

Abstract
We report here the influence of antibody immobilization strategy on protein immunosensors on screen printed carbon electrode arrays in terms of antibody binding activity, analytical sensitivity, limit of detection, and stability. Horseradish peroxidase (HRP) was the model analyte with anti-HRP immobilized on the sensors, and HRP activity was used for detection. Covalently immobilized anti-HRP antibodies on electrodes coated with chitosan, electrochemically reduced graphene oxide (rGO), and dense gold nanoparticle (AuNP) films had only 20–30% of the total immobilized antibodies active for binding. Active antibodies increased to 60% with passively adsorbed antibodies on bare electrodes, to 85 % with oriented antibodies using protein A covalently immobilized on AuNP-coated carbon electrode, and to 98% when attached to protein A passively adsorbed onto bare electrodes. Passively adsorbed antibodies on bare electrodes lost activity in 1–2 days, but antibodies immobilized using other strategies remained relatively stable after 5 days. Covalent immobilization gave limits of detection (LOD) of 40 fg mL−1, while passively adsorbed antibodies or protein A on carbon electrodes had LODs 4–8 fg mL−1, but were unstable. Sensitivity was highest for antibodies covalently attached to AuNP electrodes (2.40 nA/log pg mL−1) that also had highest antibody coverage, and decreased slightly when protein A on AuNP was used to orient antibodies. Passively adsorbed antibodies and oriented antibodies on protein A gave slightly lower sensitivities. Immobilization strategy or antibody orientation did not have a significant effect on LOD, but dynamic range increased as the number of active antibodies on sensor surfaces increased.
Graphical Abstract
The influence of antibody attachment method, orientation and two nanostructured films on immunoarray sensor performance was critically evaluated.
1. Introduction
Antibodies are major tools for ligand binding assays due to their ability to bind and target analytes in complex sample matrices with high selectivity and specificity.1 Immobilization of antibodies on a solid surface or nanoparticle is often a crucial step for developing sensors for target analytes. Immunoassay format has been adapted to a wide spectrum of diagnostic tests including enzyme linked immunosorbent assay (ELISA),2 protein microarrays,3 lateral flow assay (LFA),4 radioimmunoassay (RIA),5 and electrochemical immunosensors.6,7 Performance is predicted to depend on the method of antibody immobilization that can control surface coverage, correct orientation of antibodies to bind analytes, degree of nonspecific binding, and sensor shelf life.8
Electrochemical immunosensors are attractive due to inherent high sensitivity, simplicity of instrumentation, ease of integration into microfluidic systems, multiplexing capabilities, and possible upgrades to point-of-care (POC) devices.9,10 Microfluidic electrochemical immunosensors have been developed to detect target analytes in environmental samples,11 biological fluids,12–13, 14 and pharmaceutical preparations.15 In electrochemical immunosensors, antibodies are immobilized onto sensor electrodes of materials including metals,16,17 conductive polymers18 and different forms of carbon.19 Carbon electrodes are available at low cost and give excellent performance in sensors due to a wide range potential window, low resistance, and low residual currents.20–, 21, 22 Screen printed electrodes have been widely used in sensor applications due to their relative low-cost mass production in single sensor or array format, and ease of design and miniaturization.23
For antibody attachment, sensor electrodes have been modified with functionalized nanomaterials via drop casting, inkjet24 or manual25 deposition, electrodeposition26 and electrospray sputtering.27 Carbon nanotube forests were used on electrodes to immobilize increased amounts of antibodies on immunosensors resulting from high surface area of the nanostructured surface.28
Layer-by layer (LBL) assembly is a fast, facile technique for immobilizing polymers and nanomaterials on carbon electrodes.29–, 30, 31 Decorating carbon electrodes with a film of glutathione-coated gold nanoparticles (AuNP) is an effective antibody immobilization strategy to construct ultrasensitive immunosensors that also yields a large population of antibodies on a nanostructured surface.32 Electrochemical deposition of graphene on electrodes from graphene oxide dispersions can also improve immunosensor performance.33 Passive adsorption of antibodies on carbon electrode surface is another approach that can been utilized.34,35 Antibodies can also be oriented on electrodes by binding the Fc region onto immobilized protein A, leaving Fab regions free for antigen capture. Protein A is 42 KDa surface protein derived from Staphylococcus aureus bacteria and has five specific IgG binding domains.
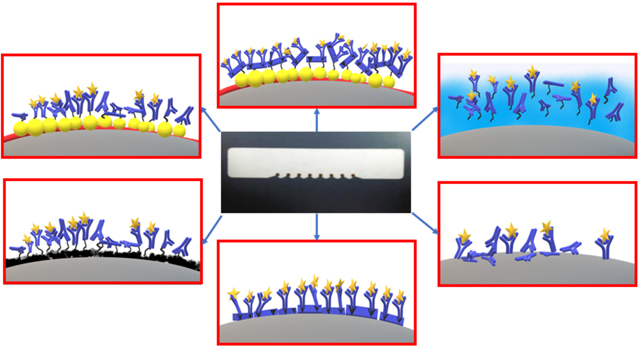
In this paper, we compare six different antibody immobilization techniques on screen printed carbon electrodes, namely, attachment to glutathione-AuNP layers, electrochemical deposition of reduced graphene oxide (rGO), chitosan films, passive antibody adsorption, passively adsorbed protein A oriented antibody immobilization and covalent immobilization of protein A on AuNP electrodes (Scheme 1). We utilized anti-horseradish peroxidase (anti-HRP) as a model antibody to quantify horseradish peroxidase (HRP) in calf serum as a human serum surrogate. Amperometric current was measured at −0.3 V against Ag/AgCl upon addition of H2O2 to activate HRP and hydroquinone (HQ) as mediator.28 Results were confirmed by measuring catalytic activity of captured HRP on sensor surfaces. Surface loading of antibody, sensitivity, limit of detection (LOD) and sensor shelf life were compared for the different immobilization strategies. High sensitivity was achieved when large amounts of active antibodies were immobilized. A high degree of antibody orientation increased dynamic range, but was not a factor in LOD or sensitivity.
Scheme 1:

Studied antibody immobilization techniques on screen printed carbon
2. Materials and methods
Materials.
All chemicals were of analytical grade and nanopure water was prepared using a Hydro® Picosystem®. Screen printed carbon electrodes featuring eight electrodes were from Kanichi®. Gold (III) chloride trihydrate (HAuCl4), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-hydroxysulfosuccinimide sodium salt (NHSS), hydroquinone (HQ), hydrogen peroxide (30%), graphite powder (99%), potassium permanganate, poly(diallyldimethylammonium chloride) (PDDA, MW 100,00–200,000, 20%) were from Sigma Aldrich®. Poly(dimethoxy)silane (PDMS) kit was from Dow Corning®. Phosphate buffer saline (PBS) pH 7.4 was 0.01 M sodium phosphate in 0.14 M NaCl, 2.7 mM KCl and phosphate buffer saline-tween20 (PBS-T20) was 0.01 M sodium phosphate in 0.14 M NaCl, 2.7 mM KCl and 0.5% Tween-20. SIGMAFAST® O-phenylenediamine (OPD) tablets and horseradish peroxidase (HRP) were from Sigma Aldrich®. Anti-horseradish peroxidase mouse monoclonal antibody (Ani-HRP) [2H11] (ab10183) and recombinant protein A (ab52953) were from Abcam®.
Instrumentation:
Microfluidic device incorporated a PDMS gasket with a flow channel in between two micromachined poly(methyl methacrylate) (PMMA) plates. The chamber is equipped with Pt counter electrode and Ag/AgCl reference electrode wires. A Kanichi® screen printed carbon electrode array featuring eight electrodes was inserted in the microfluidic chip under the PDMS flow channel. Amperometric measurements were done by applying −0.3V vs. Ag/AgCl using a multichannel CHI 1040 electrochemical workstation as reported previously in similar microfluidic arrays.,29,36(Fig. 1) while flowing a mixture of 100 μM H2O2 in 1 mM HQ in PBS buffer.
Figure 1.

Immunoassay system: (A) syringe pump, injector for samples and standards; (B) assembled detection chamber consisting of 2 machined PMMA plates, top plate holds symmetrically placed reference Ag/AgCl 0.6 mm diameter and 0.2 mm platinum counter electrode wires along the entire length of the 8-sensor array. Peek tubing is fitted to connect inlet and outlet; (C) PDMS channel 2.8 × 0.15 × 1.0 cm, volume 60 μL, placed above sensors; (D) Kanichi screen-printed carbon 8-sensor array alone.
Antibody concentration.
A relatively high concentration of anti-HRP (100 μg/mL) was used to achieve maximum surface coverage for all tested strategies. This concentration of antibodies was also used for different immobilization techniques to achieve comprehensive comparisons in which sensor performance will depend only on the immobilization technique and amounts of antibodies used in the immobilization.
Layer-by-layer (LBL) electrode modification.
LBL film growth was used for anti-HRP immobilization using alternating layers of PDDA and glutathione-gold nanoparticles (AuNP). As reported previously (See supporting Information (SI) file).32 Carboxylic acid groups on AuNP were activated by EDC/NHSS for 10 minutes, rinsed with DI water and dried with nitrogen. Anti-HRP at 100 μg mL−1 was spotted onto sensors, and arrays were left overnight at 4° C. Before use, arrays were washed with PBS-T20, incubated for 1 hour with 1% bovine serum albumin (BSA) in PBS buffer to reduce nonspecific binding.
Electrochemical deposition of graphene oxide (rGO).
The array was immersed in 4 mg/mL graphene oxide (GO) solution prepared using modified Hummer’s method37 (SI file) in 0.5 M lithium perchlorate. rGO was electrochemically deposited at −1.2 V vs. SCE for 60 s. Arrays were rinsed with water and dried with nitrogen. EDC-NHSS was used activate residual carboxylic groups on rGO for ten min, rinsed with water and dried with nitrogen, anti-HRP (100 μg mL−1) was spotted and arrays were left overnight at 4° C. Before use, arrays were washed with PBS-T20 and incubated with 1% BSA in PBS for 1 hr, and then rinsed with PBS-T20.
Antibody immobilization on chitosan.
A thin film of chitosan was formed on sensor electrodes by spotting 0.25 mg mL−1 of chitosan in 0.05 M hydrochloric acid (pH 4.5). After 1 hr incubation, arrays were dried under vacuum, and amine groups of chitosan were activated by spotting 3% glutaraldehyde in PBS (pH 7.8), incubated for 2 hr, washed with water and dried using nitrogen. Anti-HRP (100 μg mL−1) was spotted on the arrays and left overnight at 4° C, then rinsed with PBS-T20, incubated with 1% BSA for 1 hr and rinsed with PBS-T20 before use.
Passive adsorption of antibodies.
100 μg mL−1 of anti-HRP antibodies were adsorbed on sensors by incubating overnight at 4° C. Arrays were rinsed with PBS-T20, blocked with 1% BSA for 1 hr, and washed with PBS-T20 before use.
Passively adsorbed protein A oriented antibody immobilization.
25 μg mL−1 protein A were spotted on bare electrodes and allowed to incubate overnight at 4° C. Arrays were rinsed with PBS-T20 and incubated with 100 μg mL−1 anti-HRP for 3 hr, rinsed with PBS-T20, blocked with 1% BSA in PBS for 1 hr, and washed with PBS-T20 before use.
Covalent immobilization of protein A on AuNP electrode for oriented antibody immobilization.
LBL strategy was utilized to decorate electrode surface with AuNP, that was activated by incubation with EDC/NHSS for 10 minutes. Protein A (25 μg mL−1) was spotted and incubated on electrode surfaces overnight at 4°C. Arrays were rinsed with PBS-T20, and incubated for 3 hr with 100 μg mL−1 anti-HRP, rinsed with PBS-T20, blocked with 1% BSA in PBS for 1 hr and washed with PBS-T20 before use
HRP assay.
Arrays decorated with anti-HRP were inserted into the microfluidic chamber (Figure 1) where PBS buffer flowed at 100 μL min−1 flow rate using a syringe pump (New Era, NE-1000). 100 μL of standard or sample solution was loaded into the sample loop and injected using the sample injector (Rheodyne®, 9725i). Once the sample filled the microfluidic detection chamber, as determined by tests with dye solutions, flow was stopped and sample was incubated for 20 mins. Then, arrays were washed by flowing PBS-T20 for 3 min at 100 μL min−1. Flow was switched from PBS-T20 to 1 μM HQ mediator in PBS and amperometry was done at −0.3 V vs. Ag/AgCl while injecting 100 μL of 100 μM H2O2 in 1 mM HQ/PBS.
Quantitation of antibodies immobilized on electrode surface.
Bicinchoninic acid assay (BCA) was used to quantify the actual number of antibodies immobilized on the electrode surface38. Briefly, 100 μL micro BCA reagent kit (ThermoFisher® 23235) was prepared according to the vendor specification and incubated with 80 electrodes with immobilized antibodies at 37° C. BCA solution with the developed blue color was transferred to microplate and absorbance was measured at 562 nm. The concentration of immobilized anti-HRP was estimated utilizing a calibration graph constructed by running the same BCA procedures for series of standards of anti-HRP (Fig. S1, SI).
Quantitation of active antibodies:
In order to estimate the number of active antibodies immobilized on electrodes, increasing concentrations of HRP were allowed to incubate for 20 minutes with electrodes and electrochemical oxidation catalyzed by HRP was measured. Hydroquinone was used as a mediator to shuttle electrons between electrode surface and enzyme distant from the electrode surface.36 The mediator is essential to improve electrochemical signals resulting from a significant distance between enzyme and electrode surface that leads to slow direct electron transfer. Signal saturation was considered as the point where all active antibodies captured HRP molecules and further increase in HRP did not result in any increase in the signal. HRP concentration at the saturation point was equivalent to the concentration of active antibodies immobilized on electrode surface.
HRP activity.
Arrays decorated with anti-HRP captured HRP from solution using excess amount HRP (50 ng mL−1) for 20 min. Each array was then washed with PBS-T20 and incubated with 100 μL of 4 mg mL−1 O-phenylenediamine (OPD) substrate in 0.05 M phosphate-citrate buffer (pH 5.0). OPD is converted to colored product (2,3–diaminophenazine) and the solution was transferred to a micro-well plate and absorbance measured at 492 nm after stopping reaction with 25 μL 3M HCL.39 Concentration of HRP on electrodes was estimated using a calibration made with standard HRP solutions and the same protocol.
Inter-day assays.
Electrodes were prepared, blocked, rinsed with PBS-T20 and stored in PBS buffer at 4° C for up to 5 days, the tested with 5 pg mL−1 HRP over 5 days. Results were used to estimate the loss of the immobilized antibody activity.
3. Results
Nominal electrode surface area and antibody coverage.
Sensor surface area was estimated before and after surface modifications. Electrochemically active surface area of the bare electrode was estimated at 9.5±0.5 ×10−4 cm2, using the slope of cyclic voltammogram (CV) peak currents vs. square root of the scan rate (ѵ1/2) for 0.5 mM ferrocene methanol (Fc-MeOH, D: diffusion coefficient of Fc-MeOH in TEAP = 7 × 10−6 cm2 s−1)40 in 0.1 M tetra-ethyl ammonium perchlorate (TEAP, Fig. 2).41 Surface area after modification with rGO and was 2.1±0.1 ×10−3 cm2, and for AuNP 2.4±0.2 × 10−3 cm2, while chitosan electrodes had area 9.1±0.3 ×10−4 cm2. Surface areas were used to compute the theoretical monolayer coverage of antibodies using antibody dimensions (14.5 × 8.5 × 4 nm3),42 and considering side-on or end-on orientation (Table 1).43
Fig. 2.

Electrode surface area studies before and after electrode modifications using 0.5 mM Fc-MeOH in 0.1 M TEAP vs Ag/AgCl (0.14M NaCl); (A) Cyclic voltammograms at different scan rates (from 10 to 200 mV/s) for bare electrode; (B) peak current (Ip) vs. square root of the scan rate (ν1/2)for bare electrode; (C) Ip vs. ν1/2 for rGO coated electrode; (D) Ip vs. ν1/2 for AuNP electrode; (E) Ip vs. ν1/2 for chitosan electrode. (Error bars = standard deviation, n=8)
Table 1.
Actual antibody coverage from BCA assay and theoretical estimated antibody coverage based on electrode surface area and antibody dimensions
| Immobilization Strategy | Theoretical Coverage/electrode × 109 | Coverage found × 109 | |
|---|---|---|---|
| (Side-on) | (end-on) | ||
| AuNP/(EDC/NHSS) | 1.90 | 7.10 | 6.2± 0.3 |
| rGO/(EDC/NHSS) | 1.70 | 6.20 | 4.3±0.3 |
| Chitosan/Glutaraldehyde | 0.73 | 2.40 | 6.1±0.6 |
| Direct antibody Adsorption | 0.76 | 2.70 | 1.1±0.03 |
| Protein A (Passive Adsorption) | 0.24 | 0.30 | 0.26±0.01 |
| Protein A on AuNP electrodes | 0.54 | 0.64 | 6.1±0.4 |
Protein A is a 42 kDa polypeptide with two functionally distinct halves, A C-terminal half for binding to cell walls and an N-terminal half for IgG binding. The protein binding portion is composed of five semi-identical, three-helix bundles, i. e. Ab-binding domains (E-D-A-B-C) that are interconnected with highly flexible linkers.44 Average dimensions of protein A are 25–30 nm,45 so theoretical maximum surface coverage of protein A was 1.2 × 108 – 1.5 × 108 on bare electrodes and 2.7 × 108 - 3.2 × 108 on AuNP electrodes. Although protein A has five antibody binding sites, actual binding of antibodies per protein A molecule is been estimated at 2.46 Theoretical numbers of antibodies were also estimated for binding onto protein A on electrodes (Table 1).
Actual antibody coverage.
BCA assays were used to measure the actual number of bound antibodies on the electrode (Table 1). Surprisingly, chitosan electrodes had antibody coverage higher than the theoretically estimated coverage since it had 1.4±0.1 × 10−3 μg anti-HRP/electrode equivalent to 6.1±0.6 × 109 anti-HRP/electrode, this was about 3 times higher coverage than the theoretical value, probably because of the 3 dimensional chitosan hydrogen forms with an area larger than that of the electrode.47 Protein A immobilized on AuNP electrodes had approximately 10 times higher number of antibodies compared to the theoretical estimation based on calculated electrochemical active surface area of the modified electrodes, as flexible anchoring of protein A on a dense film of AuNP on electrode surface provided an excellent platform to covalently immobilize protein A that had the flexibility to bind to up to 6.1 ± 0.4 × 109 anti-HRP/electrode. Passively adsorbed Protein A had a much smaller number of immobilized anti-HRP.
Active Antibodies.
The saturation level of HRP signals in the arrays where signals level off, was used to estimate the number of active antibodies. Bulk HRP concentration at each saturation point was used to estimate the maximum number of HRP molecules that can interact with antibodies on electrode surface during incubation. Number of HRP molecules that would bind to antibodies immobilized on each electrode, from the volume of solution inside microfluidic chamber, was estimated using Einstein’s diffusion equation (Eq. 1) assuming that binding is diffusion controlled.48 For diffusion coefficient (D) of HRP 5 × 10−7 cm2 s−1,49 the distance travelled by HRP in 20 min incubation time is 0.012 mm.
| (1) |
where d is distance in cm, D is the diffusion coefficient, and t is time in sec.
The solution containing HRP available to bind to anti-HRP immobilized on each electrode is a hemisphere of radius 0.012 mm with volume 7.24 × 10−6 μL (Fig. S2, SI). The number of HRP molecules in this available volume of solution to bind anti-HRP was estimated from the saturation concetrations (Tables2 and Table S1, SI). Amounts of actively binding antibodies were relatively low except for those using an initial layer of protein A, which had 85% active antibodies for AuNP electrodes and 98% on bare carbon.,
Table 2.
Active antibodies/electrode using different immobilization strategies on screen printed carbon electrode using electrochemical assay and OPD enzyme activity assay (n=16 for electrochemical measurements and n=3 for OPD assay).
| Immobilization Strategy | No. of Active antibodies/electrode × 109 | |
|---|---|---|
| Electrochem. estimate | OPD Assay | |
| AuNP/(EDC/NHSS) | 2.56±0.20 | 2.94±0.11 |
| rGO/(EDC/NHSS) | 1.28±0.14 | 1.48±0.13 |
| Chitosan/Glutaraldehyde | 1.28±0.09 | 1.00±0.05 |
| Direct antibody Adsorption | 2.56±0.08 | 2.11±0.11 |
| Protein A (Passive Adsorption) | 0.26±0.01 | 0.23±0.01 |
| Protein A on AuNP electrodes | 5.13±0.36 | 5.86±0.41 |
Active antibodies on each electrode was also measured using the HRP enzyme activity assay for oxidaton of O-phenylenediamine (OPD). Enzymes captured on sensor arrays were incubated with OPD for 1 hr and color developed was used to estimate the number of HRP molecules that correspond to the number of active anti-HRP. Calibration using soluble HRP with was used to esimate the concenterations of HRP on arrays (Fig. S3, SI). Good agreement between results obtained from electrochemical and OPD assays was found (Table 2).
Electrode to electrode variation.
Selected concentrations of HRP were assayed on different electrode arrays on the same day to estimate intra-day signal variations for each immobilization strategy. Passive adsorption of antibodies on electrode surface and oriented antibody immobilization using passively adsorbed protein A gave relative standard deviation (RSD) less than ±5%. For covalent attachment, RSDs were ±11%, for rGo electrodes, ±8% for AuNP, and 7% for chitosan and oriented antibodies on protein A covalently immobilized on AuNP (Fig. S4, SI).
Stability.
Arrays were challenged with the same concentration of HRP over 5 days. Chitosan coated electrodes showed the best stability with only a small loss in signal over 5 days (≤ 7%). Electrodes with passively adsorbed antibodies were the fastest to deteriorate with 15–20% loss of activity each day in the first 3 days. AuNP and rGO electrodes were relatively stable for the first three days of storage (~5% decrease/day) followed by larger decrease in signal in 5th day of storage (~10%). Antibodies adsorbed on protein A electrodes had good stability over the test period with approximately 20% decrease in the overall signal after 5 days (Fig. S5, SI,Table 3).
Table 3.
Comparison of performance for HRP sensors using different antibody immobilization strategies on screen printed carbon arrays
| Immobilization strategy | LOD (fgmL−1) | Dynamic Range | Sensitivity (nA/log (pg mL−1) | No. of Ab/Electrode (×109) | Percent active antibodies | Signal loss after 5 days storage |
|---|---|---|---|---|---|---|
| Anti-HRP/AuNP | 40 | 40 fg ML−1 | 2.40 | 6.2 ± 0.3 | 21% | ≤ 30% |
| Anti-HRP/rGO | 40 | 40 fg mL−1 | 0.55 | 4.3 ± 0.3 | 30% | ≤ 35% |
| Anti-HRP/Chitosan | 40 | 40 fg mL−1 | 0.90 | 6.1 ± 0.6 | 21% | ≤ 7% |
| Anti-HRP/Protein A/AuNP | 40 | 40 fg mL−1 | 2.01 | 6.1 ± 0.4 | 85% | ≤ 20 |
| Anti-HRP/bare electrode | 4 | 8 fg raL−1 | 1.75 | 1.1 ± 0.03 | 60% | >60% |
| Anti-HRP/Protein A/bare electrode | 8 | 8 fg mL−1 | 1.62 | 0.26 ± 0.1 | 98% | ≤ 25 |
4. Discussion
Results above demonstrate the influence of antibody immobilization strategy and coatings on screen-printed carbon electrode for immunoarrays. Sufficient binding activity and availability of antibodies immobilized on sensor electrodes is a key factor for sensor performance, we found that other factors such as stability, degree of antibody coverage, orientation, and assay to assay variations also have an influence. We found that highly oriented antibody systems (using protein A) can extend the dynamic range, but have little influence on sensitivity or LOD, most easily seen by comparing AuNP-electrodes with Protein A/AuNP (Table 3). This goes against the common view that improving antibody orientation also improves sensitivity and LOD of immunoassays.7,8 Sensors with 20–30% orientation (Table 3) still have enough active antibodies to achieve good sensitivity and LOD in the lower concentration range, and had AuNP electrodes with 21% active antibodies had the best sensitivity. This result agrees with our earlier study of sensors coated with upright single-wall carbon nanotube forests having large antibody coverage, but ~30% active antibodies.50 These sensors had excellent sensitivity and LODs for prostate specific antigen and interlukin-6 proteins in serum. For sensors with 85–95% correct antibody orientation, the main effect is maintenance of sensitivity in the higher concentration range because more of the antibodies are being active, and that extends dynamic range upward. It is possible that these conclusions may depend on antigen and antibodies size.
Antibody coverage was highest when antibodies were bound to protein A/AuNP films electrodes (Table 3), although these sensors did not have the best sensitivities. In addition, nanostructured dense 5 nm AuNP packing32 and the chitosan hydrogel property both extend the surface area available for Ab binding, leading to the binding of more antibodies. Electrodes with passively adsorbed antibodies showed the highest LODs (Table 3), but were the least stable (Table 3), Instability can be attributed to documented hydrophobic interactions between antibodies and electrode which lead to denaturation and loss of binding activity.51 The hydrogel nature of chitosan causes antibody-chitosan matrix swelling in aqueous solution to more than 600% of its original mass.47 The hydrogel provides high water content that also helps preserve antibody activity during storage by stabilizing conformation.52,53
rGO coated electrodes may have mixed affinities toward antibodies through covalent binding to residual carboxylic groups after electrochemical deposition and hydrophobic adsorption to graphene. This explains the intermediate antibody activity (30%), that lies between covalent immobilization and passive adsorption, of rGO coated sensors. Sensitivities and LODs of sensors with covalently immobilized antibodies were similar with LOD 40 fg mL−1 for chitosan modified, rGO coated and AuNP electrodes (Table 3). Wider dynamic range achieved by AuNP sensors compared to other covalent immobilization techniques can be attributed to high surface coverage of antibodies compared to other sensors. Although chitosan modified electrodes had surface antibody coverage similar to AuNP films, its narrower dynamic range is due to a degree of electron transfer blockage by the chitosan (Table 2).
Protein A passively adsorbed on the sensors preserved 98% of immobilized antibody activity, and maintained up to 80% of sensor activity after 5 days storage, but had the worst dynamic range (Table 3). This is presumably due to low surface coverage of antibodies, about 10% of that of the other sensors. This was overcome by covalent immobilization of protein A onto AuNP electrodes, which offers flexible anchoring and allowed a large increase in antibody coverage while maintaining 85% activity.
5. Conclusions
Results show that ability of the immunosensors to achieve high sensitivity is mainly a function of having sufficient active antibodies immobilized and the degree of surface coverage. Protein A mediated antibody surface conjugation that allowed oriented immobilization of antibodies preserved 98% of antibody activity, but was outperformed by other sensors possessed higher surface coverage (Table 3). In general, a high degree of antibody orientation on AuNP sensor did not improve sensitivity or LOD, but extended dynamic range to higher concentrations. Our findings suggest that increasing the number of antibodies on sensors by increasing electrode surface area is key factor to improve sensitivity. In addition, stable covalent conjugation of antibodies protects them from hydrophobic interaction-induced denaturation found in passive adsorption.
Supplementary Material
Fig. 3.

Calibration for different anti-HRP immobilization strategies; (A) Example showing amperometric peaks with increasing concentrations of HRP using antibodies (Ab) passively adsorbed on bare carbon. Calibration graphs: (B) passively adsorbed Ab on bare electrodes; (C) covalently immobilized Ab on AuNP electrodes; (D) covalently immobilized Ab on rGO electrodes; (E) covalently immobilized on chitosan electrodes; (F) Ab oriented onto protein A passively adsorbed on bare electrodes; (G) Ab oriented onto protein A covalently immobilized on AuNP electrode. Logarithmic fits shown as blue dashed line; arrows indicate saturation points (Error bars = standard deviation, n=8)
Acknowledgment
This work was supported financially by an Academic Plan Grant from The University of Connecticut and in part by Grant no. EB016707 from the National Institute of Biomedical Imaging and Bioengineering (NIBIB), NIH.
References
- (1).Wei Y, Zeng Q, Huang J, Hu Q, Guo X and Wang L, Chem. Commun 2018, 54, 9163–9166. [DOI] [PubMed] [Google Scholar]
- (2).Lequin RM, Clin. Chem 2005, 51, 2415–2418. [DOI] [PubMed] [Google Scholar]
- (3).Peluso P, Wilson DS, Do D, Tran H, Venkatasubbaiah M, Quincy D, Heidecker B, Poindexter K, Tolani N, Phelan M, et al. , Anal. Biochem 2003, 312, 113–124. [DOI] [PubMed] [Google Scholar]
- (4).Koczula KM and Gallotta A, Essays Biochem. 2016, 60, 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Brock P, Eldred EW, Woiszwillo JE, Doran M and Schoemaker HJ, Clin. Chem 1978, 24, 1595–1598. [PubMed] [Google Scholar]
- (6).Rusling JF, Chem. Rec 2012, 12, 164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Shen M, Rusling J and Dixit CK, Methods, 2017, 116, 95–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sharma S, Byrne H and O’Kennedy RJ, Essays Biochem. 2016, 60, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Yáñez-Sedeño P, Campuzano S and Pingarrón JM, Chem. Commun 2019, 55, 2563–2592. [DOI] [PubMed] [Google Scholar]
- (10).Rusling JF, Anal. Chem 2013, 85, 5304–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Jokerst JC, Emory JM and Henry CS, Analyst 2012, 137, 24–34. [DOI] [PubMed] [Google Scholar]
- (12).Chikkaveeraiah BV, Mani V, Patel V, Gutkind JS and Rusling JF, Biosens. Bioelectron 2011, 26, 4477–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Carvajal S, Fera SN, Jones AL, Baldo TA, Mosa IM, Rusling JF and Krause CE, Biosens. Bioelectron 2018, 104, 158–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lee G, Lee J, Kim J, Choi HS, Kim J, Lee S and Lee H, Sci. Rep 2017, 7, 7545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bertolino FA, De Vito IE, Messina GA, Fernández H and Raba J, J. Electroanal. Chem 2011, 651, 204–210. [Google Scholar]
- (16).Uosaki K, Sato Y and Kita H, Langmuir 1991, 7, 1510–1514. [Google Scholar]
- (17).Määttänen A, Ihalainen P, Pulkkinen P, Wang S, Tenhu H and Peltonen J, ACS Appl. Mater. Interfaces 2012, 4, 955–964. [DOI] [PubMed] [Google Scholar]
- (18).Malhotra BD, Chaubey A and Singh SP, Anal. Chim. Acta 2006, 578, 59–74. [DOI] [PubMed] [Google Scholar]
- (19).Tiwari JN, Vij V, Kemp KC and Kim KS, ACS Nano 2016, 10, 46–80. [DOI] [PubMed] [Google Scholar]
- (20).Céspedes F, Martinez-Fàbregas E and Alegret S, TrAC Trends Anal. Chem 1996, 15, 296–304. [Google Scholar]
- (21).Weber SG, Anal. Chem 1989, 61, 295–302. [DOI] [PubMed] [Google Scholar]
- (22).Céspedes F and Alegret S, Food Technol. Biotechnol 1996, 34, 143–146. [Google Scholar]
- (23).Arduini F, Micheli L, Moscone D, Palleschi G, Piermarini S, Ricci F and Volpe G, TrAC Trends Anal. Chem 2016, 79, 114–126. [Google Scholar]
- (24).Cinti S, Arduini F, Moscone D, Palleschi G and Killard AJ, Sensors 2014, 14, 14222–14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Arduini F, Amine A, Majorani C, Di Giorgio F, De Felicis D, Cataldo F, Moscone D and Palleschi G, Electrochem. Commun 2010, 12, 346–350. [Google Scholar]
- (26).Nunez-Bajo E, Blanco-López MC, Costa-García A and Fernández-Abedul MT, Anal. Chem 2017, 89, 6415–6423. [DOI] [PubMed] [Google Scholar]
- (27).Hatakeyama Y, Onishi K and Nishikawa K, RSC Adv. 2011, 1, 1815–1821. [Google Scholar]
- (28).Yu X, B Munge V Patel G Jensen, Bhirde A, Gong JD, Kim SN, Gillespie J, Gutkind JS, Papadimitrakopoulos F, et al. , J. Am. Chem. Soc 2006, 128, 11199–11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Krause CE, Otieno BA, Bishop GW, Phadke G, Choquette L, Lalla RV, Peterson DE and Rusling JF, Anal. Bioanal. Chem 2015, 407, 7239–7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Lvov YM, Lu Z, Schenkman JB, Zu X and Rusling JF, J. Am. Chem. Soc 1998, 120, 4073–4080. [Google Scholar]
- (31).Lvov Y, Decher G and Moehwald H, Langmuir 1993, 9, 481–486. [Google Scholar]
- (32).Mani V, Chikkaveeraiah BV, Patel V, Gutkind JS and Rusling JF, ACS Nano 2009, 3, 585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Sharafeldin M, Bishop GW, Bhakta S, El-Sawy A, Suib SL and Rusling JF, Biosens. Bioelectron 2017, 91, 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Parkash O, Yean CY and Shueb RH, Diagnostics 2014, 4, 165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Pemberton RM and Hart JP, Preparation of Screen-Printed Electrochemical Immunosensors for Estradiol, and Their Application in Biological Fluids In Biosensors and Biodetection: Methods and Protocols: Electrochemical and Mechanical Detectors, Lateral Flow and Ligands for Biosensors; Rasooly A, Herold KE, Eds.; Methods in Molecular BiologyTM; Humana Press: Totowa, NJ, 2009; pp 85–98. [DOI] [PubMed] [Google Scholar]
- (36).Chikkaveeraiah BV, Mani V, Patel V, Gutkind JS and Rusling JF, Biosens. Bioelectron 2011, 26, 4477–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kim MI, Kim MS, Woo M-A, Ye Y, Kang KS, Lee J and Park HG, Nanoscale 2014, 6, 1529–1536. [DOI] [PubMed] [Google Scholar]
- (38).Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ and Klenk DC, Anal. Biochem 1985, 150, 76–85. [DOI] [PubMed] [Google Scholar]
- (39).Porstmann B, Porstmann T and Nugel E, J. Clin. Chem. Clin. Biochem. Z. Klin. Chem. Klin. Biochem 1981, 19, 435–439. [DOI] [PubMed] [Google Scholar]
- (40).Cannes C, Kanoufi F and Bard AJ, J. Electroanal. Chem, 2003, 547, 83–91. [Google Scholar]
- (41).Bard Allen J. and Faulkner Larry R., Electrochemical Methods: Fundamentals and Applications, New York: Wiley, 2001, 2nd ed. [Google Scholar]
- (42).Saha B, Evers TH and Prins MWJ, Anal. Chem 2014, 86, 8158–8166. [DOI] [PubMed] [Google Scholar]
- (43).Sarma VR, Silverton EW, Davies DR and Terry WD, J. Biol. Chem 1971, 246, 3753–3759. [PubMed] [Google Scholar]
- (44).Deis LN, Pemble CW, Qi Y, Hagarman A, Richardson DC, Richardson JS and Oas TG, Structure 2014, 22, 1467–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Tripathi K and Driskell JD, ACS Omega 2018, 3, 8253–8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Yang L, L.; Biswas ME and Chen P, Biophys. J 2003, 84, 509–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Sharafeldin M, Kadimisetty K, Bhalerao KR, Bist I, Jones A, Chen T, Lee NH and Rusling JF, Anal. Chem 2019, 91, 7394–7402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Wilson MS and Nie W, Anal. Chem 2006, 78, 2507–2513. [DOI] [PubMed] [Google Scholar]
- (49).Schurr JM, Biophys. J 1970, 10, 717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Malhotra R, Papadimitrakopoulos F and Rusling JF, Langmuir, 2010, 26, 15050–15056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Butler JE, Ni L, Brown WR, Joshi KS, Chang J, Rosenberg B and Voss EW, Mol. Immunol 1993, 30, 1165–1175. [DOI] [PubMed] [Google Scholar]
- (52).Higashi T, Ohshita N, Hirotsu T, Yamashita Y, Motoyama K, Koyama S, Iibuchi R, Uchida T, Mieda S and Handa K, et al. , J. Pharm. Sci 2017, 106, 1266–1274. [DOI] [PubMed] [Google Scholar]
- (53).Szenczi Á, Kardos J, Medgyesi GA and Závodszky P, Biologicals 2006, 34, 5–14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.