

Abstract
Background
Mechanical ventilation (MV) is a life‐saving measure for patients in respiratory failure. However, prolonged MV results in significant diaphragm atrophy and contractile dysfunction, a condition referred to as ventilator‐induced diaphragm dysfunction (VIDD). While there are currently no clinically approved countermeasures to prevent VIDD, increased expression of heat shock protein 72 (HSP72) has been demonstrated to attenuate inactivity‐induced muscle wasting. HSP72 elicits cytoprotection via inhibition of NF‐κB and FoxO transcriptional activity, which contribute to VIDD. In addition, exercise‐induced prevention of VIDD is characterized by an increase in the concentration of HSP72 in the diaphragm. Therefore, we tested the hypothesis that increased HSP72 expression is required for the exercise‐induced prevention of VIDD. We also determined whether increasing the abundance of HSP72 in the diaphragm, independent of exercise, is sufficient to prevent VIDD.
Methods
Cause and effect was determined by inhibiting the endurance exercise‐induced increase in HSP72 in the diaphragm of exercise trained animals exposed to prolonged MV via administration of an antisense oligonucleotide targeting HSP72. Additional experiments were performed to determine if increasing HSP72 in the diaphragm via genetic (rAAV‐HSP72) or pharmacological (BGP‐15) overexpression is sufficient to prevent VIDD.
Results
Our results demonstrate that the exercise‐induced increase in HSP72 protein abundance is required for the protective effects of exercise against VIDD. Moreover, both rAAV‐HSP72 and BGP‐15‐induced overexpression of HSP72 were sufficient to prevent VIDD. In addition, modification of HSP72 in the diaphragm is inversely related to the expression of NF‐κB and FoxO target genes.
Conclusions
HSP72 overexpression in the diaphragm is an effective intervention to prevent MV‐induced oxidative stress and the transcriptional activity of NF‐κB and FoxO. Therefore, overexpression of HSP72 in the diaphragm is a potential therapeutic target to protect against VIDD.
Keywords: HSP70, Muscle atrophy, VIDD, Respiratory, Mitochondria
Introduction
Mechanical ventilation (MV) is required for millions of patients each year to sustain adequate pulmonary ventilation during surgery and in conditions of respiratory insufficiency (e.g. coma, spinal cord injury, heart failure, and chronic obstructive pulmonary disorder).1, 2 While MV is a life‐saving modality, prolonged MV elicits diaphragm weakness, resulting in difficulties in weaning patients from the ventilator.3, 4 Specifically, prolonged MV promotes the rapid atrophy of diaphragm muscle fibres and contractile dysfunction, a condition referred to as ventilator‐induced diaphragm dysfunction (VIDD).4 Currently, there is no standard therapeutic strategy to combat VIDD. Therefore, further work is required to develop effective interventions to prevent VIDD.
Heat shock proteins (HSPs) are chaperone proteins whose expression is greatly induced in skeletal muscle in response to environmental or metabolic stress.5 Particularly, the 72 kDa HSP isoform (HSP72) is upregulated in response to exercise6, 7, 8 and heat stress.9, 10 In regard to MV, previous work from our laboratory revealed that endurance exercise training prior to the initiation of MV is sufficient to prevent VIDD,8 with evaluation of the proteomic changes occurring in the diaphragm following endurance exercise training revealing that 2 weeks of treadmill running results in a significant increase in diaphragm HSP72 levels.11 Further, repeated bouts of heat stress have also been shown to protect against VIDD, presumably as result of a progressive accumulation of HSP72 in the diaphragm.10 These findings are significant because a therapeutic benefit of HSP72 has been established in skeletal muscle during conditions that promote muscle wasting (i.e. muscle dystrophy and muscle immobilization).12, 13, 14
While the physiological significance of increased HSP72 transcription during atrophic conditions is unknown, reports indicate that increased expression of HSP72 can preserve skeletal muscle function by reducing the activity of both the forkhead boxO (FoxO) and nuclear factor kappa B (NF‐κB) transcription factors,12, 14 promoting refolding of oxidized proteins15 and/or reducing cellular oxidative stress.16 In this regard, endurance exercise training also protects against VIDD through increasing SOD2 expression,17 inhibiting oxidative stress and inhibiting the activity of FoxO and NF‐κB.8 Therefore, the mechanism responsible for endurance exercise‐induced protection against VIDD may be related to increased expression of HSP72. Moreover, the possibility exists that HSP72 overexpression independent of exercise training may have therapeutic effects to counteract the effects of MV on diaphragm muscle fibre size and function. Therefore, these experiments tested a dual hypothesis. First, we tested the hypothesis that the protective effects of exercise preconditioning against VIDD are dependent on increased levels of HSP72 in the diaphragm. Second, we tested the hypothesis that increased HSP72 expression in the diaphragm is sufficient to alleviate VIDD.
Methods
Experimental animals
Young adult (~6 months old) female Sprague–Dawley rats were used for these experiments. Female rats were selected because effects of prolonged MV on diaphragm fibres are identical in male and female rats,18, 19 and female body weights remain relatively stable from 3–8 months of age.20 Animals were housed at the University of Florida Animal Care Services Center according to guidelines set forth by the Institutional Animal Care and Use Committee. Animals were maintained on a 12:12 h light:dark cycle and provided food and water ad libitum. These experiments were performed in accordance with the guidelines for the Care and Use of Laboratory Animals. The Animal Care and Use Committee at the University of Florida approved these experiments.
Experiment one
To test the hypothesis that increased HSP72 expression is required for the exercise‐induced protection against VIDD, animals were randomly assigned to one of six experimental groups (Figure 1) (n = 10/group): (i) sedentary, treated with saline; acutely anaesthetized controls (CON‐SED‐SALINE); (ii) sedentary, treated with saline; 12 h of mechanical ventilation (MV‐SED‐SALINE); (iii) exercise trained, treated with saline; acutely anaesthetized controls (CON‐EX‐SALINE); (iv) exercise trained, treated with saline; 12 h of mechanical ventilation (MV‐EX‐SALINE); (v) exercise trained, treated with antisense oligonucleotide (AO) towards HSP72; acutely anaesthetised (CON‐EX‐HSPAO); and (vi) exercise trained, treated with AO towards HSP72; 12 h of mechanical ventilation (MV‐EX‐HSPAO).
Figure 1.
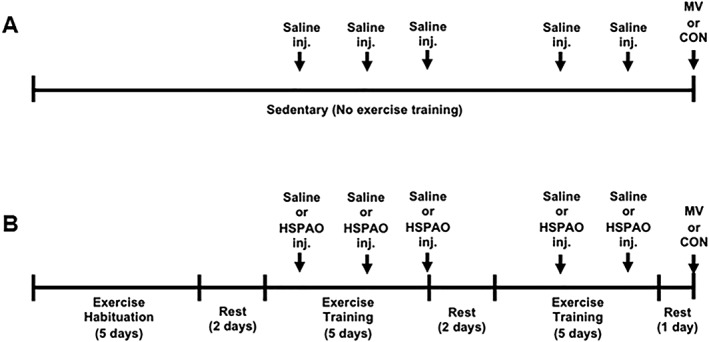
Schematic of the study design and timeline for experiment one. (A) Schematic for all sedentary groups (CON‐SED‐SALINE and MV‐SED‐SALINE). Sedentary animals received no exercise training and were administered saline (ip) at equal time points as the exercise trained animals. (B) Schematic for all exercise trained groups (CON‐EX‐SALINE, MV‐EX‐SALINE, CON‐EX‐HSPAO, and MV‐EX‐HSPAO). Exercise trained animals underwent 5 days of treadmill habituation followed by a 10‐day exercise training protocol. Saline or HSPAO was administered after Days 1, 3, 5, 7, and 9 of training. All animals were sacrificed at the same time‐point following the last injection.
Exercise training protocol
Animals assigned to the exercise‐trained groups were habituated to treadmill running over a 5‐day period, gradually increasing the duration each day (i.e. 10, 20, 30, 40, and 50 min). After habituation, animals rested for 2 days before beginning a 10‐day exercise training protocol. Animals ran for 5 days, rested for 2 days, and then completed the remaining 5 days of training. The intensity of the daily training protocol was 30 m/min at a 0% gradient for 60 min. Approximately 24 h following the final training session animals were mechanically ventilated for 12 h. Our laboratory has demonstrated that this exercise training protocol is sufficient to prevent VIDD following 12 h of MV.8
Antisense oligonucleotide administration
Exercise trained animals were either treated with an intraperitoneal (ip) injection of an AO targeted to HSP72 (12.5 mg/kg) or saline immediately following training on Days 1, 3, 5, 7, and 9. Preliminary animals were utilized to determine the dose of the AO required to reduce the exercise‐induced increase in HSP72 in the diaphragm to basal (control) levels. In addition, preliminary experiments also determined that administration of the AO‐phosphorothioate backbone, without the HSP72 targeting sequence, has no effect on diaphragm muscle size or force production (data not shown).
Experiment two
To determine if, independent of exercise training, transgene overexpression of HSP72 in the diaphragm protects against VIDD, animals were randomly assigned to one of four experimental groups (n = 8/group): (i) saline treatment, acutely anaesthetized controls (CON‐SALINE); (ii) saline treatment, 12 h of mechanical ventilation (MV‐SALINE); (iii) AAV‐HSP72 overexpression, acutely anaesthetized controls (CON‐HSP72); and (iv) AAV‐HSP72 overexpression, 12 h of mechanical ventilation (MV‐HSP72).
Packaging and purification of recombinant AAV vectors
The HSP72‐green fluorescent protein (GFP) plasmid construct has been described previously and was a gift of Dr Andrew Judge (University of Florida, Gainesville).14 Briefly, the HSP72 sequence was fused to a GFP tag. The HSP72‐GFP fused sequence was amplified and subcloned into the KpnI and NotI sites of the p43.2 empty vector. Verification of the appropriate fusion sequence was performed at the University of Florida DNA Sequencing Core facility. The AAV‐HSP72 vector was packaged by the University of Florida Powell Gene Therapy Center.
AAV delivery protocol
To increase the expression of HSP72 in the diaphragm, we delivered an adeno‐associated virus (AAV) vector to the costal diaphragm (AAV‐HSP72) via direct intramuscular injection as previously described.21, 22 Following this procedure, animals were administered buprenorphine (0.1 mg/kg) prior to awakening. Additional doses were administered as needed for 72 h. This method of gene transfer effectively transduces the rat diaphragm with no adverse side effects to the muscle.21, 22 Preliminary experiments were performed to determine the proper concentration of AAV‐HSP72 to increase HSP72 protein expression ~50% above basal (control) levels. This percentage increase was chosen to match the increase achieved by our exercise training protocol. Animals in the saline‐treated control groups underwent an identical surgical procedure and received diaphragm intramuscular injections of equal volumes of saline. Four weeks after diaphragm AAV‐HSP72 transduction or saline treatment, animals were either acutely anaesthetized or underwent 12 h of MV.
Experiment three
To determine if pharmacological‐induced overexpression of HSP72 can protect against VIDD, rats were randomly assigned to one of four experimental groups (n = 10/group): (i) saline treatment, acutely anaesthetized controls (CON‐SALINE); (ii) saline treatment, 12 h of mechanical ventilation (MV‐SALINE); (iii) BGP‐15 treatment, acutely anaesthetized controls (CON‐BGP‐15); and (iv) BGP‐15 treatment, 12 h of mechanical ventilation (MV‐BGP‐15).
BGP‐15 administration
BGP‐15 (Sigma‐Aldrich, St. Louis, MO) was administered daily (15 mg/kg) to animals by oral gavage. This dose was chosen based preliminary data demonstrating that this dose of BGP‐15 results in a similar fold increase in diaphragm HSP72 abundance compared to our exercise training protocol. BGP‐15 is a pharmacological inducer of HSP72 that has been shown to be safe and well tolerated in Phase II clinical trials for diabetes and insulin resistance.23, 24 Animals received five consecutive days of BGP‐15 treatment prior to acute anaesthesia or MV.
Experimental procedures
Acutely anaesthetized control animals
Animals in the acutely anaesthetized control groups received an ip injection of sodium pentobarbital (60 mg/kg body weight). After reaching a surgical plane of anaesthesia, the heart was removed and the costal diaphragm was removed and utilized for subsequent analyses. Note that all acutely anaesthetized control animals were sacrificed at the same time point as the mechanically ventilated animals after cessation of their experimental treatments.
Mechanical ventilation
All surgical procedures were performed using aseptic techniques. Animals in the MV groups were anaesthetized with an ip injection of sodium pentobarbital (60 mg/kg body weight), tracheostomized, and mechanically ventilated with a pressure‐controlled ventilator (Servo Ventilator 300 Siemens) for 12 h with the following settings: upper airway pressure limit: 10 cmH2O; respiratory rate: 80 bpm and; PEEP: 1 cmH2O. We estimate that these ventilator settings result in a tidal volume of ~1 mL/100 g of body weight.25
The carotid artery was cannulated to permit the continuous measurement of blood pressure and the collection of blood during the protocol. Arterial blood samples were removed periodically and analysed for arterial pO2, pCO2, and pH using an electronic blood‐gas analyser (GEM Premier 3000; Instrumentation Laboratory, Lexington, MA). Ventilator adjustments were made as necessary, and no differences existed in ventilation between groups.
A venous catheter was inserted into the jugular vein for continuous infusion of sodium pentobarbital (~10 mg/kg/h). Temperature was maintained at 37°C by use of a recirculating heating blanket, and heart rate was monitored via a lead II electrocardiograph. Continuous care during the MV protocol included lubricating the eyes, expressing the bladder, removing airway mucus, rotating the animal, and passively moving the limbs. Animals also received an intramuscular injection of glycopyrrolate (0.04 mg/kg) every 2 h during MV to reduce airway secretions. Following 12 h of MV, while the animals were at a surgical plane of anaesthesia, the heart was removed, and the costal diaphragm was removed and utilized for subsequent analyses.
Biochemical measures
HSP72
Changes in HSP72 were assessed via western blot analysis. Diaphragm tissue samples were homogenized 1:10 (wt/vol) in 5 mM Tris (pH 7.5) and 5 mM EDTA (pH 8.0) with a protease inhibitor cocktail (Sigma‐Aldrich, St. Louis. MO) and centrifuged at 1500 g for 10 min at 4°C. After collection of the resulting supernatant, diaphragmatic protein content was assessed by the method of Bradford (Sigma‐Aldrich). Proteins from the supernatant fraction were separated via polyacrylamide gel electrophoresis and transferred to nitrocellulose. Membranes were probed for HSP72 (Stressgen Biotechnologies, San Diego, CA) and α‐tubulin (loading control) (Developmental Studies Hybridoma Bank, University of Iowa) and were imaged fluorescently using the LI‐COR Odyssey CLx Imaging System (LI‐COR Biosciences, Lincoln, NE).
Myofiber cross‐sectional area
Sections from frozen diaphragm samples were cut at 10 microns using a cryotome (Shandon Inc., Pittsburgh, PA) and stained for dystrophin, myosin heavy chain Type I, and myosin heavy chain Type IIa proteins for fibre cross‐sectional area (CSA) analysis as described previously.26 CSA was determined using Scion software (NIH).
In vitro diaphragmatic contractile properties
A muscle strip, including the tendinous attachments at the central tendon and rib cage, was dissected from the midcostal region of the diaphragm. The strip was suspended vertically between two lightweight Plexiglas clamps with one end connected to an isometric force transducer (model FT‐03, Grass Instruments, Quincy, MA) within a jacketed tissue bath. The force output was recorded via a computerized data‐acquisition system (Super Scope II, GW Instruments Somerville, MA). The muscle strip was stimulated along its entire length with platinum wire electrodes (modified S48 stimulator, Grass Instruments) by using supramaximal (~150%) stimulation voltage to determine the optimum contractile length (Lo). To measure maximal isometric twitch force, each strip was stimulated supramaximally with 120‐V pulses at 160 Hz, and to measure the force frequency response, each strip was stimulated supramaximally with 120‐V pulses at 15–160 Hz.27
Permeabilized muscle fibre preparation
A portion of diaphragm muscle was dissected and placed on a plastic Petri dish filled with ice‐cold buffer X (60 mM K‐Mes, 35 mM KCl, 7.23 mM K2EGTA, 2.77 mM CaK2EGTA, 20 mM imidazole, 0.5 mM dithiothreitol, 20 mM taurine, 5.7 mM ATP, 15 mM PCr, and 6.56 mM MgCl2, pH 7.1). The muscle was gently separated to expose each fibre. Muscle fibre bundles were permeabilized in ice‐cold buffer X containing 75 μg/mL saponin for 30 min at 4°C followed by three, 5‐min washed in ice‐cold buffer Z (110 mM K‐Mes, 35 mM KCl, 1 mM EGTA, 5 mM K2HPO4, and 3 mM MgCl2, 0.005 mM glutamate, 0.02 mM malate, and 0.5 mg/mL BSA, pH 7.1).
Mitochondrial reactive oxygen species emission
Mitochondrial hydrogen peroxide emission rate was measured continuously via a spectrofluorometer (Fluorolog‐3 HORIBA Jobin Yvon, Edison, NJ, USA) at 37°C using diaphragm muscle fibre bundles in buffer Z containing Amplex Ultra Red (10 μM)/horseradish peroxidase (3 U/mL). Excitation and emission was set at 565 and 600, respectively. Blebbistatin (25 μM) prevented myofiber contraction during the measurements. Each fibre was washed and dehydrated for normalization of the measurements. The respiration rate was expressed as hydrogen peroxide emission in pmol/mg (dry wt)/min.
Mitochondrial respiration
Respiration was measured via polarography with permeabilized fibre bundles in water‐jacketed respiration chambers maintained at 37°C (Hansatech Instruments, King's Lynn, UK). Each chamber contained 1 mL of respiration buffer Z containing 20 mM creatine to saturate creatine kinase. After equilibrium was reached with 5 mM pyruvate and 5 mM malate, 0.25 mM ADP was added to stimulate State 3 respiration. State 4 was then achieved through the addition of 10 μg/mL oligomycin to inhibit ATP synthesis. The respiratory control ratio (RCR) was then calculated by dividing the State 3 by the State 4.
SOD2 protein abundance
Diaphragm samples were homogenized 1:10 (mg wt/μL buffer) in 5 mM Tris (pH 7.5) and centrifuged at 1500 g for 5 min at 4°C. Upon collection of the supernatant, protein content was assessed via Bradford method. A commercially available rat SOD2 enzyme linked immunosorbent assay (Life Span BioSciences, Seattle, WA) kit was used to determine total protein for SOD2 according to manufacturer's instructions.
SOD2 enzyme activity
Activity was measured with a commercially available enzyme activity kit according to manufacturer's instructions (Cayman Chemical, Ann Arbor, MI); 2 mM KCN was added to the reaction mixture to inhibit SOD1. Briefly, tissue samples were homogenized 1:10 (mg wt/vol buffer) in 20 mM Hepes, 1 mM EGTA, 210 mM mannitol, and 70 mM sucrose (pH 7.2) and centrifuged at 1500 g for 5 min at 4°C. Upon collection of the supernatant, protein content was assessed via Bradford method. After measurement of SOD2 activity, the activities were normalized by the protein concentrations.
PGC‐1α expression
Total RNA was isolated from muscle tissue with TRIzol Reagent (Life Technologies, Carlsbad, CA) according to the manufacturer's instructions. Total RNA and RNA content (μg/mg muscle) were evaluated by spectrophotometry. Total RNA (5 μg) was then reverse transcribed with the Superscript III First‐Strand Synthesis System for RT‐PCR (Life Technologies), using oligo (dT)20 primers and the protocol outlined by the manufacturer. One microliter of cDNA was added to a 25 μL PCR reaction for real‐time PCR using Taqman chemistry and the ABI StepOnePlus Real‐Time PCR system (ABI, Foster City, CA). Relative quantification of PGC‐1α gene expression was performed using the comparative computed tomography method (ABI, User Bulletin no. 2). β‐Glucuronidase was chosen as the reference gene based on previous work showing unchanged expression with our experimental manipulations.28, 29 mRNA transcripts were assayed using predesigned rat primer and probe sequences commercially available from Applied Biosystems (Assays‐on‐Demand).
E3 ligase expression
Relative quantification of Atrogin‐1/MaFbx and MuRF1 gene expression was performed using the comparative computed tomography method (ABI, User Bulletin no. 2) as described above. β‐Glucuronidase was chosen as the reference gene based on previous work showing unchanged expression with our experimental manipulations.28, 29 mRNA transcripts were assayed using predesigned rat primer and probe sequences commercially available from Applied Biosystems (Assays‐on‐Demand).
Statistical analysis
Sample size for each experiment was chosen based on preliminary data and statistical power calculations. The unexpected loss or necessary removal of an animal from a group was also accounted for when calculating sample size for each experiment. Between group comparisons for each dependent variable were made via two‐way analysis of variance. When appropriate, a Bonferroni test was performed post hoc. Comparisons of blood gas parameters were made using a one‐way analysis of variance for experiment one and a two‐tailed Student's t‐test for experiments two and three. Significance was established a priori at α < 0.05. Values are presented as mean ± standard deviation (Figure 1).
Results
HSP72 expression is required for exercise‐induced protection against ventilator‐induced diaphragm dysfunction
Two weeks of treadmill exercise training resulted in a ~50% increase in the abundance of HSP72 in diaphragm fibres, compared to untrained (sedentary) animals. Importantly, treatment of exercise trained animals with an AO directed towards HSP72 (HSPAO) prevented this exercise‐induced increase in HSP72 protein content in the diaphragm (Figure 2A). In spontaneously breathing animals (CON), no differences existed in the diaphragm muscle fibre CSA or specific force production between any of the treatment groups (i.e. SED‐SALINE, EX‐SALINE, and EX‐HSPAO) (Figure 2B and 2C). Compared to CON, prolonged MV resulted in significant atrophy of Types I, IIa, and IIb/x diaphragm muscle fibres and in a significant reduction in diaphragm muscle specific force production at all stimulation frequencies in sedentary animals. In contrast, endurance exercise training performed prior to MV protected the diaphragm against MV‐induced diaphragmatic atrophy and contractile dysfunction (Figure 2B and 2C). Notably, exercise trained animals that were treated with HSPAO immediately post exercise and were not protected from MV‐induced atrophy in both Types IIa and IIb/x diaphragm muscle fibres. Additionally, diaphragms from MV‐EX‐HSPAO animals showed a similar phenotype to MV‐SED‐SALINE animals, as their diaphragm specific force production was significantly reduced compared to non‐ventilated animals.
Figure 2.
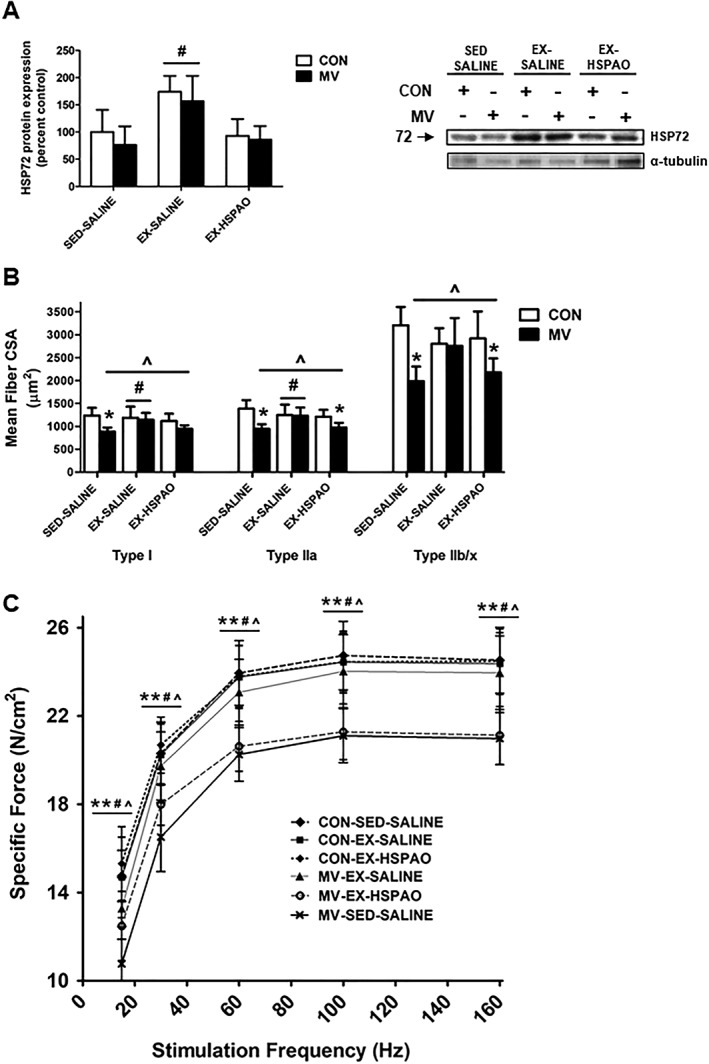
Effect of exercise‐induced HSP72 expression on VIDD. (A) HSP72 protein expression. Representative western blot images are shown to the right of the graph. (B) Diaphragm muscle cross‐sectional area (CSA) of myosin heavy chain (MHC) Types I, IIa, and IIb/x fibres. (C) Diaphragm muscle specific force production. Values are presented as mean ± SD. CON = acutely anaesthetized control; EX‐HSPAO = exercise‐trained animals treated with an antisense oligonucleotide directed against HSP72; EX‐SALINE = exercise‐trained animals treated with saline; MV = mechanical ventilation; SED‐SALINE = sedentary animals treated with saline. * = MV significantly different versus CON within treatment (P < 0.05). ^ = main effects for MV (P < 0.05). # = main effects for treatment (P < 0.05).
Exercise‐induced HSP72 expression is required to prevent markers of oxidative stress in the diaphragm
Several markers of diaphragm oxidative stress were measured to determine the role of HSP72 in reducing oxidative damage to the diaphragm during MV. Prolonged MV resulted in an elevated production of mitochondrial reactive oxygen species (ROS) and respiratory chain dysfunction (i.e. reduced RCR) in diaphragm fibres (Figure 3A and 3B). Importantly, endurance exercise training performed prior to MV prevented the MV‐induced mitochondrial damage and ROS emission. Conversely, exercise trained animals receiving HSPAO treatment were not protected from MV‐induced increased mitochondrial ROS production or reduced RCR.
Figure 3.
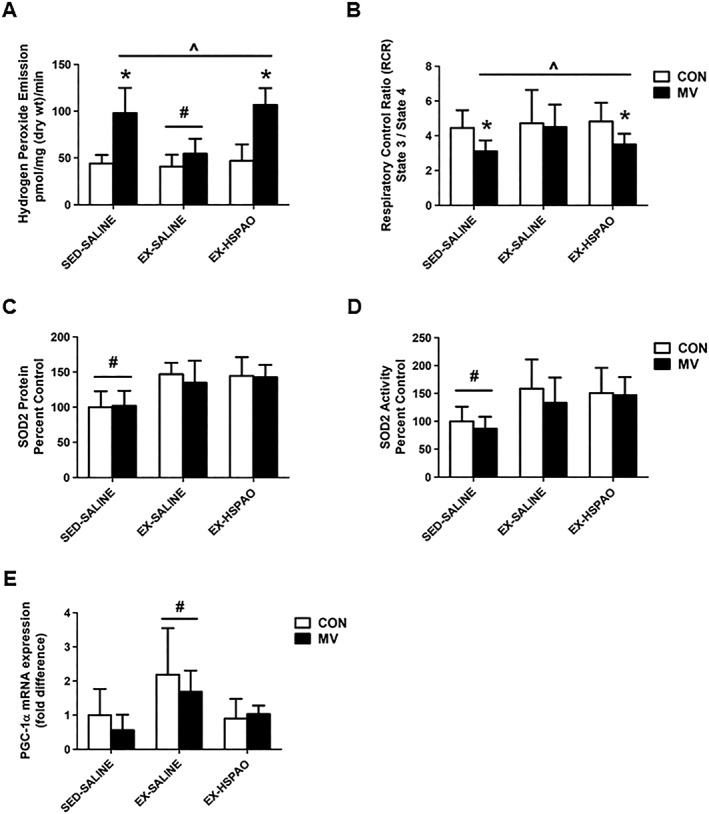
Effect of exercise‐induced HSP72 expression on markers of mitochondrial function. (A) Mitochondrial ROS emission; (B) respiratory control ratio (RCR); (C) SOD2 protein expression; (D) SOD2 activity; and (E) PGC‐1α mRNA expression. Values are presented as mean ± SD. CON = acutely anaesthetized control; MV = mechanical ventilation; EX‐HSPAO = exercise‐trained animals treated with an antisense oligonucleotide directed against HSP72; EX‐SALINE = exercise‐trained animals treated with saline; SED‐SALINE = sedentary animals treated with saline. * = MV significantly different versus CON within treatment (P < 0.05). ^ = main effects for MV (P < 0.05). # = main effects for treatment (P < 0.05).
In regard to mitochondrial antioxidant capacity and increased mitochondrial biogenesis/turnover, we show that exercise results in an increase in both the protein abundance and activity of the mitochondria‐localized antioxidant enzyme SOD2 (Figure 3C and 3D) and the mRNA expression of PGC‐1α (Figure 3E). Importantly, HSPAO treatment did not impact the exercise‐induced training response of SOD2, as protein expression and activity levels in the diaphragm were elevated compared to sedentary animals. Interestingly, HSPAO treatment in exercise trained animals was associated with a depression of the exercise‐induced increase in PGC‐1α mRNA expression.
Exercise‐induced HSP72 expression reduces the expression of proteolytic proteins
Expression of the E3 ligases Atrogin‐1/MaFbx and MurF1 were assessed as markers of FoxO and NF‐κB transcriptional activity. Assessment of Atrogin‐1/MaFbx and MuRF1 in the diaphragm demonstrated a significant increase following MV, which is prevented by exercise preconditioning (Figure 4A and 4B). In addition, our results demonstrate HSPAO administration ameliorated the exercise‐induced reduction in both Atrogin‐1/MaFbx and MuRF1 mRNA expression (Figure 4A and 4B).
Figure 4.
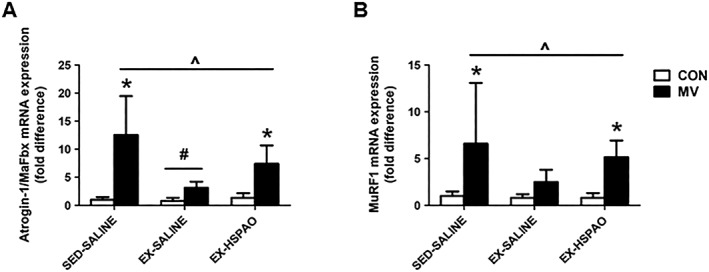
Effect of exercise‐induced HSP72 expression on mRNA expression of (A) Atrogin‐1/MaFbx and (B) MuRF1. Values are presented as mean ± SD. CON = acutely anaesthetized control; MV = mechanical ventilation; EX‐HSPAO = exercise‐trained animals treated with an antisense oligonucleotide directed against HSP72; EX‐SALINE = exercise‐trained animals treated with saline; SED‐SALINE = sedentary animals treated with saline. * = MV significantly different versus CON within treatment (P < 0.05). ^ = main effects for MV (P < 0.05). # = main effects for treatment (P < 0.05).
Overexpression of HSP72 in the diaphragm is sufficient to prevent ventilator‐induced diaphragm dysfunction
Both genetic and pharmacological overexpression of HSP72 reduced diaphragm muscle dysfunction after 12 h of MV. Specifically, both methods of HSP72 upregulation caused an ~1.5‐ to 2‐fold induction in HSP72 protein expression (Figures 5A and 6A), which resulted in significant protection against VIDD.
Figure 5.
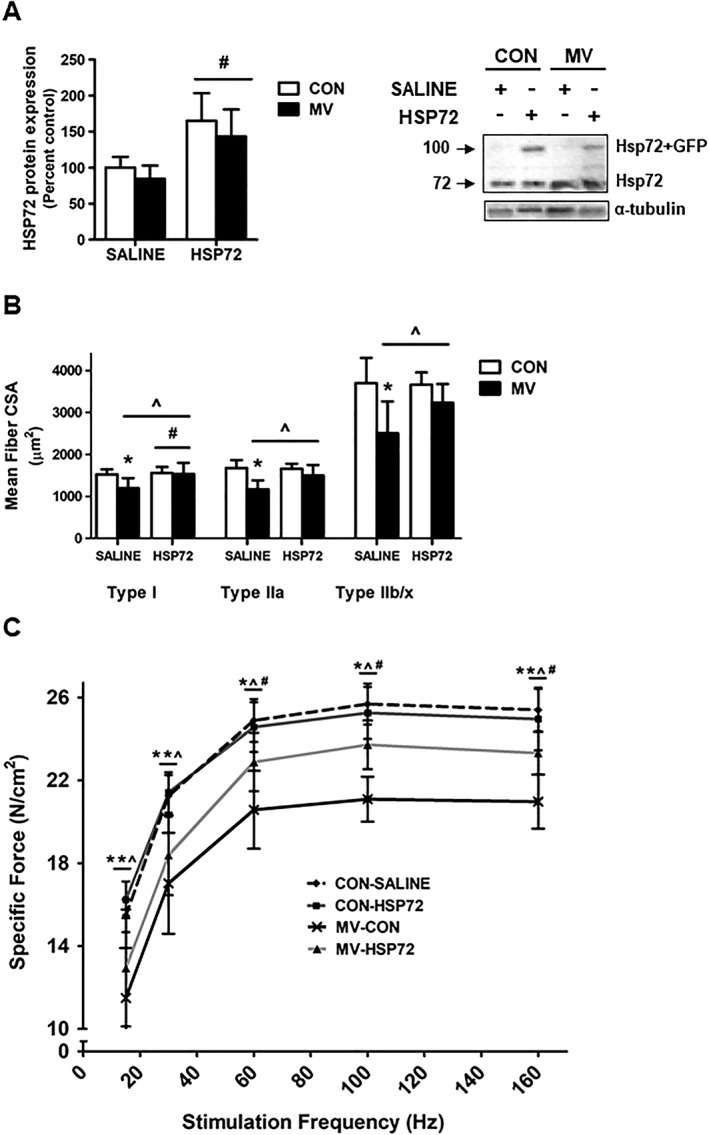
Effect of HSP72 overexpression on diaphragm function. (A) HSP72 protein expression. Representative western blot images are shown to the right of the graph. (B) Diaphragm muscle cross‐sectional area (CSA) of myosin heavy chain (MHC) Types I, IIa, and IIb/x fibres. (C) Diaphragm muscle specific force production. Values are presented as mean ± SD. CON = acutely anaesthetised control; HSP72 = AAV‐HSP72 overexpression; MV = mechanical ventilation; SALINE = saline injection. * = MV significantly different versus CON within treatment (P < 0.05). ^ = main effects for MV (P < 0.05). # = main effects for HSP72 (P < 0.05).
Figure 6.
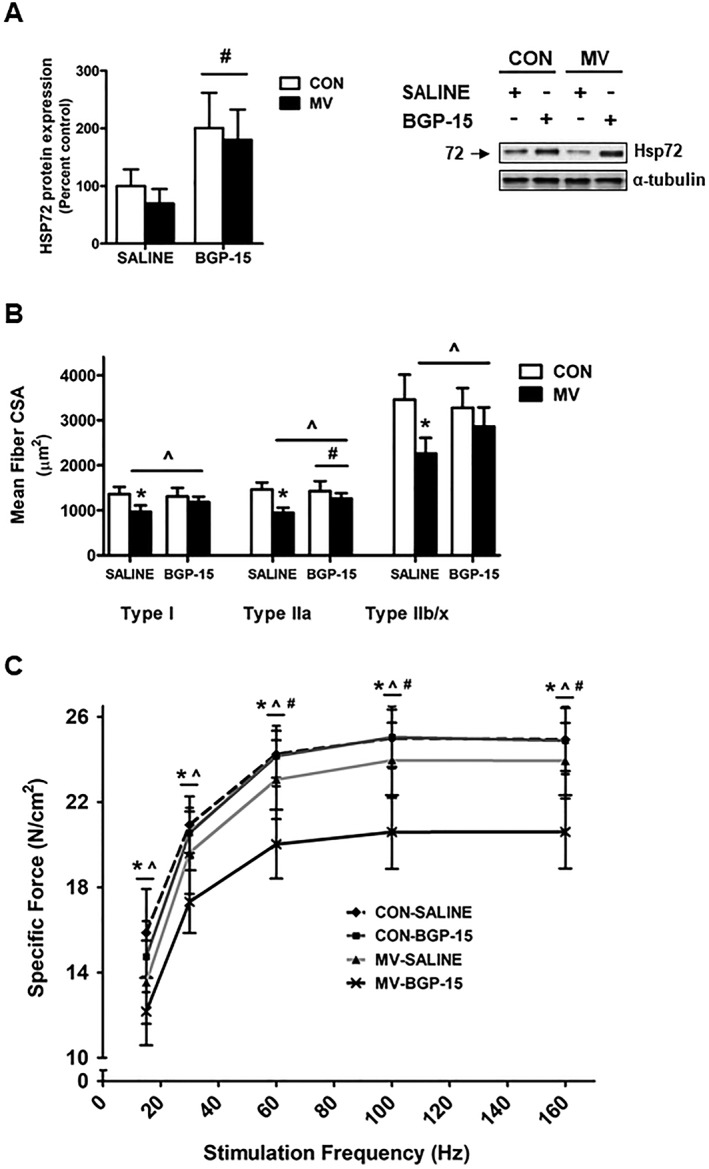
Effect of BGP‐15 administration on diaphragm function. (A) HSP72 protein expression. Representative western blot images are shown to the right of the graph. (B) Diaphragm muscle cross‐sectional area (CSA) of myosin heavy chain (MHC) Types I, IIa, and IIb/x fibres. (C) Diaphragm muscle specific force production. Values are presented as mean ± SD. CON = acutely anaesthetized control; MV = mechanical ventilation; SALINE = saline treatment; BGP‐15 = BGP‐15 treatment. * = MV significantly different versus CON within treatment (P < 0.05). ^ = main effects for MV (P < 0.05). # = main effects for BGP‐15 (P < 0.05).
In regard to MV‐induced diaphragm atrophy, overexpression of HSP72 had no effect on diaphragm fibre CSA in non‐ventilated (CON) animals (Figures 5B and 6B). In saline‐treated animals, MV resulted in significant atrophy of all diaphragm fibre types compared to control animals. Moreover, in MV animals overexpressing HSP72 via AAV‐HSP72, diaphragm fibre atrophy was significantly attenuated in Type I fibres, while in MV animals overexpressing HSP72 via BGP‐15, Type IIa fibre CSA was preserved.
Identical to experiment one, prolonged MV in saline‐treated animals resulted in a significant downward shift of the diaphragm force‐frequency curve, indicating contractile dysfunction (Figures 5C and 6C). Notably, overexpression of HSP72 in the diaphragm of mechanically ventilated animals via AAV‐HSP72 and BGP‐15 administration significantly preserved diaphragm muscle specific force production at frequencies greater than 30 Hz.
Overexpression of HSP72 prevents mechanical ventilation‐induced mitochondrial dysfunction
Our data demonstrate that overexpression of HSP72 in the diaphragm via both AAV‐HSP72 and BGP‐15 treatment prevented the MV‐induced increase in mitochondrial ROS emission (Figure 7A and 7C). In addition, HSP72 overexpression in the diaphragm also prevented the MV‐induced reduction in the mitochondrial RCR (Figure 7B and 7D). Finally, note that the HSP72‐mediated attenuation of mitochondrial dysfunction is not due to changes in SOD2 antioxidant capacity as HSP72 overexpression did not impact SOD2 protein expression or activity in the diaphragm (Figure 7E and 7F).
Figure 7.
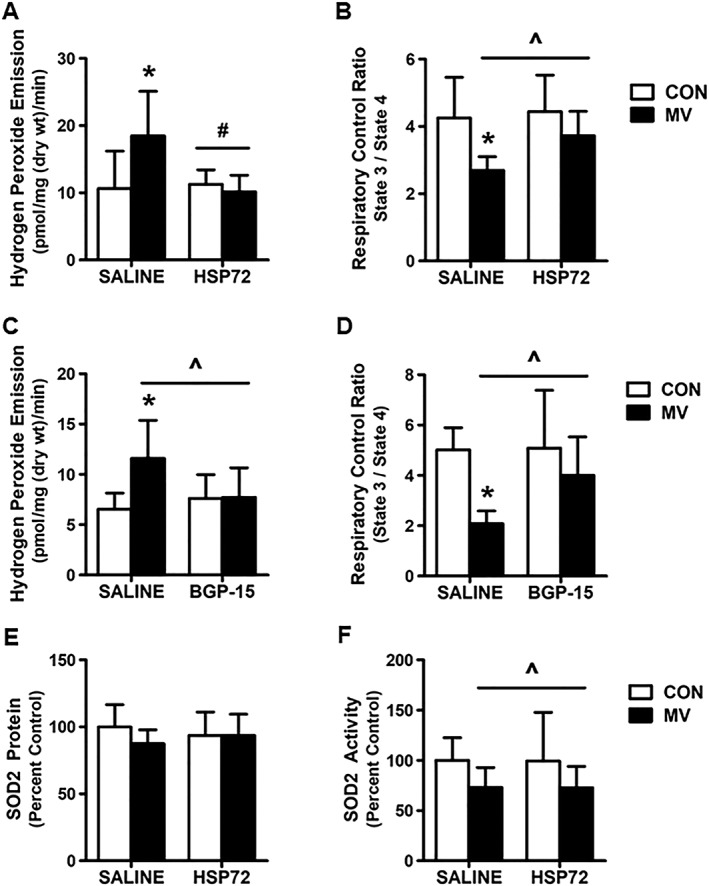
Effect of HSP72 overexpression on markers of mitochondrial function. (A) and (C) mitochondrial ROS emission; (B) and (D) respiratory control ratio (RCR); (E) SOD2 protein expression; and (F) SOD2 activity. Values are presented as mean ± SD. CON = acutely anaesthetized control; MV = mechanical ventilation; SALINE = saline treatment; HSP72 = AAV‐HSP72 overexpression; BGP‐15 = BGP‐15 treatment. * = MV significantly different versus CON within treatment (P < 0.05). ^ = main effects for MV (P < 0.05). # = main effects for HSP72 (P < 0.05).
HSP72 overexpression in the diaphragm prevents mechanical ventilation‐induced increases in E3 ligase expression
Prolonged MV promotes the increased transcription of both Atrogin‐1/MaFbx and MurF1 in the diaphragm, and overexpression of HSP72 by AAV prevented the MV‐induced increase in the mRNA expression of both of these E3 ligases (Figure 8A and 8B). Further, BGP‐15 administration significantly blunted the MV‐induced expression of both Atrogin‐1/MaFbx and MurF1 in the diaphragm. However, this treatment did not completely prevent the MV‐induced rise in the expression of these E3 ligases in the diaphragm (Figure 8C and 8D).
Figure 8.
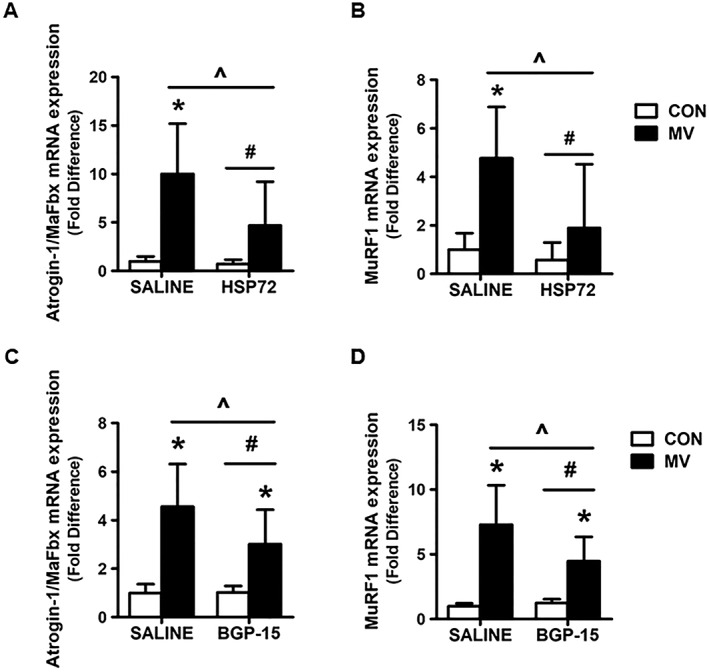
Effect of HSP72 on mRNA expression of (A) and (C) Atrogin‐1/MaFbx and (B) and (D) MuRF1. Values are presented as mean ± SD. CON = acutely anaesthetized control; MV = mechanical ventilation; SALINE = saline treatment; HSP72 = AAV‐HSP72 overexpression; BGP‐15 = BGP‐15 treatment. * = MV significantly different versus CON within treatment (P < 0.05). ^ = main effects for MV (P < 0.05). # = main effects for HSP72/BGP‐15 (P < 0.05).
Discussion
We recently explored the global proteomic changes specific to diaphragm muscle following endurance exercise training.11 Our findings revealed that following endurance exercise training, diaphragm HSP72 levels are elevated in both the cytosolic and mitochondrial protein compartments.11 This finding confirmed the previous discovery from our laboratory that 2 weeks of endurance exercise conditioning increases HSP72 expression in the diaphragm.8 This is significant because endurance exercise training is sufficient to protect against VIDD,8 and overexpression of HSP72 has been shown to protect limb muscle against disuse muscle atrophy.12, 14 Therefore, the current experiments tested the hypothesis that exercise‐induced increases in HSP72 in the diaphragm are essential to protect the diaphragm against VIDD. Notably, our results support this hypothesis and demonstrate that elevated HSP72 plays an important role in protecting the diaphragm against VIDD. Moreover, our novel results also reveal that, independent of exercise, increased expression of HSP72 in the diaphragm results in protection against VIDD. A detailed discussion of these findings follows.
Elevated HSP72 expression in the diaphragm is required for exercise‐induced protection against ventilator‐induced diaphragm dysfunction
Endurance exercise training promotes cellular adaptations to skeletal muscle, resulting in a phenotype that is resistant to atrophic conditions.30, 31, 32 This is important because it allows endurance exercise training to be used as a tool for preclinical studies to elucidate cytoprotective proteins with the potential for clinical translation. Among the modifications to diaphragm muscle protein expression occurring as a result of endurance exercise training, our laboratory determined that exercise training stimulates an increase in the abundance of electron transport chain components, anti‐apoptotic proteins, antioxidant proteins, and chaperone/stress proteins.11 Of these classes of proteins, the chaperone/stress proteins, and in particular the family member HSP72, has been demonstrated to protect against limb skeletal muscle atrophy resulting from a variety of wasting conditions.12, 13, 14 However, to date, no studies have evaluated the role of HSP72 expression in exercise‐induced protection against skeletal muscle atrophy. Indeed, the current study is the first to demonstrate that elevated HSP72 expression in the diaphragm is required to provide exercise‐induced protection against MV‐induced diaphragm atrophy and contractile dysfunction.
In regard to HSP72 overexpression independent of exercise, it is recognized that both transgenic and pharmacological overexpression of HSP72 in dystrophic mice preserves diaphragm muscle strength and morphology.13 Additionally, transgenic animals overexpressing HSP72 in skeletal muscle have been shown to have an increased rate of skeletal muscle recovery following hindlimb immobilization.33 These findings corroborated an earlier finding by Senf et al. which demonstrated that limb muscle fibres overexpressing HSP72 are protected against atrophy following 7 days of immobilization.14 Our results verify the protective effects of HSP72 overexpression on diaphragm muscle wasting as both genetically and pharmacologically induced overexpression of HSP72 protected against VIDD. Specifically, genetic and pharmacological overexpression of HSP72 both prevented diaphragm muscle force deficits at stimulation frequencies greater than 30 Hz. In addition, genetic overexpression resulted in a significant attenuation of diaphragm atrophy of the Type 1 fibres and partially protected all other fibres, while pharmacological overexpression significantly attenuated Type IIa fibre area and partially protected all others. While the mechanisms responsible for these differences are unknown, these findings demonstrate that HSP72 appears to play a critical role in regulating muscle mass and function during atrophic conditions.
The similar findings between the genetic and pharmacological methods of HSP72 are important for several reasons. Specifically, they could provide effective therapeutic benefits based on the amount of time a patient requires ventilatory support. A pharmacological intervention is ideal for patients undergoing prolonged surgeries similar to the length of MV used in these studies and due to the whole‐body effects may also be beneficial in conditions where weaning problems are exacerbated by ventilator‐induced lung injury or other co‐morbidities involving multiple organ systems. This fact could account for some of the functional and biochemical differences noted between groups. Finally, it is also acknowledged that gene therapy may have clinical significance in mechanically ventilated patients, particularly with those suffering from more long‐term respiratory dysfunction.34, 35
Exercise‐induced HSP72 is required to prevent mechanical ventilation‐induced mitochondrial dysfunction
Increased mitochondrial ROS production in diaphragm muscle fibres is associated with calcium dysregulation, myonuclear apoptosis, and activation of several key proteolytic systems, and it is established that mitochondrial ROS production is the principal oxidant‐generating pathway involved in the development of VIDD.36, 37 Endurance exercise results in changes to diaphragm muscle mitochondria, resulting in a phenotype that is more resistant to oxidative and apoptotic challenges.11 Interestingly, our data reveal that increased HSP72 expression in the diaphragm is required to reduce mitochondrial ROS emission and mitochondrial dysfunction.
One mechanism by which HSP72 is hypothesized to prevent mitochondrial ROS emission is through its chaperone activity.38, 39 Specifically, HSP72 may increase mitochondrial antioxidant capacity by transporting cytosolic proteins to the mitochondria. Specifically, SOD2 is a mitochondrial antioxidant that is synthesized in the cytosol and then transported into the mitochondria via chaperone proteins.38, 39 Once in the mitochondria, SOD2 is assembled and plays an important role in buffering mitochondrial ROS.40 While a direct interaction between SOD2 and HSP72 is not proven, studies report that increased HSP72 protein expression is associated with similar increases in SOD2 protein abundance and activity.41, 42 For example, SOD2 activity is significantly decreased in HSP72 knockout mice,43 whereas animals overexpressing HSP72 show enhanced SOD2 activity, which is associated with protection against mitochondrial dysfunction and apoptosis during myocardial ischemia–reperfusion injury.42 Therefore, during exercise, the protective effects of HSP72 may be dependent upon an exercise‐induced increase in SOD2 protein expression. Our results contradict this hypothesis as SOD2 was elevated in our MV‐EXHSPAO animals and activity was also increased, whereas there was no evidence of protection from VIDD. Therefore, elevated SOD2 alone does not appear sufficient to prevent VIDD. In addition, results from the HSP72 overexpression studies show that HSP72 is not sufficient to induce an increase in SOD2 protein expression or activity in the diaphragm.
Another potential mechanism by which HSP72 may protect against mitochondrial dysfunction is through increasing mitochondrial quality and number. Specifically, increased HSP72 expression has been shown to increase markers of mitochondrial biogenesis and mitochondrial quality control,44, 45 which may preserve mitochondrial function in skeletal muscle as a result of reduced mitochondrial lipid peroxidation, increased refolding of denatured proteins, and/or increased mitochondrial number.41, 46 In the current study, increased mitochondrial protein folding and biogenesis appears to be the more likely explanation for the HSP72‐induced preservation of mitochondrial function, as both the prevention of exercise‐induced increases in HSP72 levels and overexpression of HSP72 in the diaphragm did not influence SOD2 protein expression or activity. In contrast, HSPAO administration prevented the exercise‐induced increase in PGC‐1α expression in the diaphragm.
Overexpression of HSP72 reduces mechanical ventilation‐induced proteolysis in the diaphragm
Accelerated proteolysis in the diaphragm contributes to VIDD as a result of the increased proteolytic breakdown of key sarcomeric proteins necessary for muscle contraction.47, 48 Indeed, all four major proteolytic systems (i.e. calpain, caspase‐3, autophagy/lysosomal, and ubiquitin/proteasome) are activated in the diaphragm during MV and contribute to varying degrees to MV‐induced diaphragm atrophy and contractile dysfunction.49, 50, 51 Moreover, exercise training protects against VIDD through limiting the activation of these proteolytic systems,8 at least in part, as a result of increased diaphragm levels of HSP72. HSP72 can attenuate MV‐induced proteolysis in several different ways. Specifically, Senf et al. demonstrated that HSP72 overexpression is sufficient to prevent the transcriptional activity of both FoxO and NF‐κB in limb skeletal muscle.14 This is important because our prior work revealed that activation of FoxO and NF‐κB contribute to VIDD by regulating the transcription of several proteins required for protein breakdown via the caspase‐3, autophagy/lysosomal, and ubiquitin/proteasome proteolytic systems.22, 52 Specifically, inhibiting the activation of FoxO in the diaphragm during MV resulted in a significant attenuation of both diaphragm atrophy and contractile dysfunction.22 This is important because recent work elucidating the signalling mechanisms responsible for VIDD in patients mechanically ventilated for 18–72 h demonstrated that atrophic AKT‐FoxO signalling plays an important role in eliciting VIDD.53 Finally, inhibition of NF‐κB during MV also affords protection against VIDD as a result of inhibiting MuRF1 expression and proteasome activity.52 Therefore, interventions that target these atrophic signalling pathways may be effective in preventing VIDD.
Conclusions
Preclinical models of MV provide important information regarding the signalling pathways required for VIDD. The rat model of MV is clinically significant because it greatly mimics the diaphragm weakness observed in mechanically ventilated patients,54 including during conditions where diaphragm weakness may be exacerbated as a result of ICU‐acquired comorbidities (i.e. sepsis).55 Indeed, sepsis in critically ill patients has been demonstrated to prolong MV and prolong recovery time. Similarly, sepsis in an animal model of MV has been shown to exacerbate VIDD.56 Moreover, the rat model also allows the development of MV‐induced diaphragm weakness to be studied in a shorter time period (i.e. 12 h).
These novel results demonstrate for the first time that increased HSP72 expression is required for exercise‐induced prevention of VIDD in a preclinical model of MV. Importantly, these preclinical results also reveal that both molecular and pharmacological upregulation of HSP72 expression rescued the diaphragm from VIDD following 12 h of controlled MV. This is significant because these findings represent an important first step in identifying molecular targets with the potential to be clinically manipulated to prevent VIDD and problematic weaning in critical care patients and patients suffering from ICU‐acquired muscle weakness.
Conflict of Interest
None declared.
Funding
This work was supported by the National Institutes of Health awarded to S.K. Powers (R01 AR064189).
Acknowledgements
The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.57
Smuder A. J., Morton A. B., Hall S. E., Wiggs M. P., Ahn B., Wawrzyniak N. R., Sollanek K. J., Min K., Kwon O. S., Nelson W. B., and Powers S. K. (2019) Effects of exercise preconditioning and HSP72 on diaphragm muscle function during mechanical ventilation, Journal of Cachexia, Sarcopenia and Muscle, 10, 767–781, 10.1002/jcsm.12427.
References
- 1. Tang H, Lee M, Budak MT, Pietras N, Hittinger S, Vu M, et al. Intrinsic apoptosis in mechanically ventilated human diaphragm: linkage to a novel Fos/FoxO1/Stat3‐Bim axis. FASEB J 2011;25:2921–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wunsch H, Linde‐Zwirble WT, Angus DC, Hartman ME, Milbrandt EB, Kahn JM. The epidemiology of mechanical ventilation use in the United States. Crit Care Med 2010;38:1947–1953. [DOI] [PubMed] [Google Scholar]
- 3. Powers SK, Kavazis AN, Levine S. Prolonged mechanical ventilation alters diaphragmatic structure and function. Crit Care Med 2009;37:S347–S353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vassilakopoulos T, Petrof BJ. Ventilator‐induced diaphragmatic dysfunction. Am J Respir Crit Care Med 2004;169:336–341. [DOI] [PubMed] [Google Scholar]
- 5. Senf SM. Skeletal muscle heat shock protein 70: diverse functions and therapeutic potential for wasting disorders. Front Physiol 2013;4:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Noble EG, Ho R, Dzialoszynski T. Exercise is the primary factor associated with Hsp70 induction in muscle of treadmill running rats. Acta Physiol (Oxf) 2006;187:495–501. [DOI] [PubMed] [Google Scholar]
- 7. Smuder AJ, Kavazis AN, Min K, Powers SK. Exercise protects against doxorubicin‐induced oxidative stress and proteolysis in skeletal muscle. J Appl Physiol 2011;110:935–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Smuder AJ, Min K, Hudson MB, Kavazis AN, Kwon OS, Nelson WB, et al. Endurance exercise attenuates ventilator‐induced diaphragm dysfunction. J Appl Physiol 2012;112:501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ichinoseki‐Sekine N, Yoshihara T, Kakigi R, Sugiura T, Powers SK, Naito H. Heat stress protects against mechanical ventilation‐induced diaphragmatic atrophy. J Appl Physiol 2014;117:518–524. [DOI] [PubMed] [Google Scholar]
- 10. Yoshihara T, Ichinoseki‐Sekine N, Kakigi R, Tsuzuki T, Sugiura T, Powers SK, et al. Repeated exposure to heat stress results in a diaphragm phenotype that resists ventilator‐induced diaphragm dysfunction. J Appl Physiol 2015;119:1023–1031. [DOI] [PubMed] [Google Scholar]
- 11. Sollanek KJ, Burniston JG, Kavazis AN, Morton AB, Wiggs MP, Ahn B, et al. Global proteome changes in the rat diaphragm induced by endurance exercise training. PLoS One 2017;12:e0171007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dodd S, Hain B, Judge A. Hsp70 prevents disuse muscle atrophy in senescent rats. Biogerontology 2009;10:605–611. [DOI] [PubMed] [Google Scholar]
- 13. Gehrig SM, van der Poel C, Sayer TA, Schertzer JD, Henstridge DC, Church JE, et al. Hsp72 preserves muscle function and slows progression of severe muscular dystrophy. Nature 2012;484:394–398. [DOI] [PubMed] [Google Scholar]
- 14. Senf SM, Dodd SL, McClung JM, Judge AR. Hsp70 overexpression inhibits NF‐kappaB and Foxo3a transcriptional activities and prevents skeletal muscle atrophy. FASEB J 2008;22:3836–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reeg S, Jung T, Castro JP, Davies KJA, Henze A, Grune T. The molecular chaperone Hsp70 promotes the proteolytic removal of oxidatively damaged proteins by the proteasome. Free Radic Biol Med 2016;99:153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Broome CS, Kayani AC, Palomero J, Dillmann WH, Mestril R, Jackson MJ, et al. Effect of lifelong overexpression of HSP70 in skeletal muscle on age‐related oxidative stress and adaptation after nondamaging contractile activity. FASEB J 2006;20:1549–1551. [DOI] [PubMed] [Google Scholar]
- 17. Morton AB, Smuder AJ, Wiggs MP, Hall SE, Ahn B, Hinkley JM, et al. Increased SOD2 in the diaphragm contributes to exercise‐induced protection against ventilator‐induced diaphragm dysfunction. Redox Biol 2018;20:402–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thomas D, Maes K, Agten A, Heunks L, Dekhuijzen R, Decramer M, et al. Time course of diaphragm function recovery after controlled mechanical ventilation in rats. J Appl Physiol 2013;115:775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Powers SK, Shanely RA, Coombes JS, Koesterer TJ, McKenzie M, Van Gammeren D, et al. Mechanical ventilation results in progressive contractile dysfunction in the diaphragm. J Appl Physiol 2002;92:1851–1858. [DOI] [PubMed] [Google Scholar]
- 20. Pahl PJ. Growth curves for body weight of the laboratory rat. Aust J Biol Sci 1969;22:1077–1080. [DOI] [PubMed] [Google Scholar]
- 21. Smuder AJ, Falk DJ, Sollanek KJ, Nelson WB, Powers SK. Delivery of recombinant adeno‐associated virus vectors to rat diaphragm muscle via direct intramuscular injection. Hum Gene Ther Methods 2013;24:364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Smuder AJ, Sollanek KJ, Min K, Nelson WB, Powers SK. Inhibition of forkhead boxO‐specific transcription prevents mechanical ventilation‐induced diaphragm dysfunction. Crit Care Med 2015;43:e133–e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Literati‐Nagy B, Kulcsar E, Literati‐Nagy Z, Buday B, Peterfai E, Horvath T, et al. Improvement of insulin sensitivity by a novel drug, BGP‐15, in insulin‐resistant patients: a proof of concept randomized double‐blind clinical trial. Horm Metab Res 2009;41:374–380. [DOI] [PubMed] [Google Scholar]
- 24. Literati‐Nagy B, Peterfai E, Kulcsar E, Literati‐Nagy Z, Buday B, Tory K, et al. Beneficial effect of the insulin sensitizer (HSP inducer) BGP‐15 on olanzapine‐induced metabolic disorders. Brain Res Bull 2010;83:340–344. [DOI] [PubMed] [Google Scholar]
- 25. Falk DJ, Kavazis AN, Whidden MA, Smuder AJ, McClung JM, Hudson MB, et al. Mechanical ventilation‐induced oxidative stress in the diaphragm: role of heme oxygenase‐1. Chest 2011;139:816–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McClung JM, Kavazis AN, DeRuisseau KC, Falk DJ, Deering MA, Lee Y, et al. Caspase‐3 regulation of diaphragm myonuclear domain during mechanical ventilation‐induced atrophy. Am J Respir Crit Care Med 2007;175:150–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reid MB. Free radicals and muscle fatigue: Of ROS, canaries, and the IOC. Free Radic Biol Med 2008;44:169–179. [DOI] [PubMed] [Google Scholar]
- 28. DeRuisseau KC, Shanely RA, Akunuri N, Hamilton MT, Van Gammeren D, Zergeroglu AM, et al. Diaphragm unloading via controlled mechanical ventilation alters the gene expression profile. Am J Respir Crit Care Med 2005;172:1267–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deruisseau KC, Kavazis AN, Powers SK. Selective downregulation of ubiquitin conjugation cascade mRNA occurs in the senescent rat soleus muscle. Exp Gerontol 2005;40:526–531. [DOI] [PubMed] [Google Scholar]
- 30. Bredahl EC, Pfannenstiel KB, Quinn CJ, Hayward R, Hydock DS. Effects of exercise on doxorubicin‐induced skeletal muscle dysfunction. Med Sci Sports Exerc 2016;48:1468–1473. [DOI] [PubMed] [Google Scholar]
- 31. Nakamura K, Ohsawa I, Masuzawa R, Konno R, Watanabe A, Kawano F. Running training experience attenuates disuse atrophy in fast‐twitch skeletal muscles of rats. J Appl Physiol 2017;jap002892017. [DOI] [PubMed] [Google Scholar]
- 32. Padilha CS, Borges FH, Costa Mendes da Silva LE, Frajacomo FTT, Jordao AA, Duarte JA, et al. Resistance exercise attenuates skeletal muscle oxidative stress, systemic pro‐inflammatory state, and cachexia in Walker‐256 tumor‐bearing rats. Appl Physiol Nutr Metab 2017;42:916–923. [DOI] [PubMed] [Google Scholar]
- 33. Miyabara EH, Nascimento TL, Rodrigues DC, Moriscot AS, Davila WF, AitMou Y, et al. Overexpression of inducible 70‐kDa heat shock protein in mouse improves structural and functional recovery of skeletal muscles from atrophy. Pflugers Arch 2012;463:733–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bowles DE, McPhee SW, Li C, Gray SJ, Samulski JJ, Camp AS, et al. Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol Ther 2012;20:443–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Byrne BJ, Falk DJ, Pacak CA, Nayak S, Herzog RW, Elder ME, et al. Pompe disease gene therapy. Hum Mol Genet 2011;20:R61–R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kavazis AN, Talbert EE, Smuder AJ, Hudson MB, Nelson WB, Powers SK. Mechanical ventilation induces diaphragmatic mitochondrial dysfunction and increased oxidant production. Free Radic Biol Med 2009;46:842–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Powers SK, Hudson MB, Nelson WB, Talbert EE, Min K, Szeto HH, et al. Mitochondria‐targeted antioxidants protect against mechanical ventilation‐induced diaphragm weakness. Crit Care Med 2011;39:1749–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Beddoe T, Lithgow T. Delivery of nascent polypeptides to the mitochondrial surface. Biochim Biophys Acta 2002;1592:35–39. [DOI] [PubMed] [Google Scholar]
- 39. Glover LA, Lindsay JG. Targeting proteins to mitochondria: a current overview. Biochem J 1992;284:609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zou X, Ratti BA, O'Brien JG, Lautenschlager SO, Gius DR, Bonini MG, et al. Manganese superoxide dismutase (SOD2): is there a center in the universe of mitochondrial redox signaling? J Bioenerg Biomembr 2017;49:325–333. [DOI] [PubMed] [Google Scholar]
- 41. Afolayan AJ, Teng RJ, Eis A, Rana U, Broniowska KA, Corbett JA, et al. Inducible HSP70 regulates superoxide dismutase‐2 and mitochondrial oxidative stress in the endothelial cells from developing lungs. Am J Physiol Lung Cell Mol Physiol 2014;306:L351–L360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Suzuki K, Murtuza B, Sammut IA, Latif N, Jayakumar J, Smolenski RT, et al. Heat shock protein 72 enhances manganese superoxide dismutase activity during myocardial ischemia‐reperfusion injury, associated with mitochondrial protection and apoptosis reduction. Circulation 2002;106:I270–I276. [PubMed] [Google Scholar]
- 43. Choi S, Park KA, Lee HJ, Park MS, Lee JH, Park KC, et al. Expression of Cu/Zn SOD protein is suppressed in hsp 70.1 knockout mice. J Biochem Mol Biol 2005;38:111–114. [DOI] [PubMed] [Google Scholar]
- 44. Liu CT, Brooks GA. Mild heat stress induces mitochondrial biogenesis in C2C12 myotubes. J Appl Physiol 2012;112:354–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Henstridge DC, Bruce CR, Drew BG, Tory K, Kolonics A, Estevez E, et al. Activating HSP72 in rodent skeletal muscle increases mitochondrial number and oxidative capacity and decreases insulin resistance. Diabetes 2014;63:1881–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gonzalez B, Hernando R, Manso R. Stress proteins of 70 kDa in chronically exercised skeletal muscle. Pflugers Arch 2000;440:42–49. [DOI] [PubMed] [Google Scholar]
- 47. Shanely RA, Van Gammeren D, Deruisseau KC, Zergeroglu AM, McKenzie MJ, Yarasheski KE, et al. Mechanical ventilation depresses protein synthesis in the rat diaphragm. Am J Respir Crit Care Med 2004;170:994–999. [DOI] [PubMed] [Google Scholar]
- 48. Zergeroglu MA, McKenzie MJ, Shanely RA, Van Gammeren D, DeRuisseau KC, Powers SK. Mechanical ventilation‐induced oxidative stress in the diaphragm. J Appl Physiol 2003;95:1116–1124. [DOI] [PubMed] [Google Scholar]
- 49. Nelson WB, Smuder AJ, Hudson MB, Talbert EE, Powers SK. Cross‐talk between the calpain and caspase‐3 proteolytic systems in the diaphragm during prolonged mechanical ventilation. Crit Care Med 2012;40:1857–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Smuder AJ, Nelson WB, Hudson MB, Kavazis AN, Powers SK. Inhibition of the ubiquitin‐proteasome pathway does not protect against ventilator‐induced accelerated proteolysis or atrophy in the diaphragm. Anesthesiology 2014;121:115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Smuder AJ, Sollanek KJ, Nelson WB, Min K, Talbert EE, Kavazis AN, et al. Crosstalk between autophagy and oxidative stress regulates proteolysis in the diaphragm during mechanical ventilation. Free Radic Biol Med 2017;115:179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Smuder AJ, Hudson MB, Nelson WB, Kavazis AN, Powers SK. Nuclear factor‐kappaB signaling contributes to mechanical ventilation‐induced diaphragm weakness*. Crit Care Med 2012;40:927–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Levine S, Biswas C, Dierov J, Barsotti R, Shrager JB, Nguyen T, et al. Increased proteolysis, myosin depletion, and atrophic AKT‐FOXO signaling in human diaphragm disuse. Am J Respir Crit Care Med 2011;183:483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Berger D, Bloechlinger S, von Haehling S, Doehner W, Takala J, Z'Graggen WJ, et al. Dysfunction of respiratory muscles in critically ill patients on the intensive care unit. J Cachexia Sarcopenia Muscle 2016;7:403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schefold JC, Bierbrauer J, Weber‐Carstens S. Intensive care unit‐acquired weakness (ICUAW) and muscle wasting in critically ill patients with severe sepsis and septic shock. J Cachexia Sarcopenia Muscle 2010;1:147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Maes K, Stamiris A, Thomas D, Cielen N, Smuder A, Powers SK, et al. Effects of controlled mechanical ventilation on sepsis‐induced diaphragm dysfunction in rats. Crit Care Med 2014;42:e772–e782. [DOI] [PubMed] [Google Scholar]
- 57. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the journal of cachexia, sarcopenia and muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]