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharpless NE, Sherr CJ, Nat. Rev. Cancer 15, 397–408 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Chicas A et al. , Cancer Cell 17, 376–387 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Narita M et al. , Cell 113, 703–716 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Coppé JP et al. , PLOS Biol. 6, e301 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eggert T et al. , Cancer Cell 30, 533–547 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang TW et al. , Nature 479, 547–551 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Krizhanovsky V et al. , Cell 134, 657–667 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xue W et al. , Nature 445, 656–660 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tasdemir N et al. , Cancer Discov. 6, 612–629 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coppé JP, Desprez PY, Krtolica A, Campisi J, Annu. Rev. Pathol. 5, 99–118 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW, Cell 88, 593–602 (1997). [DOI] [PubMed] [Google Scholar]
- 25.Lowe SW, Sherr CJ, Curr. Opin. Genet. Dev. 13, 77–83 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Collado M, Serrano M, Nat. Rev. Cancer 6, 472–476 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Fridman AL, Tainsky MA, Oncogene 27, 5975–5987 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morvan MG, Lanier LL, Nat. Rev. Cancer 16, 7–19 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Sagiv A et al. , Aging (Albany NY) 8, 328–344 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Acosta JC et al. , Cell 133, 1006–1018 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J, Proc. Natl. Acad. Sci. U.S.A. 106, 17031–17036 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chien Y et al. , Genes Dev. 25, 2125–2136 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jackson EL et al. , Genes Dev. 15, 3243–3248 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Childs BG, Durik M, Baker DJ, van Deursen JM, Nat. Med. 21, 1424–1435 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.