

Abstract
DNA methylation has frequently been implicated in sex determination and differentiation in teleost species. In order to detect the DNA methylation patterns established during sexual differentiation in tiger pufferfish T. rubripes, we performed comprehensive whole genome methylation sequencing and analyses of the gonads of male, female, and pseudo male. We obtained a total of 33.12, 32.44, and 31.60 Gb clean data for male, female, and pseudo male, with a sequencing depth of 66.44×, 60.47× and 54.86×, respectively. The methylation level of cytosine (C) residues in the genomic DNA from gonads was 11.016%, 10.428%, and 11.083% in male, female, and pseudo male, respectively. More than 65% of C methylation was at CpG sites, and less than 1% was at CHG and CHH sites. In each regulatory element, there were low methylation levels on both sides of the transcription start site, and higher methylation levels in exons, introns, and downstream of genes. The highest mCpG was on chromosome 8 and the lowest mCpG was on chromosome 5. Comparisons of whole-genome DNA methylation between pairs of samples revealed that there were 3,173 differentially methylated regions (DMRs) between female and male, and 3,037 DMRs between male and pseudo male, corresponding to 0.232% and 0.223% of the length of the genome, respectively. There were only 1,635 DMRs between female and pseudo male, representing 0.127% of the length of the genome. A number of differentially methylated genes (DMGs) related to sex determination and differentiation were selected, such as amhr2 and pfcyp19a. After Bisulfite Sequencing PCR (BSP) verification, amhr2 was exhibited low methylation level in normal males and pseudo male, and high methylation level in normal females but pfcyp19a showed low methylation level in normal females and high methylation level in normal males and pseudo males. These results provide information about the molecular epigenetic mechanisms of DNA methylation during low-temperature induced masculinization of tiger pufferfish, and increase our understanding of the mechanisms of sex determination and differentiation in this important aquaculture fish species.
Introduction
Sex determination and differentiation is a fundamental developmental process that establishes the sexual genotype at the early stage of an organism and then destines the undifferentiated gonad into ovary or a testis [1, 2]. Teleost fishes with 25,000 species are the most diverse group of vertebrates and consequently have the most complicated sex determination mechanisms ranging from genetic sex determination (GSD) to environmental sex determination (ESD) [2]. In some fish species, sex is determined by genetic factor but also largely dependent on the environmental factors (eg. temperature, salinity, density, pH and social interactions) [2, 3]. This has led to desire to produce mono sex population of interest for aquaculture by artificial manipulation with examples of all male population of tilapia (Oreochromis mossambicus), yellow catfish (Pelteobagrus fulvidraco) and all female population of Japanese flounder(Paralichthys olivaceus) and chinook salmon (Oncorhynchus tshawytscha) [4–7].
The tiger pufferfish T. rubripes, commonly known as ‘fugu’, is one of the most popular aquacultural fish in China, Korea and Japan [8]. Traditionally, the testes of tiger pufferfish were considered as a delicacy and thus the male fish have a higher economic value than females [9]. Tiger pufferfish has an XY sex determination system with environmental influence, especially the temperature during the sex determination sensitive stages [10]. Therefore, all male production by treatment of the temperature was extensively performed in tiger pufferfish. Previous studies have shown that a low-temperature treatment in the 2–3 weeks after hatching significantly increased the proportion of male tiger pufferfish [11]. Recently, Zhou et al. [12] obtained a population with 75% male tiger pufferfish by incubating fertilized eggs for 45–75 days at 20 °C, and then for 35–65 days at 15–17 °C. Meanwhile, by extensive genetic analysis, a sex determination gene (amhr2) residing on the chromosome 19 was identified and a single nucleotide polymorphism (SNP) in amhr2 associated with sex determination that can be used for molecular sexing in sex reversal fish by low temperature [13]. Although the sex reversal has been successful in practical, little is known about the molecular mechanism of sex determination and differentiation under low temperature in tiger pufferfish.
Epigenetics was considered as a bridge between genetics and environment, which refers to changes in gene expression that do not involve changes in the DNA sequence [14]. DNA methylation is a major epigenetic modification and an important regulator of gene function [15]. In general, gene expression levels are negatively correlated with DNA methylation [15]. Various environmental factors, especially temperature, affect DNA methylation patterns in fish species [16–20]. Recent studies confirmed that DNA methylation play an important role in fish sex determination and differentiation. In Chinese tongue sole (Cynoglossus semilaevis), the high temperature (28 °C) can induce sex reversal of juvenile genotypic females into phenotypic pseudo-males by epigenetically regulation and more interestingly such epigenetic modification can be passed down to the ZW offspring of pseudo males in normal temperature [17]. In European sea bass (Dicentrarchus labrax), the methylation levels of the cyp19a promoter in female increased under high temperature, leading to inhibition of gene expression [18]. An increase of even 2 °C can lead to global changes in DNA methylation in European sea bass larvae [19]. High temperature has also been shown to increase gene methylation levels in Nile tilapia (Oreochromis niloticus) [20].
To date, the role of DNA methylation in the low-temperature-induced masculinization of tiger pufferfish is unclear. We inferred that low temperature treatment in tiger pufferfish may change the DNA methylation pattern and further affect gene expression levels, and finally cause significant phenotypic changes, the sex reversal. In this study, we obtained a population by low-temperature treatment containing pseudo male fish as well as female and male fish. The sex of each fish was identified by analyzing the specific SNP locus in amhr2 and by physiological analyses. We then conducted whole genome bisulfite sequencing (WGBS) of DNA from gonads of male, female, and pseudo male tiger pufferfish, and constructed a whole-genome methylation map at single-base resolution. These analyses revealed details of the epigenetic mechanism of low-temperature induced masculinization and thus provided the theoretical basis for breeding all male population of tiger pufferfish.
Materials and methods
Ethics statement
This study was performed according to the Guide for the Care and Use of Laboratory Animals in Dalian Ocean University, Dalian, China. All animal experiments were approved by the Animal Study Ethical Committee of Dalian Ocean University, and comply with Chinese laws, regulations, and ethics.
Fish sample collection
The fish samples used in this study was produced by the low-temperature treatment induced masculinization of fugu in Zhou et al. [21]. Briefly, an orthogonal test L9 (34) design was chosen with three factors at three levels: treatment starting times (days post-hatch, dph) of 20, 50, 80 dph; treatment temperatures of 13 °C, 15 °C and 17 °C; treatment duration of 30, 45, 60 days. The control group was reared at 21±1 °C. The experiments were repeated twice. Conduct vivisection until development to 230 days of age, gather the fin structure in 95% alcohol and the gonads in cryogenic vials, preserved at -80 °C. These experiments were conducted at Daheishi Aquaculture Facility of TianZheng Industrial Co., Ltd.
Screening of T. rubripes pseudo males
Pseudo males were screened in the following two groups: 1. 20 days after hatching, treating the offspring for 30 days at 13 °C; 2. 80 days after hatching, treating the offspring for 60 days at 13 °C.
To identify sex, we used the specific SNP gender identification markers developed by Kamiya [13]. Caudal fin tissue (30 mg) was ground in 95% alcohol, and high-quality DNA was extracted using the phenol/chloroform extraction method. The DNA concentration was determined by spectrophotometry, and then diluted to 50 ng/ml. The PCR primers used were SD3exon8F (ACGATGCACACAAACCACCT) and SD3exon10R (TCCCAGTGTTGCGGTATGTA). Each 50 μl PCR reaction mixture contained 5.0 μl Taq buffer, 1.0 μl dNTPs (10 mmol/L), 1.0 μl upstream and downstream primers, 5 μl MgCl2 (25 mmol/L), 0.5 μl Taq DNA polymerase (5 U/μl), 1.0 μl template DNA, and 35.5 μl double-distilled water. The PCR cycling conditions were as follows: 95 °C pre-degeneration for 3 min; then 35 cycles of 94 °C for 30 s, enaturation at 55–60 °C for 35 s, and extension at 72°C for 40–50 s; followed by final extension at 72 °C for 5–8 min. The reaction products were kept at 4 °C until further analysis.
The gender was also assessed by a tissue sectioning technique. Gonads were collected from fugu in the low-temperature treatment group and control group (30 per group) at 230 days of age. The gonads were fixed in Bouin’s solution, and then subjected to gradient alcohol dehydration before clearing and embedding in xylene paraffin. Sections (6–8 μm thick) were cut using a microtome, stained with hematoxylin-eosin, sealed in neutral rubber, and then observed and photographed under an Olympus microscope (Olympus, Tokyo, Japan).
Construction and sequencing of genome-wide bisulfite methylC-seq libraries
We extracted DNA from gonads (frozen at -80 °C) of five individuals of male (average body length: 140.35±7.68 mm, average body weight: 84.94±6.30 g), female (141.04±4.44 mm, 85.88±6.86 g), and pseudo male (121.18±6.69 mm, 50.73±10.68 g) using the phenol/chloroform method. Equal amounts of DNA samples from male, female, and pseudo male were mixed and then broken into fragments (approximately 250 bp average size) using a Bioruptor Ultrasonic Cell Disruptor (Diagenode, Sparta, NJ, USA). The 3′ ends were repaired by adding adenine with methylation connectors. The bisulfite treatment was conducted using an EZ-DNA Methylation-Gold kit (Zymo Research, Orange, CA, USA). The fragments were separated by 2% agarose gel electrophoresis, recovered for PCR amplification using a QIAquick Gel Extraction kit (Qiagen, Hilden, Germany), and used to build the methylC-seq enrichment library. When we built the methylation library, an internal reference phage DNA was added, the conversion rate could be obtained by counting the phase DNA. We used the lllumina HiSeq2000 platform for high-throughput sequencing.
Data analyses
We used the Solexa Pipeline -1.0 for basic data processing (e.g. filtering). After filtering, clean data was compared with the reference genome (Assembly accession: No. GCA_000180615.2) by BSMAP [22] to calculate the rate of bisulfite conversion. When read coverage ≥ 4, supported methylation read numbers > unsupported methylation read numbers, we could determine the methylation site. We then calculated the mthelation level for an individual CpG using the formula:
(where Nm is the read number of methylC, and Nnm is the read number of methylC at the locus) [23]. For the methylation level of a specific region, it was calculated by the sum of methylation levels of individual CpGs divided by the total number of covered CpGs in the region.
We next searched for the 500 bp sliding windows containing at least five CG (CHG or CHH) at the same position of the genome in two samples, and compared the difference in CG methylation levels in the window between the two samples. Regions showing a significant difference in methylation levels (2-times difference, Fisher’s test P value ≤ 0.05) between two samples were defined as DMRs. If a continuous region of two adjacent DMRs showed differences in methylation levels between two samples, the two DMRs were either merged as a continuous DMR or treated as two separate DMRs. We further performed the Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses of the genes in the DMRs.
Bisulfite sequencing method
The genome-wide methylation sequencing analysis of fugu revealed several genes related to gonad development. These genes were selected for further analysis by BSP. Primers were designed using Primer Premier 5.0. For the amhr2 the primers were as follows: Forward, TTTGTTATTTATGGTGTT TTTAGATG; Reverse, CAAAAAAATATCAAAACAAAAATAAAC. For the pfcyp19a the primers were as followers: Forward, TAGGAGGGATGATAGAAATATGAAG; Reverse, CAAACC AACTAAATAATAAACAATACAC. High-quality DNA was extracted from the gonads by the phenol/chloroform method. To reduce variation in the male, female, and pseudo male samples, each DNA sample was a mixture of DNA extracted from gonads of three T. rubripes individuals. The EZ-DNA Methylation-Gold kit (Zymo Research) was used for genomic DNA sulfite modification. The PCR reaction conditions were as follows: 4 min at 98 °C; 20 cycles of 45s at 94 °C, 45s at 66 °C, 1min at 72 °C, 20 cycles of 45s at 94 °C, 45s at 56 °C, 1 min at 72 °C and a final extention of 8 min at 72 °C. PCR products were detected by gel electrophoresis and recycled by SanPrep Column DNA Gel Extraction Kit (Sangon). The recycled DNA was cloned into the pUCm-T Vector and transformed in E. coil compent cells (Sangon). Then 20 clones per loci for each sample were sequenced. The sequencing results were analyzed using DNAStar software.
qPCR analysis
qPCR was used to examine the different expression quantity of amhr2 and pfcyp19a gene in female, male and pseudo male gonad. Total RNA was reverse transcribed to first-strand cDNA using RevertAid Premium Reverse Transcriptas kits (#EP0733; Thermo Scientific, Waltham, MA, USA). The reactions were performed in an ABI Step One Plus machine (Thermo Scientific). Primer Premier 5.0 was used to desgined the primers for these genes. For the amhr2 the primers were as follows: Forward, TGTGCGATTTCGGATGTTC; Reverse, CGTGCATCAGATAC CATTTGTT. For the pfcyp19a the primers were as follows: Forward, TACC GAAGGGCACAAACATC; Reverse, GAACCGAACGGCTGAAAGT. The cDNA samples diluted 10 times served as test templates. The reaction mix included SybrGreen qPCR Master Mix 10 μl, 0.4 μl fragment F (10 μM), 0.4 μl fragment R (10 μM), 7.2 μl ddH2O and 2 μl template cDNA. The following thermal cycling parameters were used: initiation at 95 °C for 3 min followed by 45 cycles of 95 °C for 7 s for denaturation, 57 °C for 10 s for annealing, and 72 °C for 15 s for extension. All experiments were conducted with three biological replicates for each sample. The relative expression levels were normalized to the endogenous control geneβ-actin, and expression ratios were calculated by using the 2-ΔΔCt method. Statistical analysis among the results of female, male and pseudo male gonad was performed using Duncan’s multiple range alignment and significance tests with SPSS version 17.0 (Statistical Product and Service Solutions Inc., Chicago, IL, USA).
Results
Sex identification
The genotypic sex of tiger pufferfish was identified by SNP analysis of amhr2 genes using PCR method. The SNP at the amhr2 loci should be C/G in male (Fig 1a), and C/C in female and pseudo male (Fig 1b). The phenotypic sex of tiger pufferfish was identified using a histological analysis. The histological sections of gonad revealed that the testes structure was similar in male and pseudo male tiger pufferfish (Fig 1c–1f). In total, 37 male, 23 female and 5 pseudo male tiger pufferfish were identified.
Fig 1. Genetic and phenotypic sex identification in T. rubripes.

a: normal male; b: normal female and pseudo male; c: normal female ovary; d pseudo male testis; e: pseudo male testis amplification; f: normal male testis; OC: oocytes; OL: ovarian lamella; PSC: primary spermatocytes; SSC: secondary spermatocytes; bars = 100 μm.
Whole genome methylation sequencing data
The DNA extracted from gonads of male, female and pseudo male was used for genome-wide methylation sequencing by illumina HiSeq2000 platform. We obtained 34.64, 33.24, and 33.83 Gb raw data for male, female, and pseudo male, respectively. After removing the low-quality data and contaminants, a total of 33.12, 32.44, and 31.60 Gb clean data for male, female, and pseudo male, with a sequencing depth of 66.44×, 60.47× and 54.86×, respectively, were generated for further analysis (Table 1). The genome coverage was higher than 95% for all samples and the mapping rate for each sample is 71.35%, 66.78% and 63.11% for male, female and pseudo male respectively. In the three different samples, the proportion of C in different forms covered by at least two reads was greater than 97% (S1 Table). The data has uploaded to NCBI Sequence Read Archive (SRA accession: PRJNA524991).
Table 1. Methylation sequencing data for male, female and pseudo male tiger pufferfish.
| Sample | Raw data(Gb) | Clean data (Gb) | Average depth (×) | Genome Coverage (%) | Mapping rate (%) | Bisulfite conversion rate (%) |
|---|---|---|---|---|---|---|
| Male | 34.64 | 33.12 | 66.44 | 95.04 | 71.35 | 99.60 |
| Female | 33.24 | 32.44 | 60.47 | 95.06 | 66.78 | 99.61 |
| Pseudo male | 33.83 | 31.60 | 54.86 | 95.03 | 63.11 | 99.58 |
Methylation features in tiger pufferfish genome
We identified an average of 13.2 million methylated cytosines (mCs) accounting for 11% of Cs in each sample (S2 Table). The classification of mCs showed a similar proportion in male, female and pseudo male genome. More than 98% of mCs were mCG type while about 0.3% and 0.9% were mCHG and mCHH types in the CpG context. Thus, it can be seen that genomic methylation of each gender occurred mainly at CG loci (Fig 2, S2 Table and S1 Fig). The mCpG (ratio of mC to CpG dinucleotide sites) was about 65% (66.009%, 65.027% and 65.945% for male, female and pseudo male respectively) (S2 Fig). We thus focus solely on CpG sites for subsequent analyses. We found the CG methylation level was between 60% and 70% in exons, introns, mRNA, and non-coding RNA, which is much higher than in tRNA (an average of 20.2%) (S3 Table). We further analyzed the methylation status of CpGs in various genomic elements. The lowest methylation level was detected around TSS but increased within the exons and introns with the peak at internal exon, which was exhibited the similar methylation level in male, female and pseudo male. (Fig 2 and S1 Fig). Furthermore, we investigated the DNA methylation on a chromosomal level. The mCpG is around 65% for each chromosome and the highest mCpG was on chromosome 8 (72.024%, 71.686%, and 73.471% for male, female and pseudo male, respectively) and the lowest mCpG was on chromosome 5 (65.985%, 60.006% and 60.659% for male, female and pseudo male, respectively) (Fig 2 and S3 Fig).
Fig 2. Methylation features in male tiger pufferfish genome.

a: Distribution ratio of C methylated of different sequence types; b: Methylation patterns of methylated C. X axis was the methylation level, from left to right is 0% ~ 100%, 10% each for a Y axis, the proportion of specific methylation level of mC for all mC, the methylation level of C base is equal to the C base sites on the support sequence of the site for effective coverage methylation sites in the proportion of the value in the sequence; c: Methylation patterns of different functional element regions in the whole genome. The dotted line between a and b is the TSS position; d: Methylation level of each chromosome.
Methylation density analysis
According to the chromosome level density distribution of methylation C bases, with 10 kb area as a unit length, the single-base resolution methylation pattern of female, male and pseudo male was been printed. In pseudo male, methylation density of both ends in some of the chromosomes were greater than the middle area, such as chromosome 1. And in few chromosome, methylation density of ends is greater than the other area density, such as chromosome 3 (S4 Fig). In male and female, the density distribution in the form of probability showed the similar situation with pseudo male.
DMRs and the corresponding genes
We made pair-wise comparisons of male, female, and pseudo male using a 10 kb sliding window method to detect the DMRs. In total, the 3,173 DMRs with a length of 898,849 bp covering 1,931 differentially methylated genes (DMGs) were identified between male and female. Besides, the 1,635 DMRs between pseudo male and female and 3,037 DMRs between pseudo male and male were identified with a length of 498,023 bp and 909,539 bp, representing 966 and 1,782 DMGs, respectively. Of note, the sex chromosome (Chr. 19) contained the highest number of DMRs (229) and 121 DMGs between male and female. Both the highest number of DMRs (256 between male and pseudo male and 177 between female and pseudo male respectively) were located on the chromosome 1. The distribution of DMRs on each chromosome are shown in Fig 3. Furthermore, the DMGs were functionally clustered via the KEGG. We observed a strong enrichment in different biological processes for DMGs in each pair-wise comparison and notably, all the DMGs in sex chromosome were enriched in functions associated with the embryonic or gonad development such as Notch signaling pathway, Wnt signaling pathway and Ovarian steroidogenesis (Fig 4).
Fig 3. The distribution of DMRs on each chromosome in tiger pufferfish.

Fig 4. The scatter plot for KEGG enrichment of DMGs on the sex chromosome of tiger pufferfish.

Rich Factor is the ratio of differentially expressed gene numbers annoted in this pathway terms to all gene numbers annoted in this pathway term. Q ≤ 0.05 as significantly enriched.
Verification of differentially methylation genes
Differential methylation genes screening several genetic variations were related to sex differentiation, such as amhr2, zglp1, nobox, dmrt1, foxl2 and pfcyp19a. Two genes (amhr2 and pfcyp19a) were selected for BSP verification and the results showed the difference between both sexes in the methylation level of promoter region of CpG island in the promoter and first exon. 40% (40/100) of the CpG sits in promoter and the first exon in amhr2 gene in female ovaries were methylated, while 26% (26/100) in normal male testis and 14% (14/100) in pseudo male testis (Fig 5a). Besides, 8.6% (12/140) of the CpG sits in promoter and the first exon in normal female ovary was methylated, and 25.7% (36/140) and 22.1% (31/140) in the testis of normal males and pseudo males, respectively in pfcyp19a gene (Fig 5b).
Fig 5. amhr2 and pfcyp19a gene promoter and the first exon of CpG island methylation analysis of T. rubripes.
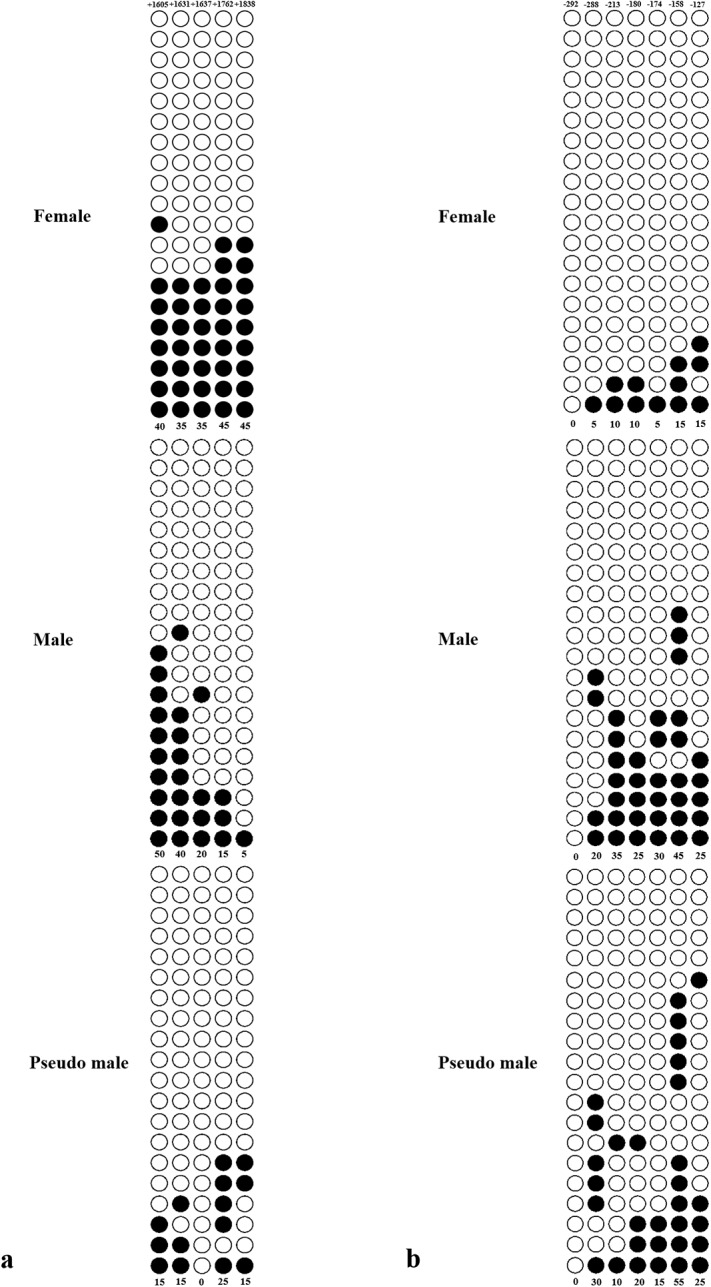
a: amhr2; b: pfcyp19a. Solid circles represent methylated C, hollow circles represent unmethylated C, without the circle line represents the missing value (may be due to sequencing errors). Numbers with a plus or minus sign indicate CpG positions with respect to the transcription starting site.
Expression of differentially methylation genes by qPCR
qPCR was performed on amhr2, pfcyp19a gene for detecting the expression pattern among female, male and pseudo male. For amhr2, the relative expression of male is higher than female and pseudo male, female is close to pseudo male (Fig 6a). For pfcyp19a, the relative expression of female is higher than male and pseudo male, male is close to pseudo male (Fig 6b).
Fig 6. qPCR expression of amhr2 and pfcp19a in female, male and pseudo male gonad.

a: amhr2; b: pfcyp19a. Different letters indicated significant differences (P < 0.05) while the same letters indicated that the differences were not significant (P > 0.05).
Discussion
With the rapid development of next generation sequencing technology, several techniques for detecting the genome-wide DNA methylation were developed including the WGBS [23], reduced representation bisulfite sequencing (RRBS) [24], methylated DNA binding domain sequencing (MBD-seq) [25] and methylated DNA immunoprecipitation sequencing (MeDIP-seq) [26]. Comparatively speaking, WGBS are more accurate and can detect the methylation status with an unbiased view and frequency of each CpG locus in the whole genome, making it better to determine the effect of DNA methylation on various aspects of growth, development, and immunity [27]. Our study presents the first whole-genome methylation map through WGBS in tiger pufferfish and gives a comparatively investigation of the DNA methylomes among sexual systems.
To date, there have been few reports on DNA methylation at a whole genome level of fish species. It was firstly reported that it has heavy methylation exclusively at CG sites in the Tetraodon nigroviridis genome [28]. Recently, the dynamics of DNA methylomes at a single-base resolution for zebrafish gametes and early embryos reaveled that the sperm DNA methylome can be inherited in zebrafish early embryos [29]. Also, comparative analysis of the kinds of gonadal DNA methylomes in Chinese tongue sole revealed that epigenetic regulation controlled temperature-related sex reversal in this fish species [17]. Another example in tilapia, a number of significant clusters of sexually dimorphic DMRs on chromosomal regions associated with sex determination were detected, suggesting the DNA methylation involving into the sex-specific phenotypes [30]. In this study, we determined the effect of temperature on the sex determination and differentiation of tiger pufferfish from an DNA methylation perspective, and revealed how environmental factors interact with the epigenetic mechanism of sex determination.
Our analysis on the male, female and pseudo male methylomes in tiger pufferfish revealed that about 11% of C is methylated in the genome, with more than 98% of C methylation at CpG context. This result is consistent with genome methylation patterns reported for above fish species. In Chinese tongue sole, about 9% of mCs in each sample while more than 99% of mCs were in the CpG context [17]. Other fish species above showed similar patterns with high mC in CpG context. This phenomenon is also observed as in other veterbrate genomes such as human [23] and chicken [31], suggesting that C methylation patterns are highly conserved in vertebrates. Besides, our analyses revealed that the methylation level was lower in controlling elements than in other gene parts. Specifically, the methylation levels were lower on both sides of TSSs, and higher in exons, introns, and downstream genes. These results are consistent with those reported for T. nigroviridis [28], Chinese tongue sole [17] and indicate that the methylation level of the promoter is negatively correlated with gene expression as reported for other vertebrates [32, 33]. This gene structure CpG methylation pattern may be explained by functional constraints within genic regions to prevent the frequency of mutations [34]. All of these observations on the distribution of DNA methylation in gene structure showed largely conserved from teleost to humans and consistent with the enriched CpG dinucleotides in functional regions (eg. promoter regions) of the genome.
DNA methylation as a mediator of environmental change is a gene expression regulatory mechanism in adaptive phenotypic variation [35]. In this study, we observed the methylation alterations in the genome level accompanying by the sex reversal of tiger pufferfish induced by low temperature. The male tiger pufferfish showed higher methylation level (66.009%) than in female (65.027%) and the pseudo male showed an increase in methylation level (65.945%) which is close to methylation level in male after low-temperature induction. This phenomenon was also observed in Chinese tongue sole and tilapia. The methylation level was consistently enhanced by 10% in pseudo male gonad compared with female gonad after high temperature induction in Chinese tongue sole [17]. Similarly, the tilapia gonad showed an increase in methylation level induced by high temperature at a genome wide [20]. Interestingly, no matter high or low temperature treatment, it can increase in methylation levels resulting in sex reversal in fish species. These results suggested that environmentally-induced DNA methylation can cause the phenotypic plasticity, the sex reversal induced by temperature for above instance, which provides a substrate for adaptive phenotypic variation triggered by epigenetic regulation.
Sexually dimorphic patterns of DNA methylation in the genomic context, particularly in sex chromosome or sex-associated regions have regularly been implicated on the sex determination and differentiation in teleost species [30]. Our analysis identified the most DMRs (3,173) between female and male and followed by 3,037 DMRs between pseudo male and male, 1,625 DMRs between pseudo male and female, which were relatively enriched in sex chromosome (229, 235, and 82). This result suggested the DMRs in sex chromosome are sensitive to environment, which play a key role in epigenetic modification on sex differentiation of tiger pufferfish. We further found enrichment in DMGs in sex chromosome in a few functionally well-characterized pathways, namely ovarian steroidogenesis. Genes associated with gonadal development were selected for BSP verification and expression analysis. Generally, the CpG island is mainly located in the promoter region and the first exon region, hypermethylation of CpG in promoter regions and enhancer regions inhibits the expression of genes [36–38]. Besides, most of sex related genes exhibited cellular-specific expression pattern during the development of gonad. amhr2 is the master male sex-determining gene in tiger pufferfish and it was expressed in the Sertoli cells and Leydig cells in the testis [39]. pfcyp19a encodes aromatase P450A that converts androgens into estrogens and it was expressed in the follicular cells and granulosa cells around oocytes [40, 41]. In this study, the amhr2 gene is hypermethylated in female but hypomethylated in male and correspondingly it shows low expression in females and high expression in males. However, the hypomethylation of this gene in pseudo male did not resulting into its high expression. These results suggest that there may be more regulatory approaches to sex-determining genes during the sex reversal of tiger pufferfish. Conversely, the patterns of negative correlation between methylation and expression in the promoter of pfcyp19a in pseudo male is possibly involved in a male transition in tiger pufferfish.
In conclusion, we performed comprehensive whole genome methylation sequencing and analyses on the gonads of tiger pufferfish and further observed a pattern of an increase in methylation level in pseudo male tiger pufferfish after low temperature treatment. We also identified the DMRs enriched in the sex chromosome and the corresponding pathways that involved in the gonad development in tiger pufferfish. These results not only contribute to knowledge about the sex control of fish, but also provide a scientific basis for the production of all-male tiger pufferfish population.
Supporting information
(XLSX)
(XLSX)
(XLSX)
(TIF)
a, d: Distribution ratio of C methylated of different sequence types; b, e: Methylation patterns of methylation; c, f: Methylation patterns of different functional element regions in the whole genome.
(TIF)
(TIF)
From left to right was the starting point to the end point of chromosomes. The vertical axis represents 10 kb for calculation of the mC window density, with blue dots represent the distribution density of mC on the chromosomes, the vertical axis represents the standard ratio of mC, said the smooth curves of different types of methylation of C bases (CG, CHG and CHH) density distribution. The black part of the cross axle indicates the centromere.
(ZIP)
Acknowledgments
We are grateful to the BGI Technologies (Shenzhen) Co., Ltd for DNA-seq and data analysis. We thank Jennifer Smith, PhD, from Liwen Bianji, Edanz Group China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Data Availability
The data of WGBS has uploaded to NCBI Sequence Read Archive (SRA accession: PRJNA524991) and the data of BSP and qPCR are available from figshare (DOI: 10.6084/m9.figshare.8150528).
Funding Statement
Funded by He Zhou, National Natural Science Foundation of China (NO.41606178), Liaoning Doctor Scientific Research Foundation Project (NO.201501185); Ya-Juan Li, Liaoning Department of Education Key Laboratory of Basic Research Project (No. LZ2015010); Shahid Mahboob, International Scientific Partnership Program ISPP at King Saud University for funding this research work through ISPP No. 0050.
References
- 1.Penman DJ, Piferrer F. Fish gonadogenesis. Part I: Genetic and environmental mechanisms of sex determination. Reviews in Fisheries Science. 2008; 16(sup 1): 16–34. 10.1080/10641260802324610 [DOI] [Google Scholar]
- 2.Devlin RH, Nagahama Y. Sex determination and sex differentiation in fish: an overview of genetic, physiological, and environmental influences. Aquaculture. 2002; 208(3–4): 191–364. 10.1016/s0044-8486(02)00057-1 [DOI] [Google Scholar]
- 3.Baroiller JF, D’cotta H. Environment and sex determination in farmed fish. Comparative Biochemistry & Physiology Part C: Toxicology & Pharmacology. 2001; 130(4): 399–409. 10.1016/S1532-0456(01)00267-8 [DOI] [PubMed] [Google Scholar]
- 4.Beardmore JA, Mair GC, Lewis RI. Monosex male production in finfish as exemplified by tilapia: applications, problems, and prospects. Aquaculture. 2001; 197(1): 283–301. 10.1016/s0044-8486(01)00590-7 [DOI] [Google Scholar]
- 5.Wang D, Mao HL, Chen HX, Liu HQ, Gui JF. Isolation of Y-and X-linked SCAR markers in yellow catfish and application in the production of all-male populations. Animal Genetics. 2009; 40(6): 978–981. 10.1111/j.1365-2052.2009.01941.x [DOI] [PubMed] [Google Scholar]
- 6.Bang IC, Kim KK, Kim Y, Kim JH. Studies on the production of all-female populations of Olive Flounder, Paralichthys olivaceus II. Progeny test from sex-reversed gynogenetc diploid male. Journal of Aquaculture. 1996; 9(2): 125–132. [Google Scholar]
- 7.Devlin RH, McNeil BK, Solar II, Donaldson EM. A rapid PCR-based test for Y-chromosomal DNA allows simple production of all-female strains of chinook salmon. Aquaculture. 1994; 128(3–4): 211–220. 10.1016/0044-8486(94)90310-7 [DOI] [Google Scholar]
- 8.Lei JL. Marine fish culture theory and technology. Beijing: China Agricultural Press; 2005; 683–702. [Google Scholar]
- 9.Ma AJ, Lu LJ, Chen C, Wang X’ A, Meng XS, L WY, et al. Breeding and genetic research of major economic species of Fugu. Marine Science. 2011; 35(11): 128–133. 10.1016/S1671-2927(11)60313-1 [DOI] [Google Scholar]
- 10.Kikuchi K, Kai W, Hosokawa A, Mizuno N, Suetake H, Asahina K, et al. The sex-determining locus in the tiger pufferfish, Takifugu rubripes. Genetics. 2007, 175(4): 2039–2042. 10.1534/genetics.106.069278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hattori G, Gong S, Sawada Y. II-3. Effective masculinization method of tiger puffer by temperature control under culture condition. Journal of the Fisheries Society of Japan. 2012; 78(1): 87 10.2331/suisan.78.87 [DOI] [Google Scholar]
- 12.Zhou H, Li JQ, Ma HY, Li YJ, Jiang ZQ, Li X, et al. Low temperature-induced masculinization and histological observation of gonadal differentiation in redfin puffer Takifugu rubripes. Journal of Dalian Ocean University, 2015; 30(1): 41–47. 10.3969/J.ISSN.2095-1388.2015.01.008 [DOI] [Google Scholar]
- 13.Kamiya T, Kai W, Tasumi S, Oka A, Matsunaga T, Mizuno N, et al. A trans-species missense SNP in Amhr2 is associated with sex determination in the tiger pufferfish, Takifugu rubripes (fugu). PLoS Genetics. 2012; 8(7): e1002798 10.1371/journal.pgen.1002798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bird A. Perceptions of epigenetics. Nature. 2007; 447(7143), 396–398. 10.1038/nature05913 [DOI] [PubMed] [Google Scholar]
- 15.Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science. 2001; 293(5532): 1068–1070. 10.1126/science.1063852 [DOI] [PubMed] [Google Scholar]
- 16.Dowen RH, Pelizzola M, Schmitz RJ, Lister R, Dowen JM, Nery JR. et al. Widespread dynamic DNA methylation in response to biotic stress. Proceedings of the National Academy of Science of the United States of America. 2012; 109(32): E2183–2191. 10.1073/pnas.1209329109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shao CW, Li QY, Chen SL, Zhang P, Lian JM, Hu QM, et al. Epigenetic modification and inheritance in sexual reversal of fish. Genome Research. 2014; 24(4): 604–615. 10.1101/gr.162172.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Navarro-Martín L, Viñas J, Ribas L, Díaz N, Gutiérrez A, Di Croce L, et al. DNA methylation of the gonadal aromatase (cyp19a) promoter is involved in temperature-dependent sex ratio shifts in the European sea bass. PLoS Genetics. 2011; 7(12): e1002447 10.1371/journal.pgen.1002447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anastasiadi D, Díaz N, Piferrer F. Small ocean temperature increase elicit stage-dependent changes in DNA methylation and gene expression in a fish, the European sea bass. Scientific Reports. 2017; 7(1): 12401 10.1038/s41598-017-10861-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun LX, Wang YY, Zhao Y, Wang H, Li N, Ji XS. Global DNA methylation changes in Nile tilapia gonads during high temperature-induced masculinization. Plos One. 2016; 11(8): e0158483 10.1371/journal.pone.0158483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou H, Zhuang ZX, Zhang R, Xu QZ, Liang YT, Jiang ZQ, et al. Temperature-control-induced masculinization in tiger puffer Takifugu rubripes. Journal of Oceanology and Limnology. 2019; 37(3): 1125–1135. 10.1007/s00343-019-7382-z [DOI] [Google Scholar]
- 22.Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics, 2009; 10(1): 232 10.1186/1471-2105-10-232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Fillippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009; 462(7271): 315–322. 10.1038/nature08514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagarajan A, Roden C. Wajapeyee N. Reduced representation bisulfite sequencing to identify global alteration of DNA methylation. Methods in Molecular Biology. 2014; 1176: 23–31. 10.1007/978-1-4939-0992-6_3 [DOI] [PubMed] [Google Scholar]
- 25.Aberg KA, McClay JL, Nerella S, Xie LY, Clerk SL, Hudson AD, et al. MBD-seq as a cost-effective approach for methylome-wide association studies: demonstration in 1500 case-control samples. Epigenomics. 2012; 4(6): 605–621. 10.1038/nprot.2012.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taiwo O, Wilson GA, Morris T, Seisenberger S, Reik W, Pearce D, et al. Methylome analysis using MeDIP-seq with low DNA concentrations. Nature Protocols. 2012; 7(4): 617–636. 10.2217/epi.12.59 [DOI] [PubMed] [Google Scholar]
- 27.Bird A. DNA methylation patterns and epigenetic memory. Genes and Development. 2002; 16(1): 6–21. 10.1101/gad.947102 [DOI] [PubMed] [Google Scholar]
- 28.Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-Wide Evolutionary analysis of eukaryotic DNA methylation. Science. 2010; 328(5980): 916–919. 10.1126/science.1186366 [DOI] [PubMed] [Google Scholar]
- 29.Jiang L, Zhang J, Wang JJ, Wang L, Zhang L, Li G, et al. Sperm, but not oocyte, DNA methylome is inherited by zebrafish early embryos. Cell. 2013; 153(4): 773–784. 10.1016/j.cell.2013.04.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wan ZY, Xia JH, Lin G, Wang L, Lin VC, Yue GH. Genome-wide methylation analysis identified sexually dimorphic methylated regions in hybrid tilapia. Scientific Reports. 2016; 6:35903 10.1038/srep35903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li QH, Li N, Hu XX, Li JX, Du Z, Chen L, et al. Genome-wide mapping of DNA methylation in chicken. PLoS ONE. 2011; 6(5), e19428 10.1371/journal.pone.0019428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reue K, Zhang PX. The lipin protein family: dual roles in lipid biosynthesis and gene expression. FEBS Lett. 2008; 582(1): 90–96. 10.1016/j.febslet.2007.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng S, Cokus SJ, Zhang X, Chen PY, Bostick M, Goll MG, et al. Conservation and divergence of methylation patterning in plants and animals. Proceedings of the National Academy of Sciences of the United States of America. 107(19), 8689–8694. 10.1073/pnas.1002720107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang N, Wang L, Chen J, Wang LW, Leach L, Luo ZW. Conserved and divergent patterns of DNA methylation in higher vertebrates. Genome Biology and Evolution. 2014; 6(11): 2998–3014. 10.1093/gbe/evu238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flores KB, Wolschin F, Amdam GV. The role of methylation of DNA in environmental adaptation. Integrative and Comparative Biology. 2013; 53(2): 359–372. 10.1093/icb/ict019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103(5): 1412–1417. 10.1073/pnas.0510310103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klose RJ and Bird AP. Genomic DNA methylation: the mark and its mediators. Trends in Biochemical Sciences. 2006; 31(2): 89–97. 10.1016/j.tibs.2005.12.008 [DOI] [PubMed] [Google Scholar]
- 38.Ball MP, Li JB, Gao Y, Lee JH, LeProust EM, Park IH, et al. Targeted and genome-scale strategiesreveal gene-body methylation signatures in human cells. Nature Biotechnology. 2009; 27(4): 361–368. 10.1038/nbt.1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu GC, Chiu PC, Lyu YS, Chang CF. The Expression of amh and amhr2 is associated with the development of gonadal tissue and sex change in the protandrous black porgy, Acanthopagrus schlegeli. Biology of Reproduction. 2010; 83(3): 443–453. 10.1095/biolreprod.110.084681 [DOI] [PubMed] [Google Scholar]
- 40.Huang W, Zhou L, Li Z, Gui JF. Expression pattern, cellular localization and promoter activity analysis of ovarian aromatase (cyp19a1a) in protogynous hermaphrodite red-spotted grouper. Molecular and Cellular Endocrinology. 2009; 307(1–2): 224–236. 10.1016/j.mce.2009.04.003 [DOI] [PubMed] [Google Scholar]
- 41.Kobayashi Y, Horiguchi R, Miura S, Nakamura M. Sex- and tissue-specific expression of P450 aromatase (cyp19a1a) in the yellowtail clownfish, Amphiprion clarkii. Comparative Biochemistry and Physiology, Part A. 2010; 155(2): 237–244. 10.1016/j.cbpa.2009.11.004 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
(XLSX)
(XLSX)
(TIF)
a, d: Distribution ratio of C methylated of different sequence types; b, e: Methylation patterns of methylation; c, f: Methylation patterns of different functional element regions in the whole genome.
(TIF)
(TIF)
From left to right was the starting point to the end point of chromosomes. The vertical axis represents 10 kb for calculation of the mC window density, with blue dots represent the distribution density of mC on the chromosomes, the vertical axis represents the standard ratio of mC, said the smooth curves of different types of methylation of C bases (CG, CHG and CHH) density distribution. The black part of the cross axle indicates the centromere.
(ZIP)
Data Availability Statement
The data of WGBS has uploaded to NCBI Sequence Read Archive (SRA accession: PRJNA524991) and the data of BSP and qPCR are available from figshare (DOI: 10.6084/m9.figshare.8150528).