

Abstract
Alzheimer’s Amyloid beta can perform a wide variety of actions that are highly concentration-dependent. This note aims to provide a framework for basic considerations on what might be considered brain-relevant concentrations of the peptide. Some implications for the therapeutic implementation of the recently emerged oligomer-to-fibril strategy are discussed.
Graphical Abstract
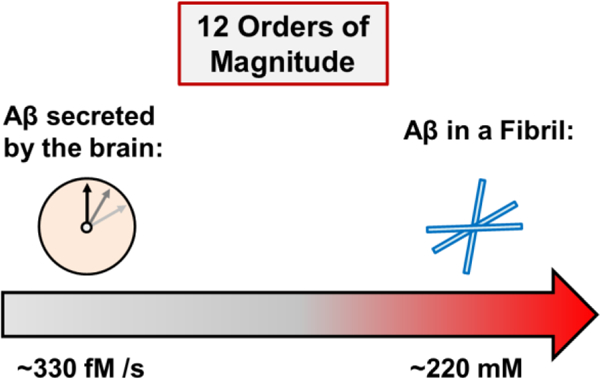
A question of concentration: Alzheimer’s Amyloid β 42 has an unusually wide range of differential activity as a function of concentration. The present analysis suggests that, in order to better understand the system, it needs to be considered over twelve orders of magnitude – from femtomolar to millimolar.
Amyloid β (Aβ) is an intrinsically disordered peptide and, in its 42 amino acid long isoform is the believed culprit of Alzheimer’s Disease.[1] Much attention has been focused on its biophysical, biological and electrophysiological properties and, depending on the context, the concentrations at which Aβ42 was studied ranged from femtomolar to millimolar, covering a remarkable twelve orders of magnitude.a
The highest concentration of Aβ42 in the human brain is found in senile plaques,[2] where the peptide is packed into a dense composite of Aβ fibrils, as well as other biomolecules.[3] As the hypothetical upper boundary of physiologically relevant Aβ42 concentration, the fibrillary state appears reasonable, although plaque heterogeneity will produce somewhat lower values (vide infra). Aβ42 has a molar mass of 4514 g/mol. Assuming fibril density of ~1 g/mL (i.e., similar to water, although likely slightly higher, since fibrils do sediment slowly in water), this yields a concentration of 221.5 millimolar (i.e., 221.5 mmol / L) Aβ42 “monomer-in-fibril”. Quantitative post mortem analyses of patient-derived senile plaques revealed Aβ (40 and 42) as their predominant components,[2] but also showed a remarkable heterogeneity of the deposits, additional plaque constituents including diverse proteins and peptides,[2b, 4] lipids,[5] RNA, carbohydrates, as well as various metal ions.[3] As a result, the Aβ42 concentration in a senile plaque is expected to be somewhat lower than 221.5 mM, but certainly still in the high mM range. In consequence of a recent paradigm shift, Aβ fibrillary deposits are no longer considered to be the culprit of Alzheimer’s Disease, but instead benign, possibly even protective, which is likely a consequence of fibril-borne Aβ42 being packaged into a relatively inert state with very low biological activity.[1b, 6] Still, because Aβ42 fibrils exist in the brain, the staggeringly high concentration of 221.5 mM of “monomer-in-fibril” is formally relevant, although clearly only so in the context of plaques. It should be considered the absolute upper boundary. Such fibrillary deposits are formed very slowly, over decades, and the more disease-relevant, soluble species of Aβ are unlikely to exist in the brain anywhere near those extreme concentrations.[1b, 7]
Toxicity of soluble Aβ42 aggregation intermediates, i.e., diffusible oligomers, can be robustly observed through exposure of cells to the peptide at low to intermediate μM concentration.[8] Assuming an IC50 concentration of Aβ42 on the order of ~10–100 μM (IC50 can vary to degree between preparation), this means that Aβ42 oligomers are highly neurotoxic at a concentration that is ~3–4 orders of magnitude lower than that calculated for Aβ42 “monomer-in-fibril”. If one hypothetically considers a human brain (~1.5 L volume) that is homogeneously filled with cerebrospinal fluid at 50 μM Aβ42 (a hypothetical neurotoxic concentration), oligomer-to-fibril conversion would condense it by a factor of 221.5 mM / 50 μM = 221500 μM / 50 μM = 4430, i.e., 0.34 mL Aβ42 fibril. Because of identical arguments applied in reverse direction, 0.34 mL reservoir of Aβ42 fibrils could, in principle, produce 1.5 L of a homogeneous, highly neurotoxic Aβ42 solution at 50 μM. This prompts a note of caution with regard to therapeutic approaches that act by solubilizing Aβ fibrils and plaques, since those approaches may result in the undesirable release of toxic entities. It is worth noting that Aβ42 is also bactericidal, and is capable of killing 80 % of various microorganisms within 6 h of application at 50 μg/mL (11 μM).[9] Whether Aβ42 has truly evolved to be part of the host immune system remains subject of active research.[10]
The Aβ42 peptide has also received considerable attention for its synaptomodulatory activity in hippocampal neuronal networks.[11] Exposure of (typically rat or mouse) hippocampal slices to oligomeric Aβ42 preparations produces robust inhibition of long-term potentiation (LTP, a well-established model for synaptic plasticity and learning).[11a] The effect is readily observed at ~50–500 nM concentration,[11b] i.e., ~2–3 orders of magnitude lower than the concentration required in order to produce neurotoxic effects. The mechanism of LTP-inhibition by Aβ42 remains subject of active research.[1b, 11d, 12] It is intriguing to note that at ~100–300 pM concentration, Aβ42 was shown to be LTP-enhancing,[11b] which was later suggested to be a consequence of positive regulation of neurotransmitter release probability at hippocampal synapses.[11c] A recent quantitative mass spectrometric study reported Aβ42 concentrations of ~0.5 ng/mL (~110 pM) in the CSF of non-diseased humans. Intriguingly, this falls into the concentration regimen of Aβ42, in which the peptide was found to positively regulate LTP.[11b, c]
Aβ42 was recently shown to be generated at a frequency of 2–4 molecules per neuron per second.[13] With the human brain containing on the order of 100 × 109 neurons, this yields ~3 × 1011 Aβ42 molecules per second for the brain. Using Avogadro’s constant of 6 × 1023, this can be converted to 330 femtomoles of Aβ42 generated by the human brain every second, or 28.5 nanomoles of it per day. This number is likely to be somewhat of an underestimation, since the human brain has a comparable number of glial cells,[14] including astrocytes, which have been found capable of producing Aβ under certain conditions, such as stress.[15] The number listed above should therefore be considered a conservative estimate, a lower boundary. Under healthy conditions, accumulation and clearance are balanced, and sleep appears to play a vital role.[16] Disruption of Aβ42 brain clearance can result in accumulation of synaptotoxic peptide concentrations (~50–500 nM) within days or weeks, if clearance is impaired. Intriguingly, if all Aβ42 produced by the human brain over the course of a year - a total of ~10.4 micromoles - were converted into pure fibrils, the resultant deposit would have a volume of 0.05 mL or 50 mm3. This is noteworthy, because, showing that large volumes of soluble Aβ42 oligomers can be compacted into very small solid deposits, it highlights the potential of the recently established oligomer-to-fibril conversion mechanism, which was proposed as an alternative approach to eliminate neurotoxic, soluble Aβ42 oligomers.[8b, c, 17]
A remarkable property of the Aβ42 system that emerges from this analysis is that, in order to better understand its action in the context of the human brain, an unusually wide range of concentrations must be considered. The purpose of this short note was to provide a set of ballpark concentration guidelines and to stimulate cross-disciplinary dialogue, which is vitally needed, so that advances can be made with regard to Aβ42 actions in Alzheimer’s Disease and beyond.
Table 1.
Ballpark concentrations of Aβ42 in a variety of physiologically relevant contexts.
| Context | Ballpark Concentration |
|---|---|
| Aβ42 “monomer-in-fibril” | 221.5 mM |
| Neurotoxicity | ~10–100 μM |
| Negative LTP modulation | ~50–500 nM |
| Positive LTP modulation | ~50–500 pM |
| Aβ42 generation by the brain | ≥330 fM s−1 |
Acknowledgements
The author thanks Dr. Jeremy C. Lee, Mr. Thomas S. Finn, Ms. Ariel J. Kuhn and Mr. Alejandro R. Foley for helpful discussions and the NIH for funding (R21AG058074).
Footnotes
For comparison, a human hair has a thickness of ~0.1 mm and the distance from Los Angeles to Beijing is ~10,000 km. This corresponds to eleven orders of magnitude, one less than the concentration range of Aβ42.
References
- [1].a) Chiti F and Dobson CM, Annu. Rev. Biochem 2006, 75, 333–366; [DOI] [PubMed] [Google Scholar]; b) Selkoe DJ and Hardy J, EMBO Mol. Med 2016, 8, 595–608; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Bilousova T, Miller CA, Poon WW, Vinters HV, Corrada M, Kawas C, Hayden EY, Teplow DB, Glabe C, Albay R 3rd, Cole GM, Teng E and Gylys KH, Am J Pathol 2016, 186, 185–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Glenner GG and Wong CW, Biochem. Biophys. Res. Commun 1984, 120, 885–890; [DOI] [PubMed] [Google Scholar]; b) Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL and Beyreuther K, Proc. Nat. Acad. Sci. USA 1985, 82, 4245–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Stewart KL and Radford SE, Biophys. Rev 2017, 9, 405–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Liao L, Cheng D, Wang J, Duong DM, Losik TG, Gearing M, Rees HD, Lah JJ, Levey AI and Peng J, J. Biol. Chem 2004, 279, 37061–37068. [DOI] [PubMed] [Google Scholar]
- [5].Gellermann GP, Appel TR, Tannert A, Radestock A, Hortschansky P, Schroeckh V, Leisner C, Lutkepohl T, Shtrasburg S, Rocken C, Pras M, Linke RP, Diekmann S and Fandrich M, Proc. Natl. Acad. Sci. USA 2005, 102, 6297–6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, Elliott JI, Van Nostrand WE and Smith SO, Nat. Struct. Mol. Biol 2010, 17, 561–U556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Querfurth HW and LaFerla FM, N. Engl. J. Med 2010, 362, 329–344. [DOI] [PubMed] [Google Scholar]
- [8].a) Ono K, Condron MM and Teplow DB, Proc. Natl. Acad. Sci. USA 2009, 106, 14745–14750; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bieschke J, Herbst M, Wiglenda T, Friedrich RP, Boeddrich A, Schiele F, Kleckers D, Lopez del Amo JM, Grüning BA, Wang Q, Schmidt MR, Lurz R, Anwyl R, Schnoegl S, Fändrich M, Frank RF, Reif B, Günther S, Walsh DM and Wanker EE, Nat. Chem. Biol 2012, 8, 93–101; [DOI] [PubMed] [Google Scholar]; c) Dutta S, Foley AR, Warner CJA, Zhang X, Rolandi M, Abrams B and Raskatov JA, Angew. Chem. Int. Ed 2017, 56, 11506–11510; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Ovchinnikova OY, Finder VH, Vodopivec I, Nitsch RM and Glockshuber R, J. Mol. Biol 2011, 408, 780–791. [DOI] [PubMed] [Google Scholar]
- [9].Spitzer P, Condic M, Herrmann M, Oberstein TJ, Scharin-Mehlmann M, Gilbert DF, Friedrich O, Grömer T, Kornhuber J, Lang R and Maler JM, Sci. Rep 2016, 6, 32228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Kumar DKV, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, Lefkowitz A, McColl G, Goldstein LE, Tanzi RE and Moir RD, Sci. Transl. Med 2016, 8, 340ra372; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) De Lorenzi E, Chiari M, Colombo R, Cretich M, Sola L, Vanna R, Gagni P, Bisceglia F, Morasso C, Lin JS, Lee M, McGeer PL and Barron AE, J. Alzheimers Dis 2017, 59, 1213–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ and Selkoe DJ, Nature 2002, 416, 535–539; [DOI] [PubMed] [Google Scholar]; b) Puzzo D, Privitera L, Leznik E, Fa M, Staniszewski A, Palmeri A and Arancio O, J. Neurosci 2008, 28, 14537–14545; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E and Slutsky I, Nat. Neurosci 2009, 12, 1567–1576; [DOI] [PubMed] [Google Scholar]; d) Orr AL, Hanson JE, Li D, Klotz A, Wright S, Schenk D, Seubert P and Madison DV, Neuron 2014, 82, 1334–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Hong S, Ostaszewski BL, Yang T, O’Malley TT, Jin M, Yanagisawa K, Li SM, Bartels T and Selkoe DJ, Neuron 2014, 82, 308–319; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Selkoe DJ, Nat. Med 2011, 17, 1060–1065; [DOI] [PubMed] [Google Scholar]; c) Selkoe DJ, Behav. Brain Res 2008, 192, 106–113; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Haass C and Selkoe DJ, Nature Reviews Molecular Cell Biology 2007, 8, 101–112. [DOI] [PubMed] [Google Scholar]
- [13].Moghekar A, Rao S, Li M, Ruben D, Mammen A, Tang X and O’Brien RJ, J. Biol. Chem 2011, 286, 15989–15997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Herculano-Houzel S, Front. Human Neurosci 2009, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Frost G R. and Li Y-M, Open Biol 7, 170228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Brower CS, Piatkov KI and Varshavsky A, Mol. Cell 2013, 50, 161–171; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Varshavsky A, Prot. Sci 2012, 21, 1634–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Sonzini S, Stanyon HF and Scherman OA, Physical Chemistry Chemical Physics 2017, 19, 1458–1465; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Raskatov JA, Chem. Eur. J 2017, 23, 16920–16923. [DOI] [PubMed] [Google Scholar]