

Abstract
gem-Disubstituted N-heterocycles are rarely found in drugs, despite their potential to improve the drug-like properties of small molecule pharmaceuticals. Linezolid, a morpholine heterocycle-containing oxazolidinone antibiotic, exhibits significant side effects associated with human mitochondrial protein synthesis inhibition. We synthesized a gem-disubstituted linezolid analogue that when compared to linezolid, maintains comparable (albeit slightly diminished) activity against bacteria, comparable in vitro physicochemical properties, and a decrease in undesired mitochondrial protein synthesis (MPS) inhibition. This research contributes to the structure-activity-relationship data surrounding oxazolidinone MPS inhibition, and may inspire investigations into the utility of gem-disubstituted N-heterocycles in medicinal chemistry.
Keywords: Antibiotic, Mitochondria, Allylic Alkylation, Heterocycle, Linezolid
Graphical Abstract
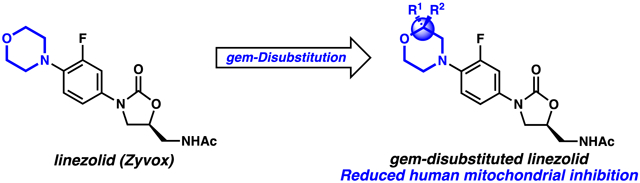
gem-Disubstituted heterocycles are rare in small molecule pharmaceuticals. However, they exhibit the potential to further enhance the drug-like properties of small molecules (Figure 1). For example, gem-disubstitution significantly increases molecular complexity, which is correlated with decreased promiscuity and enhanced binding affinity to desired targets.1-3 Furthermore, depending on the chemical identity of the substitutions, properties such as metabolic stability and polarity may also be altered (Figure 1).
Figure 1.

Biological properties altered by hypothetical gem-disubstitution of the antibiotic linezolid
Given this potential, we sought to investigate the practical utility of gem-disubstituted heterocycles in bioactive small molecules. After surveying various N-heterocyclic drugs for cases in which physicochemical attributes could be improved by heterocyclic gem-disubstitution, we selected the morpholine-containing oxazolidinone antibiotic linezolid (Zyvox) (Figure 1). Approved by the FDA in 2001, linezolid inhibits bacterial peptide synthesis. Co-crystal structures show that linezolid binds to the A-site of the 50S subunit of the ribosome, interacting with an RNA pocket at the ribosomal peptidyl transferase center.4,5 This binding mode suggests that linezolid inhibits binding of aminoacyl tRNA. Linezolid and the recently approved oxazolidinone Tedizolid are important last resort antibiotics that are active against drug-resistant pathogens like methicillin-resistant Staphylococcus aureus (MRSA),6 vancomycin-resistant Enterococcus faecalis (VRE),7 and multi-drug resistant Mycobacterium tuberculosis (MDR-TB).8
Because the bacterial and human ribosomes are highly homologous, oxazolidinone antibiotics bind a commonly conserved site and thus also inhibit mitochondrial protein synthesis (MPS).9-12 This off-target binding is thought to be responsible for linezolid’s more significant side-effects, including myelosuppression, hyperlactatemia, and peripheral neuropathy.13 Other ribosome-targeting antibiotics such as clindamycin and chloramphenicol also exhibit corresponding myelotoxic side effects.14,15 If a structure-activity-relationship (SAR) could be determined for linezolid’s mitochondrial binding ability, then new oxazolidinones could be designed with reduced MPS inhibition. Indeed, reducing myelotoxic side effects while maintaining antibacterial potency is often cited as one of the greatest challenges in new oxazolidinone design.16
Fortunately, the modular structure of oxazolidinones enables chemical modification of all three rings, A, B, and C, facilitating SAR studies for mitochondrial ribosome binding (Figure 1). Some data have already been emerged. In 2006, McKee identified several oxazolidinones that more potently inhibit MPS yet display MIC values comparable to that of linezolid (Table 1).17 In these cases, the morpholine ring was replaced with other heterocycles, suggesting the C-ring may be the greatest determinant of MPS inhibition. In spite of these initial results, there are no known reports of a potent oxazolidinone featuring reduced MPS inhibition.
Table 1.
MIC and MPS IC50 values for linezolid analogues
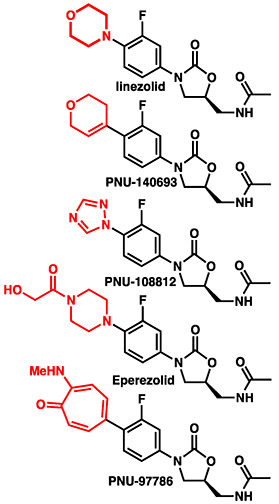
MIC (μg/mL) for S. aureus JC9213
IC50 (μg/mL) for mitochondrial protein synthesis
We thus initiated a medicinal chemistry project seeking to modify the morpholine C-ring of linezolid with the goal of reducing MPS inhibition. Importantly, crystal structure studies note that the morpholine ring does not make significant interactions with the binding pocket, suggesting that the ring can be modified without compromising binding.5 Because increased molecular complexity is associated with reduced ligand promiscuity, we hypothesized that gem-disubstitution of the morpholine ring could potentially increase selectivity for the bacterial ribosome and reduce off-target side effects such as MPS inhibition (Figure 1). To test this hypothesis, we began by identifying a modular route to enable the synthesis of a library of gem-disubstituted linezolid analogs (Scheme 1). Cross-coupling of a gem-disubstituted morpholine with aryl halide 7 was identified as an efficient route. Thus, aryl halide 7 was synthesized by adapting a patent procedure.18 Briefly, (S)-epichlorohydrin (2) was coupled with 4-chlorobenzaldehyde (1) and ammonia to give imine 3. To forge the oxazolidinone intermediate 5, imine 3 was then subjected to a base-catalyzed coupling reaction with carbamate 4,19 which itself was made by addition of benzyl chloroformate to 3-fluoroaniline. Finally, imine hydrolysis, N-acetylation, and iodination provided the aryl iodide 7. 7 was cross-coupled under copper-catalyzed Ullmann conditions20 with a variety of gem-disubstituted morpholines resulting in linezolid analogues 12-19 (Figure 2). Notably, this synthetic route will facilitate future efforts to modify the C morpholine ring of linezolid.
Scheme 1.

Synthesis of gem-disubstituted linezolid analogues via Cu-catalyzed Ullmann coupling. a. (S)-epichlorohydrin, NH4OH (aq). THF, 40 °C, 12 h, 55% yield; b. LiOt-Bu, CH2Cl2, rt to 40 °C, 87% yield; c.1N HCl, H2O/EtOAc; d. Ac2O, CH2Cl2, 96% yield over 2 steps; e. NIS, TFA, rt, 92% yield; f. Substituted morpholine 11a-p, CuBr (10 mol %), BINOL (20 mol %), K3PO4, DMF, 80 °C.
Figure 2.

gem-Disubstituted linezolid analogues synthesized via Ullmann coupling
Two analogues 13 and 14 were prepared using substituted morpholines 10 and 11, which were synthesized via a decarboxylative alkylation protocol and subsequent deprotective and reductive transformations (Scheme 2).20 Other analogues including the gem-dimethyl compound 12 and spiro compounds 15-19 were prepared from commercially available di-substituted morpholines. We note that all analogs were synthesized as racemates.
Scheme 2.

Synthesis of gem-disubstituted morpholines by benzoyl cleavage and reduction of morpholinone decarboxylative alkylation products
Additionally, analogues synthesized via Ullman coupling could be further derivatized (Scheme 3). Taking advantage of the versatile allyl handle of 13, hydroboration-oxidation afforded hydroxyl analogue 20, which could be acetyl-protected to give analogue 21. Additionally, a Lemieux-Johnson oxidation provided the aldehyde intermediate, which was reductively aminated with dimethylamine to provide analogue 23. Catalytic hydrogenation afforded the reduced analogue 22. Similarly, the benzyloxy analogue 14 could also be hydrogenated using catalytic Pd(OH)2 on carbon to provide the hydroxyl analogue 24. Finally, Boc-spiro compound 16 was deprotected using HCl resulting in spiropiperidine 25; subsequent acetyl protection gave 26. Similarly, acid-catalyzed Boc-cleavage of 15 afforded spiropyrrolidine 27.
Scheme 3.

Additional analogues synthesized by derivatization
With a diverse set of gem-disubstituted linezolid analogues in hand, we proceeded with broth microdilution assays against S. aureus to determine minimum inhibitory concentration (MIC) values (Table 2). The initial three compounds, 12, 13, 24, were noticeably less potent than linezolid, suggesting that bulky alkyl di-substitution on the morpholino ring reduces activity. Similarly, bulky hydroxyl-substituted analogue 20 and protected alcohols 21 and 14 were also inactive. In contrast, hydroxyl analogue 24 retained a moderate amount of activity, displaying 48% growth inhibition at the maximal tested concentration of 6 μg/mL. The amine-bearing derivatives, 23, 27, 17, and 25, featuring methylamino, dimethyl amino, and spiroamine functionalities, were uniformly inactive up to 32 μg/mL concentrations. Interestingly, when the basic nitrogen of 25 was masked as an amide as in 26, an 18% growth inhibition at the maximal tested concentration of 6 μg/mL was achieved, suggesting that positively charged substituted morpholines are not tolerated. Finally, we were excited to observe increased growth inhibition when the spirofuran 18 and spirotetrahydropyran 19 analogues were tested, with 19 displaying the greatest potency of all analogues examined. Since stereoisomers often exhibit differing biological activity, we used chiral HPLC to obtain both diastereomers of 19, and then assigned their absolute stereochemistry using vibrational circular dichroism (VCD) and optical rotation calculations, both of which were in agreement (see supporting information for details of synthesis and purification) (Figure 3). Notably a eudysmic difference between the two diastereomers 19a and 19b was observed, with the more active diastereomer 19a displaying an MIC of 6 μg/mL, roughly sixfold less potent than linezolid (Table 2).
Table 2.
MIC values against ATCC 8235-4 (MSSA) or ATCC 43300 (MRSA)
| Compound | MIC (μg/mL) | Compound | MIC (μg/mL) |
|---|---|---|---|
| Linezolid | 1a,b | 23 | > 32b |
| 12 | 8a | 27 | > 32b |
| 13 | 16a | 17 | > 32b |
| 22 | 16a | 25 | > 32b |
| 20 | > 32a | 26 | 6 μg/mL: 18% inhib.c |
| 21 | > 32b | 18 | 6 μg/mL: 65% inhib.c |
| 14 | > 32b | 19 | 7b |
| 24 | 6 μg/mL: 48% inhib.b,c | 19a | 6b |
| 19b | 9b |
MIC: the lowest concentration of molecule preventing visible growth
Tested against S. aureus ATCC 43300.
Tested against S. aureus ATCC 8235-4.
6 μg/mL was the maximal concentration tested.
Figure 3.

Diastereomers of analogue 19. Absolute configuration of the spirocyclic stereocenter determined by both VCD and optical rotations (See supporting information for details).
We further investigated the bioactivity of 19a against other strains of S. aureus, determining the MIC values to be consistent against a range of MSSA and MRSA strains (Table 3).
Table 3.
MIC values of lead analogue 19a against various S. aureus strains
| Strain | Linezolid MIC (μg/mL) | 19a MIC (μg/mL) |
|---|---|---|
| S. aureus ATCC 8235-4 | 1 | 6 |
| S. aureus 43300 | 1 | 5 |
| S. aureus 29213 | 1 | 5 |
| S. aureus 25923 | 1 | 5 |
We next examined the pharmacokinetic properties of 19a and 19b, determining most properties were slightly lower but comparable to that of linezolid (Table 4). For instance, 19a and 19b demonstrated slightly lower aqueous solubility and stability at low pH. Microsomal stability for was also lower than that of linezolid, perhaps due to the metabolic susceptibility of the tetrahydropyran ring. Initial safety data including cytotoxicity and cytochrome P450 isoform inhibition were satisfactory. One interesting difference was MPS inhibition, in which linezolid displayed a relatively potent 8 μM IC50. In contrast, 19a displayed an IC50 value of 30 μM. This finding correlates with the relative MIC values of linezolid and 19a; in this case, MPS inhibition is also roughly three-fold less potent, suggesting that the spirotetrahydropyran ring of 19a maintains moderate binding affinity to the bacterial ribosome while reducing inhibition of the mitochondrial ribosome.
Table 4.
Pharmacokinetic properties, inhibitory activity, and physicochemical properties
| Linezolid | 19a | 19b | |
|---|---|---|---|
| Aq. Solubility (μg/mL) | >67.47 | >52.15 | >48.49 |
| Stability at gastric pH (% remaining 24 h) | 98% | 94% | 89% |
| t1/2 microsomes (min)a | >216.8 | 170.0 | 128.3 |
| Cytotoxicity EC50 (μM)b | >30 | > 30 | >30 |
| CYP inhibition (μM)c | >100 | > 100 | >100 |
| Mt protein synthesis inhibition IC50 (μM)d | 8.19 | 30 | >30 |
Metabolic stability performed with mouse liver microsomes
Cytotoxicity against HepG2 cells using CellTiter Glo
Measured against CYP1A2, 2C9, 2C19, 2D6, 3A4
MitoBiogenesis In-Cell ELISA assay for COXI and SDH-A mitochondrial proteins
In conclusion, we identified a gem-disubstituted morpholine analogue of linezolid bearing a spirotetrahydropyran substitution, 19a, that displays slightly reduced potency compared to linezolid against various S. aureus strains while also having reduced mitochondrial inhibition. These results contribute to the existing SAR of MPS inhibition (Table 1). Although the mitochondrial and bacterial ribosomes share homology, they have structural differences that may be exploited to design molecules with reduced selectivity for the mitochondrial ribosome.9 Our research further contributes to the body of data suggesting that the morpholine ring is a key structural component whose modification can reduce mitochondrial inhibition while maintaining bacterial ribosome inhibition. Continued efforts are needed to identify a molecule as potent as linezolid but with reduced MPS inhibition. This ability of gem-disubstituted heterocycles to alter selectivity for a target such as the bacterial ribosome highlights one of the many useful properties of heterocyclic substitution. In recent years, powerful methods have been developed to stereoselectively synthesize gem-disubstituted heterocycles. For instance, our laboratory has pioneered the development of Pd-catalyzed decarboxylative asymmetric allylic alkylation methodologies to synthesize a range of gem-disubstituted lactams of ring size 5 to 7.21-24 Such lactams can be deprotected and reductively transformed into the corresponding gem-disubstituted N-heterocycles. These methods and others to access gem-disubstituted heterocycles will greatly enable the investigation of the medicinal utility of such heterocycles. Efforts, such as those underway in our laboratory to incorporate gem-disubstituted heterocycles into other small molecule scaffolds will undoubtedly shed light on the broader medicinal utility of gem-disubstituted heterocycles.
Supplementary Material
Acknowledgments
A.W.S. and B.M.S conceived of the project. A.W.S., B.M.O, M.D.B., and S.C.V. performed experimental chemistry. P.L.B., P.A.J., and WuXi AppTec performed biological assays. M.D.B. performed VCD experiments. A.W.S., P.L.B., M.D.B., P.A.J., B.M.O., J.F.M., and B.M.S. wrote the manuscript.
The NIH-NIGMS (R01GM080269), Caltech, the Paul and Daisy Soros Foundation, and the UCLA-Caltech Medical Scientist Training Program are thanked for support of our research program. (Grants R01GM080269 to B.M.S., F30GM120836 and T32GM008042 to A.W.S., F30AI118342, T32GM008042 and P.D. Soros Fellowship to P.L.B.). Dr. David VanderVelde is thanked for assistance with structural assignments via NMR analysis. Dr. Justin Hilf is thanked for helpful discussions. Professor Dianne K. Newman is thanked for MIC testing. The COADD is thanked for MIC testing. The UCLA Microbiology Laboratory is thanked for providing bacterial strains.
Footnotes
Supplementary Material
Supplementary material that may be helpful in the review process should be prepared and provided as a separate electronic file. That file can then be transformed into PDF format and submitted along with the manuscript and graphic files to the appropriate editorial office.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- (1).Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- (2).Lovering F; Bikker J; Humblet C Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- (3).Lovering F Escape from Flatland 2: Complexity and Promiscuity. Med. Chem. Commun. 2013, 4, 515–519. [Google Scholar]
- (4).Ippolito JA; Kanyo ZF; Wang D; Franceschi FJ; Moore PB; Steitz TA; Duffy EM Crystal Structure of the Oxazolidinone Antibiotic Linezolid Bound to the 50S Ribosomal Subunit. J. Med. Chem. 2008, 51, 3353–3356. [DOI] [PubMed] [Google Scholar]
- (5).Wilson DN; Schluenzen F; Harms JM; Starosta AL; Connell SR; Fucini P The Oxazolidinone Antibiotics Perturb the Ribosomal Peptidyl-Transferase Center and Effect TRNA Positioning. PNAS 2008, 105, 13339–13344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ender M; McCallum N; Adhikari R; Berger-Bächi B Fitness Cost of SCCmec and Methicillin Resistance Levels in Staphylococcus Aureus. Antimicrob. Agents Chemother. 2004, 48, 2295–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Gonzales RD; Schreckenberger PC; Graham MB; Kelkar S; DenBesten K; Quinn JP Infections Due to Vancomycin-Resistant Enterococcus Faecium Resistant to Linezolid. The Lancet 2001, 357, 1179. [DOI] [PubMed] [Google Scholar]
- (8).Schecter GF; Scott C; True L; Raftery A; Flood J; Mase S Linezolid in the Treatment of Multidrug-Resistant Tuberculosis. Clin. Infect. Dis. 2010, 50, 49–55. [DOI] [PubMed] [Google Scholar]
- (9).Leach KL; Swaney SM; Colca JR; McDonald WG; Blinn JR; Thomasco LM; Gadwood RC; Shinabarger D; Xiong L; Mankin AS The Site of Action of Oxazolidinone Antibiotics in Living Bacteria and in Human Mitochondria. Mol. Cell 2007, 26, 393–402. [DOI] [PubMed] [Google Scholar]
- (10).Sharma MR; Koc EC; Datta PP; Booth TM; Spremulli LL; Agrawal RK Structure of the Mammalian Mitochondrial Ribosome Reveals an Expanded Functional Role for Its Component Proteins. Cell 2003, 115, 97–108. [DOI] [PubMed] [Google Scholar]
- (11).Greber BJ; Bieri P; Leibundgut M; Leitner A; Aebersold R; Boehringer D; Ban N The Complete Structure of the 55S Mammalian Mitochondrial Ribosome. Science 2015, 348, 303–308. [DOI] [PubMed] [Google Scholar]
- (12).Amunts A; Brown A; Toots J; Scheres SHW; Ramakrishnan V The Structure of the Human Mitochondrial Ribosome. Science 2015, 348, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Narita M; Tsuji BT; Yu VL Linezolid-Associated Peripheral and Optic Neuropathy, Lactic Acidosis, and Serotonin Syndrome. Pharmacotherapy 2007, 27, 1189–1197. [DOI] [PubMed] [Google Scholar]
- (14).Duewelhenke N; Krut O; Eysel P Influence on Mitochondria and Cytotoxicity of Different Antibiotics Administered in High Concentrations on Primary Human Osteoblasts and Cell Lines. Antimicrob. Agents Chemother. 2007, 51, 54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Li C-H; Cheng Y-W; Liao P-L; Yang Y-T; Kang J-J Chloramphenicol Causes Mitochondrial Stress, Decreases ATP Biosynthesis, Induces Matrix Metalloproteinase-13 Expression, and Solid-Tumor Cell Invasion. Toxicol. Sci. 2010, 116, 140–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shaw KJ; Barbachyn MR The Oxazolidinones: Past, Present, and Future. Ann. N. Y. Acad. Sci. 2011, 1241, 48–70. [DOI] [PubMed] [Google Scholar]
- (17).McKee EE; Ferguson M; Bentley AT; Marks TA Inhibition of Mammalian Mitochondrial Protein Synthesis by Oxazolidinones. Antimicrob. Agents Chemother. 2006, 50, 2042–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Imbordino R; Perrault W; Reeder M Process for Preparing Linezolid. WO/2007/116284, October 19, 2007. [Google Scholar]
- (19).Yang B; Shi L; Wu J; Fang X; Yang X; Wu F Microwave-Assisted Expeditious Synthesis of 5-Fluoroalkyl-3-(Aryl/Alkyl)-Oxazolidin-2-Ones. Tetrahedron 2013, 69, 3331–3337. [Google Scholar]
- (20).Mahy W; Leitch JA; Frost CG Copper Catalyzed Assembly of N-Aryloxazolidinones: Synthesis of Linezolid, Tedizolid, and Rivaroxaban. Eur. J. Org. Chem. 2016, 1305–1313. [Google Scholar]
- (21).Sun AW; Hess SN; Stoltz BM Enantioselective Synthesis of Gem-Disubstituted N-Boc Diazaheterocycles via Decarboxylative Asymmetric Allylic Alkylation. Chem. Sci. 2019,10, 788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Behenna DC; Liu Y; Yurino T; Kim J; White DE; Virgil SC; Stoltz BM Enantioselective Construction of Quaternary N-Heterocycles by Palladium-Catalysed Decarboxylative Allylic Alkylation of Lactams. Nat. Chem. 2012, 4, 130–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Numajiri Y; Jiménez-Osés G; Wang B; Houk KN; Stoltz BM Enantioselective Synthesis of Dialkylated N-Heterocycles by Palladium-Catalyzed Allylic Alkylation. Org. Lett. 2015, 17, 1082–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Korch KM; Eidamshaus C; Behenna DC; Nam S; Horne D; Stoltz BM Enantioselective Synthesis of α-Secondary and α-Tertiary Piperazin-2-Ones and Piperazines by Catalytic Asymmetric Allylic Alkylation. Angew. Chem. Int. Ed. 2015, 54, 179–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.