

Abstract
Background
Muscle wasting, anorexia, and metabolic dysregulation are common side‐effects of cytotoxic chemotherapy, having a dose‐limiting effect on treatment efficacy, and compromising quality of life and mortality. Extracts of Cannabis sativa, and analogues of the major phytocannabinoid Δ9‐tetrahydrocannabinol, have been used to ameliorate chemotherapy‐induced appetite loss and nausea for decades. However, psychoactive side‐effects limit their clinical utility, and they have little efficacy against weight loss. We recently established that the non‐psychoactive phytocannabinoid cannabigerol (CBG) stimulates appetite in healthy rats, without neuromotor side‐effects. The present study assessed whether CBG attenuates anorexia and/or other cachectic effects induced by the broad‐spectrum chemotherapy agent cisplatin.
Methods
An acute cachectic phenotype was induced in adult male Lister‐hooded rats by 6 mg/kg (i.p.) cisplatin. In total 66 rats were randomly allocated to groups receiving vehicle only, cisplatin only, or cisplatin and 60 or 120 mg/kg CBG (po, b.i.d.). Feeding behavior, bodyweight and locomotor activity were recorded for 72 hours, at which point rats were sacrificed for post‐mortem analyses. Myofibre atrophy, protein synthesis and autophagy dysregulation were assessed in skeletal muscle, plasma metabolic profiles were obtained by untargeted 1H‐NMR metabonomics, and levels of endocannabinoid‐like lipoamines quantified in plasma and hypothalami by targeted HPLC‐MS/MS lipidomics.
Results
CBG (120 mg/kg) modestly increased food intake, predominantly at 36‐60hrs (p<0.05), and robustly attenuated cisplatin‐induced weight loss from 6.3% to 2.6% at 72hrs (p<0.01). Cisplatin‐induced skeletal muscle atrophy was associated with elevated plasma corticosterone (3.7 vs 13.1ng/ml, p<0.01), observed selectively in MHC type IIx (p<0.05) and IIb (p<0.0005) fibres, and was reversed by pharmacological rescue of dysregulated Akt/S6‐mediated protein synthesis and autophagy processes. Plasma metabonomic analysis revealed cisplatin administration produced a wide‐ranging aberrant metabolic phenotype (Q2Ŷ=0.5380, p=0.001), involving alterations to glucose, amino acid, choline and lipid metabolism, citrate cycle, gut microbiome function, and nephrotoxicity, which were partially normalized by CBG treatment (Q2Ŷ=0.2345, p=0.01). Lipidomic analysis of hypothalami and plasma revealed extensive cisplatin‐induced dysregulation of central and peripheral lipoamines (29/79 and 11/26 screened, respectively), including reversible elevations in systemic N‐acyl glycine concentrations which were negatively associated with the anti‐cachectic effects of CBG treatment.
Conclusions
Endocannabinoid‐like lipoamines may have hitherto unrecognized roles in the metabolic side‐effects associated with chemotherapy, with the N‐acyl glycine subfamily in particular identified as a potential therapeutic target and/or biomarker of anabolic interventions. CBG‐based treatments may represent a novel therapeutic option for chemotherapy‐induced cachexia, warranting investigation in tumour‐bearing cachexia models.
Keywords: Cachexia, Cisplatin, Chemotherapy, Cannabinoid, Cannabigerol, Lipoamine
Introduction
Cachexia is a multi‐factorial wasting syndrome, characterized by skeletal muscle atrophy, anorexia, and dysregulation of metabolic homeostasis. The syndrome presents as a comorbidity with many common pathologies including chronic heart failure and kidney disease, chronic obstructive pulmonary disease, and most commonly cancer, with prevalence of 50–80% in advanced cancer patients, of which 20% will die as a result.1, 2 While several decades of research have helped elucidate the pathophysiology of the cancer‐induced cachexia syndrome, less appreciated is the contributory role of cytotoxic chemotherapy regimens. In addition to the well‐documented side effects of anorexia and chemotherapy‐induced nausea and vomiting (CINV), evidence is emerging for comorbidities that are closely related to cancer‐induced cachexia, characterized by muscle atrophy and metabolic dysregulation that is qualitatively distinct from nausea and starvation‐induced pathophysiology.3, 4, 5, 6, 7, 8 This syndrome, which we and others describe as chemotherapy‐induced cachexia,9 is highly debilitating to patients, has a dose‐limiting effect on treatment efficacy, and compromises quality of life and ultimately mortality. Despite a considerable translational research effort and numerous clinical trials, to date no specific treatments for cancer‐induced cachexia are currently licensed,10 and strategies to ameliorate the side effects of chemotherapy are limited to anti‐emetics and control of acute nausea.11
Pharmacological interventions based on Cannabis sativa have been utilized clinically for decades to prevent CINV and anorexia associated with AIDS and cancer, with dronabinol, a synthetic form of Δ9‐tetrahydrocannabinol (Δ9‐THC), licensed for CINV in 1985.12, 13, 14 Coupled with the well‐documented hyperphagic effects of Δ9‐THC, this led to clinical trials of C. sativa extracts, purified Δ9‐THC, and dronabinol against cancer‐induced cachexia. However, these failed to demonstrate efficacy, in part due to dose‐limiting psychoactive side effects.15, 16 In addition to Δ9‐THC, there are now known to be over 100 other phytocannabinoids in C. sativa, the majority of which are non‐psychoactive, some of which have appetite‐modulating effects.17, 18, 19, 20 We have recently shown that cannabigerol (CBG), a non‐psychoactive phytocannabinoid, increases food intake in healthy rats without eliciting neuromotor side effects.21, 22 Given that patients undergoing chemotherapy commonly report appetite loss, altered food palatability, and early satiety,23, 24, 25, 26 CBG may have potential to attenuate these anorectic side effects. While the molecular targets of CBG are poorly characterized, it has been shown to be an agonist at PPARγ and inhibitor of anandamide cellular uptake at micromolar concentrations,27, 28 which are achieved in brain and plasma following oral administration of CBG at doses used in our studies to elicit hyperphagia.29 These pharmacological activities of CBG could conceivably mediate beneficial effects in the context of cachexia on appetite, inflammation, and multiple aspects of metabolic function.30, 31, 32
The present study was designed to test the hypothesis that CBG is able to attenuate the anorexia, weight loss, and metabolic dysregulation induced by the broad‐spectrum cytotoxic chemotherapy agent cisplatin. We utilized a previously validated rat model of cisplatin‐induced cachexia, in which an acute cisplatin dose induces progressive bodyweight loss and anorexia, which reaches a nadir 48–72 h after administration.33, 34, 35 Animals were randomly allocated to receive either saline and vehicle (CON), cisplatin and vehicle (CIS), or cisplatin and CBG at 60 or 120 mg/kg b.i.d. (CIS + CBG60 and CIS + CBG120). Food intake, bodyweight, and locomotor activity were recorded over 72 h, after which animals were sacrificed, and plasma, hypothalamus, and muscle samples were taken for ex vivo analyses. Muscle mass and molecular markers of protein synthesis and catabolic pathways were assessed by immunohistochemistry and Western blot to investigate effects of CBG on cisplatin‐induced muscle atrophy. Given the multi‐factorial nature of cachexia, and the pleiotropic activity profiles typical of phytocannabinoids, metabonomic and lipidomic profiles were obtained from plasma and hypothalamus samples to further characterize the pathophysiology of cisplatin‐induced metabolic dysregulation and any normalization due to CBG treatment. These multi‐omic datasets were interrogated to identify the salient compartments and biochemical processes associated with the therapeutic effects of CBG.
Materials and methods
Study design
The controlled laboratory experiment reported here was conducted to test the pre‐specified hypothesis that the appetite‐stimulating phytocannabinoid CBG would attenuate the anorexia and/or weight loss induced by a single 6 mg/kg dose of cisplatin in rats. Primary endpoints were cumulative food intake, bodyweight change, and muscle mass at the end of the 72 h test, a duration specified based on literature reports that this timepoint represents the nadir of cisplatin's cachectic effects.23, 33, 34, 35 Animals were randomized to one of four groups as detailed below, with sample sizes based on literature reports of this model and our own pilot studies. Additional in vitro analyses were conducted as detailed below, using post‐mortem tissue samples to investigate the effects of cisplatin, and CBG treatment, on muscle catabolism, plasma metabolic and hypothalamic and plasma lipidomic phenotypes. All post‐mortem analyses were conducted by experimenters blinded to treatment.
Animals
Male Lister hooded rats (Harlan, UK) weighing 200–225 g at the start of studies were housed in pairs in the test room on a 12/12 light cycle (white light on at 11:00) with standard laboratory chow and water available ad libitum. Animals were habituated to the test environment and handling for 7 days prior to the start of testing. Sixty‐six animals were tested in total, nine of which were excluded as asymptomatic for the cachectic effects of cisplatin (as detailed below) and a further two excluded due to technical errors with the automated food intake system. All animal protocols were approved by the University of Reading and followed ARRIVE guidelines.
Drugs
Cisplatin (cis‐diammineplatinum (II) dichloride; Sigma‐Aldrich, UK) was dissolved by sonication in sterile 0.9% saline at 1.5 mg/mL and administered intraperitoneally at a volume of 4 mL/kg, at the onset of the light period (11:00) on the first day of testing. CBG (GW Research Ltd, UK) was dissolved in sesame seed oil (by magnetic stirring at 57°C) at 120 mg/mL and administered per ora at 60 or 120 mg/kg in 1 mL/kg dose volume. Animals were administered CBG or vehicle immediately prior to cisplatin, then subsequently every 12 h at the light/dark period transition throughout the 3 day test session (Figure 1A).
Figure 1.
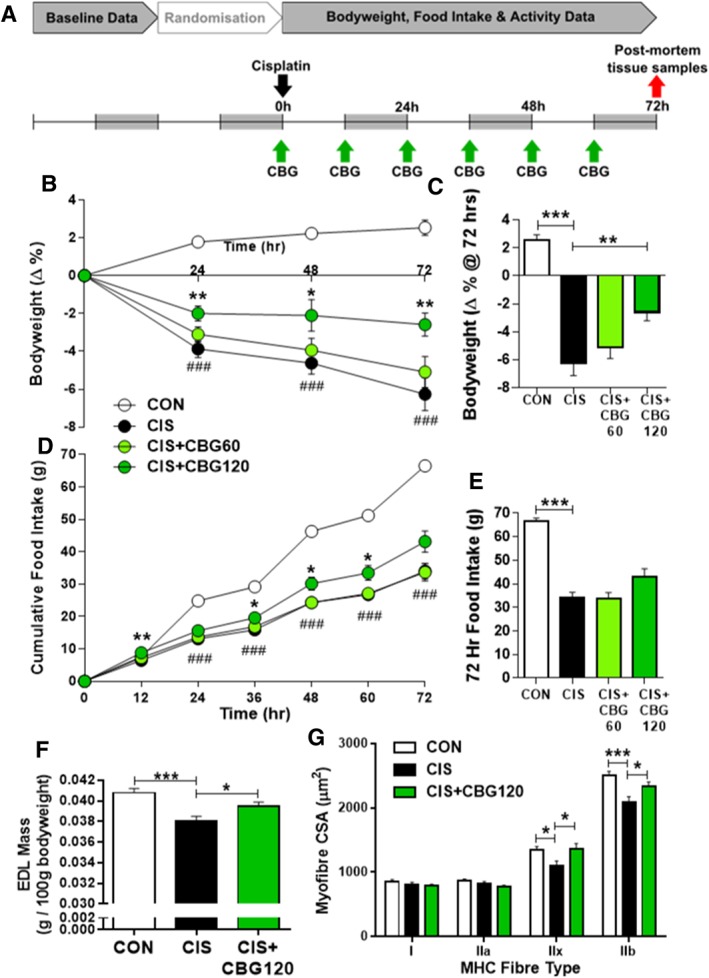
Cannabigerol attenuates cisplatin‐induced weight loss, anorexia, and myofibre hypotrophy. Experimental timeline and dosing schedule (A), bodyweight change (B, C), and cumulative food intake (D, E) prior to post‐mortem tissue harvest 72 h after administration of 6 mg/kg cisplatin. Data presented as mean ± SEM and analysed by two‐way mixed‐model ANOVA (drug × day) followed by one‐way between‐subjects ANOVA and Tukey's post hoc comparisons at each timepoint, n = 11–16. ### P < 0.0005 vs. CON, * P < 0.05 vs. CIS, ** P < 0.01 vs. CIS. Mass of isolated extensor digitorum longus (EDL) muscle 72 h after cisplatin administration (F; n = 11–15) and cross‐sectional area (CSA) of myofibres by MHC fibre type (G; n = 7–10). Data presented as mean ± SEM and analysed by one‐way between‐subjects ANOVA and planned comparisons. * P < 0.05, *** P < 0.0005.
In vivo protocol
The protocol for induction of cisplatin‐induced anorexia and fatigue was based on previously published studies using a single dose of cisplatin at 6 mg/kg.33, 34, 35 Briefly, following 1 week of habituation to handling, animals were weighed and administered sesame oil vehicle at light onset (11:00) and immediately placed in test chambers for 24 h for a habituation and baseline session. Animals were then randomly allocated to receive saline and sesame oil vehicles (CON), cisplatin and sesame oil vehicle (CIS), cisplatin and 60 mg/kg CBG (CIS + CBG 60), or cisplatin and 120 mg/kg CBG (CIS + CBG 120). Successful randomization was confirmed by a lack of significant overall analysis of variance (ANOVA) results for measures of bodyweight, 24 h food intake, or 24 h locomotor activity during baseline sessions (Supporting Information, Table S1). After 24 h in home cages following baseline sessions, animals began a 72 h test session at 11:00. Animals were administered an initial dose of CBG or vehicle, immediately prior to cisplatin or saline, and placed in test cages, then subsequently dosed with CBG or vehicle every 12 h for the remainder of the test. Health checks and bodyweight measurements were conducted every 12 h immediately before dosing. Food intake and locomotor activity were recorded automatically as previously described,21, 22, 36 with video recordings used to confirm feeding episodes. At the end of the 72 h test period, animals were euthanized by CO2 inhalation, blood samples collected by cardiac puncture, and hypothalami rapidly removed and snap frozen on liquid nitrogen. Extensor digitorum longus (EDL) muscles were removed from both hind limbs, weighed, and snap frozen.
Data processing and analysis
Pilot studies using this model revealed a considerable heterogeneity in cachectic response to cisplatin, with a discrete minority of animals displaying little or no discernible effect on bodyweight or food intake. Using pilot data, a priori exclusion criteria were determined and applied prospectively to the present study, resulting in the exclusion of nine animals as effectively asymptomatic for the cachectic effects of cisplatin (across the three groups administered cisplatin). Animals were excluded if they failed to show a minimum cachectic response of either <9 g food intake during the first dark period (12–24 h) or a loss of >3% bodyweight at 36 h (Supporting Information, Figure S2). These criteria, which were 2 SD below the mean intake and bodyweight measures of saline‐treated controls in our pilot experiments, ensured CBG efficacy was only assessed in animals showing a robust cisplatin‐induced cachectic effect consistent with previous studies. Data were analysed using two‐way mixed‐model ANOVA (drug × time) and/or one‐way ANOVA (drug), followed by Tukey's all‐pairwise comparisons for main outcome dose–response measures, or planned comparisons (CON vs. CIS and CIS vs. CIS + CBG 120) for port‐mortem analyses, as detailed in figure legends. Where assumptions of sphericity were violated (as indicated by a significant Mauchly's test result), degrees of freedom and P values from the Greenhouse–Geisser correction were utilized. Results were considered significant at α = 0.05.
Ex vivo analysis of muscle atrophy
Post‐mortem analyses of muscle fibre atrophy, and molecular markers of anabolic and catabolic signalling pathways in muscle tissue, were conducted by immunohistochemistry and Western blot, as fully detailed in the Supporting Information.
Corticosterone and cytokine analyses
Post‐mortem plasma samples were used in commercial ELISA kits to quantify concentrations of corticosterone (Enzo Life Sciences, Exeter, UK), IL‐1β (Abcam, Cambridge, UK), IL‐6 (Novex, Thermofisher, Waltham, MA), and TNFα (Thermofisher, Waltham, MA) according to manufacturers' instructions.
1H‐NMR spectroscopy‐based metabonomics analysis
An untargeted 1H‐NMR spectroscopy‐based metabonomics approach was used to determine the plasma metabolic profiles of animals in CON, CIS, and CIS + CBG120 groups 72 h after cisplatin administration. Multivariate modelling including principal component analysis (PCA) and orthogonal projection to latent structures discriminant analysis (OPLS‐DA) were performed using in‐house scripts and those provided by Korrigan Sciences Ltd, UK. Sample preparation, spectra acquisition, and data processing were conducted as previously reported37 and detailed in the Supporting Information.
HPLC–MS/MS lipidomic analysis of endocannabinoids and lipoamines
Targeted HPLC–MS/MS lipidomic analysis was performed on hypothalamus and plasma samples from animals in CON, CIS, and CIS + CBG120 groups, harvested 72 h after cisplatin administration, as previous reported38 and fully detailed in the Supporting Information. Hypothalamus samples were screened against a library of 71 lipoamines (including anandamide), four 2‐acyl‐sn‐glycerols (including 2‐AG), arachidonic and linoleic acids, and the prostaglandins PGE2 and PGF2α. Plasma samples were screened for 18 of the lipoamines (known to be detectable in plasma), in addition to the 2‐acyl‐sn‐glycerols, free fatty acids, and prostaglandins.
Results
Cannabigerol attenuates cisplatin‐induced weight loss, anorexia, and myofibre hypotrophy
Consistent with previous reports using this model of chemotherapy‐induced cachexia, administration of cisplatin resulted in progressive loss of bodyweight over the 72 h test (Figure 1B,C; drug × time F 7.3, 124.3 = 14.731, P < 0.0005). Animals administered cisplatin alone lost 6.3% bodyweight after 72 h (P < 0.0005), consistent with the clinical diagnostic criteria for cachexia of ≥5% bodyweight loss.39 While treatment with 60 mg/kg CBG did not have a significant protective effect against this loss, 120 mg/kg CBG significantly attenuated bodyweight loss at all timepoints. At 24 h, 120 mg/kg CBG reduced cisplatin‐induced weight loss by approximately half (P = 0.006), and effectively prevented any further decrease during the remaining test duration, such that this group only lost 2.6% after 72 h (P = 0.004). Cisplatin elicited a profound anorectic effect (Figure 1D,E; drug × time F 3.4, 57.0 = 26.648, P < 0.0005), apparent from 24 h onwards, with cumulative intake at 72 h of 34 g compared with 67 g in controls (P < 0.0005). This anorectic effect was due to decreased feeding frequency during animals' active (dark) phase on days 1–3, and reductions in meal size on days 2–3 (Supporting Information, Table S2). Treatment with 120 mg/kg CBG elicited a mild hyperphagic response during the first 12 h and a significant protective effect against cisplatin‐induced anorexia from 36–60 h (P = 0.018, P = 0.045, and P = 0.044, respectively; Figure 1D), with a near‐significant increased total intake over the test duration (P = 0.067). Cisplatin‐induced reductions in meal size were prevented by 120 mg/kg CBG during the dark phase of day 3 (P = 0.037). Cisplatin also induced a suppression of dark phase ambulatory locomotor activity on days 1–3, which was not significantly altered by either dose of CBG (Supporting Information, Table S3).
To investigate the extent to which cisplatin‐induced weight loss was driven by skeletal muscle atrophy, hindlimb muscle mass and myofibre hypotrophy were quantified in post‐mortem samples from CON, CIS, and CIS + CBG120 animals taken at the nadir of cisplatin's cachectic effects (72 h). Drug‐induced alterations in the mass of EDL muscle were observed (Figure 1F; F 2, 36 = 10.146, P < 0.0005), which was reduced by 6.7% by cisplatin (P < 0.0005). However, CBG attenuated this loss to 3.2% (P = 0.021). Immunohistochemical analysis of myofibre hypotrophy by MHC fibre type (Figure 1G) demonstrated cisplatin‐induced reductions of cross‐sectional area in fast glycolytic type IIx (P = 0.025) and IIb (P < 0.0005) fibres, which were attenuated by CBG treatment (P = 0.018 and P = 0.025). In contrast, no cisplatin‐induced hypotrophy was observed in slow oxidative type Ia or fast oxidative/glycolytic type IIa fibres.
Cannabigerol rescues cisplatin‐induced dysregulation of myofibre autophagy and protein synthesis
Inflammation‐mediated HPA axis activation, and subsequent chronic elevation of systemic corticosterone, has recently been demonstrated to induce myofibre atrophy following cyclophosphamide‐doxorubicin‐5‐fluorouracil chemotherapy in mice.5 Given that corticosterone‐induced atrophy selectively occurs in MHC IIb/x fibres,40, 41 resulting from dysregulated autophagy‐lysosome catabolism and inhibited muscle protein synthesis,42 we investigated these pathways in plasma and EDL muscle samples from animals administered cisplatin and treated with 120 mg/kg CBG. Plasma corticosterone concentration was increased by cisplatin 72 h after administration; however, this was not modulated by CBG treatment (Figure 2A; P = 0.002 and P = 0.754). In contrast, plasma concentrations of the pro‐inflammatory cytokines IL‐1β, IL‐6, and TNFα were below the limit of quantitation or unaffected by either cisplatin or CBG at this timepoint (data not shown).
Figure 2.
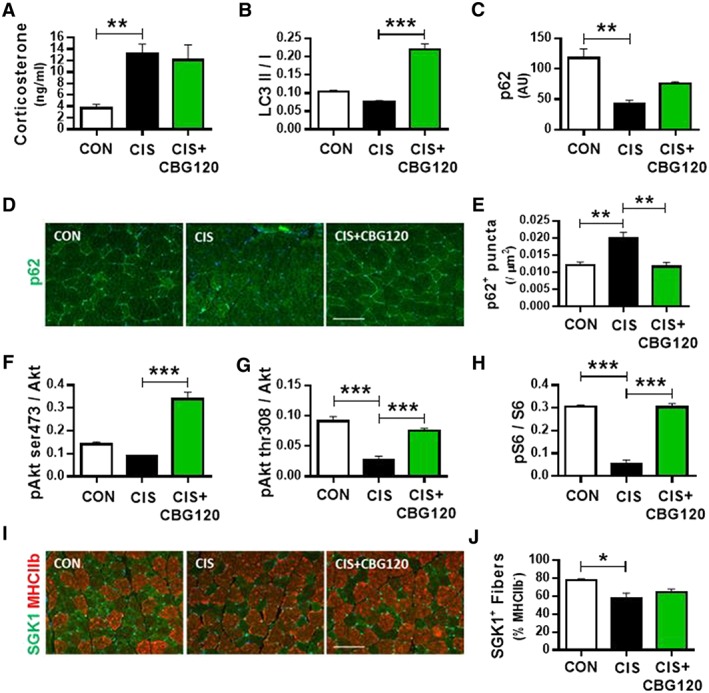
Cannabigerol rescues cisplatin‐induced dysregulation of myofibre autophagy and protein synthesis. Plasma corticosterone levels determined by ELISA (A; n = 6–9); EDL muscle tissue Western blot analysis of autophagy markers LC3 (B) and p62 (C) and immunohistochemical quantification of p62 (D, E; scale bars = 100 μm); expression of protein synthesis markers pAkt ser473 (F), pAkt thr308 (G) and pS6 (H) and immunohistochemical quantification of protein synthesis marker SGK1 (I, J; scale bars = 100 μm; all n = 3). Data presented as mean ± SEM and analysed by one‐way between‐subjects ANOVA and planned comparisons. * P < 0.05, ** P < 0.01, *** P < 0.0005.
Dysregulation of autophagic flux, a mechanism of muscle catabolism recently implicated in cachexia,43, 44 was investigated by Western blot for the ratio of active LC3 protein (LC3 II) to the inactive cleaved form of the propeptide (LC3 I; Figure 2B and Supporting Information, Figure S3), expression of p62 protein (Figure 2C and Supporting Information, Figure S3), and by immunohistochemical analysis of p62 + ve puncta density in myofibres (Figure 2D,E). Cisplatin elicited a near‐significant reduction in active LC3 ratio (P = 0.073), which was increased by CBG treatment (P < 0.0005). Expression of p62 was reduced by cisplatin (P = 0.002) and partially normalized by CBG treatment (P = 0.051). These results imply that autophagy is blunted by cisplatin but is activated by CBG. We investigated this further by quantifying p62 puncta that accumulate when autophagy is blunted. Consistent with the notion of blunted autophagy, cisplatin induced a significant increase in p62 + ve puncta density (P = 0.0029), which was normalized to control levels by CBG treatment (P = 0.004).
We next profiled key components of the protein synthesis pathway to develop a mechanistic understanding of the changes in muscle mass induced by cisplatin and CBG by profiling Akt and SGK‐1, as well as key downstream targets (Figure 2F–J and Supporting Information, Figure S3). Cisplatin elicited a near‐significant reduction in pAkt ser473 (P = 0.088), while CBG significantly increased this ratio above control levels (P < 0.0005). Phosphorylation of Akt at the thr308 residue was reduced four‐fold by cisplatin (P < 0.0005), which was normalized to near control levels by CBG (P < 0.0005). Phosphorylation of ribosomal protein S6 (the downstream target of Akt) was reduced six‐fold by cisplatin (P < 0.0005), and this reduction was normalized to control levels by CBG (P < 0.0005). In addition to the effects on the canonical Akt/S6 protein synthesis pathway, cisplatin also decreased the number of SGK1 + ve fibres in EDL muscle (P = 0.025); however, this was not significantly altered by CBG treatment (P = 0.358). These results demonstrate that suppression of normal anabolic protein synthesis contributes to cisplatin‐induced muscle atrophy and that this suppression is normalized by CBG treatment, primarily via the Akt‐S6 pathway.
Cannabigerol partially normalizes the cisplatin‐induced aberrant metabolic phenotype
HPLC and 1H‐NMR spectroscopy‐based metabonomic approaches have recently been used to investigate markers of metabolic abnormalities in urine and kidney associated with cisplatin‐induced nephrotoxicity.45, 46 Here, we report the use of untargeted 1H‐NMR spectroscopy‐based metabonomics to characterize the systemic metabolic disruptions induced by a cachectic dose of cisplatin and to identify metabolic processes involved in the anti‐cachectic effects of CBG. Biochemical profiles were acquired from the post‐mortem plasma samples of animals in CON, CIS, and CIS + CBG120 groups. PCA was used to identify metabolic variation in the plasma profiles associated with cisplatin and/or CBG treatment. From the pairwise PCA model, clear metabolic variation was observed between the CON and CIS profiles (Figure 3A). This is consistent with the widespread metabolic dysregulation that characterizes the cachectic phenotype. The pairwise PCA model comparing the plasma metabolic profiles from CIS and CIS + CBG120 groups showed a modest degree of overlap between the groups (Figure 3B), although the overall distributions were distinct. Interestingly, a greater spread was observed in the CIS + CBG120 profiles indicating a heterogeneous response to CBG treatment, consistent with the effects on main outcome measures seen in vivo. No distinct clustering was observed in the scores plot from the PCA model comparing the CON and CIS + CBG120 profiles (Figure 3C), suggesting these groups exhibit much greater homogeneity, reflecting a normalization of the cisplatin‐associated metabolic variation by CBG treatment.
Figure 3.
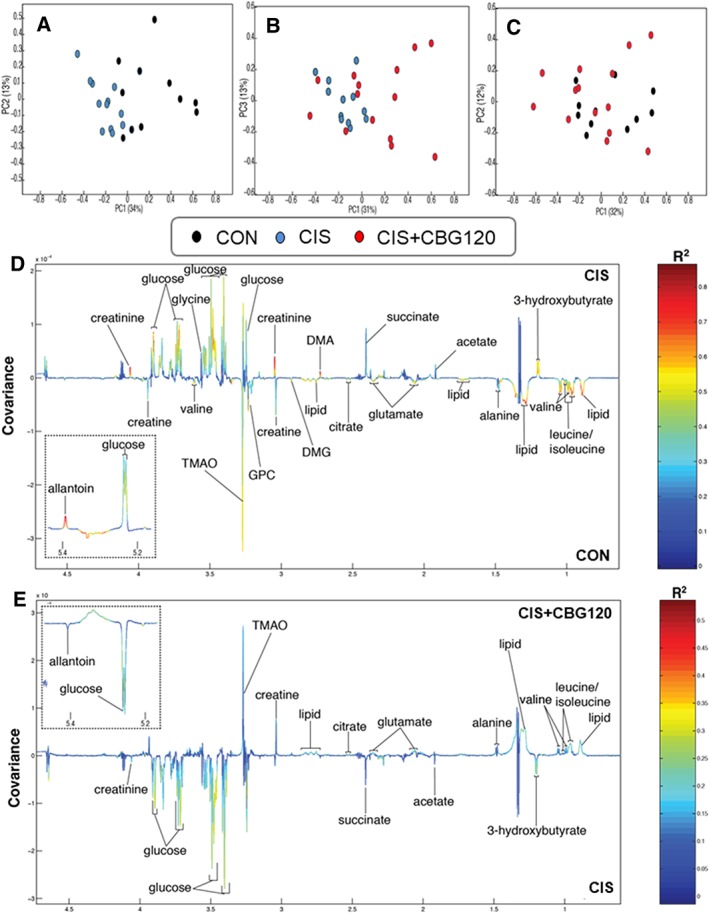
Cannabigerol partially normalizes the cisplatin‐induced aberrant metabolic phenotype. Principal component analysis scores plots (PC1 vs. PC2/3, % variance in parentheses) of the 1H‐NMR plasma metabolic profiles for pairwise comparisons of CON vs. CIS (A), CIS vs. CIS + CBG120 (B), and CON vs. CIS + CBG120 (C). Orthogonal projection to latent structures discriminant analysis (OPLS‐DA) revealed a widespread aberrant metabolic phenotype induced by cisplatin (D; Q2Ŷ = 0.5380, P = 0.001), with many of these cisplatin‐induced metabolite changes partially reversed by CBG treatment (E; Q2Ŷ = 0.2345, P = 0.01). Relative abundance plots for all individual metabolites derived from the OPLS‐DA integrals are provided in Supporting Information, Figure S4.
To identify individual metabolites driving the cisplatin‐induced alterations to metabolic phenotype, and those normalized by CBG treatment, OPLS‐DA was applied to these metabolic profiles (Figure 3D,E and Supporting Information, Figure S2; CON vs. CIS: Q2Ŷ = 0.5380, P = 0.001, CIS vs. CIS + CBG120: Q2Ŷ = 0.2345, P = 0.01). The cisplatin‐induced phenotype (Figure 3D) was characterized by hyperglycaemia and elevated creatinine, glycine, allantoin, the ketone body 3‐hydroxybutyrate, and the marker of gut microbiome choline metabolism dimethylamine. Relative to controls, cisplatin reduced creatine, citrate, the gut microbiome metabolite trimethylamine‐N‐oxide (TMAO), the branched‐chain amino acids (BCAAs) leucine, isoleucine and valine, and choline derivatives dimethylglycine and glycerophosphocholine. Pairwise comparison of CIS + CBG120 and CIS samples (Figure 3E) confirmed the partial normalization of the aberrant metabolic phenotype suggested by PCA analysis, with samples from the CIS + CBG120 group having lower levels of glucose, creatinine, allantoin, 3‐hydroxybutyrate, and higher trimethylamine‐N‐oxide and BCAAs, than those from the CIS group. No significant differences were observed between the CIS + CBG120 and CON profiles confirming the attenuation of CIS‐induced biochemical perturbations by CBG treatment.
Cisplatin‐induced cachexia is associated with extensive modulation of hypothalamic and systemic endocannabinoid and lipoamine signalling
While the molecular targets of CBG are quite poorly characterized, it has known affinities for several enzymes and receptors involved in endocannabinoid signalling.27, 28, 47, 48 The canonical endocannabinoids, N‐arachidonoyl ethanolamine (AEA / anandamide) and 2‐arachidonoyl glycerol (2‐AG), and an increasing number of the structurally analogous lipoamine and 2‐acyl‐sn‐glycerol signalling lipids, have established modulatory roles in many metabolic processes dysregulated in cachectic pathophysiology.49, 50 Furthermore, recent metabonomic studies of cisplatin toxicity have reported alterations to pathways involved in amino acid and lipid metabolism, involving the substrates for endocannabinoid and lipoamine synthesis.46 We thus conducted targeted HPLC–MS/MS lipidomic analyses of hypothalamus and plasma samples from animals administered cisplatin and CBG, to test the hypotheses that cisplatin‐induced cachexia is associated with alterations to signalling lipids and that the anti‐cachectic effects of CBG involve normalization of such signalling.
Of the 79 lipids screened in hypothalami (Supporting Information, Table S4), cisplatin significantly altered the concentrations of 29 lipoamines/2‐acyl‐sn‐glycerols from 11 subfamilies, and 11 of the 26 lipids screened for in plasma (Table 1 and Supporting Information, Table S5). Of particular note are the 1.5‐fold to three‐fold decreases in all six hypothalamic N‐acyl ethanolamines (NAEs), including anandamide, and similar decreases in plasma NAEs. Reductions were also seen in hypothalamic concentrations of various N‐acyl GABAs, ‐leucines, ‐prolines, ‐serines, ‐taurines, and ‐valines. In contrast, concentrations of several different N‐acyl phenylalanines and ‐tyrosines were increased by cisplatin, but 2‐acyl‐sn‐glycerols (including 2‐AG), free fatty acids, and prostaglandins were unaffected. In hypothalami, CBG treatment generally had a minimal effect on cisplatin‐induced alterations; however, increases in N‐palmitoyl proline, N‐stearoyl proline, and N‐stearoyl tyrosine were reversed. Plasma concentrations of linoleic and arachidonic acids were also increased by cisplatin and unaffected by CBG treatment. At the post‐mortem time of sampling, these data suggest that the anti‐cachectic effects of CBG are not mediated by the normalization of wider endocannabinoid‐like lipid signalling in central metabolic control regions. However, in contrast to the modest CBG‐induced alterations in the central lipidomic profile, plasma concentrations of the N‐acyl glycine subfamily exhibited a remarkably consistent association with the anti‐cachectic effects of CBG treatment (Table 1, Figure 4, and Supporting Information, Figure S5). With the exception of N‐docosahexaenoyl glycine, concentrations of all lipoamines in this subfamily were significantly increased by cisplatin (P < 0.05–0.001; Figure 4A–E), and CBG attenuated the increases in N‐palmitoyl glycine (P < 0.05), N‐stearoyl glycine (P < 0.01), and N‐oleoyl glycine (P < 0.05).
Table 1.
Single‐dose cisplatin administration extensively modulates hypothalamic anandamide and lipoamine concentrations. Targeted HPLC–MS/MS lipidomic analysis was used to screen hypothalamic samples for 71 lipoamine and four 2‐acyl‐sn‐glycerol lipoamines in addition to linoleic and arachidonic acids and the prostaglandins PGE2 and PGF2α, with plasma samples screened for 18 of the lipoamine lipids, as detailed in Supporting Information, Table S3 .
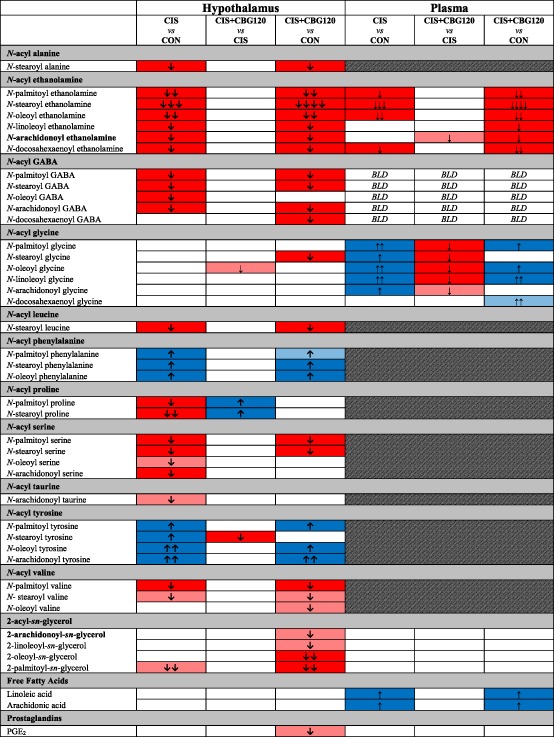
Boxes in solid blue or red denote lipids significantly increased or decreased, respectively, with shaded boxes denoting alterations that were near significance (P < 0.01). BLD, below limit of detection. Number of arrows indicates the magnitude of difference (one arrow = one‐fold to 1.49‐fold change; two arrows = 1.5‐fold to 1.99‐fold change; three arrows = two‐fold to 2.99‐fold change; four arrows = three‐fold to 9.99‐fold change). The canonical endocannabinoids N‐arachidonoyl ethanolamine (anandamide/AEA) and 2‐arachidonoyl‐sn‐glycerol (2‐AG) are highlighted in bold. Only lipoamines for which at least one significant comparison was observed are presented here; concentrations of all lipoamines screened in hypothalamus and plasma samples are provided in Supporting Information, Table S5. All data analysed by one‐way between‐subjects ANOVA and post hoc Fisher's LSD tests where appropriate, n = 11–16.
Figure 4.
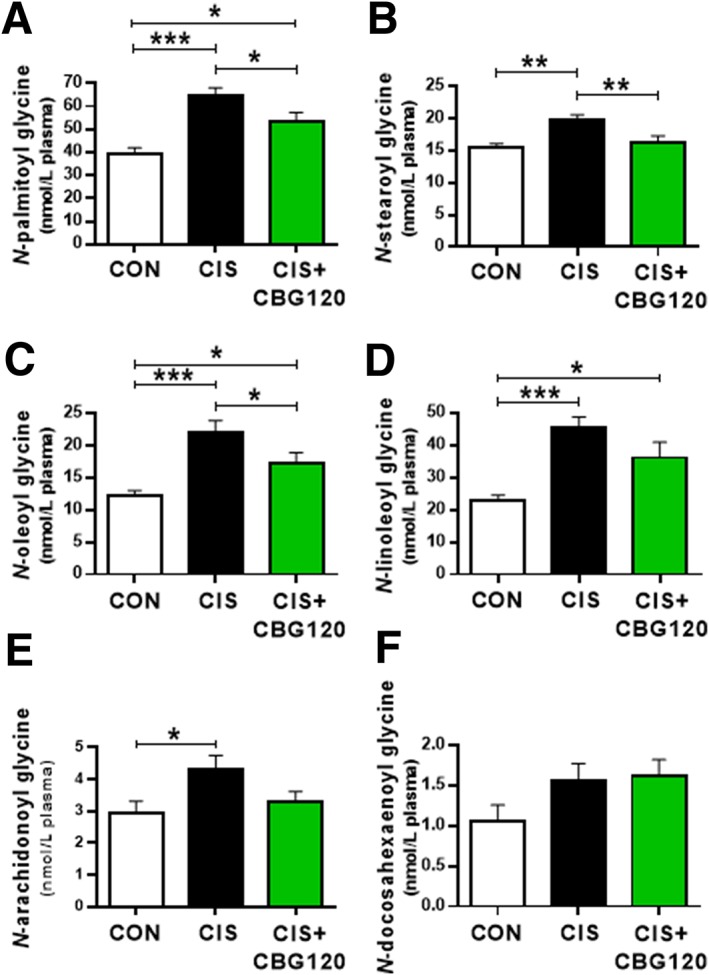
Cannabigerol attenuates cisplatin‐induced increases in plasma N ‐acyl glycine concentrations. All data analysed by one‐way between‐subjects ANOVA and post hoc Fisher's LSD tests where appropriate, n = 11–16. * P < 0.05, ** P < 0.01, *** P < 0.0005.
Cannabigerol has a prohomeostatic effect on systemic cachectic phenotype
Given the complex and diverse phenotypic alterations observed in this model, cluster and correlational analyses were performed on the metabonomic and lipidomic datasets to clarify the important compartments and pathways involved in the prohomeostatic effect of CBG. Cluster analyses were used to determine the similarity between CON, CIS, and CIS + CBG120 groups at the levels of metabonomic phenotype (Figure 5A), hypothalamic lipidomic phenotype (Figure 5B), and the combined systemic phenotype (Figure 5C), comprising all metabolites, plasma lipoamines, and primary outcome measures of bodyweight, food intake, and EDL muscle mass. Consistent with PCA and OPLS‐DA analyses, cluster analysis of the metabonomic phenotype demonstrated the greatest similarity between CON and CIS + CBG120 groups, supporting the notion that CBG treatment partially normalizes plasma metabolic phenotype. In contrast, a substantial degree of similarity was observed in the hypothalamic lipidomic phenotype between CIS and CIS + CBG120 groups, confirming the qualitative observation that CBG has little effect on cisplatin‐induced alterations to central lipoamines. Most notably, the combined systemic phenotype of animals treated with CBG was most similar to control animals, confirming a wide‐ranging prohomeostatic effect of CBG treatment.
Figure 5.
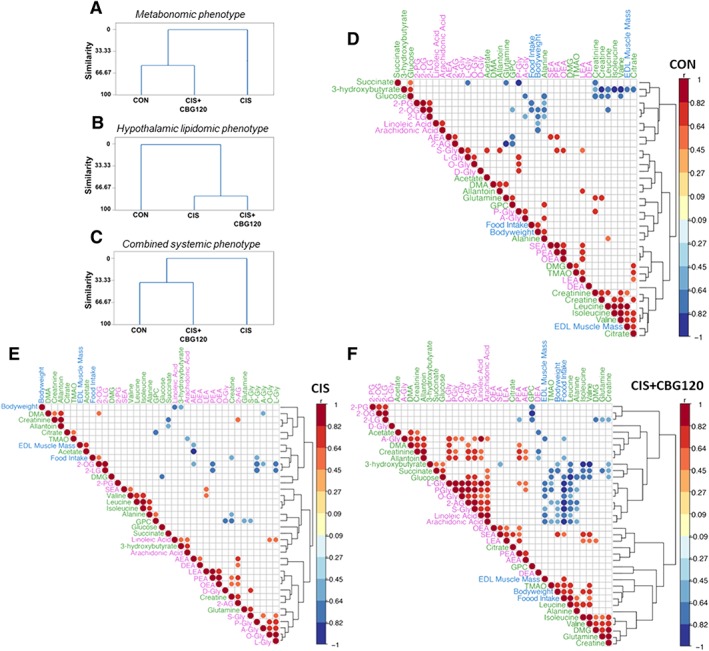
Cannabigerol has a prohomeostatic effect on systemic cachectic phenotype. cluster analyses of metabolite and lipid concentrations between three treatment groups for metabonomic phenotype (A), hypothalamic lipidomic phenotype (B) and combined systemic phenotype comprising all plasma metabolites, plasma lipoamines and main outcome measures of bodyweight, food intake, and EDL muscle mass (C). Dendrograms were produced using the single‐linkage method and Euclidean distance measure. The similarity level at any step is the percentage of the minimum distance at that step relative to the maximum inter‐observation distance in the data. Correlograms of all significant Spearman's rho correlations between all individual metabolites, lipoamines and outcome measures comprising the combined systemic phenotype, for animals in CON (D), CIS (E), and CIS + CBG120 (F) groups. Colour intensity and size of circles are proportional to correlation coefficients.
Correlational analyses were then conducted between all individual plasma metabolites, plasma lipoamines/2‐acyl‐sn‐glycerols, and main outcome measures in each of CON, CIS, and CIS + CBG120 groups (Figure 5D–F). Of note, the negative correlation between plasma arachidonic acid and bodyweight in control animals was lost in those administered cisplatin but re‐established with CBG treatment. More strikingly, strong negative correlations were observed in CBG‐treated animals between EDL muscle mass and plasma concentrations of N‐stearoyl glycine (r = −0.7477, R 2 = 0.5591, P = 0.0033; Supporting Information, Figure S5A), N‐oleoyl glycine (r = −0.6971, R 2 = 0.486, P = 0.0081; Supporting Information, Figure S5B), N‐palmitoyl glycine (r = −0.6068, R 2 = 0.3682, P = 0.0279; Supporting Information, Figure S5C), N‐arachidonoyl glycine (r = −0.6523, R 2 = 0.4255, P = 0.0157; Supporting Information, Figure S5D), and N‐linoleoyl glycine (r = −0.6203, R 2 = 0.3848, P = 0.0237; Supporting Information, Figure S5E). These data provide further evidence for the association between systemic N‐acyl glycine signalling and the anti‐cachectic effects of CBG treatment and suggest that these lipoamines may represent novel biomarkers of anabolic treatment efficacy.
Discussion
The data presented here demonstrate that CBG (at 120 mg/kg, b.i.d.) has protective effects against multiple components of chemotherapy‐induced cachexia pathophysiology, including anorexia, weight loss, muscle atrophy, and metabolic dysregulation. In addition, it provides the first evidence that cisplatin administration profoundly alters hypothalamic and systemic levels of many endocannabinoid‐like signalling lipids and that the beneficial effects of CBG in this model are associated with modulation of the N‐acyl glycine lipoamine subfamily.
Cannabigerol produced a robust therapeutic effect on total bodyweight change, with the magnitude of cisplatin‐induced weight loss reduced by approximately 60%. CBG exerted a considerable protective effect within 24 h and prevented further weight loss over the remaining experimental timecourse. Previous studies in this model over a longer duration have shown the effects of acute cisplatin on bodyweight reach a nadir 48–72 h after administration, after which they slowly return to near baseline levels after 16 days.23, 33, 34, 35 The present data thus strongly suggest that CBG is able to robustly protect against the most detrimental phase of cisplatin‐induced weight loss. Somewhat in contrast to this robust effect, CBG produced only a modest attenuation of cisplatin‐induced anorexia, predominantly during the 36–60 h period. This cisplatin‐induced anorexia was characterized by decreases in feeding frequency and meal size, suggesting a suppression of consummatory feeding processes, with CBG significantly increasing meal size during the third dark period only. These observations suggest that the protective effect of CBG against cisplatin‐induced weight loss was not solely due to attenuation of anorexia, and indeed appears to be largely independent of its hyperphagic effects. This apparent decoupling of the hyperphagic effects of CBG from its therapeutic effects against muscle catabolism further supports the contention that cisplatin does indeed elicit a bona fide chemotherapy‐induced cachectic pathophysiology—a syndrome more physiologically complex than simply starvation due to nausea and vomiting. Segregation of the starvation‐induced components of the observed pathophysiology by incorporation of a pair‐fed group was beyond the scope of the present study. However, similar analyses have recently been conducted in mouse models of cisplatin‐induced cachexia, which reported marked differences in hindlimb muscle mass, myofibre hypotrophy, molecular markers for muscle atrophy, and lipolysis and β‐oxidation between cisplatin and pair‐fed (and water‐restricted) groups.4, 7
To investigate the putative anti‐cachectic effect of CBG on muscle homeostasis, a detailed investigation of these drugs' effects on muscle catabolism mechanisms was conducted. Cisplatin caused substantial loss of skeletal muscle mass after 72 h, due to selective atrophy of fast glycolytic type IIx and IIb myofibres consistent with hyperactivation of endogenous glucocorticoid (GC) signalling,40, 41, 51 and indeed, the first observations of cachexia‐associated elevations in systemic corticosterone following cisplatin administration are reported here. The failure to detect elevations in pro‐inflammatory cytokines is perhaps surprising, and may be due to the relatively late timepoint at which plasma samples were obtained. However, recent studies suggest that, in contrast to their established role in the pathophysiology of cancer‐induced cachexia, they may not have such physiological relevance in the chemotherapy‐induced aspects of this syndrome.4, 48 Further evidence for a central role of hyperactivated GC signalling in cisplatin‐induced muscle catabolism, and the prohomeostatic effects of CBG in this process, was obtained from the plasma metabonomic profiles, characterized by cisplatin‐induced hyperglycaemia and elevated creatinine, which were partially normalized by CBG. The cisplatin‐induced metabolic phenotype was further characterized by decreased levels of the BCAAs leucine, isoleucine, and valine and elevated 3‐hydroxybutyrate, all of which were normalized by CBG. While beyond the scope of the present study, the extent to which normalization of these metabolites merely reflects attenuation of anorexia, and the functional consequences of their normalization in the context of muscle homeostasis (and tumour cell metabolism), warrants further investigation.
At the molecular level, inhibition of Akt/mTOR‐mediated muscle protein synthesis is a well‐established hallmark of both cancer—and chemotherapy‐induced cachexia.52, 53 In the present study, a clear cisplatin‐induced suppression of Akt activation was observed, as determined by its lower phosphorylation at serine 473 and threonine 308 residues. This effect was particularly apparent from the six‐fold decrease in the phosphorylation ratio of ribosomal protein S6, the downstream target of Akt that controls transcriptional initiation of muscle protein synthesis. CBG treatment resulted in a robust reversal of the inhibition of Akt/S6‐mediated protein synthesis, indicating that at least part of its beneficial effect against muscle fibre atrophy was due to rescue of the anabolic component of muscle homeostasis. CBG also exhibited beneficial effects against hyperactivated muscle catabolism, via a normalizing effect on dysregulated autophagic flux, a target with emerging importance in cachexia.43, 54, 55 Cisplatin increased the density of p62 puncta (intracellular receptors that target proteins and organelles for autophagic degradation), accumulation of which is indicative of dysregulated formation and turnover of autophagosomes. Consistent with this dysregulation, synthesis of both the LC3 autophagosome membrane component and the p62 proteins was decreased by cisplatin, further supporting a pathological suppression of effective turnover. CBG treatment reversed the effects of cisplatin on LC3 and p62 expression and p62 puncta density, indicative of a rescue of normal homeostatic autophagy function, which is necessary to prevent pathological muscle catabolism.56
Upstream pathological processes that drive chemotherapy‐induced alterations in muscle anabolism and catabolism are less clear but have been suggested to involve NFκB signalling.6 However, a recent study has demonstrated the critical role of GC signalling, as chemotherapy‐induced cachexia was abolished in transgenic mice lacking muscle GC receptors.5, 51 That study demonstrated that cyclophosphamide‐doxorubicin‐5‐fluorouracil chemotherapy elicited rapid systemic inflammation (within 4 h), leading to HPA axis activation and subsequent elevation in systemic corticosterone levels. Consistent with this mechanism, corticosterone levels were elevated 72 h after cisplatin administration in the present study; however, it is interesting to note that this was not modulated by CBG treatment. These data therefore suggest that CBG likely exerts its protective effects downstream of muscle GC receptors, via action on some component of the intracellular signal transduction pathways that mediate GC‐induced muscle atrophy. A plausible candidate is PGC‐1α, the PPARγ nuclear hormone receptor coactivator 1α, which has been shown to control autophagy and protein synthesis processes selectively in glycolytic MHC IIx/b fibres,40, 41, 57 which exhibited selective atrophy here. Given that CBG has recently been identified as a moderately potent agonist of PPARγ,27, 58 and synthetic PPARγ agonists have shown efficacy against cachectic weight loss,59 substantiation of this putative mechanism would be of considerable value.
While a PPARγ‐mediated mechanism for the protective effect of CBG on muscle homeostasis has been posited here, phytocannabinoids typically have pleiotropic pharmacological activities, and indeed, CBG has a number of identified activities, including modulation of endocannabinoid system components.28 Both the canonical endocannabinoids, and an increasing number of endocannabinoid‐like signalling lipids, have established modulatory roles in many physiological processes involved in metabolism and dysregulated in cachexia.49, 50, 60 We thus performed what is, to the best of our knowledge, the first comprehensive lipidomic analysis of this class of lipids in hypothalamic and plasma samples from cachectic animals. Due to limitations in the number of lipidomic analyses feasible in the present study, we chose to focus on the hypothalamus, being the primary brain region involved in metabolic homeostasis, and for which there exists a considerable body of evidence for neuromodulatory roles by endocannabinoids in the physiology and pathophysiology of feeding.28 However, further lipidomic studies would be of interest in brain regions containing nausea and anorexia circuits activated by cisplatin, particularly the nucleus of the solitary tract, lateral parabrachial nucleus, and central amygdala.49 In hypothalami, cisplatin administration elicited extensive alterations to levels of lipoamines in 11 amide‐conjugate subfamilies, including a profound suppression of all ethanolamine‐conjugated lipoamines (NAEs, which include anandamide). These observations bear considerable similarity to the recently characterized brain lipidome of a NAPE‐PLD KO mouse,38 suggesting that cisplatin may have an inhibitory effect on the predominant biosynthetic enzyme for NAEs. While the lipidome of other brain regions was not investigated in the present study, broadly consistent reductions in NAE levels were observed across eight major regions in the NAPE‐PLD KO mouse, indicating that cisplatin's effects may not be restricted to the hypothalamus. Consistent with this possibility, cisplatin also decreased plasma levels of NAEs, the only screened subfamily for which a decrease was observed in this compartment. Given the diverse central and peripheral roles of anandamide, oleoylethanolamide and palmitoylethanolamide in modulating appetite and metabolic processes,49, 61 this observation provides evidence for hitherto unrecognized mechanisms by which cisplatin may exert many of its cachectic effects. Furthermore, if cisplatin does have a similar effect on NAEs as NAPE‐PLD KO, which decreased levels in hippocampus, cortex, and striatum, this raises the intriguing possibility that such a mechanism may be involved in the phenomenon of chemotherapy‐induced cognitive impairment, also known as ‘chemo brain’.62 Surprisingly, CBG treatment was not associated with any normalization of cisplatin's effects on NAE levels, which could be considered consistent with the relatively modest effects on cisplatin‐induced anorexia. However, given that the lipodomic profile was only determined at a single timepoint, an NAE‐mediated mechanism for CBG in this model cannot be ruled out. In contrast, arguably the most salient feature of the lipidomic characterization of this model is the observation that plasma levels of all but one of the N‐acyl glycine subfamily of lipoamines were elevated by cisplatin and reversed by CBG. The association between the anti‐cachectic effect of CBG and systemic N‐acyl glycines was further supported by the significant negative correlations between these lipoamines and EDL muscle mass in animals treated with CBG. Given the recently emerging evidence for the role of this lipoamine subfamily in modulating inflammation, adipogenesis, insulin release, and several GPR18‐mediated metabolic processes,63, 64, 65, 66 their functional involvement in the therapeutic effects of CBG, and potential as biomarkers for such anabolic interventions, warrants urgent investigation.
A very limited number of pre‐clinical studies have attempted to investigate the pathophysiology of chemotherapy‐induced cachexia, and the efficacy of anti‐cachectic interventions, in tumour‐bearing models.8, 67, 68 However, the number of experimental groups necessitated by this approach limits the power of such designed studies to investigate the chemotherapy‐induced cachectic phenotype in detail. The present study was specifically designed to overcome such limitations, and the inherent impossibility of administering chemotherapy agents to cancer‐naïve human subjects, such that it had the power to provide a comprehensive multi‐omic characterization of cisplatin‐induced cachexia, and assess the efficacy of CBG at multiple doses. The dose of cisplatin used induced a level of weight loss and anorexia highly consistent with previous reports and was intended to replicate a typical cycle of cisplatin chemotherapy in humans that results in a similarly mild but significant loss of bodyweight,7, 22, 32, 33, 34 rather than high and/or prolonged dosing regimens that elicit a magnitude of weight loss (≥20%) that would not be tolerated clinically.4, 65 Similarly, the two doses of CBG were chosen based on our previous studies in cisplatin‐naïve animals, which were well tolerated and elicited a modest but significant hyperphagia, which were thus considered translationally appropriate. Due to the modest effects of CBG administration in cisplatin‐naïve animals in pilot studies (Supporting Information, Figure S1), the use of considerable analytical resources for the inclusion of such groups in the main experimental design was not considered justified. Similarly, having determined that the lower dose of CBG had limited, if any, protective effects against cisplatin‐induced weight loss or anorexia, follow‐up ex vivo mechanistic analyses of samples from this group was considered unnecessary. While investigation of pharmacokinetic parameters was beyond the scope of the present study, previously reported plasma and brain half‐lives for CBG of 100 and 96 min,28 suggest that CBG may not have reached steady‐state before the onset of cisplatin's cachectic effects. Further studies may thus be warranted to determine whether prophylactic doses of CBG given at an earlier timepoint (relative to cisplatin) elicit a greater anti‐cachectic effect.
Given that many components of chemotherapy‐induced cachectic pathophysiology attenuated by CBG are common to cancer‐induced cachexia and that CBG has recently been shown to inhibit colon carcinogenesis and cancer progression in vivo,69 it will be of great interest to investigate CBG as an adjunct treatment in tumour‐bearing animals receiving cisplatin treatment. Given that the doses of CBG utilized in the present study exceed levels typically ingested from medicinal (or recreational) C. sativa use, phase I safety trials in humans of purified CBG would be required for progression of this treatment, should the anti‐cachectic efficacy of CBG be demonstrated in pre‐clinical cancer models. Demonstration of efficacy in pre‐clinical tumour studies, and safety in human trials, is particularly pressing for such a compound, given the increasingly widespread interest and use of C. sativa‐based treatments (particularly for oncology indications) outside of the clinical setting, as a result of relaxation of legal restrictions in the USA and other countries.
The data reported here reveal a number of novel aspects of chemotherapy‐induced cachexia pathophysiology, particularly regarding the involvement of endocannabinoid‐like signalling lipids, which may provide novel targets for therapeutic interventions against the metabolic and cognitive side effects of chemotherapy. As a non‐psychoactive phytocannabinoid that robustly attenuates chemotherapy‐induced muscle atrophy, CBG may represent a valuable adjunct treatment with significant implications for the treatment efficacy, quality of life, and mortality of the many patients receiving cisplatin treatment.
Author contributions
D.I.B., C.M.W., B.J.W., K.P., J.R.S., and H.B.B. conceived and designed the studies. D.I.B. conducted in vivo experiments and data analysis. J.R.H. and B.A.D.M. conducted immunohistochemistry and western blot experiments. E.L. and H.B.B. conducted lipidomic experiments. N.G. and J.R.S. conducted metabonomic experiments. A.E.R. and N.G. conducted cluster and correlational analyses. D.I.B. and C.M.W. wrote the manuscript with assistance from all other authors.
Conflict of interest
The work reported was funded in part by grants to B.J.W. and C.M.W. from GW Research Ltd and Otsuka Pharmaceuticals. The original study concept was discussed with the sponsor (GW Research Ltd), although all subsequent study design, data collection, analysis, and interpretation were conducted independently by the authors. The report was approved by the sponsor company prior to submission, and the authors retain full control of all primary data.
Data and materials availability
CBG material was supplied by GW Research Ltd. Requests for CBG material should be directed to GW Research Ltd, Sovereign House, Vision Park, Histon Cambridge CB24 9BZ, United Kingdom, and should be accompanied by an outline explanation of the intended use. Material for approved requests will be supplied under Material Transfer Agreement.
Supporting information
Figure S1. Semi‐chronic cannabigerol administration has limited effects on food intake in cisplatin‐naive free‐feeding rats.
Figure S2. Asymptomatic animals which do not exhibit a cachectic response to cisplatin were excluded from analyses.
Figure S3. Primary Western blots for protein synthesis and autophagy markers in EDL muscle samples.
Figure S4. Relative abundance integral plots of individual plasma metabolites.
Figure S5. Plasma concentrations of N‐acyl glycine subfamily lipoamines negatively correlate with muscle mass in cannabigerol‐treated cachectic animals.
Table S1. Baseline intake, bodyweight and activity did not differ between groups post‐randomization.
Table S2. Cisplatin‐induced anorexia is driven by reductions in both feeding frequency and meal size.
Table S3. Cisplatin suppresses dark photoperiod ambulatory locomotor and rearing activity.
Table S4. Lipids in HPLC/MS/MS screening library with parent ion and fragment ion masses.
Table S5. Concentrations of N‐acyl amide and lipids in hypothalamus and plasma, arranged by amide family.
Acknowledgements
The authors would like to thank Sarah Turner, Thomas Hill, Robert Mitchell, and Henry Collins‐Hooper for their invaluable technical assistance and advice. The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.70
This research was supported by grants to C.M.W. and B.J.W. by GW Research Ltd and Otsuka Pharmaceuticals and in part by the University of Reading Research Endowment Trust Fund to D.I.B.
Brierley D. I., Harman J. R., Giallourou N., Leishman E., Roashan A. E., Mellows B. A. D., Bradshaw H. B., Swann J. R., Patel K., Whalley B. J., and Williams C. M. (2019) Chemotherapy‐induced cachexia dysregulates hypothalamic and systemic lipoamines and is attenuated by cannabigerol, Journal of Cachexia, Sarcopenia and Muscle, 10, 844–859, doi: 10.1002/jcsm.12426.
References
- 1. von Haehling S, Anker SD. Prevalence, incidence and clinical impact of cachexia: facts and numbers‐update 2014. J Cachexia Sarcopenia Muscle 2014;5:261–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Argilés JM, Busquets S, Stemmler B, López‐Soriano FJ. Cancer cachexia: understanding the molecular basis. Nat Rev Cancer 2014;14:754–762. [DOI] [PubMed] [Google Scholar]
- 3. Hajjaji N, Couet C, Besson P, Bougnoux P. DHA effect on chemotherapy‐induced body weight loss: an exploratory study in a rodent model of mammary tumors. Nutr Cancer 2012;64:1000–1007. [DOI] [PubMed] [Google Scholar]
- 4. Sakai H, Sagara A, Arakawa K, Sugiyama R, Hirosaki A, Takase K, et al. Mechanisms of cisplatin‐induced muscle atrophy. Toxicol Appl Pharmacol 2014;278:190–199. [DOI] [PubMed] [Google Scholar]
- 5. Braun TP, Szumowski M, Levasseur PR, Grossberg AJ, Zhu X, Agarwal A, et al. Muscle atrophy in response to cytotoxic chemotherapy is dependent on intact glucocorticoid signaling in skeletal muscle. PLoS ONE 2014;9:e106489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Damrauer J, Stadler M, Acharyya S. Chemotherapy‐induced muscle wasting: association with NF‐κB and cancer cachexia. Basic Appl Myol 2008;18:139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Garcia JM, Scherer T, Chen J‐A, Guillory B, Nassif A, Papusha V, et al. Inhibition of cisplatin‐induced lipid catabolism and weight loss by ghrelin in male mice. Endocrinology 2013;154:3118–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen M‐C, Hsu W‐L, Hwang P‐A, Chen Y‐L, Chou T‐C. Combined administration of fucoidan ameliorates tumor and chemotherapy‐induced skeletal muscle atrophy in bladder cancer‐bearing mice. Oncotarget 2016;7:51608–51618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hulmi JJ, Nissinen TA, Räsänen M, Degerman J, Lautaoja JH, Hemanthakumar KA, et al. Prevention of chemotherapy‐induced cachexia by ACVR2B ligand blocking has different effects on heart and skeletal muscle. J Cachexia Sarcopenia Muscle 2018;9:417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mueller TC, Bachmann J, Prokopchuk O, Friess H, Martignoni ME. Molecular pathways leading to loss of skeletal muscle mass in cancer cachexia—can findings from animal models be translated to humans? BMC Cancer 2015;16:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Janelsins MC, Tejani MA, Kamen C, Peoples AR, Mustian KM, Morrow GR. Current pharmacotherapy for chemotherapy‐induced nausea and vomiting in cancer patients. Expert Opin Pharmacother 2013;14:757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Engels FK, de Jong FA, Mathijssen RHJ, Erkens JA, Herings RM, Verweij J. Medicinal cannabis in oncology. Eur J Cancer 2007;43:2638–2644. [DOI] [PubMed] [Google Scholar]
- 13. Smith LA, Azariah F, Lavender VT, Stoner NS, Bettiol S. Cannabinoids for nausea and vomiting in adults with cancer receiving chemotherapy. Cochrane Database Syst Rev 2015;11:CD009464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. May MB, Glode AE. Dronabinol for chemotherapy‐induced nausea and vomiting unresponsive to antiemetics. Cancer Manag Res 2016;8:49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jatoi A, Windschitl HE, Loprinzi CL, Sloan JA, Dakhil SR, Mailliard JA, et al. Dronabinol versus megestrol acetate versus combination therapy for cancer‐associated anorexia: a North Central Cancer Treatment Group study. J Clin Oncol 2002;20:567–573. [DOI] [PubMed] [Google Scholar]
- 16. Strasser F, Luftner D, Possinger K, Ernst G, Ruhstaller T, Meissner W, et al. Comparison of orally administered cannabis extract and delta‐9‐tetrahydrocannabinol in treating patients with cancer‐related anorexia‐cachexia syndrome: a multicenter, phase III, randomized, double‐blind, placebo‐controlled clinical trial from the cannabis‐in‐cachexia‐study‐group. J Clin Oncol 2006;24:3394–3400. [DOI] [PubMed] [Google Scholar]
- 17. Farrimond JA, Whalley BJ, Williams CM. Non‐Δ9tetrahydrocannabinol phytocannabinoids stimulate feeding in rats. Behav Pharmacol 2012;23:113–117. [DOI] [PubMed] [Google Scholar]
- 18. Farrimond JA, Whalley BJ, Williams CM. Cannabinol and cannabidiol exert opposing effects on rat feeding patterns. Psychopharmacology (Berl) 2012;223:117–129. [DOI] [PubMed] [Google Scholar]
- 19. Farrimond JA, Mercier MS, Whalley BJ, Williams CM. Cannabis sativa and the endogenous cannabinoid system: therapeutic potential for appetite regulation. Phytother Res 2011;25:170–188. [DOI] [PubMed] [Google Scholar]
- 20. Farrimond JA, Hill AJ, Whalley BJ, Williams CM. Cannabis constituents modulate δ9‐tetrahydrocannabinol‐induced hyperphagia in rats. Psychopharmacology (Berl) 2010;210:97–106. [DOI] [PubMed] [Google Scholar]
- 21. Brierley DI, Samuels J, Duncan M, Whalley BJ, Williams CM. Cannabigerol is a novel, well‐tolerated appetite stimulant in pre‐satiated rats. Psychopharmacology (Berl) 2016;233:3603–3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brierley DI, Samuels J, Duncan M, Whalley BJ, Williams CM. A cannabigerol‐rich Cannabis sativa extract, devoid of [INCREMENT]9‐tetrahydrocannabinol, elicits hyperphagia in rats. Behav Pharmacol 2017;28:280–284. [DOI] [PubMed] [Google Scholar]
- 23. Liu Y‐L, Malik NM, Sanger GJ, Andrews PLR. Ghrelin alleviates cancer chemotherapy‐associated dyspepsia in rodents. Cancer Chemother Pharmacol 2006;58:326–333. [DOI] [PubMed] [Google Scholar]
- 24. Sakai H, Kai Y, Takase K, Sato K, Kimura M, Tabata S, et al. Role of peptide YY in 5‐fluorouracil‐induced reduction of dietary intake. Clin Exp Pharmacol Physiol 2016;43:753–759. [DOI] [PubMed] [Google Scholar]
- 25. Bennani‐Baiti N, Walsh D. Animal models of the cancer anorexia‐cachexia syndrome. Support Care Cancer 2011;19:1451–1463. [DOI] [PubMed] [Google Scholar]
- 26. Wilken MK, Satiroff BA. Pilot study of “miracle fruit” to improve food palatability for patients receiving chemotherapy. Clin J Oncol Nurs 2012;16:E173–E177. [DOI] [PubMed] [Google Scholar]
- 27. Granja AG, Carrillo‐Salinas F, Pagani A, Gómez‐Cañas M, Negri R, Navarrete C, et al. A cannabigerol quinone alleviates neuroinflammation in a chronic model of multiple sclerosis. J Neuroimmune Pharmacol 2012;7:1002–1016. [DOI] [PubMed] [Google Scholar]
- 28. De Petrocellis L, Ligresti A, Moriello AS, Allarà M, Bisogno T, Petrosino S, et al. Effects of cannabinoids and cannabinoid‐enriched cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br J Pharmacol 2011;163:1479–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deiana S, Watanabe A, Yamasaki Y, Amada N, Arthur M, Fleming S, et al. Plasma and brain pharmacokinetic profile of cannabidiol (CBD), cannabidivarine (CBDV), Δ9‐tetrahydrocannabivarin (THCV) and cannabigerol (CBG) in rats and mice following oral and intraperitoneal administration and CBD action on obsessive‐compulsive behavi. Psychopharmacology (Berl) 2012;219:859–873. [DOI] [PubMed] [Google Scholar]
- 30. Larsen TM, Toubro S, Astrup A. PPARgamma agonists in the treatment of type II diabetes: is increased fatness commensurate with long‐term efficacy? Int J Obes (Lond) 2003;27:147–161. [DOI] [PubMed] [Google Scholar]
- 31. O'Sullivan SE, Kendall DA. Cannabinoid activation of peroxisome proliferator‐activated receptors: potential for modulation of inflammatory disease. Immunobiology 2010;215:611–616. [DOI] [PubMed] [Google Scholar]
- 32. O'Sullivan SE. An update on peroxisome proliferator‐activated receptor (PPAR) activation by cannabinoids. Br J Pharmacol 2016;173:1899–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rudd JA, Yamamoto K, Yamatodani A, Takeda N. Differential action of ondansetron and dexamethasone to modify cisplatin‐induced acute and delayed kaolin consumption (‘pica’) in rats. Eur J Pharmacol 2002;454:47–52. [DOI] [PubMed] [Google Scholar]
- 34. Malik NM, Moore GBT, Smith G, Liu Y‐L, Sanger GJ, Andrews PLR. Behavioural and hypothalamic molecular effects of the anti‐cancer agent cisplatin in the rat: a model of chemotherapy‐related malaise? Pharmacol Biochem Behav 2006;83:9–20. [DOI] [PubMed] [Google Scholar]
- 35. Malik NM, Liu Y‐L, Cole N, Sanger GJ, Andrews PLR. Differential effects of dexamethasone, ondansetron and a tachykinin NK1 receptor antagonist (GR205171) on cisplatin‐induced changes in behaviour, food intake, pica and gastric function in rats. Eur J Pharmacol 2007;555:164–173. [DOI] [PubMed] [Google Scholar]
- 36. Brierley DI, Samuels J, Duncan M, Whalley BJ, Williams CM. Neuromotor tolerability and behavioural characterisation of cannabidiolic acid, a phytocannabinoid with therapeutic potential for anticipatory nausea. Psychopharmacology (Berl) 2016;233:243–254. [DOI] [PubMed] [Google Scholar]
- 37. Beckonert O, Keun HC, Ebbels TMD, Bundy J, Holmes E, Lindon JC, et al. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat Protoc 2007;2:2692–2703. [DOI] [PubMed] [Google Scholar]
- 38. Leishman E, Mackie K, Luquet S, Bradshaw HB. Lipidomics profile of a NAPE‐PLD KO mouse provides evidence of a broader role of this enzyme in lipid metabolism in the brain. Biochim Biophys Acta 2016;1861:491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Evans WJ, Morley JE, Argilés J, Bales C, Baracos V, Guttridge D, et al. Cachexia: a new definition. Clin Nutr 2008;27:793–799. [DOI] [PubMed] [Google Scholar]
- 40. Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, et al. PGC‐1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy‐specific gene transcription. Proc Natl Acad Sci U S A 2006;103:16260–16265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shimizu N, Yoshikawa N, Ito N, Maruyama T, Suzuki Y, Takeda S, et al. Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab 2011;13:170–182. [DOI] [PubMed] [Google Scholar]
- 42. Norton LE, Layman DK. Leucine regulates translation initiation of protein synthesis in skeletal muscle after exercise. J Nutr 2006;136:533S–537S. [DOI] [PubMed] [Google Scholar]
- 43. Penna F, Baccino FM, Costelli P. Coming back: autophagy in cachexia. Curr Opin Clin Nutr Metab Care 2014;17:241–246. [DOI] [PubMed] [Google Scholar]
- 44. Fanzani A, Zanola A, Rovetta F, Rossi S, Aleo MF. Cisplatin triggers atrophy of skeletal C2C12 myotubes via impairment of Akt signalling pathway and subsequent increment activity of proteasome and autophagy systems. Toxicol Appl Pharmacol 2011;250:312–321. [DOI] [PubMed] [Google Scholar]
- 45. Portilla D, Li S, Nagothu KK, Megyesi J, Kaissling B, Schnackenberg L, et al. Metabolomic study of cisplatin‐induced nephrotoxicity. Kidney Int 2006;69:2194–2204. [DOI] [PubMed] [Google Scholar]
- 46. Zhang P, Chen J‐Q, Huang W‐Q, Li W, Huang Y, Zhang Z‐J, et al. Renal medulla is more sensitive to cisplatin than cortex revealed by untargeted mass spectrometry‐based metabolomics in rats. Sci Rep 2017;7:44804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. De Petrocellis L, Orlando P, Moriello AS, Aviello G, Stott C, Izzo AA, et al. Cannabinoid actions at TRPV channels: effects on TRPV3 and TRPV4 and their potential relevance to gastrointestinal inflammation. Acta Physiol (Oxf) 2012;204:255–266. [DOI] [PubMed] [Google Scholar]
- 48. Cascio MG, Gauson LA, Stevenson LA, Ross RA, Pertwee RG. Evidence that the plant cannabinoid cannabigerol is a highly potent α2‐adrenoceptor agonist and moderately potent 5HT1A receptor antagonist. Br J Pharmacol 2010;159:129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hansen HS, Diep TA. N‐acylethanolamines, anandamide and food intake. Biochem Pharmacol 2009;78:553–560. [DOI] [PubMed] [Google Scholar]
- 50. Silvestri C, Di Marzo V. The endocannabinoid system in energy homeostasis and the etiopathology of metabolic disorders. Cell Metab 2013;17:475–490. [DOI] [PubMed] [Google Scholar]
- 51. Braun TP, Marks DL. The regulation of muscle mass by endogenous glucocorticoids. Front Physiol 2015;6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schmidt K, von Haehling S, Doehner W, Palus S, Anker SD, Springer J. IGF‐1 treatment reduces weight loss and improves outcome in a rat model of cancer cachexia. J Cachexia Sarcopenia Muscle 2011;2:105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen JL, Walton KL, Winbanks CE, Murphy KT, Thomson RE, Makanji Y, et al. Elevated expression of activins promotes muscle wasting and cachexia. FASEB J 2014;28:1711–1723. [DOI] [PubMed] [Google Scholar]
- 54. Pigna E, Berardi E, Aulino P, Rizzuto E, Zampieri S, Carraro U, et al. Aerobic exercise and pharmacological treatments counteract cachexia by modulating autophagy in colon cancer. Sci Rep 2016;6:26991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Musolino V, Palus S, Tschirner A, Drescher C, Gliozzi M, Carresi C, et al. Megestrol acetate improves cardiac function in a model of cancer cachexia‐induced cardiomyopathy by autophagic modulation. J Cachexia Sarcopenia Muscle 2016;7:555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stacchiotti A, Rovetta F, Ferroni M, Corsetti G, Lavazza A, Sberveglieri G, et al. Taurine rescues cisplatin‐induced muscle atrophy in vitro: a morphological study. Oxid Med Cell Longev 2014;2014:840951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Romanino K, Mazelin L, Albert V, Conjard‐Duplany A, Lin S, Bentzinger CF, et al. Myopathy caused by mammalian target of rapamycin complex 1 (mTORC1) inactivation is not reversed by restoring mitochondrial function. Proc Natl Acad Sci U S A 2011;108:20808–20813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Valdeolivas S, Navarrete C, Cantarero I, Bellido ML, Muñoz E, Sagredo O. Neuroprotective properties of cannabigerol in Huntington's disease: studies in R6/2 mice and 3‐nitropropionate‐lesioned mice. Neurotherapeutics 2015;12:185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Trobec K, Palus S, Tschirner A, von Haehling S, Doehner W, Lainscak M, et al. Rosiglitazone reduces body wasting and improves survival in a rat model of cancer cachexia. Nutrition 2014;30:1069–1075. [DOI] [PubMed] [Google Scholar]
- 60. Ligresti A, De Petrocellis L, Di Marzo V. From phytocannabinoids to cannabinoid receptors and endocannabinoids: pleiotropic physiological and pathological roles through complex pharmacology. Physiol Rev 2016;96:1593–1659. [DOI] [PubMed] [Google Scholar]
- 61. Hansen HS. Role of anorectic N‐acylethanolamines in intestinal physiology and satiety control with respect to dietary fat. Pharmacol Res 2014;86:18–25. [DOI] [PubMed] [Google Scholar]
- 62. Ahles TA, Saykin AJ. Candidate mechanisms for chemotherapy‐induced cognitive changes. Nat Rev Cancer 2007;7:192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang S, Xu Q, Shu G, Wang L, Gao P, Xi Q, et al. N‐Oleoyl glycine, a lipoamino acid, stimulates adipogenesis associated with activation of CB1 receptor and Akt signaling pathway in 3T3‐L1 adipocyte. Biochem Biophys Res Commun 2015;466:438–443. [DOI] [PubMed] [Google Scholar]
- 64. McHugh D, Roskowski D, Xie S, Bradshaw HB. Δ(9)‐THC and N‐arachidonoyl glycine regulate BV‐2 microglial morphology and cytokine release plasticity: implications for signaling at GPR18. Front Pharmacol 2014;4:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hanuš L, Shohami E, Bab I, Mechoulam R. N‐Acyl amino acids and their impact on biological processes. Biofactors 2014;40:381–388. [DOI] [PubMed] [Google Scholar]
- 66. Rajaraman G, Simcocks A, Hryciw DH, Hutchinson DS, McAinch AJ. G protein‐coupled receptor 18: a potential role for endocannabinoid signalling in metabolic dysfunction. Mol Nutr Food Res 2016;60:92–102. [DOI] [PubMed] [Google Scholar]
- 67. Hatakeyama S, Summermatter S, Jourdain M, Melly S, Minetti GC, Lach‐Trifilieff E. ActRII blockade protects mice from cancer cachexia and prolongs survival in the presence of anti‐cancer treatments. Skelet Muscle 2016;6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen M‐C, Chen Y‐L, Lee C‐F, Hung C‐H, Chou T‐C. Supplementation of magnolol attenuates skeletal muscle atrophy in bladder cancer‐bearing mice undergoing chemotherapy via suppression of FoxO3 activation and induction of IGF‐1. PLoS ONE 2015;10:e0143594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Borrelli F, Pagano E, Romano B, Panzera S, Maiello F, Coppola D, et al. Colon carcinogenesis is inhibited by the TRPM8 antagonist cannabigerol, a cannabis‐derived non‐psychotropic cannabinoid. Carcinogenesis 2014;35:2787–2797. [DOI] [PubMed] [Google Scholar]
- 70. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Semi‐chronic cannabigerol administration has limited effects on food intake in cisplatin‐naive free‐feeding rats.
Figure S2. Asymptomatic animals which do not exhibit a cachectic response to cisplatin were excluded from analyses.
Figure S3. Primary Western blots for protein synthesis and autophagy markers in EDL muscle samples.
Figure S4. Relative abundance integral plots of individual plasma metabolites.
Figure S5. Plasma concentrations of N‐acyl glycine subfamily lipoamines negatively correlate with muscle mass in cannabigerol‐treated cachectic animals.
Table S1. Baseline intake, bodyweight and activity did not differ between groups post‐randomization.
Table S2. Cisplatin‐induced anorexia is driven by reductions in both feeding frequency and meal size.
Table S3. Cisplatin suppresses dark photoperiod ambulatory locomotor and rearing activity.
Table S4. Lipids in HPLC/MS/MS screening library with parent ion and fragment ion masses.
Table S5. Concentrations of N‐acyl amide and lipids in hypothalamus and plasma, arranged by amide family.