

Abstract
Background
Histamine is an immune modulator, neuroprotective, and remyelinating agent, beneficially acting on skeletal muscles and promoting anti‐inflammatory features in amyotrophic lateral sclerosis (ALS) microglia. Drugs potentiating the endogenous release of histamine are in trial for neurological diseases, with a role not systematically investigated in ALS. Here, we examine histamine pathway associations in ALS patients and the efficacy of a histamine‐mediated therapeutic strategy in ALS mice.
Methods
We adopted an integrative multi‐omics approach combining gene expression profiles, copy number variants, and single nucleotide polymorphisms of ALS patients. We treated superoxide dismutase 1 (SOD1)‐G93A mice that recapitulate key ALS features, with the brain‐permeable histamine precursor histidine in the symptomatic phase of the disease and analysed the rescue from disease pathological signs. We examined the action of histamine in cultured SOD1‐G93A motor neuron‐like cells.
Results
We identified 13 histamine‐related genes deregulated in the spinal cord of two ALS patient subgroups, among which genes involved in histamine metabolism, receptors, transport, and secretion. Some histamine‐related genes overlapped with genomic regions disrupted by DNA copy number and with ALS‐linked pathogenic variants. Histidine treatment in SOD1‐G93A mice proved broad efficacy in ameliorating ALS features, among which most importantly lifespan, motor performance, microgliosis, muscle atrophy, and motor neurons survival in vivo and in vitro.
Conclusions
Our gene set/pathway enrichment analyses and preclinical studies started at the onset of symptoms establish that histamine‐related genes are modifiers in ALS, supporting their role as candidate biomarkers and therapeutic targets. We disclose a novel important role for histamine in the characterization of the multi‐gene network responsible for ALS and, furthermore, in the drug development process.
Keywords: ALS, Histamine, Motor neurons, Microglia, Skeletal muscles, SOD1‐G93A
Introduction
Amyotrophic lateral sclerosis (ALS) is a late‐onset neurodegenerative disease that primarily affects motor neurons especially in the spinal cord and evolves in progressive muscular paralysis leading to death within 2–5 years of diagnosis. About 90% of ALS cases are classified as sporadic (sALS), while the remaining 10% have a familial origin. Among the familial ALS cases, about 20% possess at least 100 different mutations in the gene coding for the copper‐zinc superoxide dismutase 1 (SOD1) enzyme,1 recognized to promote a gain of neurotoxic function when mutated. Although the SOD1‐G93A mutation represents only 2% of all ALS cases, the SOD1‐G93A mouse resembling human pathological features is the most widely used model to test new mechanisms and drugs. A number of different genes, aberrant cellular pathways, and molecular mechanisms are involved in ALS, and strong evidence points to the impairment of the neuro‐immune axis in causing motor neuron death and muscular wasting in both animal models and patients. However, ALS appears as a composite disease, and we still have much to learn concerning the potential therapeutic strategies to be used to halt its progression.
Among a vast range of constitutively bioactive molecules with pleiotropic regulatory roles in neurotransmission and immune responses, histamine is produced exclusively by decarboxylation of histidine by histidine decarboxylase (HDC) and is catabolized by histamine N‐methyltransferase (HNMT) or by diamine oxidase 1 (DAO1). These enzymes, together with the four G‐protein‐coupled receptors H1R‐H4R, are ubiquitous in the CNS and mediate histaminergic signalling. In the central nervous system (CNS), histamine regulates motor circuits and neuro‐immune functions, acts as a neuroprotective agent,2 and exerts beneficial effects on microglia under in vitro/in vivo inflammatory conditions.3, 4 In support of this, a condition of impaired histamine production as that occurring in HDC knockout mice decreases M2‐like markers and the neuroprotective role of microglia in vivo.5 Histamine signalling also promotes remyelination,6 protects skeletal muscles against exercise‐induced fatigue,7 and reduces muscle injury by improving motor performance in Duchenne dystrophic mice.8 Additionally, drugs enhancing histamine transmission, such as histidine or H3R antagonists, exhibit beneficial effects in animal models of Parkinson's disease, attention‐deficit/hyperactivity disorders, schizophrenia, dementia, and depression2 and are under scrutiny in clinical trials for Alzheimer's disease and multiple sclerosis. Their efficacy in affecting ALS disease progression has still to be proven.
The aim of this study is to establish if histamine is a disease modifier in ALS and a novel target for therapy. We adopted an integrative multi‐omics approach, combining copy number variants (CNVs), single nucleotide polymorphisms (SNPs), and gene expression profiles of ALS patients, to identify histamine markers in ALS pathogenesis. We evaluated the preclinical efficacy of histamine on disease progression in SOD1‐G93A mice, by a sustained treatment with the brain‐penetrant histamine precursor histidine provided from the onset of disease symptoms. We investigated the biological actions of histamine in cultured motor neuron cells expressing the SOD1‐G93A mutation.
Our work discloses a novel important role for histamine in characterizing the complex multi‐gene interactions responsible for the disease and, further, in developing future effective therapeutics.
Methods
Multi‐omics profiling of histamine‐related genes in sporadic amyotrophic lateral sclerosis patients
For this study, we referred to our CNV profile data set (available at NCBI's Gene Expression Omnibus through GEO Series accession number 107375) and transcriptomic analyses4, 9 performed in the post‐mortem motor cortex (Brodmann areas 4) and spinal cord (lumbar tract L1) from 31 patients with clear sALS diagnosis and 10 control individuals collected as previously described.9 Control samples (mean subject age of 55 years) were obtained from individuals who died from a non‐neurological disease (causes of death: myocardial infarction, renal failure, and pulmonary embolism). Informed consent and approval from the relevant local ethical committees for medical research were obtained for the use of brain and spinal tissues and for access to medical records for research purposes. Detailed information related to the origin, source code, age, gender, race, disease state, survival time from diagnosis date, and post‐mortem interval of patient samples is given in Supporting Information, Table S1.
In previous works, we used an unsupervised hierarchical algorithm to cluster 41 motor cortex samples from control and sALS patients on the basis of their similarities measured over the most ‘hypervariable genes’ (9.646 genes with a standard deviation > 1.5). The transcriptome profiles enabled to differentiate controls from sALS patients and clearly distinguished two sALS subgroups (termed sALS1 and sALS2), each associated with specific genomic aberrations, differentially expressed genes and biological pathways.9, 10, 11 In this work, we referred to this molecular‐based classification for the subsequent analysis. We thus filtered out our genome‐wide ‘omics’ data sets by focusing only on genes implicated in histamine cascades, by using histamine metabolism/signalling query keywords in Gene Ontology database (http://www.geneontology.org/). To better investigate the contribution of altered histamine signalling in the complex genetic structure of ALS and identify new histamine‐related loci associated with disease susceptibility, we also collected genome‐wide association studies data sets from the ALSOD and ALSGene databases (http://www.alsod.iop.kcl.ac.uk; http://www.alsgene.org). Only SNP/indel variants previously reported as ‘pathogenic’ or ‘likely pathogenic’ and with a minor allele frequency ≥ 0.1% in the 1000 Genomes Project were selected for further analysis. Benign intronic and synonymous variants were filtered out.
Meta‐analysis of gene expression data
Gene expression data from isolated motor neuron and spinal cord of wild‐type (WT) littermates and SOD1‐G93A mice at various disease stages (pre‐symptomatic, symptomatic, and terminal stage) were downloaded from the public data repositories NCBI GEO (http://www.ncbi.nlm.nih.gov/geo/) and ArrayExpress (http://www.ebi.ac.uk/arrayexpress/), using the following search terms and/or their combinations: ‘amyotrophic lateral sclerosis’, ‘motor neuron disease’, ‘SOD1 mouse model’, ‘SOD1‐G93A transgenic mouse’, and ‘expression profiling’. Data sets included in the analysis had to meet the following criteria: (i) data were acquired using a genome‐wide gene expression microarray platform with accessible and clear probe‐to‐gene mapping annotations and (ii) there were ≥3 replicates for each experimental condition. For each mouse data set, raw intensity values were thresholded to 1, log2‐transformed, normalized, and baselined to the median of all samples by using GeneSpring GX v13.1 software package (Agilent Technologies). Affymetrix gene expression data were Robust Multichip Average processed using quantile normalization, while Agilent and Illumina data were normalized to the 75th percentile.12, 13 Finally, data were integrated by using homemade functions implemented in PostgreSQL database system environment (version 9.4.5; http://www. postgresql.org/). We performed statistical analysis by using GeneSpring GX v13.1 software package (Agilent Technologies). Genes with a corrected P value <0.05 (one‐way analysis of variance followed by the Benjamin–Hochberg false discovery rate and the Tukey's post hoc test) were deemed to be statistically significant and subsequently filtered for genes implicated in histamine cascades.
Pathway‐based functional integration of multi‐omics data
To obtain a comprehensive mapping of functional associations of histamine‐related gene products, we incorporated our multi‐omic data and gene–pathway associations from the Metacore™ database (Clarivate Analytics, http://clarivate.com/life‐sciences/discovery‐and‐preclinicalresearch/ metacore/). This is a manually curated repository of molecular interactions of different types, pathways, network models, and functional ontologies covering human, mouse, and rat genes.
Superoxide dismutase 1‐G93A transgenic mice and treatments
Animal procedures performed according to European Guidelines (2010/63/EU) and Italian law requirements (D.L. 26/2014) were approved by Animal Welfare Office, Department of Public Health and Veterinary, Nutrition and Food Safety, General Management of Animal Care, and Veterinary Drugs of Italian Ministry of Health and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. Efforts were made to minimize animal suffering and number of animals necessary to produce reliable results. Adult B6.Cg‐Tg (SOD1‐G93A)1Gur/J mice expressing high copy number of mutant hSOD1 with a G93A substitution (SOD1‐G93A) and originally obtained from Jackson Laboratories (USA) were bred in our animal facility and housed as described.14 Transgenic hemizygous SOD1‐G93A males were crossbred with C57BL/6 females, both maintained on C57BL/6 genetic background. Transgenic progeny was genotyped as described.15 To overcome sex‐mixed results with low reproducibility, we used a single gender because of differences reported in male/female mice under pharmacological treatments. Female mice were randomly grouped into vehicle (0.9% NaCl) and histidine treatment groups at different doses (50, 100, and 250 mg/kg), based on previous results.4, 16, 17, 18 Mice were injected i.p. every 48 h from disease onset (110 days,15, 19) until 150 days of age or terminal stage.
Mice survival, behavioural scores, and motor studies
Behavioural scores and body weights were assessed starting at 60 days of age, in order to monitor disease progression. Behavioural score adopted a rating scale from 5 (healthy without symptoms of paralysis) to 1 (full paralysis of hind limbs, animals not able to straighten up within 30 s after being turned on the back).15 After reaching a score of 1, animals were euthanized according to guidelines for preclinical testing.20 Motor performance was tested on rotarod apparatus (Ugo Basile 7650 model).15
Reagents
Reagents were purchased from Sigma (Italy). Antibodies raised in rabbit: anti‐arginase 1 (ARG1) (1:100, Abcam, UK); anti‐CD163 (1:100, Santa Cruz Biotechnology, USA); anti‐choline acetyltransferase (1:500, AbCam); anti‐DAO1 (1:200, Bioss, USA); anti‐H1R (1:200, Alomone, Israel); anti‐H2R (1:200, MyBioSource, USA); anti‐H3R (1:200, Alomone); anti‐H4R (1:200, Santa Cruz Biotechnology); anti‐HDC (1:20, AbCam); anti‐HNMT (1:200, Sigma); anti‐inducible nitric oxide synthase (iNOS) (1:1000, CST, USA); anti‐myelin basic protein (MBP) (1:1000 CST); anti‐nuclear factor‐kappa B (NF‐κB) p65 (1:500, CST); anti‐phospho‐AKT (1:500, CST); anti‐phospho‐p44/42 MAPK (ERK1/2) (Thr202/Tyr204) (1:1000, CST); and anti‐phospho‐NF‐κB p65 (Ser536) (1:500, CST). Antibodies raised in mouse: anti‐AKT (1:500, Santa Cruz Biotechnology); anti‐GAPDH (1:2500, Calbiochem, USA); anti‐gp91phox (1:1000, BD Transduction Laboratories, USA); anti‐SMI32 (1:1000, Covance, USA); and anti‐p44/42 MAPK (ERK1/2) (L34F12) (1:1000, CST). Antibodies raised in rat: anti‐CD11b (1:200, AbD Serotec, UK).
Histamine assay by HPLC
Superoxide dismutase 1‐G93A mice were sacrificed at 2 h after an acute i.p. histidine treatment (100 mg/kg), spinal cords were isolated, homogenized with 3% perchloric solution, and centrifuged at 12 000 g, 4 °C for 20 min. Supernatants were collected and analysed for histamine content by HPLC, according to a previous study.17
Western blotting
Protein lysates were obtained by homogenization of mice lumbar spinal cords or sciatic nerves.14 Cells undergoing pharmacological treatments were washed with phosphate buffer saline (PBS) and lysed in RIPA buffer. Proteins were separated by SDS–PAGE and transferred onto nitrocellulose membranes (Amersham Biosciences, IT). Blots were probed overnight at 4 °C with the specified antibody and signals detected using ECL Advance western blotting detection kit (Amersham Biosciences). Quantitative analysis was performed by Kodak Image Station.
Nissl staining
Mice spinal cords perfused with PBS were processed as described.14 Serial 20 μm sections (one every 10 sections) from lumbar spinal cord segments (L3‐L5) were stained with 1% cresyl violet to detect the Nissl substance of neuronal cells. A minimum of six sections per mouse were photographed at ×20 magnification using Axioskop 2 (Zeiss, Germany) microscope and analysed with Neurolucida software (MBF Bioscience, USA). Motor neurons count was performed as described.15
Motor neuron cultures
Motor neuron‐enriched primary cultures from embryonic spinal cords were prepared as described.21, 22 Briefly, SOD1‐G93A mice (embryonic day 14) were sacrificed and, after removing the meninges, spinal cords were minced and digested with 0.01% trypsin. After dissociation, cells were cultured in Neurobasal media (Gibco, UK), plus B‐27 on poly‐D‐lysine‐coated plates. The medium was replaced every 3 days. Mixed primary motor neuron cultures (~55% motor neurons/total neurons) were used after 6–7 days in culture. NSC‐34 motor neurons are a mouse hybrid cell line produced by fusion of motor neuron‐enriched embryonic mouse spinal‐cord neurons with mouse neuroblastoma cells, and resemble motor neurons in many ways.23 These immortalized cells have been widely used as a motor neuron‐like model of ALS. NSC‐34 cells stably transfected with WT or mutant hSOD1‐G93A DNA24 were maintained in Dulbecco's modified eagle medium (DMEM)/F12 medium with 10% fetal bovine serum (FBS) (Invitrogen) and 1% penicillin–streptomycin. Cells were differentiated in DMEM/F12 supplemented with 1% FBS, 1% penicillin–streptomycin, and 1% modified Eagle's medium non‐essential amino acids. For cell viability, differentiated NSC‐34 cells were plated into 24‐well and cultured under serum deprivation conditions for 24 h. Cell viability was measured by counting intact nuclei.25
Immunofluorescence and confocal analysis
Cells in culture were fixed in 4% paraformaldehyde (PFA), permeabilized in PBS 0.1% Triton X‐100, then incubated overnight with the specified antibody at 4 °C in 1% bovine serum albumin (BSA) in PBS, and stained with secondary antibodies. Nuclei were stained with Höechst 33258 and cells mounted with gel/mount antifading (Biomeda, USA). Spinal cord tissues were processed as described.4 Immunofluorescence analysis was performed by confocal laser scanning microscope (Zeiss, Germany). Brightness and contrast were adjusted with Zen software, after background subtraction. Quantification of CD11b immunoreactivity was performed on lumbar spinal cord sections (at least six sections for each animal in each group were analysed), using the ImageJ software (version 1.47, National Institutes of Health, USA), after background subtraction. Data are expressed as arbitrary units with respect to WT mice.
RT‐PCR and qPCR
Total RNA from Tibialis Anterior (TA) skeletal muscle tissue was extracted with Trizol using a standard protocol. RNA was retro‐transcribed with Taqman reverse transcription kit (Applied Biosystems) following the manufacturer's indication. qRT‐PCR was performed using PrecisionPLUS qPCR Master Mix (Primerdesign, UK) following manufacturer indications. Relative expression values were normalized to the housekeeping gene ActB. The primer used was ActB_FW: CACTGTCGAGTCGCGTCC, ActB_RV: TCATCCATGGCGAACTGGTG; Hrh1_FW: ACGAGTGAAACCCGATGCTT, Hrh1_RV: CTCCCTCCCTCGGTCTCTG; Hrh2_FW: GCAGCACCAGCTCCTATGAC, Hrh2_RV: AGGCTGTCATGTCCTTTTCCA; Hrh3_FW: CTTCCTCGTGGGTGCCTTC, Hrh3_RV: TCTACCACCAGCCACAGCTT; Hnmt_FW: ACTAAGATACTAAGGCAGCGAACT, Hnmt_RV: TGCCATATTGGGTTCTCAGCA; Dao_FW: GCTGAACTGGGCTGCTGT, Dao_RV: TCTTCATGTGCAGTGGCTGT; and Hdc_FW: GGAGGAGCAATCCAAGGGAG, Hdc_RV: CATCTCTTTCCCTCTAGCTCGG.
Morphometric analysis of muscles and muscle denervation
Tibialis anterior muscles were dissected out and snap‐frozen in isopentane cooled in liquid nitrogen. Serial transverse 8 μm cryosections were fixed and permeabilized with 100% acetone for 1 min at RT. Sections were blocked for 1 h with a solution containing 4% BSA in PBS, stained using the anti‐laminin (Sigma, 1:400), and antibody binding specificity was revealed using secondary antibodies coupled to Alexa Fluor 488. For nuclear staining, sections were incubated with DAPI in PBS for 5 min, washed in PBS, and mounted with glycerol 3:1 in PBS.
For haematoxylin/eosin staining, serial transverse cryosections were fixed in 4% PFA for 10 min, washed in PBS, and then stained in haematoxylin for 8 min and eosin for 1 min, as described.26 All sections were visualized on a Zeiss confocal microscope. Myofibers cross‐sectional area was quantified using ImageJ® software. The entire muscle section was analysed.
For neuromuscular innervation analysis, longitudinal sections were stained with mouse anti‐synaptic vesicle protein (SV2; 1:100, DSHB) and anti‐neurofilament (2H3; 1:50, DSHB), with α‐bungarotoxin (BTX; 1:500, Invitrogen), and occupied endplates were quantified as described.27
Mitochondrial oxygen consumption
Evaluation of mitochondrial bioenergetics was performed by Seahorse XF96 Extracellular Flux Analyzer (Seahorse Bioscience‐Agilent, USA). Multiple parameters of mitochondrial function were measured with Stimulus XF Stress Test Kit (Seahorse Bioscience).28 Briefly, differentiated NSC‐34 cells were seeded (7 × 104 cells/well) into micro‐lysine‐coated XF96 microplates and incubated overnight in 5% CO2, 37 °C. To perform Cell Mito Stress Test, medium was replaced with XF Base supplemented with 1 mM pyruvate, 2 mM glutamine, 10 mM glucose, and cells were incubated at 37 °C for 45 min in a CO2‐free incubator, prior to oxygen consumption ratio analysis by Seahorse XF96. ATP production was measured by injection of oligomycin, while maximal respiration and spare respiration capacity were tested in the presence of carbonyl cyanide‐4‐(trifluoromethoxy)phenylhydrazone.
Data analysis
Data are presented as mean ± standard error of the mean. Kaplan–Meier analysis, one‐way analysis of variance, Student's t‐test, or Mann–Whitney test was performed using MedCalc (Medcalc Software, Mariakerke, Belgium) followed by individual post hoc comparisons (Fisher's PLSD). *P < 0.05 was considered significant.
Results
Differential expression analysis reveals alterations in histamine‐related genes in post‐mortem spinal cord samples from sporadic amyotrophic lateral sclerosis patients
To explore the potential involvement of the histaminergic system in ALS patients, we examined expression changes of genes implicated in histamine signalling and metabolism occurring in post‐mortem spinal cord samples from two transcriptome‐based subgroups of sALS patients (sALS1 and sALS2). Our analysis revealed a total of 13 histamine‐related genes differentially deregulated in sALS1 and sALS2, compared with control individuals. We identified genes encoding histamine receptors (HRH1 and HRH3), proteins involved in histamine metabolism (HNMT and PRG3), histamine transport and secretion (ADCYAP1, BTK, CCKBR, EDN1, SLC22A2, SNX4, SNX6, VAMP8, and YWHAZ), as well as multiple proteins involved in intracellular signal transduction cascades (i.e. MAPKs, p38MAPK, NF‐κB, and AKT) (Table 1). Interestingly, some of these genes were previously identified as differentially deregulated also in the motor cortex of the same sALS patient cohorts, supporting their role as potential biomarkers and therapeutic targets.4, 9
Table 1.
Transcriptome profiling of histamine‐related and signal transduction genes in post‐mortem spinal cords from sALS patient subgroups
| Histamine‐related process | Gene symbol human | sALS1 vs. Ctrl+ | sALS2 vs. Ctrl+ |
|---|---|---|---|
| Histamine receptors response | HRH1 | −1.3657919 | −3.3676426 |
| HRH3 | 3.3078184 | 3.2435715 | |
| Histamine transport/secretion | EDN1 | −1.3455846 | — |
| ADCYAP1 | 2.1645012 | 1.2958059 | |
| BTK | 1.620403 | — | |
| CCKBR | 2.0207274 | 1.3815044 | |
| SLC22A2 | — | −1.7292576 | |
| SNX4 | −1.4973569 | −2.0508032 | |
| SNX6 | −1.3490248 | −1.2742385 | |
| VAMP8 | −1.3397137 | — | |
| YWHAZ | 1.652496 | −1.4955612 | |
| Histamine metabolic process | HNMT | — | 1.2507114 |
| PRG3 | — | −1.3007661 | |
| Intracellular signal transduction | ADCY2 | 1.376056 | 1.6595138 |
| ADCY3 | −1.3174565 | — | |
| ADCY6 | 1.3229696 | — | |
| ADCY9 | — | 2.8711603 | |
| AKT1 | 1.2297583 | 1.4288182 | |
| AKT2 | −1.4742545 | — | |
| CREB1 | 1.403151 | 2.7520528 | |
| CREM | −1.9194763 | −1.822243 | |
| DRD4 | −1.7610718 | −1.7517854 | |
| GABRA1 | 1.3790314 | −1.9063799 | |
| GABRB1 | −1.3748144 | −1.313528 | |
| GABRB2 | −1.3764367 | −2.2042353 | |
| GABRG2 | — | −2.3212576 | |
| GNA11 | 1.2675852 | −1.2588073 | |
| GNAI1 | — | −1.2425077 | |
| GNAO1 | 2.0851884 | — | |
| GNAQ | 1.2491798 | — | |
| GNAS | −1.7503345 | −1.7303176 | |
| GNB1 | −1.3101016 | −1.4926312 | |
| GNB2 | 1.6231925 | — | |
| GNB4 | −1.5696274 | 1.3077484 | |
| GNG10 | −1.5117173 | — | |
| GNG11 | −1.661936 | — | |
| GNG13 | −1.6040359 | −1.5928824 | |
| GNG2 | — | −1.5869129 | |
| GNG3 | 4.5467305 | 2.6538045 | |
| GNG4 | — | 1.2450533 | |
| GNGT1 | 2.855986 | 1.8022374 | |
| IL10 | — | −1.382579 | |
| IL12B | 1.3056774 | — | |
| IL1B | — | −1.3257394 | |
| IL6 | — | −1.5675232 | |
| IRX3 | −1.2770016 | — | |
| ITPR1 | — | −1.5479783 | |
| MAP 2K2 | 1.297921 | — | |
| MAPK12 | 1.3404667 | — | |
| MAPK13 | 1.6014148 | 2.256803 | |
| MAPK14 | — | 1.7909265 | |
| MMP13 | — | 2.749664 | |
| MMP9 | — | −1.5295984 | |
| NFATC1 | −1.3547095 | −1.3149108 | |
| NFATC2 | −1.2919623 | −1.6587437 | |
| NFKB1 | −1.6059966 | −1.6690269 | |
| NFKB2 | — | −1.433936 | |
| PLCB1 | −1.3287859 | −1.4977577 | |
| PLCB3 | 1.526242 | — | |
| PRKACA | 1.3877281 | −1.8401401 | |
| PRKACB | — | −1.270291 | |
| PRKAR1A | 1.4079843 | −1.8058393 | |
| PRKAR2A | −1.447215 | −1.5083927 | |
| PRKCA | −1.3950349 | −1.2679613 | |
| RAF1 | −1.2660495 | −1.447958 | |
| RELA | −1.3689778 | −1.7076415 | |
| TNF | 1.350179 | — | |
| VCAM1 | −1.4938694 | — |
The table lists the principal histamine‐related and intracellular signal transduction genes differentially expressed in the spinal cord of two molecularly distinct subgroups of sALS patients,9 in comparison with individual controls. Expression levels are indicated as fold‐change values. Official Entrez Gene Symbols are used. HNMT, histamine N‐methyltransferase; sALS, sporadic amyotrophic lateral sclerosis.
Multi‐omic profiling defines core histamine‐related genomic alterations in amyotrophic lateral sclerosis
To obtain a genome‐wide view of multiple histamine‐related genomic aberrations in ALS and determine their effects on gene expression, we integrated data from different genomic levels (CNVs and SNPs), mainly focusing on genes associated with histamine metabolism, receptors, transport, and secretion. A complete overview of CNVs, SNPs/indels, and the combination of these events in ALS patients is investigated. Based on ongoing studies performed in our laboratories, we first analysed histamine‐related DNA copy number alterations occurring in previously characterized transcriptome‐based sALS patient subgroups9 (Morello et al., in press), in order to identify CNV‐associated differentially expressed genes. Our analysis revealed that some histamine‐related genes overlapped with genomic regions disrupted by a CNV, some of which in a subgroup‐patient specific manner (Figure 1). In particular, we observed the selective amplification of ADCYAP1 in sALS1 patients, the selective amplification of DAO1 and HRH2 in sALS2 patients, as well as duplication of HRH3 and CCKBR in both sALS subgroups (Figure 1). Interestingly among these, ADCYAP1, CCKBR, and HRH3 showed a significant, positive correlation with gene expression changes, supporting the idea that genes displaying the same tendencies between CNV status and gene expression levels may represent candidate driver genes in ALS etiopathogenesis (Table 1).
Figure 1.
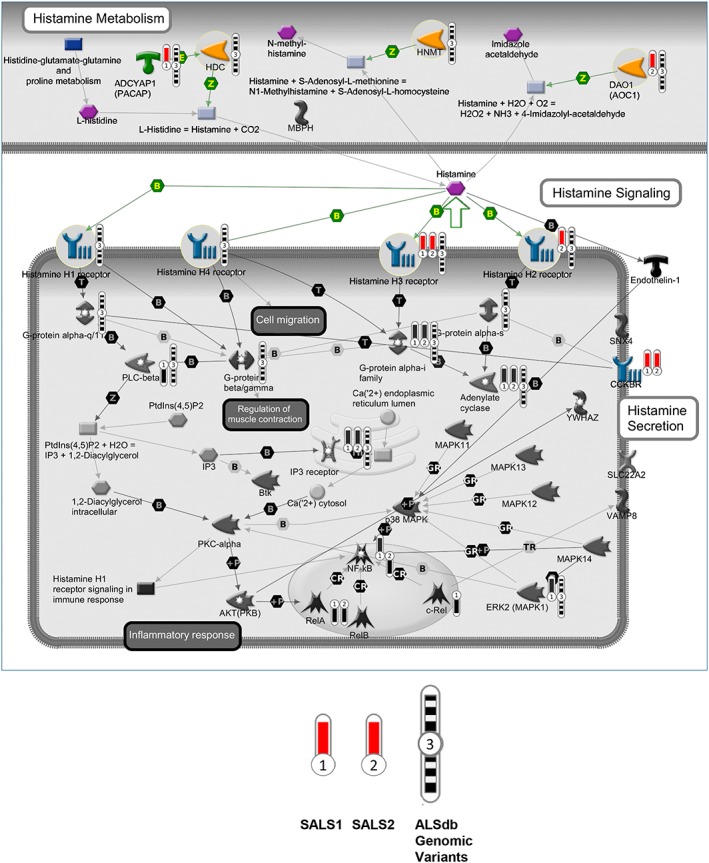
CNVs, SNVs/indels on histamine‐related metabolism and signalling pathways. The interaction pathway map represents the functional correlation between multi‐omic data (CNV, SNV/Indel) from sALS affecting histamine‐related genes and their signalling cascades. The map was created using the MetaCore pathway map Creator tool (GeneGo). Network objects related to genes specifically involved in histamine receptors, metabolism, and transport were reported as coloured objects. CNV values are presented on the map as ‘thermometer‐like’ symbols with sALS1 patients data represented as thermometer #1 and sALS2 patients #2. Genes associated with CNV gain regions are labelled with red dots. The thermometers with black/white stripes and #3 are the ALS genomic variants. ALS, amyotrophic lateral sclerosis; CNV, copy number variant; sALS, sporadic ALS; SNV, single nucleotide variant.
To provide a comprehensive multi‐omics view of the histamine‐related events occurring in ALS pathology, we then studied the pairwise overlap between the top histamine‐related genes identified from the transcriptome and CNV analyses and the corresponding ALS‐linked pathogenic variants from ALSdb, a database of exome sequencing data from mostly sALS patients. Our results demonstrated that there is a clear homogeneity between the histamine‐related genes driven by gene expression data, CNV, and SNP, supporting their role as candidate biomarkers and target genes in ALS (Figure 1, Table 2).
Table 2.
Potential pathological variants of histamine‐related and NF‐κB complex genes identified in ALS patients (ALS data Browser_ALSdb)
| Gene symbol | Chr | Position | Reference | Alternate variant | Variant_type | Codon_change | Amino acid change | Consequence | Global_MAFa (Exac) | Case_MAFb |
|---|---|---|---|---|---|---|---|---|---|---|
| HNMT | 2 | 138727773 | T | C | snv | aTa/aCa | I59T | Missense | 0 | 1.93E‐04 |
| 2 | 138738921 | G | A | snv | — | — | Intron | 0 | 1.90E‐04 | |
| 2 | 138771465 | A | G | snv | tAt/tGt | Y215C | Missense | 0 | 2.01E‐04 | |
| 2 | 138771650 | G | A | snv | Ggg/Agg | G277R | Missense | 0 | 2.06E‐04 | |
| 2 | 138771651 | GGA | G | indel | — | 277–278 | Frameshift | 0 | 2.06E‐04 | |
| 2 | 138771659 | C | CT | indel | — | 280 | Frameshift | 0 | 2.07E‐04 | |
| 2 | 138738967 | C | T | snv | — | — | Intron | 8.25E‐06 | 1.90E‐04 | |
| 2 | 138738829 | G | A | snv | — | — | Intron | 3.30E‐05 | 1.90E‐04 | |
| 2 | 138771459 | T | G | snv | cTt/cGt | L213R | Missense | 4.13E‐05 | 2.02E‐04 | |
| 2 | 138722075 | T | C | snv | aTg/aCg | M5T | Missense | 4.95E‐05 | 2.06E‐04 | |
| 2 | 138771586 | AC | A | indel | — | 256 | Frameshift | 8.25E‐05 | 4.02E‐04 | |
|
ADCYAP1 (PACAP) |
18 | 905450 | C | G | snv | agC/agG | S21R | Missense | 0 | 1.90E‐04 |
| 18 | 905450 | C | G | snv | — | — | Intron | 0 | 1.90E‐04 | |
| 18 | 905450 | C | G | snv | — | — | Noncoding exon | 0 | 1.90E‐04 | |
| 18 | 907767 | C | T | snv | — | — | Upstream | 0 | 3.55E‐04 | |
| 18 | 907767 | C | T | snv | — | — | Upstream | 0 | 3.55E‐04 | |
| 18 | 908290 | C | T | snv | — | — | Upstream | 9.40E‐04 | 0.0017 | |
| 18 | 908290 | C | T | snv | — | — | Upstream | 9.40E‐04 | 0.0017 | |
| 18 | 908345 | C | T | snv | Ctc/Ttc | L108F | Missense | 8.65E‐06 | 1.92E‐04 | |
| 18 | 908345 | C | T | snv | — | — | Upstream | 8.65E‐06 | 1.92E‐04 | |
| 18 | 908345 | C | T | snv | — | — | Upstream | 8.65E‐06 | 1.92E‐04 | |
| 18 | 905450 | C | G | snv | agC/agG | S21R | Missense | 0 | 1.90E‐04 | |
| DAO1 (AOC1) | 7 | 150554456 | C | T | snv | Ccc/Tcc | P300S | Missense | 0 | 1.94E‐04 |
| 7 | 150555081 | A | G | snv | aAc/aGc | N508S | Missense | 0 | 1.90E‐04 | |
| 7 | 150557540 | G | A | snv | — | — | Intron | 0 | 1.90E‐04 | |
| 7 | 150558299 | C | T | snv | — | — | 3′ UTR | 0 | 1.91E‐04 | |
| 7 | 150557639 | G | T | snv | aGc/aTc | S636I | Missense | 8.28E‐06 | 1.90E‐04 | |
| 7 | 150557621 | C | T | snv | tCg/tTg | S630L | Missense | 8.29E‐06 | 1.90E‐04 | |
| 7 | 150554763 | A | G | snv | cAc/cGc | H402R | Missense | 8.30E‐06 | 1.90E‐04 | |
| 7 | 150555105 | A | G | snv | tAc/tGc | Y516C | Missense | 9.24E‐06 | 3.79E‐04 | |
| 7 | 150555113 | G | C | snv | Gac/Cac | D519H | Missense | 9.28E‐06 | 1.90E‐04 | |
| 7 | 150556095 | GC | G | indel | — | 606 | Frameshift | 1.09E‐05 | 1.93E‐04 | |
| 7 | 150555050 | G | A | snv | Ggc/Agc | G498S | Missense | 2.62E‐05 | 1.90E‐04 | |
| 7 | 150554423 | G | A | snv | Ggg/Agg | G289R | Missense | 2.97E‐05 | 1.95E‐04 | |
| 7 | 150558196 | C | T | snv | Cgg/Tgg | R719W | Missense | 4.97E‐05 | 1.90E‐04 | |
| 7 | 150554459 | C | T | snv | Cgc/Tgc | R301C | Missense | 1.46E‐04 | 7.77E‐04 | |
| 7 | 150554147 | C | T | snv | Cgc/Tgc | R197C | Missense | 1.49E‐04 | 1.90E‐04 | |
| 7 | 150556052 | G | A | snv | cGc/cAc | R591H | Missense | 1.93E‐04 | 1.92E‐04 | |
| 7 | 150554538 | G | A | snv | cGg/cAg | R327Q | Missense | 2.09E‐04 | 1.93E‐04 | |
| 7 | 150558115 | G | A | snv | Gtg/Atg | V692M | Missense | 2.24E‐04 | 1.90E‐04 | |
| 7 | 150554114 | G | A | snv | Gat/Aat | D186N | Missense | 2.65E‐04 | 5.69E‐04 | |
| 7 | 150554937 | A | G | snv | aAt/aGt | N460S | Missense | 2.99E‐04 | 3.79E‐04 | |
| 7 | 150555982 | C | T | snv | Cgc/Tgc | R568C | Missense | 3.15E‐04 | 3.81E‐04 | |
| 7 | 150557707 | A | C | snv | Aac/Cac | N659H | Missense | 0.002 | 3.79E‐04 | |
| HDC | 15 | 50534538 | G | T | snv | ttC/ttA | F636L | Missense | 0 | 1.90E‐04 |
| 15 | 50534882 | T | G | snv | Atc/Ctc | I522L | Missense | 0 | 1.93E‐04 | |
| 15 | 50540515 | C | T | snv | cGg/cAg | R356Q | Missense | 0 | 3.95E‐04 | |
| 15 | 50540542 | T | C | snv | — | — | Upstream/split acceptor | 0 | 2.02E‐04 | |
| 15 | 50544830 | T | C | snv | cAt/cGt | H310R | Missense | 0 | 1.90E‐04 | |
| 15 | 50546389 | G | A | snv | Cga/Tga | R220b | Stop gain | 0 | 1.90E‐04 | |
| 15 | 50549623 | T | C | snv | cAg/cGg | Q147R | Missense; splice region | 0 | 1.90E‐04 | |
| 15 | 50546392 | G | A | snv | Ctc/Ttc | L219F | Missense | 8.24E‐06 | 1.90E‐04 | |
| 15 | 50546857 | G | A | snv | aCg/aTg | T149M | Missense | 8.26E‐06 | 1.90E‐04 | |
| 15 | 50540499 | T | C | snv | — | — | Upstream | 8.31E‐06 | 1.96E‐04 | |
| 15 | 50540509 | C | T | snv | cGc/cAc | R358H | Missense | 1.67E‐05 | 1.97E‐04 | |
| 15 | 50534613 | C | A | snv | agG/agT | R611S | Missense | 2.47E‐05 | 1.91E‐04 | |
| 15 | 50534990 | G | A | snv | Cgg/Tgg | R486W | Missense | 4.14E‐05 | 1.93E‐04 | |
| 15 | 50540475 | G | A | snv | — | — | Upstream | 6.65E‐05 | 3.92E‐04 | |
| 15 | 50535405 | T | C | snv | Aga/Gga | R393G | Missense | 9.89E‐05 | 3.83E‐04 | |
| 15 | 50535151 | C | T | snv | cGt/cAt | R432H | Missense | 1.34E‐04 | 1.92E‐04 | |
| HRH1 | 3 | 11300898 | C | T | snv | Cac/Tac | H59Y | Missense | 0 | 3.83E‐04 |
| 3 | 11301096 | C | A | snv | Cgc/Agc | R125S | Missense | 0 | 3.94E‐04 | |
| 3 | 11301711 | C | T | snv | Cag/Tag | Q330b | Stop gain | 0 | 1.93E‐04 | |
| 3 | 11302074 | A | G | snv | Atg/Gtg | M451V | Missense | 0 | 2.00E‐04 | |
| 3 | 11301963 | G | A | snv | Gcc/Acc | A414T | Missense | 8.25E‐06 | 2.08E‐04 | |
| 3 | 11301157 | C | T | snv | tCg/tTg | S145L | Missense | 2.47E‐05 | 1.92E‐04 | |
| 3 | 11301988 | C | T | snv | gCa/gTa | A422V | Missense | 4.95E‐05 | 2.05E‐04 | |
| 3 | 11301394 | G | A | snv | cGg/cAg | R224Q | Missense | 2.89E‐04 | 8.20E‐04 | |
| 3 | 11301672 | A | T | snv | Agt/Tgt | S317C | Missense | 3.13E‐04 | 1.93E‐04 | |
| 3 | 11301810 | T | C | snv | Tca/Cca | S363P | Missense | 6.85E‐04 | 0.0019 | |
| HRH2 | 5 | 175111224 | C | T | snv | Cga/Tga | R330b | Stop gain | 0 | 1.90E‐04 |
| 5 | 175110385 | G | A | snv | cGc/cAc | R50H | Missense | 8.24E‐06 | 1.90E‐04 | |
| 5 | 175110408 | G | A | snv | Gtg/Atg | V58M | Missense | 9.06E‐05 | 3.80E‐04 | |
| HRH3 | 20 | 60791717 | C | T | snv | cGc/cAc | R228H | Missense | 8.51E‐06 | 1.91E‐04 |
| 20 | 60793668 | C | T | snv | cGc/cAc | R99H | Missense | 8.63E‐06 | 1.93E‐04 | |
| 20 | 60793573 | C | T | snv | Gac/Aac | D131N | Missense | 8.94E‐06 | 1.96E‐04 | |
| 20 | 60793548 | G | A | snv | gCg/gTg | A139V | Missense; splice region | 9.58E‐06 | 1.97E‐04 | |
| 20 | 60791138 | C | T | snv | cGg/cAg | R421Q | Missense | 3.68E‐05 | 1.94E‐04 | |
| 20 | 60793653 | C | T | snv | cGg/cAg | R104Q | Missense | 1.64E‐04 | 1.93E‐04 | |
| 20 | 60793657 | C | T | snv | Ggc/Agc | G103S | Missense | 3.27E‐04 | 1.93E‐04 | |
| HRH4 | 18 | 22056997 | G | A | snv | gGa/gAa | G215E | Missense | 0 | 2.13E‐04 |
| 18 | 22057449 | CA | C | indel | — | 366 | Frameshift | 8.28E‐06 | 1.95E‐04 | |
| 18 | 22048893 | G | A | snv | cGa/cAa | R112Q | Missense | 8.34E‐06 | 1.94E‐04 | |
| 18 | 22057369 | G | A | snv | tGg/tAg | W339b | Stop gain | 1.65E‐05 | 1.93E‐04 | |
| 18 | 22040738 | G | C | snv | Gtt/Ctt | V16L | Missense | 1.65E‐05 | 2.02E‐04 | |
| 18 | 22056759 | A | T | snv | Atg/Ttg | M136L | Missense | 8.16E‐04 | 0.0025 | |
| NFKB1 (NF‐κB complex) | 4 | 103488212 | G | A | snv | — | — | Upstream | 0 | 1.95E‐04 |
| 4 | 103501792 | G | T | snv | caG/caT | Q277H | Missense | 0 | 1.95E‐04 | |
| 4 | 103514659 | G | A | snv | Gga/Aga | G382R | Missense | 8.24E‐06 | 1.93E‐04 | |
| 4 | 103518688 | C | G | snv | Cta/Gta | L503V | Missense | 8.29E‐06 | 1.95E‐04 | |
| 4 | 103514641 | G | A | snv | Ggc/Agc | G376S | Missense | 4.12E‐05 | 1.92E‐04 | |
| 4 | 103500158 | G | A | snv | cGc/cAc | R231H | Missense | 4.12E‐05 | 1.95E‐04 | |
| 4 | 103498185 | G | A | snv | cGg/cAg | R187Q | Missense | 4.16E‐05 | 2.07E‐04 | |
| 4 | 103537644 | C | T | snv | Cgc/Tgc | R935C | Missense | 4.94E‐05 | 1.93E‐04 | |
| 4 | 103533719 | C | T | snv | Cgg/Tgg | R850W | Missense | 5.77E‐05 | 3.81E‐04 | |
| 4 | 103534740 | T | TAA | indel | — | — | Splice region | 6.15E‐05 | 1.92E‐04 | |
| 4 | 103488146 | C | A | snv | — | — | Upstream | 1.57E‐04 | 1.98E‐04 | |
| 4 | 103505915 | G | A | snv | cGg/cAg | R335Q | Missense | 1.57E‐04 | 1.90E‐04 | |
| 4 | 103518782 | G | A | snv | cGc/cAc | R534H | Missense | 4.46E‐04 | 8.02E‐04 | |
| 4 | 103522150 | G | A | snv | aGa/aAa | R579K | Missense | 0.0018 | 5.86E‐04 | |
| NFKB2 (NF‐κB complex) | 10 | 104159374 | C | T | snv | — | — | Downstream | 0 | 0.0013 |
| 10 | 104159386 | C | A | snv | — | — | Downstream | 0 | 0.0014 | |
| 10 | 104160046 | C | T | snv | — | — | Downstream | 0 | 3.82E‐04 | |
| 10 | 104160123 | G | A | snv | cGg/cAg | R558Q | Missense | 0 | 2.07E‐04 | |
| 10 | 104160175 | TGAGCTGCTGCGTGCACTGCTTCAGAGTG | T | indel | — | 576–585 | Frameshift | 0 | 2.29E‐04 | |
| 10 | 104160458 | C | T | snv | — | — | Downstream | 0 | 1.93E‐04 | |
| 10 | 104161009 | C | T | snv | tCg/tTg | S715L | Missense | 0 | 1.91E‐04 | |
| 10 | 104161893 | CCCGAGA | C | indel | acCCGAGAc/acc | TRD852‐854T | Inframe deletion | 0 | 1.95E‐04 | |
| 10 | 104160802 | G | A | snv | — | — | Downstream | 8.42E‐06 | 1.92E‐04 | |
| 10 | 104160777 | C | T | snv | cCg/cTg | P681L | Missense | 2.55E‐05 | 1.92E‐04 | |
| 10 | 104160150 | C | T | snv | gCg/gTg | A567V | Missense | 3.33E‐05 | 2.20E‐04 | |
| 10 | 104160070 | G | A | snv | — | — | Downstream | 6.64E‐05 | 3.86E‐04 | |
| 10 | 104157142 | G | A | snv | cGc/cAc | R160H | Missense | 6.66E‐05 | 1.90E‐04 | |
| 10 | 104155730 | A | G | snv | tAc/tGc | Y5C | Missense | 6.66E‐05 | 3.80E‐04 | |
| 10 | 104161557 | C | T | snv | — | — | Downstream | 1.02E‐04 | 1.91E‐04 | |
| 10 | 104160707 | C | T | snv | Cgg/Tgg | R658W | Missense | 1.75E‐04 | 3.88E‐04 | |
| 10 | 104158210 | G | A | snv | — | — | Downstream | 6.63E‐04 | 3.80E‐04 | |
| REL (NF‐κB complex) | 2 | 61145580 | T | A | snv | tTt/tAt | F231Y | Missense | 0 | 1.96E‐04 |
| 2 | 61145646 | T | C | snv | aTc/aCc | I253T | Missense | 0 | 2.00E‐04 | |
| 2 | 61147583 | A | G | snv | Acc/Gcc | T330A | Missense | 0 | 7.00E‐04 | |
| 2 | 61147587 | T | C | snv | tTg/tCg | L331S | Missense | 0 | 2.15E‐04 | |
| 2 | 61149189 | C | T | snv | gCt/gTt | A460V | Missense | 1.65E‐05 | 3.94E‐04 | |
| 2 | 61149451 | C | T | snv | — | — | Upstream | 2.47E‐05 | 1.92E‐04 | |
| 2 | 61145679 | G | A | snv | cGg/cAg | R264Q | Missense | 2.47E‐05 | 2.00E‐04 | |
| 2 | 61149544 | G | A | snv | — | — | Upstream | 4.13E‐05 | 1.96E‐04 | |
| 2 | 61149514 | T | C | snv | — | — | Upstream | 6.60E‐05 | 1.96E‐04 | |
| 2 | 61149373 | C | T | snv | — | — | Upstream | 1.07E‐04 | 3.95E‐04 | |
| RELA (NF‐κB complex) | 11 | 65422000 | C | T | snv | cGc/cAc | R502H | Missense | 0 | 1.90E‐04 |
| 11 | 65422263 | G | A | snv | — | — | Downstream | 0 | 2.42E‐04 | |
| 11 | 65422321 | G | C | snv | cCt/cGt | P395R | Missense | 0 | 2.11E‐04 | |
| 11 | 65429260 | G | T | snv | aCc/aAc | T78N | Missense | 0 | 1.91E‐04 | |
| 11 | 65429561 | T | C | snv | — | — | Split acceptor | 0 | 2.21E‐04 | |
| 11 | 65426257 | A | G | snv | gTg/gCg | V199A | Missense | 4.94E‐05 | 1.90E‐04 | |
| 11 | 65421966 | T | C | snv | — | — | Downstream | 9.39E‐05 | 7.59E‐04 | |
| 11 | 65423384 | C | T | snv | — | — | Downstream | 9.89E‐05 | 3.82E‐04 | |
| 11 | 65423327 | G | A | snv | — | — | Downstream | 1.24E‐04 | 3.81E‐04 | |
| 11 | 65423400 | C | T | snv | cGt/cAt | R295H | Missense | 1.73E‐04 | 3.82E‐04 | |
| 11 | 65422425 | A | G | snv | — | — | Downstream | 3.63E‐04 | 3.89E‐04 | |
| RELB (NF‐κB complex) | 19 | 45515316 | G | C | snv | Ggc/Cgc | G96R | Missense | 0 | 3.59E‐04 |
| 19 | 45532206 | C | T | snv | aCc/aTc | T316I | Missense | 0 | 1.90E‐04 | |
| 19 | 45540874 | G | C | snv | — | — | Upstream | 0 | 1.92E‐04 | |
| 19 | 45540892 | C | A | snv | — | — | Upstream | 0 | 1.93E‐04 | |
| 19 | 45540935 | G | A | snv | Ggc/Agc | G543S | Missense | 0 | 1.93E‐04 | |
| 19 | 45540702 | ACTT | A | indel | gaCTTc/gac | DF465‐466D | Inframe deletion | 8.66E‐06 | 1.90E‐04 | |
| 19 | 45540825 | C | A | snv | cCc/cAc | P506H | Missense | 8.75E‐06 | 1.91E‐04 | |
| 19 | 45541015 | C | T | snv | — | — | Upstream | 1.95E‐05 | 2.38E‐04 | |
| 19 | 45540877 | C | T | snv | — | — | Upstream | 2.79E‐05 | 5.77E‐04 | |
| 19 | 45535998 | C | T | snv | Cgc/Tgc | R400C | Missense | 3.74E‐05 | 1.90E‐04 | |
| 19 | 45537531 | C | T | snv | Cgg/Tgg | R413W | Missense | 4.15E‐05 | 3.79E‐04 | |
| 19 | 45537720 | A | G | snv | Atc/Gtc | I430V | Missense | 8.57E‐05 | 1.90E‐04 | |
| 19 | 45540789 | C | T | snv | aCg/aTg | T494M | Missense | 6.67E‐04 | 0.0017 |
Sequence reads were aligned to the GRCh37 human reference genome and official gene symbols were used. Chr, chromosome; SNV, single nucleotide variant; Indel, insertion and deletion variant; MAF, minor allele frequency.
Golbal MAF from Exome Aggregation Consortium (ExAC).
Case MAF from 2800 Caucasian patients recruited and sequenced for their diagnosis of amyotrophic lateral sclerosis and reported in ALSdb.
Gene expression profiles of superoxide dismutase 1‐G93A mouse spinal cords at pre‐symptomatic, symptomatic, and terminal disease stages
To obtain a clearer picture of the transcriptomic changes associated with the dysregulation of the histamine signalling during ALS progression and evaluate whether these changes were cause or consequence of motor neuron degeneration, we analysed histamine‐associated genes in SOD1‐G93A mice at pre‐symptomatic, symptomatic, and terminal stages (Table 3). Remarkably, our results showed that in the majority of cases, the deregulation of histamine‐related genes (e.g. Hnmt, Hrh1, and Hrh3) had already started in ALS mice long before symptom onset. This suggests their possible role as target genes for ALS diagnosis and treatment in the early stages of the disease. Moreover, it is interesting to note that the overwhelming majority of histamine‐related genes that emerged as deregulated in terminal stages of SOD1‐G93A mice were also found deregulated in at least one of sALS patient subgroups (e.g. Hrh1 and Hrh3), thus offering a rationale for the selection and prioritization of histamine genes as potential biomarkers and targets for patient‐oriented ALS care (Tables 1 and 3).
Table 3.
Gene expression profiles of SOD1‐G93A spinal cords at pre‐symptomatic, symptomatic, and terminal disease stage
| Histamine‐related process | Gene symbol mouse | Pre‐symptomatic stage | Symptomatic stage | Terminal stage |
|---|---|---|---|---|
| Histamine receptors response | Hrh1 | −1.003499128 | — | −1.003597844 |
| Hrh3 | −1.046221992 | −1.075912684 | −1.050912759 | |
| Histamine transport/secretion | Edn1 | −1.375929264 | −1.057040107 | 1.333098846 |
| Adcyap1 | — | 1.201892853 | — | |
| Btk | −1.290913424 | — | — | |
| Cckbr | −1.167878414 | −1.070903032 | — | |
| Slc22a2 | −1.038570786 | — | — | |
| Snap23 | −1.03638262 | −1.025148052 | — | |
| Snx4 | 1.236540934 | — | −1.079338049 | |
| Snx6 | — | −1.203377265 | −1.093475719 | |
| Vamp7 | 1.340461968 | −1.038865474 | −1.052208506 | |
| Vamp8 | −1.059845532 | −1.061651987 | 1.241167685 | |
| Ywhaz | −1.098709306 | −1.350732188 | −1.02076357 | |
| Histamine metabolic process | Hnmt | 1.444280445 | — | — |
| Prg3 | −1.470871012 | — | −1.015267987 | |
| Intracellular signal transduction | Adcy2 | 2.023873708 | −1.048152441 | −1.802825559 |
| Adcy3 | — | −1.002578776 | −1.116612498 | |
| Adcy6 | −1.028682464 | −1.031914462 | 1.246062931 | |
| Adcy9 | 1.266598061 | −1.035777163 | −1.405373387 | |
| Akt1 | −1.226973947 | −1.052315605 | 1.209025549 | |
| Akt2 | −1.226090177 | −1.160688229 | — | |
| Akt3 | −1.171859879 | −1.027665214 | — | |
| Creb1 | −1.131029919 | — | — | |
| Crem | — | −1.038302234 | — | |
| Drd4 | −1.308482844 | −1.114190513 | — | |
| Gabra1 | −1.071315243 | — | — | |
| Gabrb1 | 2.755654323 | −1.116099761 | −1.815309844 | |
| Gabrb2 | — | −1.19872974 | — | |
| Gabrg2 | −1.146330802 | −1.097362011 | −1.020976133 | |
| Gna11 | — | −1.017849586 | 1.203291897 | |
| Gnai1 | — | — | −1.029191512 | |
| Gnao1 | −1.279205386 | −1.049786529 | — | |
| Gnaq | −1.077554095 | −1.042284401 | −1.139593361 | |
| Gnas | ‐ | −1.030436247 | — | |
| Gnb1 | 1.206852499 | — | — | |
| Gnb2 | −1.149063261 | −1.162953351 | −1.132948762 | |
| Gnb4 | — | — | −1.219119728 | |
| Gng10 | 1.232431089 | 1.239336488 | −1.168181168 | |
| Gng11 | — | — | −1.115660465 | |
| Gng13 | −1.359367344 | −1.555262502 | — | |
| Gng2 | −1.042849815 | −1.080945773 | 1.238813943 | |
| Gng3 | −1.297109187 | −1.05246234 | −1.097358904 | |
| Gng4 | −1.106026584 | −1.033424615 | −1.041158526 | |
| Gngt1 | −1.215627453 | −1.067192339 | — | |
| Icam1 | −1.093483742 | −1.041303247 | 1.337188809 | |
| Il10 | −1.03237061 | — | −1.054267745 | |
| Il12b | −1.160343914 | −1.059477718 | −1.069307231 | |
| Il1b | 1.002576004 | −1.179173693 | 1.075892325 | |
| Il6 | −1.197945784 | — | −1.038636007 | |
| Irx3 | −1.338152986 | −3.86809662 | −2.369452938 | |
| Itpr1 | 1.255453987 | −1.12400873 | −1.27371253 | |
| Map 2k2 | −1.197195917 | −1.063642196 | — | |
| Mapk1 | — | −1.080685442 | −1.206047606 | |
| Mapk11 | −1.144428024 | −1.181041741 | — | |
| Mapk13 | −1.236037351 | −1.231334227 | −1.082872971 | |
| Mapk14 | −1.026578354 | — | −1.176410554 | |
| Mmp13 | −1.059520405 | −1.064706356 | −1.001979303 | |
| Mmp1a | −1.140824834 | — | −1.020986832 | |
| Mmp9 | −1.184670887 | −1.126900884 | −1.609441965 | |
| Nfatc1 | −1.076125405 | −1.097443244 | −1.02229657 | |
| Nfatc2 | −1.16285032 | −1.13673478 | — | |
| Nfkb1 | — | −1.032778375 | −1.199717176 | |
| Nfkb2 | −1.363125323 | −1.02698758 | 1.415161791 | |
| Nos2 | −1.212861572 | −1.093178806 | — | |
| Plcb1 | −1.175530771 | −1.090801363 | — | |
| Plcb3 | −1.398547724 | −1.136613821 | −1.04356683 | |
| Prkaca | −1.091370655 | −1.07263985 | −1.388131461 | |
| Prkacb | — | −1.138967544 | — | |
| Prkar1a | 1.607723299 | — | −1.933887991 | |
| Prkar2a | — | −1.060189746 | — | |
| Prkca | −1.073991432 | −1.066174143 | — | |
| Raf1 | −1.112701164 | — | −1.026474401 | |
| Rel | −1.173363768 | −1.134546121 | — | |
| Rela | — | −1.009506984 | — | |
| Relb | −1.058592695 | — | — | |
| Tnf | −1.279347739 | — | −1.027736516 | |
| Vcam1 | −1.138593267 | — | — |
The table lists the principal histamine‐related and intracellular signal transduction genes differentially expressed in the spinal cord of SOD1‐G93A mice at different stages of the disease, in comparison with relative littermate controls. Expression levels are indicated as fold‐change values. Genes similarly deregulated in ALS patients are marked in grey. Official Entrez Gene Symbols are used. Microarray gene expression data sets have been downloaded from the NCBI Gene Expression Omnibus repository (http://www.ncbi.nlm.nih.gov/geo/) under accession code GSE10953. ALS, amyotrophic lateral sclerosis; SOD1, superoxide dismutase 1.
Enhanced histaminergic signalling improves motor performance and lifespan in superoxide dismutase 1‐G93A mice
Because histamine cannot penetrate the brain, we tested the efficacy of a histaminergic strategy in SOD1‐G93A mice, by using the histamine precursor histidine that is able to increase the central histamine levels17 by crossing the blood–brain barrier and being rapidly converted into histamine.29 Histidine (50–100–250 mg/kg) was injected in mice from the onset of clinical symptoms. At 100 and 250 mg/kg, histidine improved behavioural scores (Figure 2A) and motor performance (Figure 2B) and delayed disease progression, as assessed by the time taken to reach the clinical score 3 (Figure 2C), and the 10%, 50%, and 100% of rotarod impairment (Figure 2D and 2F). The dose of 50 mg/kg resulted ineffective in ameliorating mice general conditions and motor performance (data not shown). As shown in Figure 2G, histidine 100 mg/kg increased median survival of SOD1‐G93A mice (158 saline, 171 His100), whereas the dose of 250 mg/kg showed only a trend in increasing survival. Furthermore, while both histidine 100 and 250 mg/kg statistically delayed disease duration to terminal endpoint in SOD1‐G93A mice, as assessed by age at terminal endpoint (158.8 ± 1.7 saline; 171.7 ± 2.1 His100, 172.6 ± 6.4 His250), only histidine 100 mg/kg increased disease duration after the onset of first symptoms (48.8 ± 1.3 saline; 62 ± 4.8 His100, Figure 2I).
Figure 2.
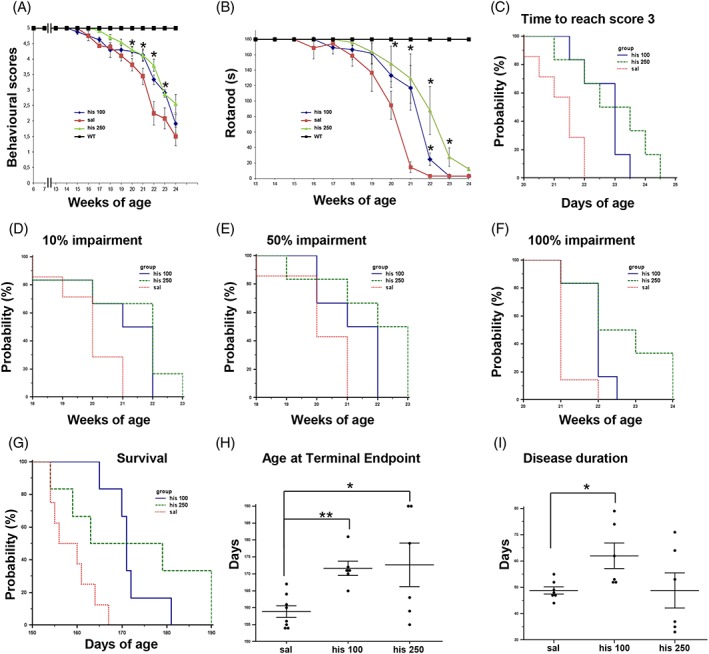
Histidine ameliorates disease progression and improves life span and motor performance in SOD1‐G93A mice. Behavioural scores (A) and rotarod performance (B) were significantly ameliorated in histidine 100 mg/kg (n = 6, blue) and histidine 250 mg/kg (n = 6, green) compared with saline‐treated SOD1‐G93A mice (n = 12, red). WT in black, n = 4. Kaplan–Meier analysis of time to reach behavioural score 3 (C) and of time to reach 10% (D), 50% (E), and 100% (F) of rotarod impairment in saline (n = 12, red), histidine 100 mg/kg (n = 6, blue), and histidine 250 mg/kg (n = 6, green) SOD1‐G93A‐treated mice. (G) Kaplan–Meier survival curve of SOD1‐G93A mice showing increased survival following histidine 100 mg/kg treatment (n = 6, blue) as compared with saline‐treated mice (n = 8, red), while histidine 250 mg/kg shows only a trend. (H) The terminal endpoint was significantly delayed in the histidine 100 mg/kg‐treated and histidine 250 mg/kg‐treated SOD1‐G93A mice compared with saline. (I) Disease duration slowed down significantly in histidine 100 mg/kg group, as indicated by increased days from clinical score 4.75 to terminal endpoint. Data represent means ± SEM. Statistical significance was calculated by analysis of variance referred to saline SOD1‐G93A, * P < 0.05, ** P < 0.01. SEM, standard error of the mean; SOD1, superoxide dismutase 1; WT, wild‐type.
At these doses, histidine showed no evidence of toxic effects in mice, as evaluated by body weight measurement, and histidine‐treated mice presented no gross pathology on necropsy compared with saline‐treated mice.
Histaminergic signalling protects motor neurons and axonal degeneration in superoxide dismutase 1‐G93A mice
Guided by the earlier results, we then adopted the most effective dose of histidine 100 mg/kg for further evaluations. HPLC analysis revealed that histamine content in the spinal cord of SOD1‐G93A mice was 45.8 ± 6.4 ng/g (n = 6), which increased to 93.6 ± 14.7 ng/g (n = 6) (P = 0.014) after histidine treatment. In SOD1‐G93A spinal cord at terminal phase of disease with respect to the saline group, histidine partially protected motor neurons from death, as observed by Nissl staining (Figure 3A). Likewise, choline acetyltransferase protein was significantly decreased in SOD1‐G93A with respect to WT mice (Supporting Information, Figure S1A) and histidine preserved its expression (Figure 3B). Moreover, in SOD1‐G93A with respect to WT mice, we observed a strong reduction in SMI32 expression (Supporting Information, Figure S1A). In histidine‐treated mice, we observed statistically significant preservation of SMI32‐positive fibres in the ventral horns of spinal cord sections (Figure 3C), and this result was confirmed by western blot analysis (Figure 3D). Because oligodendrocytes losing their myelinating and metabolic functions participate to the degeneration of ALS motor neurons,30 we examined MBP expression in SOD1‐G93A mice, where it was shown to be significantly decreased compared with WT mice (Supporting Information, Figure S1A). As indicated in Figure 3E, MBP immunoreactivity was increased in ventral grey and white horns of histidine‐treated compared with the saline spinal cord. The preservation of MBP expression was also confirmed by immunoblotting (Figure 3F).
Figure 3.
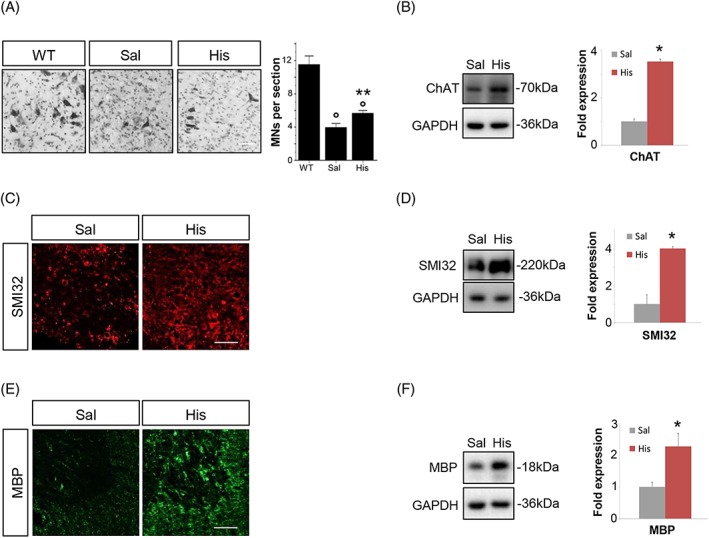
Histidine partially rescues motor neurons loss, axonal degeneration, and demyelination in SOD1‐G93A mice. (A) Nissl‐stained spinal cord sections of WT (n = 3, ~150 days) and terminal stage SOD1‐G93A mice after saline or histidine 100 mg/kg treatment (n = 4/5). Scale bar: 100 μm. Quantification of motor neuron (MNs) numbers (%) in sections of terminal SOD1‐G93A mice is provided. Representative western blots and quantification of ChAT (B), SMI32 (D), and MBP (F) in saline‐treated (n = 4) and 100 mg/kg histidine‐treated (n = 5) SOD1‐G93A mice. GAPDH was used as a loading control. Data represent means ± SEM. Statistical significance was calculated by analysis of variance or Mann–Whitney test referred to saline SOD1‐G93A group, * P < 0.05 or to WT group, ° P < 0.05. Representative immunofluorescence of SMI32 (C) and MBP (E) in saline‐treated (n = 4) and 100 mg/kg histidine‐treated (n = 5) SOD1‐G93A mice. Scale bar: 100 μm. ChAT, choline acetyltransferase; MBP, myelin basic protein; SEM, standard error of the mean; SOD1, superoxide dismutase 1; WT, wild‐type.
Histaminergic treatment decreases microgliosis and modulates pro‐inflammatory/anti‐inflammatory markers in the lumbar spinal cord of superoxide dismutase 1‐G93A mice
To investigate if histidine treatment in SOD1‐G93A mice can modify ALS affected pathways in spinal cord, we analysed tissues at symptomatic 150 days of age. We first evaluated the extent of microgliosis and the expression of microglia‐related markers. Immunofluorescence analysis of lumbar spinal cord demonstrated a remarkable increase of CD11b positive microglia in SOD1‐G93A with respect to WT mice, which was partially reverted by histidine treatment (100 mg/kg) (Figure 4A and 4B). Next, we measured the protein levels of microglial M2‐like markers ARG1 and CD163 and microglial M1‐like marker iNOS, demonstrating that in SOD1‐G93A mice, ARG1 and CD163 expressions were decreased and iNOS increased, compared with WT mice (Supporting Information, Figure S1B). Histidine was able to significantly increase ARG1 and CD163 and decrease iNOS protein expression in SOD1‐G93A mice with respect to saline‐treated mice (Figure 4C and 4D).
Figure 4.
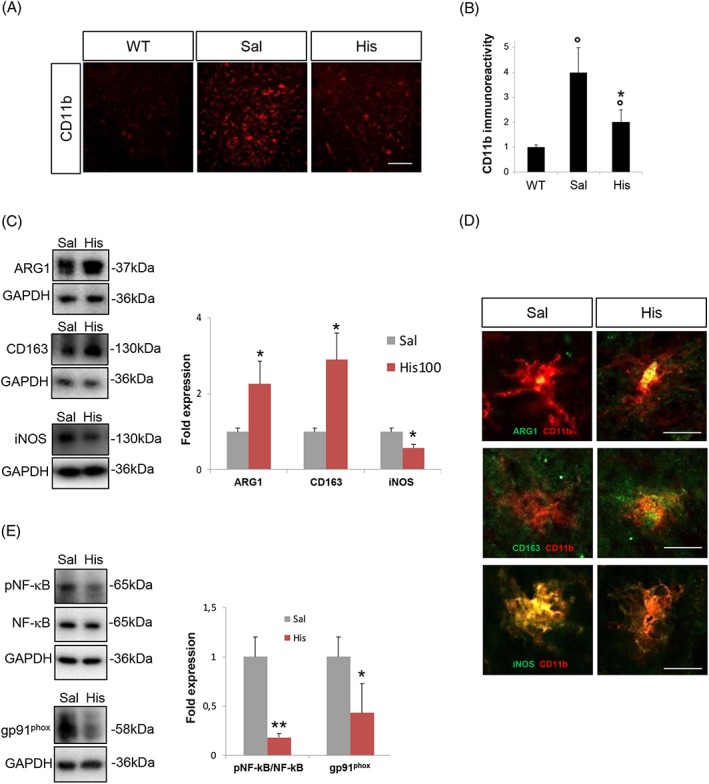
Histidine modulates microgliosis and pro‐inflammatory/anti‐inflammatory markers in the lumbar spinal cord of SOD1‐G93A mice. (A) Representative confocal images of lumbar spinal cord sections of WT, saline‐treated and 100 mg/kg histidine‐treated SOD1‐G93A mice immunolabelled with CD11b (red). Scale bar: 100 μm. (B) Quantification of CD11b staining in hemisections of WT, saline‐treated and 100 mg/kg histidine‐treated SOD1‐G93A mice. Data are expressed as means ± SEM (six sections for each animal). (C) Representative western blots and quantification of ARG1, CD163, and iNOS in saline‐treated (n = 4) and 100 mg/kg histidine‐treated (n = 5) SOD1‐G93A mice. (D) Representative confocal images of microglia cells labelled with CD11b (red) and ARG1, CD163, or iNOS (green) in saline‐treated and 100 mg/kg histidine‐treated SOD1‐G93A mice. Scale bar: 10 μm. (E) Representative western blots and quantification of pNF‐κB/NF‐κB and gp91phox in saline‐treated (n = 4) and 100 mg/kg histidine‐treated (n = 5) SOD1‐G93A mice. GAPDH was used as a loading control. Data represent means ± SEM. Statistical significance was calculated by analysis of variance, Student's t‐test, or by Mann–Whitney test referred to saline‐SOD1‐G93A group, * P < 0.05, ** P < 0.01, or to WT group, ° P < 0.05. ARG1, arginase 1; iNOS, inducible nitric oxide synthase; NF‐κB, nuclear factor‐kappa B; SEM, standard error of the mean; SOD1, superoxide dismutase 1; WT, wild‐type.
We finally measured the expression of NF‐κB and gp91phox, key inflammatory mediators that are deregulated in ALS and were previously shown to be down‐regulated by histamine in SOD1‐G93A microglia.4 SOD1‐G93A mice showed up‐regulated levels of both NF‐κB and gp91phox compared with WT mice (Supporting Information, Figure S1C). In histidine‐treated (100 mg/kg) mice respect to the saline‐treated group, we observed a statistically significant reduction of phosphorylated NF‐κB and gp91phox proteins (Figure 4E).
When we next analysed SOD1‐G93A spinal cord tissue at the end stage of disease, we observed no statistical changes in ARG1 (saline = 1 ± 0.2 vs. histidine = 1.2 ± 0.1) and gp91phox (saline = 1 ± 0.2 vs. histidine = 0.7 ± 0.1), but a significant down‐regulation of phosphorylated NF‐κB (saline = 1 ± 0.2 vs. histidine = 0.3 ± 0.06; P < 0.05), in histidine‐treated mice compared with saline‐treated mice.
Histaminergic signalling affects MAPKs and decreases denervation muscle atrophy in superoxide dismutase 1‐G93A mice
Because deviation from the strict control of MAPK signalling pathways particularly in SOD1‐G93A mice is implicated in ALS disease progression, we next analysed ERK1/2 and AKT pro‐survival kinases after histidine treatment. Compared with WT mice, SOD1‐G93A mice expressed similar levels of both phosphorylated ERK1/2 and AKT in the spinal cord (Supporting Information, Figure S1D) and robust down‐regulation of phosphorylated AKT in the sciatic nerves (Supporting Information, Figure S1E). As shown in Figure 5A, in the lumbar spinal cord, the levels of phosphorylated ERK1/2 and AKT were statistically increased in the histidine group, with respect to the saline‐treated group. Up‐regulation of pERK1/2 in SOD1‐G93A motor neurons was confirmed also by immunofluorescence (Figure 5B). We moreover observed activation of the AKT pro‐survival pathway also in sciatic nerves of histidine‐treated SOD1‐G93A mice compared with saline‐treated mice, as shown in Figure 5C. To establish if the effects in the spinal cord were accompanied by an improvement in the muscle phenotype, TA muscles were analysed. RT‐qPCR analysis demonstrated the expression of Hrh1, Hrh2, and Hrh3 (Hrh4 was undetectable) receptors and Hdc, Hnmt, and Dao1 histaminergic enzymes transcripts in SOD1‐G93A TA muscle. No statistically significant differences were found compared with WT tissues in the expression of receptors. A down‐regulation of Hnmt expression (Supporting Information, Figure S2A–F) was observed in SOD1‐G93A compared with WT muscle. Moreover, Hdc was the only transcript to be up‐regulated by histidine treatment, suggesting that histidine might cause an increase of histamine content also in the periphery of treated SOD1‐G93A mice (Supporting Information, Figure S2D).
Figure 5.
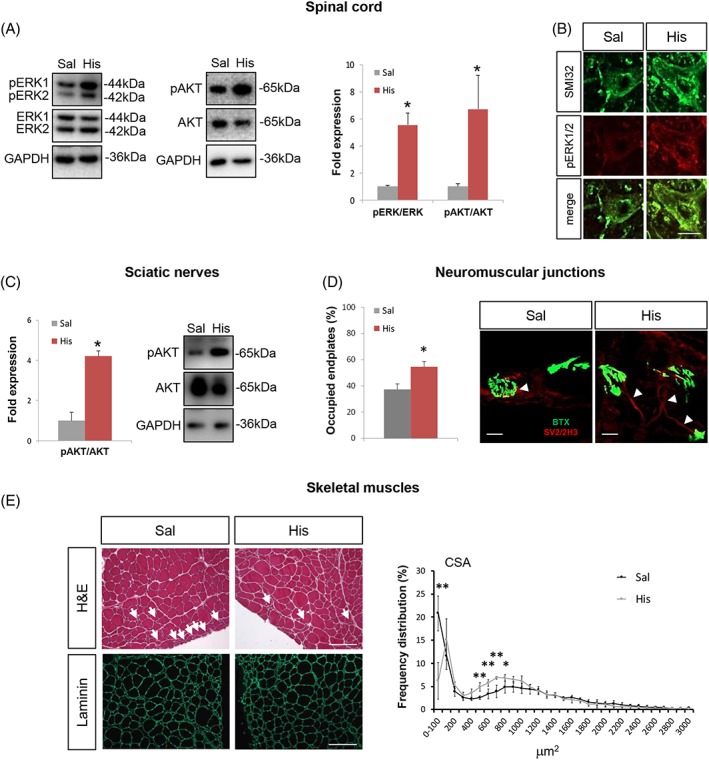
Histidine affects pro‐survival pathways in the spinal cord and sciatic nerves and decreases denervation atrophy in skeletal muscles of SOD1‐G93A mice. Representative western blots and quantification of pERK/ERK and pAKT/AKT (A), in saline‐treated (n = 4) and 100 mg/kg histidine‐treated (n = 5) SOD1‐G93A mice spinal cord. GAPDH was used as a loading control. (B) Representative confocal images of motor neurons labelled with SMI32 (green) and pERK (red) in saline‐treated and 100 mg/kg histidine‐treated SOD1‐G93A mice. Scale bar: 10 μm. (C) Representative western blots and quantification of pAKT/AKT in saline (n = 4) and 100 mg/kg histidine‐treated (n = 5) SOD1‐G93A mice sciatic nerves. GAPDH was used as a loading control. (D) Representative confocal images of BTX (green, postsynaptic domain) and SV2/2H3 (red, presynaptic terminals) in tibialis anterior (TA) muscle of saline‐treated and histidine‐treated SOD1‐G93A mice. For each mouse group, the percentage of occupied endplates was calculated on the basis of the overlay between SV2/2H3 and BTX. Arrowheads indicate innervated neuromuscular junctions. Scale bar: 20 μm. (E) Representative confocal images of TA muscle cryosections stained with haematoxylin and eosin (H&E) and labelled with anti‐laminin (green) antibody. Arrows indicate smaller atrophic fibres. Frequency distribution of fibre cross‐sectional area (CSA) is shown in the right panel. Scale bar: 100 μm. Data represent means ± SEM. Statistical significance was calculated by Student's t‐test or by Mann–Whitney test referred to saline‐SOD1‐G93A, * P < 0.05, ** P < 0.01. BTX, bungarotoxin; SEM, standard error of the mean; SOD1, superoxide dismutase 1; SV2, synaptic vescicle 2.
Moreover, neuromuscular junctions in the TA muscles showed less marked denervation in histidine‐treated mice with respect to saline‐treated mice (Figure 5D). Cross‐sectional area of muscle fibres was also measured, revealing a partial prevention of muscle atrophy in histidine‐treated mice. In particular, the number of smaller fibres (less than 100 μm2) was reduced while an increase in intermediate fibres (>500 and <1000 μm2) was observed (Figure 5E).
Histamine protects NSC‐34 superoxide dismutase 1‐G93A motor neurons through AKT/ERK1/2 and rescues mitochondrial metabolism
In order to dissect the motor neuron response to histaminergic signalling, we first evaluated the expression of histamine receptors and metabolic enzymes in mixed motor neuron primary cultures from the spinal cord of SOD1‐G93A mice and in differentiated NSC‐34 motor neuron‐like cells expressing the SOD1‐G93A mutation (NSC‐G93A). Primary (Figure 6A and 6C, left panels) and differentiated NSC‐G93A cells (Figure 6A and 6C, right panels) immunostained with the neuronal marker SMI32, both expressed histamine receptors H1R, H2R, and H3R and metabolic enzymes HDC, HNMT, and DAO1. Next, in order to evaluate differences in the regulation of histamine receptors and enzymes in NSC‐34 motor neuron cells expressing WT (NSC‐WT) or mutant SOD1, we analysed H1‐H4R and HDC, HNMT, and DAO1 by western blotting. Similarly to what previously observed in the lumbar spinal cord of SOD1‐G93A mice during disease progression compared with WT mice,4 we demonstrated decreased levels of H1R and H3R (Figure 6B) and increased levels of all histamine enzymes (Figure 6D) in NSC‐G93A with respect to NSC‐WT cells.
Figure 6.
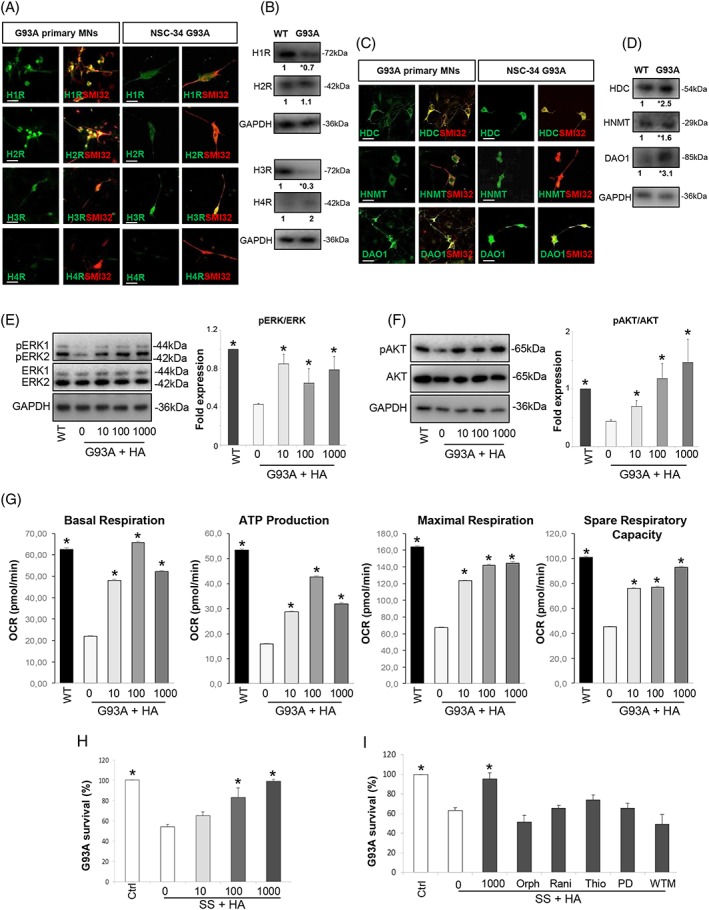
Histamine protects NSC‐34 G93A motor neurons via AKT/ERK1/2 pathways and rescues mitochondrial function. Representative confocal images of primary as well as differentiated NSC‐G93A motor neurons stained with H1‐H4R (green, A) or HDC, HNMT, DAO1 (green, C), and with SMI32 (red). Scale bar: 100 μm. Representative western blots and fold expression values of H1‐H4R (B) and HDC, HNMT, DAO1 (D) in NSC‐WT differentiated and ‐G93A differentiated cells. Values represent means from n = 3 independent experiments. Statistical significance was calculated by Student's t‐test referred to NSC‐ WT cells, * P < 0.05. Representative western blots and quantification of pERK/ERK (E) and pAKT/AKT (F) in NSC‐WT and ‐G93A differentiated cells under serum starvation and in the absence or presence of histamine (HA, 10–1000 μM for 24 h). GAPDH was used as a loading control. Data represent means ± SEM. Statistical significance was calculated by analysis of variance (ANOVA) referred to NSC‐G93A untreated cells, * P < 0.05, n = 4 replicates. (G) Measurement of the rate of oxygen consumption ratio (OCR) in differentiated NSC‐WT and ‐G93A cell lines cultured without serum and treated with different concentrations of histamine (10–1000 μM for 24 h). Individual parameters for basal respiration, ATP production, maximal respiration, and spare respiratory capacity are indicated. Each data point represents an OCR measurement. Data represent means ± SEM. Statistical significance was calculated by ANOVA referred to untreated NSC‐G93A cells, * P < 0.05, n = 4 each performed in sextuplicate. Cell death in NSC‐G93A differentiated cells was determined under serum starvation in the absence or presence of histamine (10–1000 μM for 24 h) (H) or in cells exposed to histamine 1000 μM for 24 h and orphenadrine (H1R antagonist, 10 μM), ranitidine (H2R antagonist, 10 μM), thioperamide (H3R antagonist, 5 μM), PD98059 (500 nM), or wortmannin (10 nM) (I). Data represent means ± SEM. Statistical significance was calculated by ANOVA referred to cells in serum starvation medium, * P < 0.05, n = 4 performed in triplicate. DAO1, diamine oxidase 1; HDC, histidine decarboxylase; HNMT, histamine N‐methyltransferase; SEM, standard error of the mean; SOD1, superoxide dismutase 1; WT, wild‐type.
We next investigated if histamine directly targets the AKT/ERK1/2 pro‐survival pathway after serum starvation as neuronal stressor inducing cell death. Differentiated NSC‐G93A compared with NSC‐WT showed a significant down‐regulation of ERK1/2 and AKT phosphorylations, which were instead significantly increased by histamine in a dose‐dependent manner in NSC‐G93A (Figure 6E and 6F).
We next examined the effects of histamine on bioenergetic capacity of NSC‐G93A cells under serum starvation, using the Seahorse XF96 Bioanalyser. Our results clearly showed mitochondrial damages and massive levels of energy deficit induced by expressing mutant SOD1‐G93A, as observed by a decline in basal respiration, ATP production, maximal respiration, and spare respiratory capacity (Figure 6G). The failure in maximal respiration indicated alterations in the electron transport chain, while the decrease of spare respiratory capacity suggested that mutant SOD1 inhibits the rapid adaptation of NSC‐G93A motor neurons to metabolic changes. Importantly, under serum deprivation conditions in NSC‐G93A cells, histamine with a maximal effect at 100 μΜ reverted the impaired mitochondrial metabolism (Figure 6G).
We finally evaluated the NSC‐G93A cell death induced by serum starvation for 24 h. In a dose‐dependent manner (10–1000 μM), histamine totally rescued about 35% death induced in NSC‐G93A cells by serum starvation (Figure 6H). The histamine effect was abolished by antagonists selective for H1R, H2R, and H3R and, most importantly, by PD98059 and wortmannin, inhibitors respectively of ERK1/2 and AKT (Figure 6I), thus ascribing this neuroprotective effect to AKT/ERK axis.
Discussion
In terms of disease‐modifying treatment options, several drugs have been tested in large multicentre trials for ALS,31 but albeit encouraging results, only riluzole and edaravone are FDA‐approved therapeutics available to patients.32 The present study establishes that the administration of histidine during the symptomatic phase enhancing histaminergic transmission in the SOD1‐G93A transgenic mouse model mitigates a broad range of disease phenotypes associated with ALS. This builds on our recent work showing that histamine drives an anti‐inflammatory phenotype in SOD1‐G93A microglia and that the histaminergic system is dysregulated in ALS mice and patients.4 Previous transcriptome analysis from two subgroups of sALS patients has indeed highlighted a differential expression of several histamine‐related genes in post‐mortem cortex samples. Here, we adopted an integrative multi‐omics approach combining gene expression profiles, CNVs, and SNPs of ALS patients for capturing histamine pathway associations in ALS and providing a comprehensive understanding of histamine‐related molecular mechanism that might contribute to the disease. Our genome‐wide analysis of multiple genomic aberrations occurring in sALS patients identified CNV‐affected histamine‐related genes that were differentially expressed in one or both sALS patient subgroups, among which for instance HRH3, CCKBR, and ADCYAP1 showed a positive correlation with transcriptomic changes. By integrating our transcriptomic and genomic data with the ALS‐linked pathogenic variants from ALSdb database, we also identified multiple coding variants in these genes, supporting those as candidate driver genes in ALS etiopathogenesis and thus establishing histamine as a gene modifier in ALS and a candidate therapeutic target.
The meta‐analysis approach that we have next adopted to compare gene expression profiles in spinal cord samples from sALS patients with those obtained in SOD1‐G93A mice demonstrated that many histamine‐related genes were deregulated in both patients and mice, confirming those as promising biomarkers/therapeutic targets and indicating that histaminergic molecular mechanisms are shared by both species. Despite the fact that transcriptome analysis of human tissues is generally considered as a consequence of disease progression (because most CNS specimen refer to post‐mortem tissues), the deregulation for instance of Hnmt, Hrh1, and Hrh3 genes occurred in mice long before symptom onset, suggesting these as potential early stage biomarkers and targets for treating ALS.
Enhancement of endogenous histaminergic transmission can be achieved in mice by either intracerebroventricular administration of histamine16 that has however only a limited translational value or by intraperitoneal administration of the histamine precursor histidine able to increase the histamine levels in the spinal cord,17 as we have confirmed here in SOD1‐G93A mice. We proved here that according to our pharmacological regimen, a sustained administration of histidine after symptoms insurgence ameliorated a wide range of molecular, neuropathological, and behavioural disease‐associated features. Most importantly, histidine delayed disease progression, increased life span, improved motor performance, while reduced microgliosis and denervation muscle atrophy, and attenuated motor neuron and axonal loss, moreover demyelination and neuroinflammation in the spinal cord. Remarkably, the high dose (250 mg/kg) of histidine was not as effective as the lower dose (100 mg/kg) with regard to overall survival, while it ameliorated rotarod performance and behavioural scores. Although histidine is well tolerated in human and rodents, its high intake is known to induce hyperlipidaemia, hypercholesterolemia, and copper deficiency in plasma and liver.33 Thus, we cannot exclude that high‐dosage and prolonged treatment with histidine up to the terminal stage of the disease might produce metabolic side effects that might, in turn, reduce the efficacy of the treatment itself. Moreover, it is reported that histamine at high doses down‐regulates in the brain its own synthesis by a feedback inhibition of HDC activity mediated by H3R.34 Therefore, the 250 mg/kg treatment might have likely produced lower levels of histamine in the spinal cord with respect to the 100 mg/kg treatment, and this might contribute to explain the reduced efficacy of the higher histidine dose. Further experiments will surely shed light on this issue.
Several studies now describe that not only motor neuron loss but also non‐neuronal cell dysfunctions act as major contributors to the progressive symptoms of ALS.1 The result that histidine can target several key cell phenotypes of ALS, that is, motor neurons, glial cells, and skeletal muscles as shown here, might thus explain why histidine exerted beneficial effects in SOD1‐G93A mice. The histidine treatment is particularly encouraging in symptomatic mice and makes the approach potentially more valuable for later translation, as most patients suffering from sporadic forms are diagnosed only at the symptomatic stage of the disease. In a different way, our recent studies exploring the actions of another histaminergic compound, clemastine, proved that also an anti‐histaminergic strategy reversing the inflammatory phenotype of microglia can exert positive outcomes in extending survival of ALS mice, but only when provided before the insurgence of symptoms.14 This may suggest that the therapeutic potential of central histamine in ALS mice shows a dual time window, as also demonstrated in a model of neuropathic pain.17 This is in line with the recognized dual role that histamine plays for instance in microglia, that is, detrimental under healthy conditions and beneficial within inflammatory environments.3 It might also be explained by histaminergic drugs acting on cell types that per se possess dual roles in ALS pathogenesis, for instance microglia and immune cells preventing disease progression at early stages while inducing pathogenic responses at later stages.35, 36, 37
Among the pathways involved in the protective effects of histidine in SOD1‐G93A mice, here, we demonstrated a decreased level of some key pro‐inflammatory gene modifiers of ALS, that is, gp91phox, NF‐κB,38, 39 and M1‐like microglia marker iNOS, parallel to an increase in M2‐like microglia markers ARG1 and CD163, which we previously demonstrated to be downstream pathways of histamine in SOD1‐G93A microglia.4 Interestingly, NF‐κB that was recognized to be a histamine‐related gene complex by our multi‐omics pathway analysis was also found dysregulated in our cohort of sALS patients, thus making it a likely target of histamine intervention. Because activation of NF‐κB during the early phases of ALS is sufficient to prolong survival in mice, whereas activation in glia in the late phases decreases survival,40 a therapy able to inhibit NF‐κB at symptomatic phase is predicted to be beneficial, as indeed we obtained here with histidine. The levels of phosphorylated AKT in motor neurons are decreased in ALS patients and SOD1‐G93A mice.41, 42 Known to exert a protective action on ALS neuronal cells,43 the up‐regulation of pAKT and pERK in the spinal cord of histidine‐treated SOD1‐G93A mice would thus suggest that the pro‐survival response sustained by histidine proceeds through activation of ERK and PI3K/AKT signalling. In agreement with this hypothesis, the neuroprotection of ALS motor neuron cells in vitro by histamine was dependent on AKT/ERK1/2 pathway activation, thus confirming previous results about the AKT/ERK1/2 pro‐survival axis in ALS motor neuron.44, 45, 46
The presence of mutant SOD1 in motor neurons causes mitochondrial dysfunction,24 and the mitochondrial apoptotic pathway is crucial in motor neuron death and ALS pathogenesis.47 Here, we demonstrated a role for histamine in rescuing mitochondrial metabolism and ATP production in motor neuron cells. The beneficial effects observed on mitochondrial metabolism and integrity that are critical in regulating cellular health reinforce the hypothesis that histamine treatment could be helpful in ALS.
While previous pharmacological approaches showing promise against ALS have not always reached sufficient efficacy to support translation into therapy, histamine drugs display a clear value in several animal models of neurological diseases. Furthermore, H3R antagonists increasing histamine neuronal release are in clinical trials for Alzheimer's disease and multiple sclerosis and are already approved in Europe for narcolepsy.2 Our results now add ALS disease to the growing promise of histaminergic strategies. By proposing the histamine modulation as a candidate mechanism to be further challenged in ALS, we trust not only that histamine will answer to the many questions still unsolved about ALS but also that a viable histamine‐related therapy might be proven to work for patients. Our further trials will validate H3 antagonists against ALS progression.
Author contributions
S.Ap. and C.V. presented the study concept and design. S.Ap., S.Am., P.F., G.M., A.G.S., E.C.L., I.S., D.P., A.F., L.M., S.P.‐A., S.C., and C.V. presented the data acquisition and analysis. S.Ap. and C.V. wrote the manuscript. All authors edited and approved the final version of the manuscript.
Conflict of interest
S.Ap., S.Am., P.F., G.M., A.G.S., E.C.L., I.S., D.P., A.F., L.M., S.P.‐A., S.C., and C.V. declare that they have no conflict of interest.
Supporting information
Figure S1. Dysregulation of spinal cord and sciatic nerve parameters in SOD1‐G93A mice at 150 days of age. Representative western blots and quantification of: ChAT, SMI32 and MBP (A); ARG1, CD163 and iNOS (B); pNF‐κB/NF‐κB and gp91phox (C); pERK/ERK and pAKT/AKT (D) in WT (n = 3) and SOD1‐G93A (n = 4) mice lumbar spinal cords, and of pAKT/AKT (E) in WT (n = 3) and SOD1‐G93A (n = 4) mice sciatic nerves. GAPDH was used as a loading control. Data represent means ± S.E.M. Statistical significance was calculated by student's t‐test or Mann–Whitney test referred to SOD1‐G93A group, *P < 0.05.
Figure S2. Expression of histamine receptors and enzymes in Tibialis Anterior muscle of SOD1‐G93A mice at 150 days of age. Real‐time PCR for Hrh1 (A), Hrh2 (B), Hrh3 (C), Hdc (D), Hnmt (E) and Dao1 (F) in the tibialis anterior muscle of WT (n = 3), saline‐treated (n = 4), histidine‐treaded SOD1‐G93A mice (n = 5). Data are normalized to β‐actin and expressed as means ± S.E.M. Statistical significance was calculated by ANOVA referred to WT group, *P < 0.05, **P < 0.01 or to saline‐treated SOD1‐G93A mice °°P < 0.01.
Table S1. General information of patients and individual controls.
Acknowledgements
The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.48
This work was supported by Italian Ministry for Education, University and Research through grant Flagship Project NanoMAX (grant number B81J13000310005) (C.V.), grant number CTN01_00177_817708 (S.C.) and by the international PhD programmes in Neuroscience and Computer Science of the University of Catania, moreover by the Italian Ministry of Health through Ricerca Corrente and grant number GR‐2013‐02356592 (L.M.) and by AriSLA through HyperALS project (A.F.). We thank Dr R. Coccurello for critical reading of the manuscript. We thank the Netherlands ALS Foundation and particularly Professor E. Aronica (Amsterdam UMC, University of Amsterdam, Department of (Neuro)Pathology, Amsterdam Neuroscience, Amsterdam, the Netherland), Professor D. Troost and Professor M. de Visser who contributed to the establishment of the Dutch ALS Tissue Bank. This work is dedicated to the memory of our wonderful colleague Maria Teresa Carrì, for having shared inspiring scientific discussions and friendship.
Apolloni S., Amadio S., Fabbrizio P., Morello G., Spampinato A. G., Latagliata E. C., Salvatori I., Proietti D., Ferri A., Madaro L., Puglisi‐Allegra S., Cavallaro S., and Volonté C. (2019) Histaminergic transmission slows progression of amyotrophic lateral sclerosis, Journal of Cachexia, Sarcopenia and Muscle, 10, 872–893, doi: 10.1002/jcsm.12422.
Contributor Information
Savina Apolloni, Email: s.apolloni@hsantalucia.it.
Cinzia Volonté, Email: cinzia.volonte@cnr.it.
References
- 1. Poppe L, Rue L, Robberecht W, Van Den Bosch L. Translating biological findings into new treatment strategies for amyotrophic lateral sclerosis (ALS). Exp Neurol 2014;262 Pt B:138–151. [DOI] [PubMed] [Google Scholar]
- 2. Hu W, Chen Z. The roles of histamine and its receptor ligands in central nervous system disorders: an update. Pharmacol Ther 2017;175:116–132. [DOI] [PubMed] [Google Scholar]
- 3. Barata‐Antunes S, Cristovao AC, Pires J, Rocha SM, Bernardino L. Dual role of histamine on microglia‐induced neurodegeneration. Biochim Biophys Acta 2017;1863:764–769. [DOI] [PubMed] [Google Scholar]
- 4. Apolloni S, Fabbrizio P, Amadio S, Napoli G, Verdile V, Morello G, et al. Histamine regulates the inflammatory profile of SOD1‐G93A microglia and the histaminergic system is dysregulated in amyotrophic lateral sclerosis. Front Immunol 2017;8:1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Frick L, Rapanelli M, Abbasi E, Ohtsu H, Pittenger C. Histamine regulation of microglia: gene‐environment interaction in the regulation of central nervous system inflammation. Brain Behav Immun 2016;57:326–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schwartzbach CJ, Grove RA, Brown R, Tompson D. Then Bergh F, Arnold DL. Lesion remyelinating activity of GSK239512 versus placebo in patients with relapsing‐remitting multiple sclerosis: a randomised, single‐blind, phase II study. J Neurol 2017;264:304–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Niijima‐Yaoita F, Tsuchiya M, Ohtsu H, Yanai K, Sugawara S, Endo Y, et al. Roles of histamine in exercise‐induced fatigue: favouring endurance and protecting against exhaustion. Biol Pharm Bull 2012;35:91–97. [DOI] [PubMed] [Google Scholar]
- 8. Gurel V, Lins J, Lambert K, Lazauski J, Spaulding J, McMichael J. Serotonin and histamine therapy increases tetanic forces of myoblasts, reduces muscle injury, and improves grip strength performance of Dmd (mdx) mice. Dose Response 2015;13:1559325815616351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aronica E, Baas F, Iyer A, ten Asbroek AL, Morello G, Cavallaro S. Molecular classification of amyotrophic lateral sclerosis by unsupervised clustering of gene expression in motor cortex. Neurobiol Dis 2015;74:359–376. [DOI] [PubMed] [Google Scholar]
- 10. Morello G, Cavallaro S. Transcriptional analysis reveals distinct subtypes in amyotrophic lateral sclerosis: implications for personalized therapy. Future Med Chem 2015;7:1335–1359. [DOI] [PubMed] [Google Scholar]
- 11. Morello G, Spampinato AG, Cavallaro S. Molecular taxonomy of sporadic amyotrophic lateral sclerosis using disease‐associated genes. Front Neurol 2017;8:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dagata V, Cavallaro S. Parkin transcript variants in rat and human brain. Neurochem Res 2004;29:1715–1724. [DOI] [PubMed] [Google Scholar]
- 13. Cavallaro S, Dagata V, Alkon DL. Programs of gene expression during the laying down of memory formation as revealed by DNA microarrays. Neurochem Res 2002;27:1201–1207. [DOI] [PubMed] [Google Scholar]
- 14. Apolloni S, Fabbrizio P, Amadio S, Volonté C. Actions of the antihistaminergic clemastine on presymptomatic SOD1‐G93A mice ameliorate ALS disease progression. J Neuroinflammation 2016;13:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Apolloni S, Amadio S, Montilli C, Volonté C, D'Ambrosi N. Ablation of P2X7 receptor exacerbates gliosis and motoneuron death in the SOD1‐G93A mouse model of amyotrophic lateral sclerosis. Hum Mol Genet 2013;22:4102–4116. [DOI] [PubMed] [Google Scholar]
- 16. Verma L, Jain NS. Central histaminergic transmission modulates the ethanol induced anxiolysis in mice. Behav Brain Res 2016;313:38–52. [DOI] [PubMed] [Google Scholar]
- 17. Yu J, Tang YY, Wang RR, Lou GD, Hu TT, Hou WW, et al. A critical time window for the analgesic effect of central histamine in the partial sciatic ligation model of neuropathic pain. J Neuroinflammation 2016;13:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liao RJ, Jiang L, Wang RR, Zhao HW, Chen Y, Li Y, et al. Histidine provides long‐term neuroprotection after cerebral ischemia through promoting astrocyte migration. Sci Rep 2015;5:15356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pasetto L, Pozzi S, Castelnovo M, Basso M, Estevez AG, Fumagalli S, et al. Targeting extracellular cyclophilin A reduces neuroinflammation and extends survival in a mouse model of amyotrophic lateral sclerosis. J Neurosci 2017;37:1413–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ludolph AC, Bendotti C, Blaugrund E, Chio A, Greensmith L, Loeffler JP, et al. Guidelines for preclinical animal research in ALS/MND: a consensus meeting. Amyotroph Lateral Scler 2010;11:38–45. [DOI] [PubMed] [Google Scholar]
- 21. Spalloni A, Albo F, Ferrari F, Mercuri N, Bernardi G, Zona C, et al. Cu/Zn‐superoxide dismutase (GLY93‐‐>ALA) mutation alters AMPA receptor subunit expression and function and potentiates kainate‐mediated toxicity in motor neurons in culture. Neurobiol Dis 2004;15:340–350. [DOI] [PubMed] [Google Scholar]
- 22. Carunchio I, Mollinari C, Pieri M, Merlo D, Zona C. GAB(A) receptors present higher affinity and modified subunit composition in spinal motor neurons from a genetic model of amyotrophic lateral sclerosis. Eur J Neurosci 2008;28:1275–1285. [DOI] [PubMed] [Google Scholar]
- 23. Cashman NR, Durham HD, Blusztajn JK, Oda K, Tabira T, Shaw IT, et al. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev Dyn 1992;194:209–221. [DOI] [PubMed] [Google Scholar]
- 24. Ferri A, Cozzolino M, Crosio C, Nencini M, Casciati A, Gralla EB, et al. Familial ALS‐superoxide dismutases associate with mitochondria and shift their redox potentials. Proc Natl Acad Sci U S A 2006;103:13860–13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Volonté C, Ciotti MT, Battistini L. Development of a method for measuring cell number: application to CNS primary neuronal cultures. Cytometry 1994;17:274–276. [DOI] [PubMed] [Google Scholar]
- 26. Madaro L, Passafaro M, Sala D, Etxaniz U, Lugarini F, Proietti D, et al. Denervation‐activated STAT3‐IL‐6 signalling in fibro‐adipogenic progenitors promotes myofibres atrophy and fibrosis. Nat Cell Biol 2018;20:917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nardo G, Trolese MC, Verderio M, Mariani A, de Paola M, Riva N, et al. Counteracting roles of MHCI and CD8(+) T cells in the peripheral and central nervous system of ALS SOD1(G93A) mice. Mol Neurodegener 2018;13:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Salvatori I, Ferri A, Scaricamazza S, Giovannelli I, Serrano A, Rossi S, et al. Differential toxicity of TAR DNA‐binding protein 43 isoforms depends on their submitochondrial localization in neuronal cells In J Neurochem, Vol. 146; 2018. p 585–597. [DOI] [PubMed] [Google Scholar]
- 29. Komori H, Nitta Y, Ueno H, Higuchi Y. Structural study reveals that Ser‐354 determines substrate specificity on human histidine decarboxylase. J Biol Chem 2012;287:29175–29183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Philips T, Bento‐Abreu A, Nonneman A, Haeck W, Staats K, Geelen V, et al. Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 2013;136:471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dorst J, Ludolph AC, Huebers A. Disease‐modifying and symptomatic treatment of amyotrophic lateral sclerosis. Ther Adv Neurol Disord 2018;11:1756285617734734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dash RP, Babu RJ, Srinivas NR. Two decades‐long journey from riluzole to edaravone: revisiting the clinical pharmacokinetics of the only two amyotrophic lateral sclerosis therapeutics. Clin Pharmacokinet 2018;57:1385–1398. [DOI] [PubMed] [Google Scholar]
- 33. Garlick PJ. The nature of human hazards associated with excessive intake of amino acids. J Nutr 2004;134:1633S–9S; discussion 64S‐66S, 67S‐72S–1639S. [DOI] [PubMed] [Google Scholar]
- 34. Hough L. Dynamics of Histamine in the Brain. 6th edition ed. Basic Neurochemistry: Molecular, Cellular and Medical Aspects. Philadelphia: Lippincott‐Raven; 1999. [Google Scholar]
- 35. Geloso MC, Corvino V, Marchese E, Serrano A, Michetti F, D'Ambrosi N. The dual role of microglia in ALS: mechanisms and therapeutic approaches. Front Aging Neurosci 2017;9:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Beers DR, Zhao W, Appel SH. The role of regulatory T lymphocytes in amyotrophic lateral sclerosis. JAMA Neurol 2018;75:656–658. [DOI] [PubMed] [Google Scholar]
- 37. Volonté C, Parisi C, Apolloni S. New kid on the block: does histamine get along with inflammation in amyotrophic lateral sclerosis? CNS Neurol Disord Drug Targets 2015;14:677–686. [DOI] [PubMed] [Google Scholar]
- 38. Seredenina T, Nayernia Z, Sorce S, Maghzal GJ, Filippova A, Ling SC, et al. Evaluation of NADPH oxidases as drug targets in a mouse model of familial amyotrophic lateral sclerosis. Free Radic Biol Med 2016;97:95–108. [DOI] [PubMed] [Google Scholar]
- 39. Frakes AE, Ferraiuolo L, Haidet‐Phillips AM, Schmelzer L, Braun L, Miranda CJ, et al. Microglia induce motor neuron death via the classical NF‐κB pathway in amyotrophic lateral sclerosis. Neuron 2014;81:1009–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ouali Alami N, Schurr C, Olde Heuvel F, Tang L, Li Q, Tasdogan A, et al. NF‐κB activation in astrocytes drives a stage‐specific beneficial neuroimmunological response in ALS. EMBO J 2018;37:e98697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vallarola A, Sironi F, Tortarolo M, Gatto N, De Gioia R, Pasetto L, et al. RNS60 exerts therapeutic effects in the SOD1 ALS mouse model through protective glia and peripheral nerve rescue. J Neuroinflammation 2018;15:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dewil M, Lambrechts D, Sciot R, Shaw PJ, Ince PG, Robberecht W, et al. Vascular endothelial growth factor counteracts the loss of phospho‐Akt preceding motor neurone degeneration in amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol 2007;33:499–509. [DOI] [PubMed] [Google Scholar]
- 43. Lee SH, Choi NY, Yu HJ, Park J, Choi H, Lee KY, et al. Atorvastatin protects NSC‐34 motor neurons against oxidative stress by activating PI3K, ERK and free radical scavenging. Mol Neurobiol 2016;53:695–705. [DOI] [PubMed] [Google Scholar]
- 44. Petrozziello T, Secondo A, Tedeschi V, Esposito A, Sisalli M, Scorziello A, et al. ApoSOD1 lacking dismutase activity neuroprotects motor neurons exposed to beta‐methylamino‐L‐alanine through the Ca(2+)/Akt/ERK1/2 prosurvival pathway. Cell Death Differ 2017;24:511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ono Y, Tanaka H, Takata M, Nagahara Y, Noda Y, Tsuruma K, et al. SA4503, a sigma‐1 receptor agonist, suppresses motor neuron damage in in vitro and in vivo amyotrophic lateral sclerosis models. Neurosci Lett 2014;559:174–178. [DOI] [PubMed] [Google Scholar]
- 46. Wang T, Cheng J, Wang S, Wang X, Jiang H, Yang Y, et al. α‐Lipoic acid attenuates oxidative stress and neurotoxicity via the ERK/Akt‐dependent pathway in the mutant hSOD1 related Drosophila model and the NSC34 cell line of amyotrophic lateral sclerosis. Brain Res Bull 2018;140:299–310. [DOI] [PubMed] [Google Scholar]
- 47. Moller A, Bauer CS, Cohen RN, Webster CP, De Vos KJ. Amyotrophic lateral sclerosis‐associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels. Hum Mol Genet 2017;26:4668–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Dysregulation of spinal cord and sciatic nerve parameters in SOD1‐G93A mice at 150 days of age. Representative western blots and quantification of: ChAT, SMI32 and MBP (A); ARG1, CD163 and iNOS (B); pNF‐κB/NF‐κB and gp91phox (C); pERK/ERK and pAKT/AKT (D) in WT (n = 3) and SOD1‐G93A (n = 4) mice lumbar spinal cords, and of pAKT/AKT (E) in WT (n = 3) and SOD1‐G93A (n = 4) mice sciatic nerves. GAPDH was used as a loading control. Data represent means ± S.E.M. Statistical significance was calculated by student's t‐test or Mann–Whitney test referred to SOD1‐G93A group, *P < 0.05.
Figure S2. Expression of histamine receptors and enzymes in Tibialis Anterior muscle of SOD1‐G93A mice at 150 days of age. Real‐time PCR for Hrh1 (A), Hrh2 (B), Hrh3 (C), Hdc (D), Hnmt (E) and Dao1 (F) in the tibialis anterior muscle of WT (n = 3), saline‐treated (n = 4), histidine‐treaded SOD1‐G93A mice (n = 5). Data are normalized to β‐actin and expressed as means ± S.E.M. Statistical significance was calculated by ANOVA referred to WT group, *P < 0.05, **P < 0.01 or to saline‐treated SOD1‐G93A mice °°P < 0.01.
Table S1. General information of patients and individual controls.