

Abstract
Hemorrhagic stroke is a devastating disease with high morbidity and mortality. There is still a lack of effective ther-apeutic approach. The recent studies have shown that the innate immune system plays a significant role in hemorrhagic stroke. Microglia, as major components in innate immune system, are activated and then can release cytokines and chemo-kines in response to hemorrhagic stroke, and ultimately led to neuroinflammation and brain injury. The NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome is predominantly released by microglia and is believed as the main contributor of neuroinflammation. Several studies have focused on the role of NLRP3 inflammasome in hemorrhagic stroke-induced brain injury, however, the specific mechanism of NLRP3 activation and regulation remains unclear. This re-view summarized the mechanism of NLRP3 activation and its role in hemorrhagic stroke and discussed the translational sig-nificance.
Keywords: NLRP3 inflammasome, hemorrhagic stroke, neuroinflammation, treatment, pathophysiology, innate immune
1. INTRODUCTION
Stroke is the second leading cause of death worldwide. It can be divided into two types, ischemic stroke and hemorrhagic stroke [1]. While ischemic stroke accounts for the majority of strokes, hemorrhagic stroke has higher mortality and morbidity. Intracerebral hemorrhage (ICH) and subarachnoid hemorrhage (SAH) are the subtypes of hemorrhagic stroke. Brain injury after a hemorrhagic stroke causes neural function defects and the mechanisms contributing to brain injury are related to many aspects which include inflammation, oxidative stress, mitochondrial dysfunction, apoptosis and blood-brain barrier disruption. Evidence has shown that neuroinflammation is a key contributor to brain injury [2]. Recently, NLRP3 has been recognized as an important participant in neuroinflammation, and its functions in proinflammatory cytokines secretion [3-5] and subsequent inflammatory events have been demonstrated in hemorrhagic stroke, and inhibition of NLRP3 provided protective effects in hemorrhagic stroke-induced brain injury [3, 6-8]. In this review, we discussed the important role of NLRP3 inflammasome in hemorrhagic stroke.
2. THE CONCEPT OF INNATE IMMUNITY
The innate immune system plays a critical role in response to brain injury. The components of the innate immune system mainly include microglia, macrophages and astrocytes. Among them, microglia are the primary responder in the central nervous system (CNS) [9]. Classical microglia activation is induced by innate immune signals, such as Toll-like receptors, and by the cytokine interferon (IFN)-γ. Microglia can be activated by two different pathways that serve distinct functions responding to CNS damage. The first types are classically activated microglia called M1 and the second are alternatively activated microglia called M2. M1 are involved in destroying microbes and triggering inflammation. Alternative microglia activation occurs in the absence of strong Toll-like receptors signals and is induced by the cytokines Interleukin (IL)-4 and IL-13. In addition, M2 appears to be more important in tissue repair and anti-inflammatory function [10]. In the normal CNS, the microglia surveillance monitors neuronal firing activity and synaptic function to maintain homeostasis. Accumulation and activation of microglia have been observed after experimental SAH [11-13], polarization towards M1 phenotype microglia is also seen in the early stage of ICH [14]. In addition, M1 microglia serve to exacerbate neuronal damage after ICH due to their secretion of pro-inflammatory molecules [14]. When activated, microglia can release cytokines such as IL-1β, IL-6, tumor necrosis factor (TNF)-α,IFN-γ as well as cytotoxic factors, including superoxide and nitric oxide. Furthermore, activated microglia can release glutamate, which binds to N-methyl-D-aspartic acid receptor and mediates neural injury [15].
IL-1β plays a pivotal role in peripheral and central inflammation following brain injury [16]. Studies have reported that IL-1β is up-regulated after brain injury,and it can induce neutrophil infiltration, blood-brain barrier disruption and neovascularization [17]. TNF-α is a pro-inflammatory cytokine that can destroy the blood-brain barrier, exacerbate brain edema and induce apoptosis [18]. Elevated serum TNF-α protein level has been observed in SAH and ICH [19, 20]. The up-regulation of TNF-α impaired blood-brain barrier integrity, induced the release and inhibited the uptake of glutamate, and increased the expression of Aminomethylphosphonic Acid (AMPA) as well as N-Methyl-D-aspartic acid or N-Methyl-D-aspartate (NMDA) receptors in ICH [21]. Additionally, TNF-α increase also relates to cerebral aneurysms growth, rupture and delayed cerebral infarction after SAH [20, 22, 23]. IL-6 is a cytokine that exerts both pro-inflammatory and anti-inflammatory effects [24-26]. Studies have demonstrated that IL-6 is associated with the development and progression of SAH, also, it is a potential biomarker for the diagnosis and monitoring of SAH [22]. As an anti-inflammatory cytokine, IFN-β can provide a protective effect on brain injury by reducing inflammation [27]. Its protective effect was due to IL-10 production and iNOS inhibition [28].
As another vital component of the innate immune system, pattern recognition receptors are observed to be involved in the secondary brain injury. Both Toll-like receptors and Nod-like receptors have been reported to contribute to the pathological progression of cerebral injury caused by ischemic or hemorrhagic stroke.
3. CANONICAL NLRP3 INFLAMMASOME
As the most intensively characterized inflammasome in the NLR family, NLRP3 is widely involved in the host defense response. It is involved in the pathogenesis of various diseases, including autoinflammatory syndromes, Alzheimer’s disease, asbestosis and silicosis, gout, atherosclerosis and type 2 diabetes [29]. NLRP3 is composed of an N-terminal PYD, a central NACHT domain, and a C-terminal domain. A lot of stimuli can trigger the activation of NLRP3, ranging from pathogen-derived ligands, nucleic acid, pore forming toxins, crystalline particles such like silica, asbestos, alum, as well as endogenous signals like ATP, uric acid crystals, and changes in glucose and lipid metabolism [30, 31]. In addition, blood component, such as heme, is supposed to be a NLRP3 activator [32]. After recognition of these varied substances, or perhaps some common chemical alteration induced by these substances, NLRP3 oligomerizes with an adaptor protein ASC and the inactive form of the enzyme caspase-1. Once recruited, caspase-1 is activated and cleaves a precursor form of the cytokine IL-1β and IL-18 to generate biologically active IL-1β and IL-18.
Though plenty of studies have focused on the molecular mechanism of NLRP3 inflammasome activation, the exact mechanism remains unclear. However, it is known to require two distinct steps in canonical NLRP3 inflammasome activation, priming (signal-1) and activation (signal-2) (Fig. 1). The priming is often mediated by TLRs and TNFR and leads to activation of NF-κB. Then, NF-κB binds with DNA after its translocation into the nucleus, and induces the expression of NLRP3. Caspase-8 and FAS-associated death domain protein (FADD) also play roles in the priming step via the regulation of NF-κB pathway [33]. A second signal is provided by a NLRP3-activating-agent (Pathogen Associated Molecular Patterns (PAMPs) or Damaged Associated Molecular Patterns(DAMPs)), and promotes caspase-1 processing and cleaving the cytokines IL-1β and IL-18 into their mature forms [34]. It is widely accepted that NLRP3 cannot directly detect DAMPs or PAMPs and bind to the ligands. Instead, multiple ligands induce a set of cellular events that culminate the activation of NLRP3. The well-accepted events, which can activate NLRP3 inflammasome, including potassium efflux via the purinergic receptor P2X7R, leakage of cathepsin B resulting from lysosomal disruption, reactive oxygen species, the release of mitochondria DNA or the mitochondria phospholipids cardiolipin, calcium influx, changes in cell volume [31, 35-37]. A universal requirement for potassium efflux in NLRP3 activation is widely accepted, however, a recent study has shown that potassium efflux is dispensable for NLRP3 activation by imiquimod and CL097 [38].
Fig. (1).
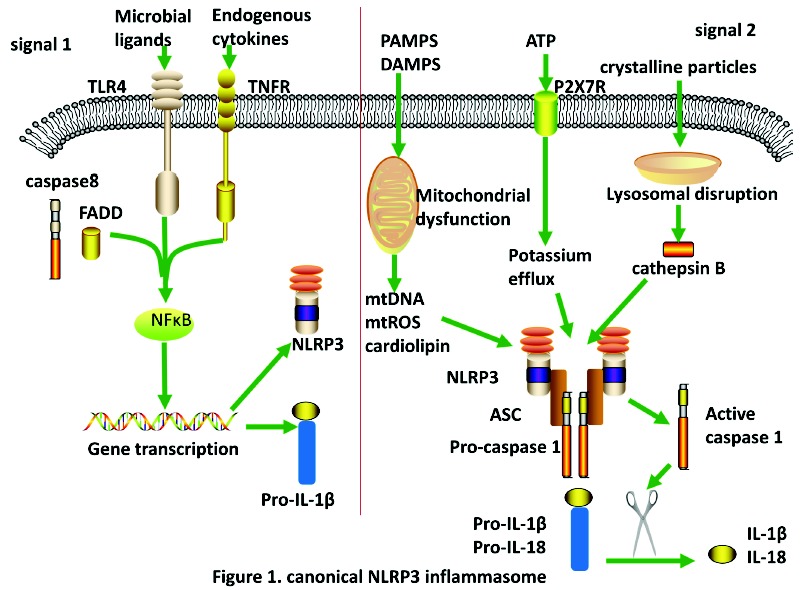
Schematic of canonical activation of NLRP3 inflammasome. Signal 1: the priming of NLRP3 inflammasome. Upon engagement of microbial ligands or endogenous cytokines, TLR4 or TNFR along with caspase8 and FADD activate NF-κB, which leads to the transcription and translation of NLRP3 and pro-IL-1β. Signal2: the activation of NLRP3 inflammasome. After phagocytosed, PAMPs and DAMPs cause mitochondrial dysfunction, which then release mtDNA, mtROS and cardiolipin. Crystalline particles will cause lysosomal disruption which release cathepsin B, otherwise ATP engage with P2X7R leads to patassium efflux, which in turn induces NLRP3 activation. Upon activation of NLRP3 inflammasome, active caspase 1 can cleave pro-IL-1βand pro-IL-18 into their mature forms. (The color version of the figure is available in the electronic copy of the article).
4. NONCANONICAL NLRP3 INFLAMMASOME
Gram-negative bacteria introduce LPS into the host cytoplasm during infection and engage non-canonical activation of the NLRP3 inflammasome via caspase-11 (Fig. 2). Caspase-11, a murine inflammatory caspase, is required for noncanonical NLRP3 activation. In human, caspase-4 and -5 are orthologs of caspase-11 [39]. Caspase-11 recognizes LPS induced into the host cytoplasm, and induces caspase-11-dependent cleavage of the pro-pyroptotic factor gasdermin-D. The N-terminal fragment of gasdermin D mediates pore formation on the cell membrane that leads to pyroptosis and activation of caspase-1 via the NLRP3 inflammasome [40]. Caspase-11 can cleave and activate pannexin-1 channel to induce ATP, which in turn activate the purinergic receptor P2X7R and thus activate NLRP3. Studies indicate that type I interferon is a requirement for the expression of caspase-11 via mediating STAT1-IRF9 signaling cascade [39, 40]. However, another study showed that type I interferon signaling is dispensable in caspase-11 expression [41]. A study observed that AIM2 cooperates with caspase-11 to trigger host resistance in response to Legionella. AIM2 engages active but unprocessed caspase-1 to trigger pore formation and potassium efflux, which leads to NLRP3 activation [42].
Fig. (2).
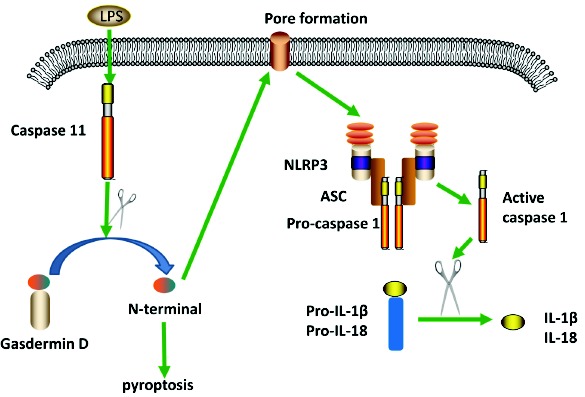
Schematic of non-canonical NLRP3 inflammasome. LPS enters into cytosol independent of TLR4, induces caspase 11 to cleave gasdermin D. N-terminal of gasdermin D leads to cell pyroptosis and pore formation on the cell membrane, which results in the formation of NLRP3 inflammasome. Formation of the NLRP3 inflammasome leads to the release of IL-1β and IL-18 by similar mechanisms to the canonical pathway. (The color version of the figure is available in the electronic copy of the article).
In human blood monocytes, there is a noncanonical NLRP3 inflammasome activation pattern called alternative NLRP3 inflammasome activation. Human blood monocytes are able to secrete active IL-1β in respond to Toll-like receptors’ ligands without a second signal opposing to macrophages. LPS stimulation can induce endogenous ATP in monocytes, which engage with P2X7 receptor to activate NLRP3 inflammasome [43]. Using oxidized ATP to block the interaction between ATP and P2X7 receptor significantly reduced the release of IL-1β [43]. In human monocytes, an alternative pathway for NLRP3 activation is also observed. Although this pathway secreted caspase-1 and IL-1β,it does not require potassium efflux compared with classical NLRP3 activation [44]. In addition, the alternative pathway is able to activate NLRP3 inflammasome in the absence of pyroptosome formation and pyroptosis [45]. A TRIF-RIPK-FADD-CASP8 signaling cascade in the alternative NLRP3 inflammasome activation was identified [44] (Fig. 3).
Fig. (3).
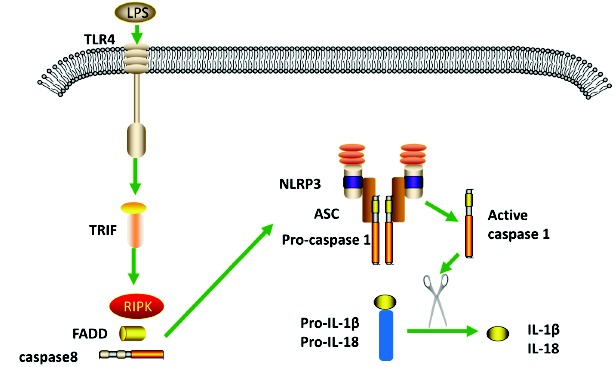
Schematic diagram of alternative NLRP3 inflammasome. In human monocytes, the engagement of LPS with TLR4 causes the formation of NLRP3 and subsequently release of IL-1β and IL-18. TRIF, RIPK, FADD and caspase 8 play roles in this pathway. (The color version of the figure is available in the electronic copy of the article).
5. THE REGULATION MECHANISMS OF NLRP3 INFLAMMASOME
Thioredoxin binding protein (TXNIP), one of the α-arrestin protein family, is associated with NLRP3 activation. TXNIP binds to endogenous inhibitor thioredoxin (TRX) in the resting state, and dissociates with TRX and binds to NLRP3 when responding to oxidative stress, leading to NLRP3 inflammasome activation and inflammatory cytokines release [46]. A study demonstrated that cyclosporine A, a MPT pore inhibitor, efficiently suppressed the NLRP3 activation, therefore we can infer that mitochondrial permeability formation is assumptive a requisite for NLRP3 activation [47]. miR-223 plays a role in the inflammatory signaling by binding to 3′-UTR of RhoB RNA, and leads to the regulation of TLR signaling through NF-κB and MAPK signaling pathways [48]. Once cytosolic double-stranded RNA is recognized by acid-inducible gene 1 and melanoma differentiation-associated protein 5, it induces membrane permeabilization and potassium efflux via common adaptor mitochondrial antiviral signaling protein, subsequently triggers NLRP3 inflammasome activation [49].
In addition, complement, as the essential part of the innate immune system, has brought attention regarding activation of NLRP3 inflammasome. When TLR4 is activated by LPS, the C3aR-driven signal will then produce C3a, and lead to ATP efflux via the ERK1/2-mediated signals, subsequently, ATP efflux activates NLRP3 and cleaves pro-caspase-1 through P2X7 signaling [50]. C5a has been identified to regulate NLRP3 activation, the mechanism is likely via increased generation of reactive oxygen species (ROS). However, in macrophages and monocytes, C5a showed an opposite regulation in NLRP3 activation [51]. Conversely, C1q exhibits negative regulation of NLRP3 activation by driving NLRP12 mRNA expression and inducing IL-10 secretion [52]. Studies have reported that scavenger receptor CD36 (cluster of differentiation) can provide NLRP3 activation in 2 signals. First, oxidized LDL captured by CD36 induces TLR4-TLR6 heteromerization, which then leads to upregulation of NLRP3. CD36 uptakes oxidized LDL and delivers it into lysosomal compartment, and consequently causes lysosomal rupture and NLRP3 activation [53]. Studies also indicated that NLRP3 deubiquitination is required for its activation [29].
The member of NIMA-related kinases, NEK7, is certified to be involved in the activation and assembly of NLRP3 inflammasome. In an experiment where FLAG-tagged wild-type or mutant NLRP3 was expressed in HEK293T cells, Nek7 was identified to interact with the C-terminal leucine-rich repeats, and the N-terminal Pyrin domain showed function in mediating the interaction between Nek7 and NLRP3. In Nek7−/− bone marrow-derived macrophages, caspase-1 activation and IL-1β release were abrogated in respond to NLRP3 stimuli including ATP, negiricin and toxin gramicidin, however, in AIM2 and NLRC4 inflammasome, caspase-1 activation and IL-1β release were not affected. Using short hairpin RNAs to knockdown Nek7 reduced caspase-1 activation and IL-1β release. Furthermore, NLRP3-Nek7 interaction was dependent on potassium efflux and ROS-induced NEK7 phosphorylation [54, 55]. In addition, NEK7 was also a requirement in mitosis, and it served as a switch between NLRP3 inflammasome and mitosis [54]. The roles of double-stranded RNA-dependent protein kinase and guanylate-binding protein 5 in NLRP3 inflammasome activation remain to be controversial [56-59].
6. THE ROLE OF NLRP3 INFLAMMASOME IN HEMORRHAGIC STROKE
Mounting evidence has shown that inflammation plays an important role in hemorrhagic stroke [60]. The pathophysiology of ICH include vessel rupture, initial hematoma growth and secondary hematoma expansion [61]. NLRP3 inflammasome is up-regulated evidently in the microglia of mouse ICH model. The NLRP3 activation and NF-κB upregulation is predominant in ICH-induced inflammatory response. P2X7R, NLRP3, caspase-1, IL-1β and IL-18 were increasingly expressed in the microglia after collagenase-induced ICH. P2X7R siRNA treatment significantly reduced the expression of NLRP3 and downstream cytokines, subsequently improved neurologic outcome. In addition, treatment with P2X7R inhibitor suppressed the expressions of NOX-2, iNOS and ONOO-. ONOO- decomposition catalyst FeTPPS downregulated levels of NLRP3, ASC and cleaved caspase-1 after ICH, the mechanism of ONOO- may be as follows: (1) the nitrification of ONOO- dissociates TXNIP with TRX, following TXNIP binding with NLRP3. (2) The damage of mitochondria results in the release of mitochondria DNA [62], which induces the activation of NLRP3 [63, 64]. H2S serves as an anti-neuroinflammatory gas transmitter in microglia, and it attenuates secondary brain injury through the P2X7R/NLRP3 inflammasome signaling pathway after experimental ICH according to a recent study [65]. Thus, P2X7R plays an important role in the NLRP3 activation of ICH. N-methyl-D-aspartic acid receptor 1 (NMDAR1) was up-regulated after ICH. Treatment with its channel blocker, MK801, attenuated the expression of NLRP3 inflammasome and IL-1β production in mouse ICH model, indicating NMDAR1 involvement in NLRP3 pathway activation by synergic activation [66]. Nuclear factor E2-related factor-2(Nrf2) is shown to scavenge reactive oxygen species(ROS) by triggering NQO1 expression, thus suppresses the expression of NLRP3 and subsequent caspase-1 cleavage. A recent study found that Nrf is downregulated in the experimental rat ICH model compared to the control group, treatment with Isoliquiritigenin(ILG) increased Nrf2 expression and alleviated neurologic deficits, also, NF-κB p65 and NLRP3 inflammasome increased after administration with Nrf2 siRNA, and ILG alleviated these effects. Taken together, the beneficial effect of isoliquiritigen in ICH may be involved with NLRP3 activation through Nrf2 and NF-κB signaling [6]. Silymarin shows similar protection in ICH model by inhibiting NLRP3 activation via Nrf2 signaling [67]. As we mentioned above, complement has gained its attention in NLRP3 inflammasome regulation. A recent study found that complement 3a and complement 5a increased significantly in rat ICH model, and ablation of C3 attenuated ICH-induced IL-1β production. This indicates that complement-induced neuroinflammation is involved in NLRP3 signaling pathway in ICH. Nevertheless, mechanism remains unclear [68]. microRNAs are endogenous noncoding RNAs,and miRNAs have been found involved in regulating innate immune responses. miR-223 can bind the 3’UTR sites of NLRP3 mRNA, subsequently down-regulates NLRP3 expression and attenuates inflammation, thus producing protective effects in rat ICH model [69].
SAH, another type of hemorrhagic stroke, has high mobility and mortality [70]. Pathophysiology of SAH is involved with vasoconstriction, blood-brain barrier disruption, increased microthrombi, brain edema, apoptosis, neurovascular coupling inversion and inflammation [71-73]. Predominantly caused by aneurysm rupture, recent researches of SAH emphasize the importance of inflammation on the formation and rupture of aneurysms [74]. One recent study observed that treatment with melatonin in mouse SAH model attenuated early brain injury via the inhibition of NLRP3 and apoptosis [75]. Furthermore, the inhibitory function of melatonin in NLRP3 activation is through increasing mitophagy proteins expression and reducing ROS generation. However, the correlation between mitophagy and ROS reduction needs further studies [76]. A previous study made an observation that P2X7R/NLRP3 inflammasome axis plays a role in SAH [77]. The receptor-interacting proteins RIP1, RIP3 and DRP1 were up-regulated after experimental SAH. Treatment with Necrostatin-1 (an inhibitor of RIP1) reduced those factors, downregulated the expression of NLRP3 inflammasome and inhibited inflammation after SAH, indicating RIP1-RIP3-DRP1 pathway mediates NLRP3 activation in SAH [78].
CONCLUSION
Hemorrhagic stroke is a commonly encountered condition. Mounting evidence has suggested that inflammation is the principal contributor to secondary brain injury. The inflammation is initiated by activation of microglia and release of proinflammatory cytokines and chemokines. A unified mechanism in NLRP3 inflammasome still needs to be elucidated by further studies. The activation of P2X7R may be one of the multiple mechanisms in NLRP3 inflammasome activation. Other than NLRP3 inflammasome signaling, miR-223 may also mediate other inflammatory responses after ICH. The RIP1-RIP3-DRP1 pathway may be a potential therapeutic target to improve the outcome of SAH. NLRP3 inflammasome inhibition can reduce inflammatory reaction, subsequently improve prognosis. However, whether excessive inhibition may affect the ability of tissue repair and the possibility of infection is unknown. Different targets of NLRP3 inflammasome signaling in hemorrhagic stroke need to be verified.
ACKNOWLEDGEMENTS
Declared none.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This study was supported by the National Natural Science Foundation of China (81500992), Natural Science Foundation of Zhejiang (LQ16H090002), Medical and health key project of Zhejiang Province (2016RCA015), and the Fundamental Research Funds for the Central Universities.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Broderick J.P., Adeoye O., Elm J. Evolution of the modified rankin scale and its use in future Stroke Trials. Stroke. 2017;48(7):2007–2012. doi: 10.1161/STROKEAHA.117.017866. [http://dx.doi.org/10.1161/strokeaha.117.017866]. [PMID:28626052]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou Y., Wang Y., Wang J., Anne S.R., Yang Q.W. Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Prog. Neurobiol. 2014;115:25–44. doi: 10.1016/j.pneurobio.2013.11.003. [http://dx.doi.org/10.1016/j.pneurobio.2013.11.003]. [PMID:24291544]. [DOI] [PubMed] [Google Scholar]
- 3.Ren H. Kong. Y., Liu, Z., Zang, D., Yang, X., Wood, K., Li, M. Liu, Q. Selective NLRP3 (Pyrin domain-containing protein 3) inflammasome inhibitor reduces brain injury after intracerebral hemorrhage. Stroke. 2018;49(1):184–192. doi: 10.1161/STROKEAHA.117.018904. [http://dx.doi.org/10.1161/strokeaha.117.018904]. [PMID:29212744]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang S.J., Shao G.F., Chen J.L., Gong J. The NLRP3 inflammasome: An important driver of neuroinflammation in hemorrhagic stroke. Cell. Mol. Neurobiol. 2018;38(3):595–603. doi: 10.1007/s10571-017-0526-9. [http://dx.doi.org/10.1007/s10571-017-0526-9]. [PMID:28752408]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heneka M.T., McManus R.M., Latz E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018 doi: 10.1038/s41583-018-0055-7. [http://dx.doi.org/10.1038/s41583-018-0055-7]. [PMID:30206330]. [DOI] [PubMed] [Google Scholar]
- 6.Zeng J., Chen Y., Ding R., Feng L., Fu Z., Yang S., Deng X., Xie Z., Zheng S. Isoliquiritigenin alleviates early brain injury after experimental intracerebral hemorrhage via suppressing ROS- and/or NF-kappaB-mediated NLRP3 inflammasome activation by promoting Nrf2 antioxidant pathway. J. Neuroinflam. 2017;14(1):119. doi: 10.1186/s12974-017-0895-5. [http://dx.doi.org/10.1186/s12974-017-0895-5]. [PMID:28610608]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng Y., Wei Y., Yang W., Song Y., Shang H., Cai Y., Wu Z., Zhao W. Cordycepin confers neuroprotection in mice models of intracerebral hemorrhage via suppressing NLRP3 inflammasome activation. Metab. Brain Dis. 2017;32(4):1133–1145. doi: 10.1007/s11011-017-0003-7. [http://dx.doi.org/10.1007/s11011-017-0003-7]. [PMID:28401330]. [DOI] [PubMed] [Google Scholar]
- 8.Shao B., Cao Z. Q., Liu, C. Targeting NLRP3 Inflammasome in the treatment of CNS diseases. Front. Mol. Neurosci. 2018;11:320. doi: 10.3389/fnmol.2018.00320. [http://dx.doi.org/10.3389/fnmol.2018.00320]. [PMID:30233319]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Becker K.J. Strain-related differences in the immune response: Relevance to human stroke. Transl. Stroke Res. 2016;7(4):303–312. doi: 10.1007/s12975-016-0455-9. [http://dx.doi.org/10.1007/s12975-016-0455-9]. [PMID:26860504]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi H., Zheng K., Su Z. Su, H. Zhong., M., He, X., Zhou, C., Chen, H., Xiong, Q. Zhang, Y. Sinomenine enhances microglia M2 polarization and attenuates inflammatory injury in intracerebral hemorrhage. J. Neuroimmunol. 2016;299:28–34. doi: 10.1016/j.jneuroim.2016.08.010. [http://dx.doi.org/10.1016/j.jneuroim.2016.08.010]. [PMID:27725118]. [DOI] [PubMed] [Google Scholar]
- 11.Schneider U., Davids C., Brandenburg A.M., Muller S., Elke A., Magrini A., Atangana S., Turkowski E.K., Finger T., Gutenberg A., Gehlhaar C., Bruck W., Heppner F.L., Vajkoczy P. Microglia inflict delayed brain injury after subarachnoid hemorrhage. Acta Neuropathol. 2015;130(2):215–231. doi: 10.1007/s00401-015-1440-1. [http://dx.doi.org/10.1007/s00401-015-1440-1]. [PMID:25956409]. [DOI] [PubMed] [Google Scholar]
- 12.Li R., Liu W., Yin J., Chen Y., Guo S., Fan H. Li. X., Zhang, X, He, X., Duan, C. TSG-6 attenuates inflammation-induced brain injury via modulation of microglial polarization in SAH rats through the SOCS3/STAT3 pathway. J. Neuroinflam. 2018;15(1):231. doi: 10.1186/s12974-018-1279-1. [http://dx.doi.org/10.1186/s12974-018-1279-1]. [PMID:30126439]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pang J., Peng J., Matei N., Yang P. Kuai, L., Wu, Y., Chen, L., Vitek., M.P., Li, F., Sun, X., Zhang, J.H., Jiang, Y. Apolipoprotein E exerts a whole-brain protective property by promoting M1? microglia quiescence after experimental subarachnoid hemorrhage in mice. Transl. Stroke Res. 2018 doi: 10.1007/s12975-018-0665-4. [http://dx.doi.org/10.1007/s12975-018-0665-4]. [PMID:30225551]. [DOI] [PubMed] [Google Scholar]
- 14.Zhao H., Garton T., Keep R.F., Hua Y., Xi G. Microglia/macrophage polarization after experimental intracerebral hemorrhage. Transl. Stroke Res. 2015;6(6):407–409. doi: 10.1007/s12975-015-0428-4. [http://dx.doi.org/10.1007/s12975-015-0428-4]. [PMID:26446073]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas A.G., O’Driscoll C.M., Bressler J., Kaufmann W., Rojas C.J., Slusher B.S. Small molecule glutaminase inhibitors block glutamate release from stimulated microglia. Biochem. Biophys. Res. Commun. 2014;443(1):32–36. doi: 10.1016/j.bbrc.2013.11.043. [http://dx.doi.org/10.1016/j.bbrc.2013.11.043]. [PMID:24269238]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim T.C., Spector M. Biomaterials for enhancing CNS repair. Transl. Stroke Res. 2017;8(1):57–64. doi: 10.1007/s12975-016-0470-x. [http://dx.doi.org/10.1007/s12975-016-0470-x]. [PMID:27251413]. [DOI] [PubMed] [Google Scholar]
- 17.Denes A., Pinteaux E. Rothwell., N.J., Allan, S.M. Interleukin-1 and stroke: biomarker, harbinger of damage, and therapeutic target. Cerebrovasc. Dis. 2011;32(6):517–527. doi: 10.1159/000332205. [http://dx.doi.org/10.1159/000332205]. [PMID:22104408]. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Z., Liu Y., Huang Q., Su Y., Zhang Y. Wang, G. Li, F. NF-kappaB activation and cell death after intracerebral hemorrhage in patients. Neurol. Sci. 2014;35(7):1097–1102. doi: 10.1007/s10072-014-1657-0. [http://dx.doi.org/10.1007/s10072-014-1657-0]. [PMID:24510152]. [DOI] [PubMed] [Google Scholar]
- 19.Yang G., Shao G.F. Elevated serum IL-11, TNF alpha, and VEGF expressions contribute to the pathophysiology of hypertensive intracerebral hemorrhage (HICH). Neurol. Sci. 2016;37(8):1253–1259. doi: 10.1007/s10072-016-2576-z. [http://dx.doi.org/10.1007/s10072-016-2576-z]. [PMID:27115896]. [DOI] [PubMed] [Google Scholar]
- 20.Young A.M., Karri S.K., You W., Ogilvy C.S. Specific TNF-alpha inhibition in cerebral aneurysm formation and subarachnoid hemorrhage. Curr. Drug Saf., 2012;7(3):190–6. doi: 10.2174/157488612803251315. 22950379] [DOI] [PubMed] [Google Scholar]
- 21.Behrouz R. Re-exploring tumor necrosis factor alpha as a target for therapy in intracerebral hemorrhage. Transl. Stroke Res. 2016;7(2):93–96. doi: 10.1007/s12975-016-0446-x. [http://dx.doi.org/10.1007/s12975-016-0446-x]. [PMID:26762364]. [DOI] [PubMed] [Google Scholar]
- 22.Wu W., Guan Y., Zhao G., Fu X.J., Guo T.Z., Liu Y.T., Ren X.L., Wang W., Liu H.R., Li Y.Q. Elevated IL-6 and TNF-alpha levels in cerebrospinal fluid of subarachnoid hemorrhage patients. Mol. Neurobiol. 2016;53(5):3277–3285. doi: 10.1007/s12035-015-9268-1. [http://dx.doi.org/10.1007/s12035-015-9268-1]. [PMID:26063595]. [DOI] [PubMed] [Google Scholar]
- 23.Starke R.M., Raper D.M., Ding D., Chalouhi N., Owens G.K., Hasan D.M., Medel R., Dumont A.S. Tumor necrosis factor-alpha modulates cerebral aneurysm formation and rupture. Transl. Stroke Res. 2014;5(2):269–277. doi: 10.1007/s12975-013-0287-9. [http://dx.doi.org/10.1007/s12975-013-0287-9]. [PMID:24323710]. [DOI] [PubMed] [Google Scholar]
- 24.Dziedzic T., Bartus S., Klimkowicz A., Motyl M., Slowik A., Szczudlik A. Intracerebral hemorrhage triggers interleukin-6 and interleukin-10 release in blood. Stroke. 2002;33(9):2334–2335. doi: 10.1161/01.str.0000027211.73567.fa. [12215608]. [DOI] [PubMed] [Google Scholar]
- 25.Tanaka T., Narazaki M., Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014;6(10):a016295. doi: 10.1101/cshperspect.a016295. [http://dx.doi.org/10.1101/cshperspect.a016295]. [PMID:25190079]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Armstead W.M., Hekierski H. Pasto, P. Yarovoi, S., Higazi, A. A., Cines. D.B. Release of IL-6 after stroke contributes to impaired cerebral autoregulation and hippocampal neuronal necrosis through NMDA receptor activation and upregulation of ET-1 and JNK. Transl. Stroke Res. 2018 doi: 10.1007/s12975-018-0617-z. [http://dx.doi.org/10.1007/s12975-018-0617-z]. [PMID:29476447]. [DOI] [PubMed] [Google Scholar]
- 27.Owens T., Khorooshi R., Wlodarczyk A., Asgari N. Interferons in the central nervous system: a few instruments play many tunes. Glia, 2014;62(3):339–55. doi: 10.1002/glia.22608. 24588027] [DOI] [PubMed] [Google Scholar]
- 28.Mohsenzadegan M., Fayazi M.R., Abdolmaleki M., Bakhshayesh M., Seif F., Mousavizadeh K. Direct immunomodulatory influence of IFN-beta on human astrocytoma cells. Immunopharmacol. Immunotoxicol. 2015;37(2):214–219. doi: 10.3109/08923973.2015.1014559. [http://dx.doi.org/10.3109/08923973.2015.1014559]. [PMID:25689952]. [DOI] [PubMed] [Google Scholar]
- 29.Juliana C., Fernandes-Alnemri T., Kang S., Farias A., Qin F., Alnemri E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012;287(43):36617–36622. doi: 10.1074/jbc.M112.407130. [http://dx.doi.org/10.1074/jbc.M112.407130]. [PMID:22948162]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Braga T.T., Forni M.F., Correa-Costa M. Ramos., R.N., Barbuto, J.A., Branco, P., Castoldi., A. Hiyane., M.I., Davanso., M.R., Latz, E., Franklin, B.S., Kowaltowski, A.J., Camara, N.O. Soluble Uric Acid Activates the NLRP3 Inflammasome. Sci. Rep. 2017;7:39884. doi: 10.1038/srep39884. [http://dx.doi.org/10.1038/srep39884]. [PMID:28084303]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hornung V., Bauernfeind F., Halle A., Samstad E.O., Kono H., Rock K.L., Fitzgerald K.A., Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008;9(8):847–856. doi: 10.1038/ni.1631. [http://dx.doi.org/10.1038/ni.1631]. [PMID:18604214]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dutra F.F., Alves L.S., Rodrigues D., Fernandez P.L., de Oliveira R.B., Golenbock D.T., Zamboni D.S., Bozza M.T. Hemolysis-induced lethality involves inflammasome activation by heme. Proc. Natl. Acad. Sci. USA. 2014;111(39):E4110–E4118. doi: 10.1073/pnas.1405023111. [http://dx.doi.org/10.1073/pnas.1405023111]. [PMID:25225402]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gurung P., Paras K., Anand P.K., Malireddi S. R.K., Vande, W.L., Van Opdenbosch, N., Christopher, P. D., Weinlich, R., Douglas, R. G., Lamkanfi., M., Kanneganti, T.D. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 2014;192(4):1835–1846. doi: 10.4049/jimmunol.1302839. [http://dx.doi.org/10.4049/jimmunol.1302839]. [PMID:24453255]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lamkanfi M., Dixit V.M. Mechanisms and functions of inflammasomes. Cell. 2014;157(5):1013–1022. doi: 10.1016/j.cell.2014.04.007. [http://dx.doi.org/10.1016/j.cell.2014.04.007]. [PMID:24855941]. [DOI] [PubMed] [Google Scholar]
- 35.Munoz-Planillo R., Kuffa P., Martinez-Colon G., Smith B.L., Rajendiran T.M., Nunez G.K. (+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38(6):1142–1153. doi: 10.1016/j.immuni.2013.05.016. [http://dx.doi.org/10.1016/j.immuni.2013.05.016]. [PMID:23809161]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou R. Yazdi., A.S., Menu, P. Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi: 10.1038/nature09663. [http://dx.doi.org/10.1038/nature09663]. [PMID:21124315]. [DOI] [PubMed] [Google Scholar]
- 37.Iyer S.S., He Q., Janczy J.R., Elliott E.I., Zhong Z., Olivier A.K., Sadler J.J., Knepper-Adrian V., Han R., Qiao L., Eisenbarth S.C., Nauseef W.M., Cassel S.L., Sutterwala F.S. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39(2):311–323. doi: 10.1016/j.immuni.2013.08.001. [http://dx.doi.org/10.1016/j.immuni.2013.08.001]. [PMID:23954133]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gross C.J. Mishra., R., Schneider., K.S., Medard, G., Wettmarshausen, J., Dittlein., D.C., Shi, H., Gorka, O., Koenig., P.A., Fromm, S., Magnani, G., Cikovic, T., Hartjes, L., Smollich, J., Robertson A.A.B., Cooper, M.A., Schmidt-Supprian, M., Schuster, M. Schroder, K., Broz, P., Traidl-Hoffmann, C., Beutler, B. Kuster., B. Ruland, J, Schneider, S. Perocchi, F., Gross, O. K(+) efflux-independent NLRP3 inflammasome activation by small molecules targeting mitochondria. Immunity. 2016;45(4):761–773. doi: 10.1016/j.immuni.2016.08.010. [http://dx.doi.org/10.1016/j.immuni.2016.08.010]. [PMID:27692612]. [DOI] [PubMed] [Google Scholar]
- 39.Casson C.N. Copenhaver., A.M., Zwack, E.E., Nguyen, H.T., Strowig, T., Javdan, B., Bradley, W.P. Fung, T.C., Flavell, R.A., Brodsky, I.E., Shin, S. Caspase-11 activation in response to bacterial secretion systems that access the host cytosol. PLoS Pathog. 2013;9(6):e1003400. doi: 10.1371/journal.ppat.1003400. [http://dx.doi.org/10.1371/journal.ppat.1003400]. [PMID:23762026]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He W.T. Wan. H., Hu, L., Chen, P., Wang, X., Huang, Z., Zhang—Hua, Y., Zhong, C.Q., Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015;25(12):1285–1298. doi: 10.1038/cr.2015.139. [http://dx.doi.org/10.1038/cr.2015.139]. [PMID:26611636]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rathinam V.A., Vanaja S.K., Waggoner L. Sokolovska, A., Becker, C., Stuart,L.M., Leong, J.M., Fitzgerald, K.A. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. 2012;150(3):606–619. doi: 10.1016/j.cell.2012.07.007. [http://dx.doi.org/10.1016/j.cell.2012.07.007]. [PMID:22819539]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cunha L.D., Silva A.L.N. Ribeiro., J.M., Mascarenhas, D.P.A., Quirino, G.F.S., Santos, L.L., Flavel, R.A., Zamboni, D.S. AIM2 engages active but unprocessed caspase-1 to induce noncanonical activation of the NLRP3 inflammasome. Cell Rep. 2017;20(4):794–805. doi: 10.1016/j.celrep.2017.06.086. [http://dx.doi.org/10.1016/j.celrep.2017.06.086]. [PMID:28746866]. [DOI] [PubMed] [Google Scholar]
- 43.Netea M.G., Nold-Petry C.A., Nold M.F., Joosten L.A., Opitz B., van der Meer J.H., van de Veerdonk F.L., Ferwerda G., Heinhuis B., Devesa I., Funk C.J., Mason R.J., Kullberg B.J., Rubartelli A., van der Meer J.W., Dinarello C.A. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood. 2009;113(10):2324–2335. doi: 10.1182/blood-2008-03-146720. [http://dx.doi.org/10.1182/blood-2008-03-146720]. [PMID:19104081]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gaidt M.M., Ebert T.S., Chauhan D., Schmidt T., Schmid-Burgk J.L., Rapino F., Robertson A.A., Cooper M.A., Graf T., Hornung V. Human monocytes engage an alternative inflammasome pathway. Immunity. 2016;44(4):833–846. doi: 10.1016/j.immuni.2016.01.012. [http://dx.doi.org/10.1016/j.immuni.2016.01.012]. [PMID:27037191]. [DOI] [PubMed] [Google Scholar]
- 45.Gaidt M.M., Hornung V. Alternative inflammasome activation enables IL-1beta release from living cells. Curr. Opin. Immunol. 2017;44:7–13. doi: 10.1016/j.coi.2016.10.007. [http://dx.doi.org/10.1016/j.coi.2016.10.007]. [PMID:27842238]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ye X., Zuo D., Yu L., Zhang L., Tang J., Cui C., Bao L., Zan K., Zhang Z., Yang X., Chen H., Tang H., Zu J., Shi H., Cui G. ROS/TXNIP pathway contributes to thrombin induced NLRP3 inflammasome activation and cell apoptosis in microglia. Biochem. Biophys. Res. Commun. 2017;485(2):499–505. doi: 10.1016/j.bbrc.2017.02.019. [http://dx.doi.org/10.1016/j.bbrc.2017.02.019]. [PMID:28202418]. [DOI] [PubMed] [Google Scholar]
- 47.Allam R., Lawlor K.E., Yu E.C., Mildenhall A.L., Moujalled D.M. Lewis., R.S., Ke, F., Mason, K.D., White, M.J., Stacey, K.J., Strasser, A., O’Reilly, L.A., Alexander, W., Kile., B.T., Vaux, D.L., Vince, J.E. Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep. 2014;15(9):982–990. doi: 10.15252/embr.201438463. [http://dx.doi.org/10.15252/embr.201438463]. [PMID:24990442]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang N., Fu L., Bu Y., Yao Y., Wang Y. Downregulated expression of miR-223 promotes Toll-like receptor-activated inflammatory responses in macrophages by targeting RhoB. Mol. Immunol. 2017;91:42–48. doi: 10.1016/j.molimm.2017.08.026. [http://dx.doi.org/10.1016/j.molimm.2017.08.026]. [PMID:28881218]. [DOI] [PubMed] [Google Scholar]
- 49.Franchi L., Eigenbrod T., Munoz-Planillo R., Ozkurede U., Kim Y.G. Arindam, C., Gale, M. Jr., Silverman, R.H., Colonna, M., Akira, S., Nunez, G. Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ efflux. J. Immunol. 2014;193(8):4214–4222. doi: 10.4049/jimmunol.1400582. [http://dx.doi.org/10.4049/jimmunol.1400582]. [PMID:25225670]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Asgari E., Le Friec G., Yamamoto H., Perucha E., Sacks S.S., Kohl J., Cook H.T., Kemper C. C3a modulates IL-1beta secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood. 2013;122(20):3473–3481. doi: 10.1182/blood-2013-05-502229. [http://dx.doi.org/10.1182/blood-2013-05-502229]. [PMID:23878142]. [DOI] [PubMed] [Google Scholar]
- 51.Haggadone M.D., Grailer J.J., Fattahi F., Zetoune F.S., Ward P.A. Bidirectional crosstalk between C5a receptors and the NLRP3 inflammasome in macrophages and monocytes. Mediators Inflamm. 2016;2016:1340156. doi: 10.1155/2016/1340156. [http://dx.doi.org/10.1155/2016/1340156]. [PMID:27382187]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arbore G., Kemper C. A novel “complement-metabolism-inflammasome axis” as a key regulator of immune cell effector function. Eur. J. Immunol. 2016;46(7):1563–1573. doi: 10.1002/eji.201546131. [http://dx.doi.org/10.1002/eji.201546131]. [PMID:27184294]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oury C. CD36: linking lipids to the NLRP3 inflammasome, atherogenesis and atherothrombosis. Cell. Mol. Immunol. 2014;11(1):8–10. doi: 10.1038/cmi.2013.48. [http://dx.doi.org/10.1038/cmi.2013.48]. [PMID:24097033]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shi H., Wang Y., Li X., Zhan X., Tang M., Fina M., Su L. Pratt, D., Bu, C.H., Hildebrand, S. Lyon., S., Scott, L., Quan, J., Sun, Q., Russell, J., Arnett, S., Jurek, P., Chen, D., Kravchenko, V.V., Mathison, J.C., Moresco, E.M., Monson, N.L., Ulevitch, R.J., Beutler, B. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2016;17(3):250–258. doi: 10.1038/ni.3333. [http://dx.doi.org/10.1038/ni.3333]. [PMID:26642356]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.He Y., Zeng M.Y., Yang D., Motro B., Nunez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530(7590):354–357. doi: 10.1038/nature16959. [http://dx.doi.org/10.1038/nature16959]. [PMID:26814970]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.He Y., Franchi L., Nunez G. The protein kinase PKR is critical for LPS-induced iNOS production but dispensable for inflammasome activation in macrophages. Eur. J. Immunol. 2013;43(5):1147–1152. doi: 10.1002/eji.201243187. [http://dx.doi.org/10.1002/eji.201243187]. [PMID:23401008]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoshida K., Okamura H., Hiroshima Y., Abe K., Kido J.I., Shinohara Y., Ozaki K. PKR induces the expression of NLRP3 by regulating the NF-kappaB pathway in porphyromonas gingivalis-infected osteoblasts. Exp. Cell Res. 2017;354(1):57–64. doi: 10.1016/j.yexcr.2017.03.028. [http://dx.doi.org/10.1016/j.yexcr.2017.03.028]. [PMID:28341446]. [DOI] [PubMed] [Google Scholar]
- 58.Shenoy A.R., Wellington D.A., Kumar P., Kassa H., Booth C.J., Cresswell P., MacMicking J.D. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science. 2012;336(6080):481–485. doi: 10.1126/science.1217141. [http://dx.doi.org/10.1126/science.1217141]. [PMID:22461501]. [DOI] [PubMed] [Google Scholar]
- 59.Meunier E., Dick M.S., Dreier R.F., Schurmann N., Kenzelmann B.D., Warming S., Roose-Girma M., Bumann D., Kayagaki N., Takeda M., Yamamoto K., Broz P. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature. 2014;509(7500):366–370. doi: 10.1038/nature13157. [http://dx.doi.org/10.1038/nature13157]. [PMID:24739961]. [DOI] [PubMed] [Google Scholar]
- 60.Xiong X.Y., Yang Q.W. Rethinking the roles of inflammation in the intracerebral hemorrhage. Transl. Stroke Res. 2015;6(5):339–341. doi: 10.1007/s12975-015-0402-1. [http://dx.doi.org/10.1007/s12975-015-0402-1]. [PMID:25940771]. [DOI] [PubMed] [Google Scholar]
- 61.Schlunk F., Greenberg S.M. The Pathophysiology of intracerebral hemorrhage formation and expansion. Transl. Stroke Res. 2015;6(4):257–263. doi: 10.1007/s12975-015-0410-1. [http://dx.doi.org/10.1007/s12975-015-0410-1]. [PMID:26073700]. [DOI] [PubMed] [Google Scholar]
- 62.Baxter P., Chen Y., Xu Y., Swanson R.A. Mitochondrial dysfunction induced by nuclear poly(ADP-ribose) polymerase-1: a treatable cause of cell death in stroke. Transl. Stroke Res. 2014;5(1):136–144. doi: 10.1007/s12975-013-0283-0. [http://dx.doi.org/10.1007/s12975-013-0283-0]. [PMID:24323707]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shimada K., Crother T.R., Karlin J., Dagvadorj J., Chiba N. Chen. S., Ramanujan, V.K., Wolf., A.J., Vergnes, L., Ojcius, D.M., Rentsendorj, A., Vargas, M., Guerrero, C., Wang, Y., Fitzgerald K.A., Underhill, D.M., Town, T., Arditi, M. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36(3):401–414. doi: 10.1016/j.immuni.2012.01.009. [http://dx.doi.org/10.1016/j.immuni.2012.01.009]. [PMID:22342844]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Feng L., Chen Y., Ding R., Fu Z., Yang S., Deng X., Zeng J. P2X7R blockade prevents NLRP3 inflammasome activation and brain injury in a rat model of intracerebral hemorrhage: involvement of peroxynitrite. J. Neuroinflam. 2015;12:190. doi: 10.1186/s12974-015-0409-2. [http://dx.doi.org/10.1186/s12974-015-0409-2]. [PMID:26475134]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao H., Pan P., Yang Y., Ge H., Chen W., Qu J., Shi J., Cui G., Liu X., Feng H., Chen Y. Endogenous hydrogen sulphide attenuates NLRP3 inflammasome-mediated neuroinflammation by suppressing the P2X7 receptor after intracerebral haemorrhage in rats. J. Neuroinflammation. 2017;14(1):163. doi: 10.1186/s12974-017-0940-4. [http://dx.doi.org/10.1186/s12974-017-0940-4]. [PMID:28821266]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weng X., Tan Y., Chu X. Wu., X.F., Liu, R., Tian., Y., Li, L., Guo, F., Ouyang, Q., Li, L. N-methyl-D-aspartic acid receptor 1 (NMDAR1) aggravates secondary inflammatory damage induced by hemin-NLRP3 pathway after intracerebral hemorrhage. Chin. J. Traumatol., 2015;18(5):254–8. doi: 10.1016/j.cjtee.2015.11.010. 26777707] [DOI] [PubMed] [Google Scholar]
- 67.Yuan R., Fan H., Cheng S., Gao W., Xu X., Lv S., Ye M., Wu M., Zhu X., Zhang Y. Silymarin prevents NLRP3 inflammasome activation and protects against intracerebral hemorrhage. Biomed. Pharmacother. 2017;93:308–315. doi: 10.1016/j.biopha.2017.06.018. [http://dx.doi.org/10.1016/j.biopha.2017.06.018]. [PMID:28651232]. [DOI] [PubMed] [Google Scholar]
- 68.Yao S.T., Cao F., Chen J.L., Chen W., Fan R.M., Li G., Zeng Y.C., Jiao S., Xia X.P., Han C., Ran Q.S. NLRP3 is required for complement-mediated caspase-1 and IL-1beta activation in ICH. J. Mol. Neurosci. 2017;61(3):385–395. doi: 10.1007/s12031-016-0874-9. [http://dx.doi.org/10.1007/s12031-016-0874-9]. [PMID:27933491]. [DOI] [PubMed] [Google Scholar]
- 69.Yang Z., Zhong L., Xian R., Yuan B. MicroRNA-223 regulates inflammation and brain injury via feedback to NLRP3 inflammasome after intracerebral hemorrhage. Mol. Immunol. 2015;65(2):267–276. doi: 10.1016/j.molimm.2014.12.018. [http://dx.doi.org/10.1016/j.molimm.2014.12.018]. [PMID:25710917]. [DOI] [PubMed] [Google Scholar]
- 70.Suzuki H., Shiba M., Nakatsuka Y., Nakano F., Nishikawa H. Higher cerebrospinal fluid pH may contribute to the development of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Transl. Stroke Res. 2017;8(2):165–173. doi: 10.1007/s12975-016-0500-8. [http://dx.doi.org/10.1007/s12975-016-0500-8]. [PMID:27623837]. [DOI] [PubMed] [Google Scholar]
- 71.Tso M.K., Macdonald R.L. Subarachnoid hemorrhage: a review of experimental studies on the microcirculation and the neurovascular unit. Transl. Stroke Res. 2014;5(2):174–189. doi: 10.1007/s12975-014-0323-4. [http://dx.doi.org/10.1007/s12975-014-0323-4]. [PMID:24510780]. [DOI] [PubMed] [Google Scholar]
- 72.Mathur A., Hayward J.A., Man S.M. molecular mechanisms of inflammasome signaling. J. Leukoc. Biol. 2018;103(2):233–257. doi: 10.1189/jlb.3MR0617-250R. [http://dx.doi.org/10.1189/jlb.3MR0617-250R]. [PMID:28855232]. [DOI] [PubMed] [Google Scholar]
- 73.Pang J.Y., Chen L., Kuai P., Yang J., Peng Y., Wu Y., Chen M., Vitek P., Chen L., Sun X., Jiang Y. Inhibition of blood-brain barrier disruption by an apolipoprotein e-Mimetic peptide ameliorates early brain injury in experimental subarachnoid hemorrhage. Transl. Stroke Res. 2017;8(3):257–272. doi: 10.1007/s12975-016-0507-1. [http://dx.doi.org/10.1007/s12975-016-0507-1]. [PMID:27796945]. [DOI] [PubMed] [Google Scholar]
- 74.Hosaka K., Hoh B.L. Inflammation and cerebral aneurysms. Transl. Stroke Res. 2014;5(2):190–198. doi: 10.1007/s12975-013-0313-y. [http://dx.doi.org/10.1007/s12975-013-0313-y]. [PMID:24323732]. [DOI] [PubMed] [Google Scholar]
- 75.Dong Y.C., Fan W., Hu S., Jiang Z., Ma X., Yan C., Deng S., Di Z., Xin G., Wu Y., Yang R., Reiter J., Liang G. Melatonin attenuated early brain injury induced by subarachnoid hemorrhage via regulating NLRP3 inflammasome and apoptosis signaling. J. Pineal Res. 2016;60(3):253–262. doi: 10.1111/jpi.12300. [http://dx.doi.org/10.1111/jpi.12300]. [PMID:26639408]. [DOI] [PubMed] [Google Scholar]
- 76.Cao S., Shrestha S., Li J., Yu X., Chen J., Yan F., Ying G., Gu C., Wang L., Chen G. Melatonin-mediated mitophagy protects against early brain injury after subarachnoid hemorrhage through inhibition of NLRP3 inflammasome activation. Sci. Rep. 2017;7(1):2417. doi: 10.1038/s41598-017-02679-z. [http://dx.doi.org/10.1038/s41598-017-02679-z]. [PMID:28546552]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen S., Ma Q., Krafft P.R., Hu Q., Rolland W., II, Sherchan P., Zhang J., Tang J., Zhang J.H. P2X7R/cryopyrin inflammasome axis inhibition reduces neuroinflammation after SAH. Neurobiol. Dis. 2013;58:296–307. doi: 10.1016/j.nbd.2013.06.011. [http://dx.doi.org/10.1016/j.nbd.2013.06.011]. [PMID:23816751]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhou K., Shi L., Wang Z., Zhou J. Manaenko, A., Reis, C., Chen. S., Zhang, J. RIP1-RIP3-DRP1 pathway regulates NLRP3 inflammasome activation following subarachnoid hemorrhage. Exp. Neurol. 2017;295:116–124. doi: 10.1016/j.expneurol.2017.06.003. [http://dx.doi.org/10.1016/j.expneurol.2017.06.003]. [PMID:28579326]. [DOI] [PubMed] [Google Scholar]