

Abstract
Traumatic brain injury (TBI) is the main reason of lifelong disability and casualty worldwide. In the United State alone, 1.7 million traumatic events occur yearly, out of which 50,000 results in deaths. Injury to the brain could alter various biological signaling pathways such as excitotoxicity, ionic imbalance, oxidative stress, inflammation, and apoptosis which can result in various neurological disorders such as Psychosis, Depression, Alzheimer disease, Parkinson disease, etc. In lit-erature, various reports have indicated the alteration of these pathways after traumatic brain injury but the exact mechanism is still unclear. Thus, in the first part of this article, we have tried to summarize TBI as a modulator of various neuronal signal-ing pathways. Currently, very few drugs are available in the market for the treatment of TBI and these drugs only provide the supportive care. Thus, in the second part of the article, based on TBI altered signaling pathways, we have tried to find out potential targets and promising therapeutic approaches in the treatment of TBI.
Keywords: Oxidative stress, excitotoxicity, apoptosis, inflammation, traumatic brain injury, mTOR pathways
1. INTRODUCTION
Traumatic Brain Injury (TBI) is an intracranial injury, which can result in the motor and cognitive dysfunction, disability and death [1, 2]. This injury could happen due to a violent blow or jolt to the head or an object incisive to the skull such as a bullet or a sharp piece of an object [3]. The etiology of TBI is multifactorial that consists of travel accidents, wounds due to gunshots, sports and fight related events [4]. Initially, injury to the brain, due to shearing, tearing and stretching, leads to mechanical-focal brain damage. If the injury is terrible, then trauma can cause damage to blood-brain barrier (BBB) which results in the outflow of molecules, ions, amino acids, proteins, which contribute to secondary injury [5].
Secondary injury happens from hours to days or months and causes various neurochemical [Monoamine oxidase (MAO), dopamine (DA), serotonin, norepinephrine], metabolic (glycolysis and oxidative metabolism in astrocytes) and cellular changes (mitochondrial morphology alteration), which results in neuronal apoptosis [6]. The delayed character of the secondary injury indicates the possible therapeutic window to avoid progressive neuronal apoptosis, which results in the alteration of the cognitive and motor functions [7].
Traumatic brain injury (TBI) is not a precisely defined condition and is characterized by broad changes in signaling mechanism. Generally, most of the individuals ignore the consequences or impact on behavior after brain injury. TBI results in deaths, injuries, disabilities in all age groups but more in the young and productive person [8]. According to the Centers for Disease Control and Prevention (CDC), in the US alone, approximately 2.5 million peoples were affected in 2010 due to TBI associated hospitalizations, or deaths [9]. The worldwide incidence rate of TBI was estimated at 200 per 100,000 people per year [10].
Various new chemical entities (NCE) are under drug discovery and preclinical phase and some of them are under clinical trials for the treatment of TBI [2]. However, despite a decade of preclinical and clinical research, there is not yet an established treatment for TBI and treatment remains restricted to supportive care particularly in case of secondary injury. Thus, there is an urgent need to develop new drugs for the treatment of TBI induced the motor and cognitive dysfunction [11]. The treatment of TBI varies from individual case to case as it depends on various factors such as age, gender, genetics, etc. However, in most of the individuals, there are some commonly altered signaling pathways after TBI [12]. Thus, there is an urgent need to understand these common pathways which will help researchers to develop new drugs in the treatment of TBI, specifically in secondary injury. Thus, in this review, we have tried to summarize TBI altered the common signaling pathways. Further, on the basis of these pathways, we have tried to highlight some potential targets in the treatment of TBI. The relevant articles related to traumatic brain injury altered normal brain signaling pathways have been collected from various sources like Pubmed, google scholar etc.
2. PRIMARY AND SECONDARY EVENTS DURING TRAUMATIC BRAIN INJURY (TBI)
In TBI, primary injury leads to shearing or tearing of blood vessels and tissue deformation which results in systemic complications like decrease cerebral blood flow (CBF), increased intracranial pressure (ICP), hemorrhage and edema [13]. The primary injury further leads to activation of various cellular injury mechanisms such as mitochondrial dysfunction, inflammation, excitotoxicity, calcium overload and oxidative stress which results in necrosis, ischemia, blood-brain barrier (BBB) damage and altered cerebral blood flow [14] as shown in Fig. (1).
Fig. (1).
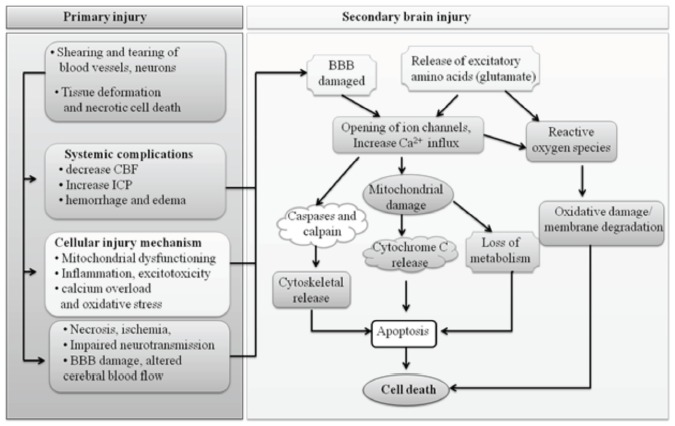
Primary and Secondary events during Traumatic brain injury (TBI).
In most cases, primary injury events further lead to secondary injury. In secondary injury, various apoptotic signaling pathways are activated such as oxidative stress, caspase-dependent and independent, GABA and glutamate signaling pathways which finally result in the neuronal cell death [14] (Fig. 1).
3. TBI ALTERED NORMAL BRAIN SIGNALING PATHWAYS
Injury to the brain disrupts normal signaling pathways which result in altered functions of a brain. Normally, in the Central nervous system (CNS), signals are transmitted by various neurotransmitters such as γ-aminobutyric acid (GABA), glutamate, glycine, norepinephrine, dopamine, serotonin, etc. TBI could alter the level of these neurotransmitters which ultimately disrupts the normal functioning of the brain. TBI altered signaling pathways are discussed below.
3.1. TBI Altered Glutamate and GABA Signaling Pathways
The Glutamate (excitatory) and γ-aminobutyric acid (GABA; inhibitory) neurotransmitters in CNS play a major role in normal neurological function. Glutamate is synthesized from glutamine in presynaptic glutamatergic neurons and then stored in presynaptic vesicles [1]. The major source of glutamine is glutamic acid, which is obtained from food (Spirulina, Cabbage, Asparagus). The excitatory signal promotes the entry of calcium via voltage-gated calcium channel into the presynaptic cell, which results in the release of glutamate into the synaptic cleft where it acts on its receptors.
Glutamate acts via two classes of receptors, i.e. ligand-gated ion channels receptors (ionotropic) and G-protein coupled receptors (metabotropic). The ionotropic receptors include N-methyl-d-aspartate (NMDA), alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA), and kainite whereas metabotropic receptors include L-2-amino-4-phosphonobutyric acid (L-AP4), 1-Amino-1, 3-dicarboxy-cyclopentane (ACPD), and L-quisqualic acid (L-QA). The glutamate acts on these receptors which result in the activation of various signaling cascades [1, 15] as shown in Fig. (2).
Fig. (2).
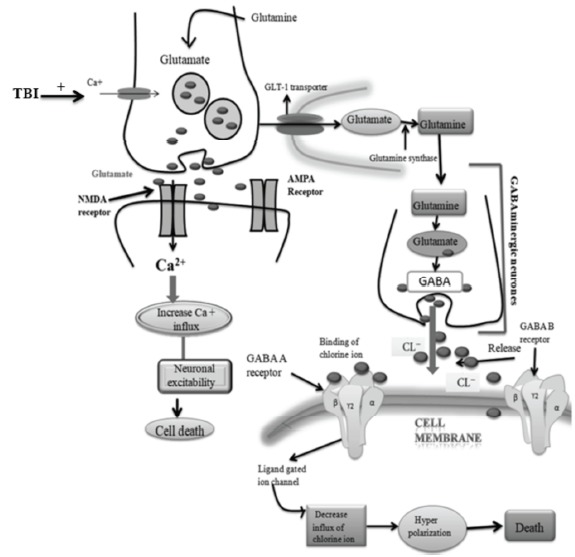
TBI altered Glutamate and GABA signaling pathways.
In the normal brain signaling, glutamate is taken up (from the synaptic cleft) by nearby astrocytes through glutamate transporter-1 (GLT-1). Further in astrocytes, glutamate is converted to glutamine by glutamine synthase and restored back to the presynaptic neuron and adjacent GABAergic interneuron’s for change to GABA via glutamine synthetase and glutamate decarboxylase (GAD) [16] as shown in Fig. (2). GABA is released from local interneurons and acts on GABA-A and GABA-B receptors. GABA-A receptors are post-synaptic ionotropic receptors that cause the opening of Cl− channels and lead to hyperpolarization of the postsynaptic cell. GABA-A receptors may be either synaptic or extra-synaptic. GABA-B receptors are metabotropic, G-protein coupled receptors that act via a second messenger cascade. GABA-B receptors may be post-synaptic or pre-synaptic and lead to the opening of K+ channels, which result in the presynaptic terminal limits GABA release. Post-synaptically, K+ leads to even more pronounced hyperpolarization than Cl−, lasting longer than the action of GABA-A receptors. Cl− and K+ enter the presynaptic pyramidal cell restoring the cell membrane to its resting state [17].
In TBI, the level of GABA and glutamate will be disturbed, which results in alteration of normal brain signaling. Guerriero et al. (2015) have reviewed the role of glutamate and GABA imbalance in traumatic brain injury and concluded that GABA-A subunits (α1, γ2, α4, δ1) modulate neuronal signals via phasic and tonic inhibition after TBI [1]. The glutamate receptor i.e. N-methyl-D-aspartate receptor (NMDAR) has a significant role in use-dependent synaptic plasticity, particularly long-term potentiation [18]. However, it also plays a major role in various neurodegenerative diseases. The NMDAR contains NR1, NR2, NR3 subunits. NR2 is further categorized into NR2A and NR2B. In TBI research, NR1, NR2A, NR2B play a major role. The glycine (inhibitory neurotransmitter) binds with NR1 and result in deactivation [19]. NR2A is usually localized with NR1, mostly at the synapse and activation of these receptors strengthens synapses and induces pro-plasticity signals [pErk, phosphorylation of cAMP-response element-binding protein (pCREB) and Brain-derived neurotrophic factor (BDNF)]. Conversely, NR2B containing NMDARs which are localized extrasynaptically which results in activation of injurious signals to the cell. These injurious signals lead to more influx of calcium which results in subsequent mitochondrial dysfunction and activation of caspase-dependent apoptotic signaling pathways [20].
In TBI, glutamate level is increased which results in activation of NR2B. The activated NR2B further increases the Ca2+ influx. The increased level of Ca2+ results in the neuronal excitability and death [1] as shown in Fig. (2). In literature, various reports have indicated the function of GABA and glutamate in traumatic brain injury. Hovda et al. (1990) [21] have reported an excess release of excitatory neurotransmitters after brain injury. Guerriero et al. (2015) [1] also demonstrated the increased level of glutamate after traumatic brain injury.
Glutamate, glutamine and GABA depend on inter-mediators from tricarboxylic acid (TCA) cycle. Thus, the decreased production of neurotransmitters happens when cellular energy metabolism will become deficit or ineffective (patients who have compromised tissue perfusion) [22].
In TBI research, the GABA-A subunits have received more attention. These subunits α1, γ2, α4 and δ1modulate neuronal signal through phasic and tonic inhibition. When GABA is released rapidly from presynaptic vesicles, it diffuses quickly across the synaptic space and acts on α1 and γ2 containing GABA-A receptors leading to phasic inhibition. Alternatively, lower concentrations of ambient or released GABA act on extra-synaptic receptors containing α4 and δ1 subunits. In TBI, the level of GABA is decreased in the synaptic cleft due to the decreased influx of Cl- ions. The decreased Cl- ions result in decreased hyperpolarization of the cell which further leads to neuronal cell death [23] as shown in Fig. (2).
3.2. TBI Induced Apoptotic Signaling Pathways in Neuronal Cells
Apoptosis or programmed cell death is one of the main factor affecting the outcome and prognosis of TBI. TBI induced neuronal apoptosis is well known, but the mechanism by which TBI induce apoptogenic signaling pathways is still unclear [24]. In literature, various reports have indicated the activation of extrinsic and intrinsic signaling pathways such as calcium signaling, p53 signaling or oxidative stress pathways depending upon the cells, which leads to cell death.
3.2.1. TBI Induced Extrinsic Signaling Pathways
The extrinsic signaling pathways of apoptosis are activated by tumor necrosis factor (TNF) and extracellular ligands (Fas-Fas ligand). The TNF and extracellular ligand bind with cell surface death receptor and formed death-inducing signaling complexes (DISC). The activated macrophages produce various cytokines including TNF-α, which is the major mediator of apoptosis. TNF-α acts on TNFR1 and TNFR2 which results in activation of initiator caspases i.e. caspases 8 and caspase 10. Caspases are a family of protease enzyme initially produced as inactive monomeric procaspases and they can play a crucial role in the apoptosis and inflammation [25]. Mammalian caspases can be divided into initiator caspases (caspase 2, 8, 9), executioner caspases (caspase 3, 6, 7) and inflammatory caspases (caspase 1, 4,5,11 and 12) which results in DNA fragmentation. Fas ligand (FasL, CD95L) is a type-II membrane protein result in apoptotic cell death mediated by caspases activation [26]. In TBI, activated macrophages produced TNF-alpha which is a major mediator of the extrinsic pathway of apoptosis. In TBI, macrophages become activated which results in the production of TNF- α. The activated TNF- α further activates the caspase-dependent apoptogenic signaling pathways [27].
TBI also leads to activation of these extrinsic signaling pathways. In literature, Saurav et al. (2017) [28] demonstrated the role of intrinsic and extrinsic apoptotic signaling pathways after traumatic brain injury. Leonardo et al. (2017) [29] reported the apoptosis of neuronal cells through the activation of extrinsic signaling pathways. Huang et al. (2017) [30] suggested the role of caspases in traumatic brain injury induced apoptotic signaling pathways. Jianhua et al. (2002) [31] demonstrated that the role of extrinsic signaling pathways of apoptosis after TBI.
Overall, it can be concluded that TBI could also lead to neuronal cell death through the activation of extrinsic signaling pathways (Fig. 3).
Fig. (3).
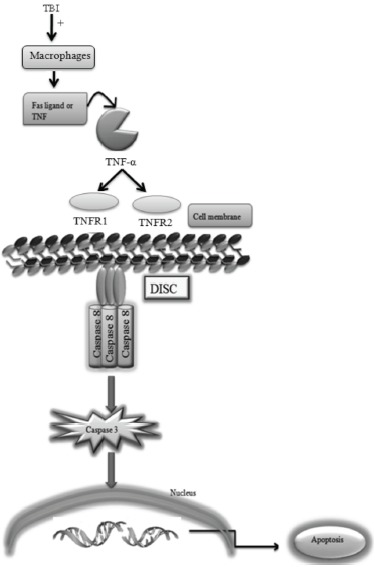
TBI altered extrinsic signaling pathways.
3.2.2. TBI Induced Intrinsic Signaling Pathways
The intrinsic apoptosis signaling pathways that initiate apoptosis involve a diverse array of non-receptor-mediated stimuli that produce intracellular signals that act directly on targets within the cell and are mitochondrial-initiated events. The intrinsic pathway is initiated by stress on cellular organelles [29]. TBI could also lead to neuronal cell death through alteration of these signaling pathways which are discussed below.
3.2.2.1. TBI Induced Calcium Signaling Pathways
Calcium is an important transduction molecule which is important for the activation of various enzymes involved in the normal cellular and physiological process. The normal level of calcium (100 nM) plays a major role in various cellular activities of the nervous system [32] but when the level of calcium is increased from its normal level (1000 nM), it leads to activation of various apoptotic signaling pathways. The various normal activity of the cell (from fertilization to death) is controlled by the level of calcium [33]. However, increased level of calcium lead to activation of various apoptotic signaling pathways.
In literature, few reports are available which indicated the activation of Ca2+ signaling pathway after traumatic brain injury. Ghazizadeh et al. (2014) [34] reported the alteration of calcium signaling pathways after TBI. Long et al. (2017) [35] reported that a high influx of Ca2+ after TBI, results in neuroinflammation and cell death by activation of apoptotic signaling pathways.
a). TBI Altered Calcium Dependent-phospholipase C Pathways
TBI may induce apoptosis by activating phospholipase C pathway. Phospholipase C (PLC) hydrolyzes the membrane phospholipid Phosphatidylinositol 4, 5 bisphosphates (PIP2) to form Inositol trisphosphate (IP3) and diacylglycerol (DAG). The DAG further activates the various types of protein kinases which finally results in apoptosis. The IP3 acts on its IP3 receptor results in the release of calcium. The release Ca2+ further activates the calcium-calmodulin-dependent and caspase-dependent pathways of apoptosis [36] as shown in (Fig. 4).
Fig. (4).
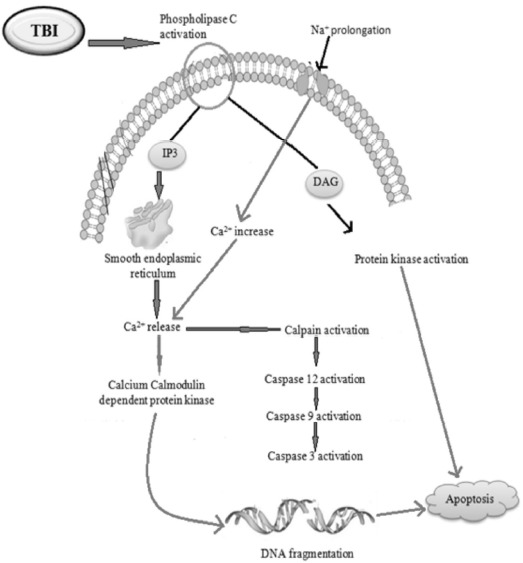
TBI altered Ca2+ dependent phospholipase C and ER stress pathways.
In TBI, the mechanical forces of brain trauma cause an increased intracranial pressure, which results in rupture of micro-vessels. These ruptured micro-vessels can release cytotoxic levels of iron into the brain parenchyma which promotes Ca2+-dependent mechanisms for cell survival but in case of severe TBI, the increased calcium level will lead to neuronal cell death as presented in (Fig. 4).
In literature, only one report is available which indicated the activation of calcium dependent-phospholipase C pathway after TBI. Abdul-Muneer et al. (2017) [35] have suggested the role of Ca2+ and caspase-dependent intrinsic apoptotic pathways in neuronal injury/traumatic brain injury. Ryan et al. (2000) [37] indicated the importance of calcium-dependent phospholipase C pathway in response to mechanical stimulation.
b). TBI Induced Calcium-dependent Endoplasmic Reticulum (ER) Stress Pathways
Endoplasmic reticulum (ER) stress pathway can also contribute to the beginning of apoptosis. To maintain intracellular calcium homeostasis, ER plays the main role, but an increased discharge of calcium can lead to activation of caspase-12. The caspase- 12 further activates the caspase-9 and caspase-3. The caspase-3 attack on DNA and results in its fragmentations which is a hallmark of apoptosis [38].
In the literature, few reports are available which indicated the activation of calcium-dependent endoplasmic reticulum stress pathway after traumatic brain injury. Sun et al. (2016) [39] reported the role of caspase-12-mediated endoplasmic reticulum (ER) stress pathways and may play a vital role in the pathophysiology of secondary brain injury. Wang et al. (2018) [40] suggested the therapeutic role of resveratrol in ER stress-associated neuronal injury. Stephen et al. (2004) [41] demonstrated the role of caspase-12-mediates ER apoptotic pathway in TBI. Weber et al. (2012) [42] also indicated the role of calcium signaling pathways in neuronal apoptosis and dysfunction after traumatic brain injury.
Traumatic brain injury induces calcium signaling pathways by activating phospholipase C which may activate IP3 pathways leading to alteration in calcium-calmodulin-dependent protein kinase which results in cell death. It also activates the ER stress pathway by increasing intracellular calcium leading to calpain activation followed by caspases activation which results in DNA fragmentation as shown in Fig. 4.
3.2.2. Activation of Oxidative Stress Signaling Pathways
Oxidative stress plays a key role in TBI [43]. Oxidative stress is the term used to describe an imbalance between reactive oxygen species (ROS) production and antioxidant enzyme system which leads to lipid peroxidation (LPO) in the cellular, mitochondrial and nuclear membranes, along with degradation of cytosolic proteins and damage to DNA. The generation of free oxygen radicals, superoxide, hydrogen peroxide, nitric oxide, and peroxy-nitrite causes excitotoxicity and impairs the energy metabolism of the cells [44]. The endogenous antioxidant system (i.e., glutathione peroxidase, superoxide dismutase, catalase, and uric acid) aims to convert/neutralize these ROS to less toxic derivatives, thus preventing binding of these to the macromolecules like DNA, RNA, or proteins. However, the excessive amount of ROS produced depletes the endogenous antioxidants and increased the peroxidation of membrane lipids or oxidation of proteins which result in DNA fragmentation and inhibition of the mitochondrial electron transport system [45].
In TBI, particularly in secondary injury, the excess of ROS such as superoxide, nitric oxide radicals is formed which impairs cerebral vascular functions.
In literature, various reports have indicated the role of oxidative stress in TBI. Lutton et al. (2017) [46] demonstrated the use of a targeted antioxidant enzyme in the treatment of TBI. The increased ROS level was observed in the various animal model of TBI which result in the alteration of mitochondrial membrane potential (MMP) and activation of caspase-dependent and independent pathways. In the caspase-independent pathway, apoptosis-inducing factor (AIF) and Endo G are released and result in cell death whereas, in the caspase-dependent pathway, cytochrome C, apoptotic protease-activating factor-1 (Apaf-1) and caspase-9 are activated which further results in activation of caspase-3. The activated caspase-3 results in DNA fragmentation [47]. Bains et al. (2012) [48] indicated the role of ROS or reactive nitrogen species (RNS) and their derived oxygen free radicals in the secondary injury. Fredrik et al. (2004) [49] also demonstrated the role of reactive oxygen species in the traumatic brain injury-induced cell death. Oxidative stress also plays a key role in traumatic brain injury induced various neuropathological conditions (Ryan et al. 2010) [37]. The oxidative stress induced by TBI can result in the activation of caspase-dependent and caspase-independent pathways of apoptosis as shown in Fig. 5.
Fig. (5).
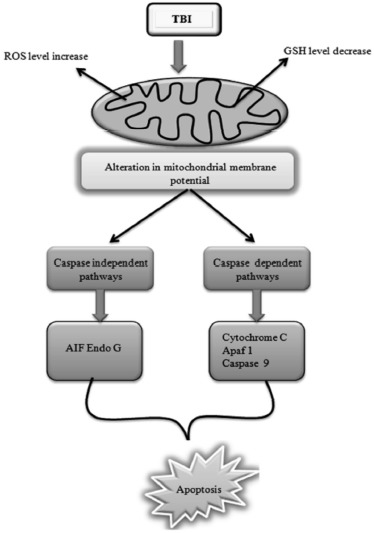
TBI altered oxidative stress signaling pathways.
3.2.3. TBI Induced p53 Signaling Pathways
In response to cellular stress, the transcriptional factor, p53, plays various distinct roles. However, inappropriate p53 activation leads to various apoptogenic signaling pathways in neurological disease and brain injuries.
In TBI, p53 is released from damaged DNA which further alters the level of Bcl-2, Bax and Bad. Bcl-2 blocks the apoptosis whereas Bax induces the apoptosis [50]. The altered level of Bcl-2 and Bax results in the release of cytochrome C. The release cytochrome further releases the Apaf-1, and results in the formation of the apoptosome. Further, caspases are activated which results in apoptosis. TBI may induce apoptosis by directly activating p53 signaling pathways or indirectly by increasing the expression of pro-apoptotic proteins (Bad, Bax) in the mitochondria [51, 52] as shown in Fig. 6.
Fig. (6).
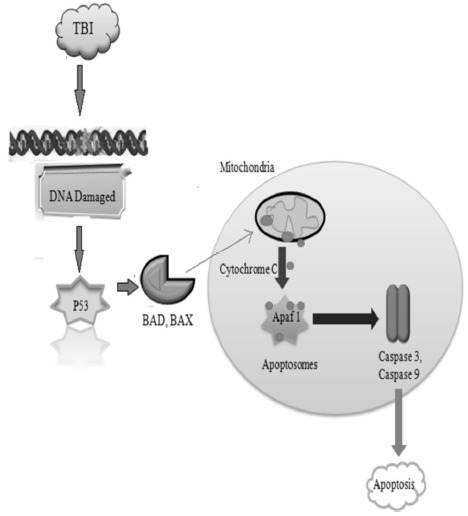
TBI altered p53 signaling pathways.
In literature, various reports indicated the role of p53, Bax, Bcl-2 in TBI induced neuronal cell death. Kinoshita et al. (2000) [53] demonstrated that increased levels of the p53 protein are linked with neuronal damage and cell death. Sabirzhanov et al. (2014) [54] indicated the down-regulation of pro-apoptotic Bcl-2 family members after TBI. Tehranian et al. (2008) [55] observed the role of Bax expression in activating the intrinsic pathway of mitochondrial apoptosis in neurons. It can be concluded that TBI induces apoptosis in neuronal cells by activating p53, oxidative stress and calcium signaling pathways as shown in (Fig. 6).
3.3. Activation of Inflammatory Signaling Pathways
Inflammation is necessary for the removal of damaged cells by phagocytosis which helps in the maintenance of homeostasis [56]. In CNS, both microglia and astrocytes act as a neuroprotective cell against injury. These cells clear the damaged tissue by the process of phagocytosis [57].
After TBI, robust inflammatory responses develop which are characterized by the activation of resident cells, migration and recruitment of peripheral leukocytes, and the release of various inflammatory mediators. Cellular damage associated with the mechanical impact also results in the release of a number of endogenous factors such as RNA, DNA, heat shock proteins, and HMGB1 (high mobility group box 1). These endogenous factors act as damage-associated molecular patterns (DAMPs). The DAMPs bind to Toll-like receptors (TLRs), which activate myeloid differentiation primary response 88 (MYD88) dependent pathways. MYD88 dependent pathway is further categorized into NFκB and Mitogen-activated protein kinase (MAPK) pathways which result in transcription of a large number of downstream genes. These downstream genes leading to the release of a variety of pro-inflammatory factors including cytokines (IL-1β, IL-6), chemokines, and immune receptors [58] as shown in Fig. 7.
Fig. (7).
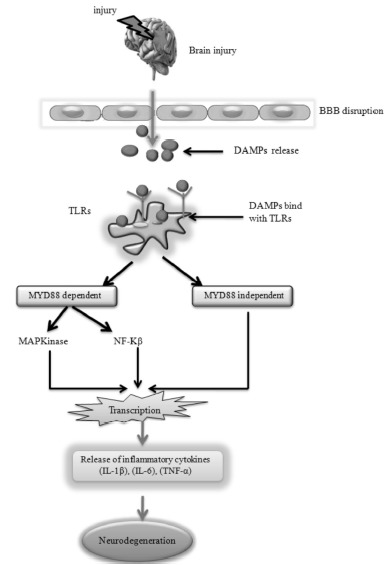
TBI altered inflammatory signaling pathways.
In literature, various reports have indicated the activation of the inflammatory signaling pathway after TBI. Atkins et al. (2007) [59] demonstrated that the activation of cAMP-PKA inflammatory signaling pathways after TBI. Donat et al. (2017) [60] reported the neuroinflammation after traumatic brain injury by activating various cytokines such as (IL-1β), (IL-6) etc. Overall, from the literature, it can be concluded that TBI leads to activation of various inflammatory signaling pathways in neuronal cells which results in damage to CNS (Fig. 7).
3.4. TBI Altered mTOR Pathways
Mammalian target of rapamycin (mTOR) has a significant role in various physiological functions of the nervous system such as neuronal cell growth, survival, dendritic development during differentiation, and synaptic plasticity [61]. The mammalian target of rapamycin (mTOR) is a kinase that is encoded by the mTOR gene in humans involves physiological functions, including cell growth, proliferation, metabolism, protein synthesis and autophagy. Normally, growth factors, hormones and receptor tyrosine kinase bind with growth hormone receptor which results in activation of mitogen-activated protein kinase (MAPK) and phosphatidylinositide 3-kinases (PI3K). The pathway is antagonized by various factors including Phosphatase and tensin homolog (PTEN), Glycogen synthase kinase 3 beta (GSK3B), https://en.wikipedia.org/wiki/PI3K/AKT/mTOR_pathway - cite_note-10 and homeobox gene HB9 [62]. MAPK results in activation of various downstream signaling cascade i.e (MEK) and extracellular signal-regulated kinase (ERK) which leads to apoptosis. In the other position, PI3K further activates serine/threonine-specific protein kinase or protein kinase B (AKT) which results in the activation of mTOR. Further, activated mTOR leads to the destruction of cells (autophagy). mTOR-dependent physiological functions are also important during CNS repair and regeneration; therefore, m TOR is likely to have an instrumental role in the functional recovery process following a traumatic CNS injury [63].
In TBI, growth factor binds with growth hormone receptor which results in the activation of MAPK and PI3K. MAPK results in activation of various downstream signaling cascade i.e. the level of MEK is increased which results in the activation of ERK (extracellular signal-regulated kinase). After the injury, ERK level is also increased which results in apoptosis. On the other side, the PI3K level is decreased which results in the activation of PDK1 (Phosphoinositide-dependent kinase-1) and they can be antagonized by PTEN (Phosphatase and tensin homolog). Further, PI3K activates the AKT (protein kinase B) and the level of AKT is also decreased which results in the activation of m TOR (mammalian target of rapamycin). Further activated m TOR leads to autophagy [64].
A role for mTOR signaling has been identified in traumatic brain injury (TBI). As in hypoxic-ischaemic injury, mTOR could have disparate roles in cell death and neuroprotection: some studies suggested that mTOR inhibition prevented neuronal injury and death following TBI, whereas others suggested that increased post-injury mTOR signaling promoted regeneration and recovery of function.
In literature, Xiaoting et al. (2016) [65] suggested the role of mTOR in neural-stem-cell (NSC) proliferation after injury. After TBI, extracellular signal-regulated kinase (ERK) level is increased, whereas, the PI3K level is decreased which results in the activation of various kinases like (protein kinase B) AKT [66]. Thus, mTOR might be a potential therapeutic target in the treatment of secondary injury due to TBI as shown in Fig. 8.
Fig. (8).
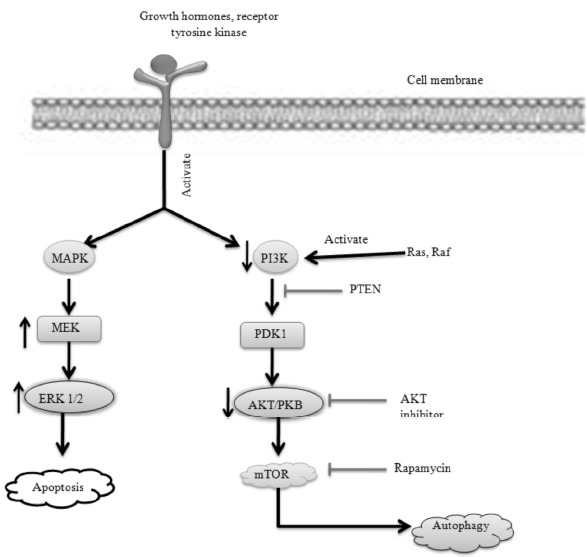
TBI altered mTOR signaling pathways.
4. COMPLICATIONS AFTER TBI, DIAGNOSIS AND CURRENT TREATMENT OPTIONS
In TBI, various changes occur in the brain according to the type and severity of an injury. If an injury will happen in the frontal lobe, then various alterations occur such as loss of body movement, changes in social behavior, mood swings and difficulties with problem-solving. If injury will happen to parietal lobe then there will be a lack of awareness and neglection of certain body parts, loss of sensation, problems with writing and reading, etc. In the occipital lobe such as blurring of vision, hallucination and problem with writing and reading will be observed [67]. If an injury will occur in the temporal lobe, then there will be a disturbance of auditory sensation and perception, difficulty to identify the object, difficulty in understanding language and speaking or increase the aggressive behavior. If the injury will happen in the cerebellum, then various alterations occur such as difficulty in walking, dizziness, tremors, blurred vision etc. If an injury will happen in the brain stem, then there will be a problem with balance and movements, changes in breathing, dizziness, nausea. TBI patients tend to have an increased metabolic rate, which leads to an excessive amount of heat produced within the body. The brain swelling occurs secondary to TBI and contributes to increased intracranial pressure as a result of cerebral vasodilatation and increased cerebral blood flow [68] (Fig. 9).
Fig. (9).
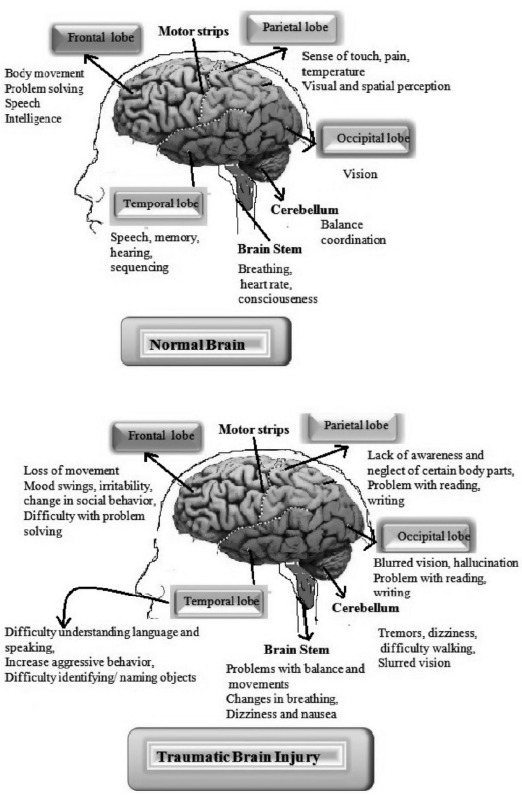
Short-term and long-term effects after TBI.
The diagnosis of brain injury could be done by using various methods such as Computed tomography (CT) scan (for detection of structural damage and abnormalities), Glasgow coma scale (level of consciousness) and Cerebrospinal fluid (CSF) biomarkers (Neurofilament light protein, IL-6, IL-8 and IL-10) [69-71].
The treatment of TBI varies from case to case. Currently, the case of TBI is managed by giving specialized prehospital care, intensive clinical care and long-term rehabilitation. Further, to protect the patient from secondary injury, various neuroprotective agents are prescribed. Currently, various classes of drugs are available in the market such as anti-psychotic (quetiapine, olanzapine, and clozapine), anti-convulsant (sodium valproate, topiramate) and anti-depressant (paroxetine, amitriptyline) in the treatment of secondary injury caused due to TBI. These classes of drugs only provide the symptomatic relief but do not modify the disease progression. Moreover, the available drug therapies are also associated with various side effects of dryness of mouth, dizziness, insomnia, gastrointestinal disturbance, etc. Thus, there is a need for novel approaches in the treatment of TBI associated complications [72].
5. PROMISING APPROACHES FOR THE TREAT-MENT OF TRAUMATIC BRAIN INJURY (TBI)
On the basis of TBI altered various neurological signaling pathways, various approaches can be used to reduce the complications after TBI. Various classes of drugs have been tested for their therapeutic benefit in the various types of animal models of TBI by targeting the mechanism of secondary injury such as calcium channel blockers, corticosteroids, antioxidants etc. The various promising approaches are described below and compiled in Fig. 10.
Fig. (10).
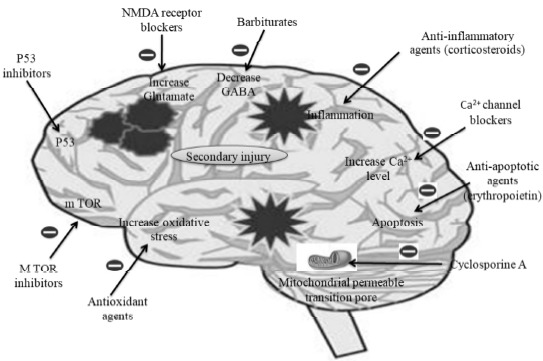
Promising approaches for the treatment of traumatic brain injury (TBI).
5.1. Antioxidants
The antioxidant is a promising approach particularly in the treatment of secondary injury, due to activation of oxidative stress signaling pathway after TBI. Antioxidant, can either act by enzymatic scavenging of free radicals or by inhibition of lipid peroxidation. Several researchers have described the role of various antioxidants such as polyethylene glycol-conjugated superoxide dismutase (PEG-SOD), the lipid peroxide inhibitor tirilazad in animal models of TBI [73].
In literature, Yang et al. (2014) [74] observed the neuroprotective role of Resveratrol (5 μM, 100 mg/kg) due to its antioxidant activity in traumatic brain injury (TBI) under in-vitro and in-vivo conditions. Recently, Venegoni et al. (2017) [75] suggested that Coenzyme Q10 as an antioxidant could reduce the magnitude of secondary injury in traumatic brain injury. In literature, Muhammad et al. (2011) [76] suggested the neuroprotective role of glutathione against oxidative damage and cell death induced by TBI in rats. Due to encouraging preclinical results, the antioxidants could play a key role in the management of TBI. However, the combination of two or more antioxidants (different mechanism) may improve the neuroprotective efficacy. Thus, these types of combination should be tested in preclinical TBI models.
5.2. Anti-inflammatory Approaches
Traumatic brain injury, particularly in the secondary injury, led to activation of various types of inflammatory signaling pathways. Thus, anti-inflammatory agents could act as a neuroprotective agent in the treatment of TBI. Glucocorticoids have broad anti-inflammatory activity. All glucocorticoids act by inhibiting the production of prostaglandins, leukotrienes, histamine, bradykinin and platelet activating factor [58]. In literature, various animal studies of TBI have shown the reduction in inflammation of neurons after treatment of glucocorticoids. However, none of the studies have shown improvement in brain function after treatment with glucocorticoids.
Non-steroidal anti-inflammatory drugs (NSAIDs) are well-known analgesic and antipyretic drugs. They also have anti-inflammatory activity due to inhibition of COX-1 and COX-2 enzymes. After TBI, these both enzymes (COX-1 and COX-2) become active and result in the synthesis of prostaglandins. The NSAIDs (both selective and nonselective) have been tested in the various animal model of TBI and depending upon the animal model, NSAIDs produced anti-inflammatory activity against experimental TBI. However, the anti-inflammatory effect produced by the NSAIDs is not sufficient to prevent tissue damage and functional impairments [77].
After TBI, TNF-α (proinflammatory cytokines) is released which further activate various inflammatory signaling pathways and results in neuronal cell death. Thus, TNF-α inhibitors could play an important role in the treatment of TBI. The two well-known TNF-α antagonists are etanercept and 3, 6 dithiothalidomide. After TBI, etanercept decreased TNF-α, IL-1β, IL-6, at 3 days post-injury. Thus, from the literature, it can be concluded that early antagonism of TNF-α could be a promising treatment for the TBI [78].
The transient increase of cAMP level in experimental TBI rats arises the attention regarding the use of phosphodiesterase inhibitors. Rolipram, a specific inhibitor of phosphodiesterase IV showed the neuroprotective effect by decreasing the level of cAMP, IL-1β and TNF-α. However, the simple reduction in proinflammatory cytokines is not predictive of a favorable histological outcome. In the brain, multiple isoforms of phosphodiesterase are present and expression of these isoforms changes rapidly after TBI [79]. Thus, more study is required to develop phosphodiesterase for the treatment of TBI.
Minocycline is a well-known antibiotic belong to the family of tetracycline. Apart from antimicrobial action, several studies have indicated the anti-inflammatory activity of minocycline after brain injury. Various drugs have also tested to limit neuroinflammation after TBI [80]. Progesterone is a gonadal hormone and has multiple anti-inflammatory properties. Progesterone inhibited IL-1β at 4h post injury and TNF-α at 12 h post injury [81]. Statins are well-known lipid-lowering drugs which act by inhibition of HMG CoA reductase, an enzyme responsible for the synthesis of cholesterol. The inhibition of cholesterol synthesis underlines the anti-inflammatory activity of statins [82]. In literature, various studies have shown the anti-inflammatory activity of statins drugs in experimental TBI animals.
In literature, Hoane et al. (2013) [83] observed that Nicotinamide (500 mg/kg or 150 mg/kg/day) reduced the excitotoxicity due to its anti-inflammatory properties. Cheng et al. (2017) [84] reported the neuroprotective effect of atorvastatin due to its anti-inflammatory property in TBI induced mice. Homsi et al. (2011) [85] demonstrated the neuroprotective role of minocycline (90 mg/kg, i.p) due to its anti-inflammatory activity in traumatic brain injury. The TBI induced complications can be reduced by modulating chemokine signaling especially CCL2/CCR2.
5.3. m TOR Pathways Inhibitors
mTOR pathways are activated after TBI. Thus, m TOR inhibitors could play an important role in the treatment of TBI. Rapamycin is a macrolide antibiotic and has potential to inhibit m TOR pathway [86]. Erlich et al. (2007) [87] suggested that rapamycin could be developed as the neuroprotective agent in TBI. However, further studies are required to consider it as neuroprotective agents in TBI. m TOR inhibition after TBI could represent a new avenue for therapeutic intervention. In literature, various reports have indicated the potential of mTOR targeting agents in the treatment of TBI. Zhu et al. (2014) [88] suggested a therapeutic role of Akt/mTOR signaling in Traumatic brain injury. Yuan et al. (2011) [89] suggested that phosphatase and tensin homolog deleted on the chromosome 10 (PTEN) prevented the neuronal cell death and its neuroprotective effects were mediated by increasing the injury-induced mTOR phosphorylation. Wanchun et al. (2016) [90] demonstrated that the rapamycin and AZD8055 could reduce the development of early brain injury (EBI), possibly through inhibition of the activated microglia by the mTOR pathway.
5.4. Neuroprotective Approaches
5.4.1. Calcium Channel Blockers
Calcium signaling pathways have been activated after TBI which results in neuronal cell death. Thus, calcium channel blockers could act as a neuroprotective agent in TBI. However, the exact role of Ca2+channel blockers in TBI still needs further investigation. Calcium channel blockers (calcium antagonists) have been tried to prevent cerebral vasospasm after injury. Further, they are also used to maintain blood flow to the brain, and in the prevention of further damage [32]. Calcium channel blockers increased intracellular calcium is a very important element in the cascade of the cellular damage after TBI. Using 2 types of calcium channel blockers (L-type and N-type) to neutralize intracellular calcium has shown benefits in preventing TBI-induced cellular death. Gurkoff et al. (2013) [91] suggested that the development of neuronal N-type calcium channel antagonists is useful therapeutic agents in the treatment of TBI.
5.4.2. Corticosteroids
Corticosteroids have been used to treat head injuries due to their ability in the reduction of intracranial pressure (ICP) for more than 3 decades. Dexamethasone and methylprednisolone are well-known corticosteroids, which can be used in the treatment of neuroinflammation due to traumatic brain injury [92].
5.5. GABA Minergic or Antiepileptic Drugs
GABA is the major inhibitory neurotransmitters in the brain. Thus, changes in the level of GABA in the brain can result in major consequences. The decreased level of GABA can cause seizures and could be detrimental to the patients. Indeed, loss of GABAergic neurons after TBI may be responsible for post-traumatic epilepsy. Drugs which facilitate GABAergic neurotransmission are widely used in TBI [93]. For example, Baclofen (GABAB agonist) is used to treat spasticity whereas clonazepam and diazepam are used to suppress the seizures and anxiety. Thus, drugs which facilitate the GABAergic transmission could be developed as promising approaches in TBI.
5.6. Glycinergic Drugs
The glycine is also inhibitory neurotransmitters which act through a ligand-gated chloride channel [94]. Currently, no drug is available that modulate neurotransmission. However, in the future, glycinergic drugs might be helpful. To the best of our knowledge, currently, there is no information on whether or not glycinergic drugs would be useful in TBI patient.
5.7. Anti-Glutamatergic Drugs
In TBI, particularly in secondary injury, glutamate (excitatory amino acid) has long been known to produce excitotoxic damage to neurons and glial cells. Thus, treatment with glutamate antagonist in early hours after TBI has the potential to limit the damage and facilitate recovery. The glutamate antagonists such as memantine have been tested in the animal model of TBI and have shown reduced neuronal cell loss in the hippocampus of rats [95]. However, to the best of our knowledge, whether it would benefit for TBI patients is still unclear.
5.8. Opioids
In literature, it has been suggested that endogenous peptides may be detrimental to the recovery of function following TBI. In TBI, an increase in dynorphin (kappa agonist) has been observed which results in increase neurologic deficits. Thus, Kappa agonists could play important role in the treatment of TBI. However, activation of Mu (µ) and delta (δ) opioid receptors may be neuroprotective rather than neurotropic [96]. Thus, Mu agonist could be beneficial in the reduction of neurological damage associated with TBI. However, further studies are required to confirm their clinical role.
5.9. p53 Inhibitors
P53 inhibitors [pifithrin-α oxygen analog (PFT-α (O)] are used to reduce hippocampal neuronal loss and improve cognitive deficits after experimental traumatic brain injury. Plesnila et al. (2007) [97] observed that p53 inhibition provides a promising approach for the treatment of acute brain injury by blocking apoptotic pathways. Yang et al. (2016) [98] suggested that PFT-α and especially PFT-α (O) significantly reduce hippocampal neuronal degeneration and ameliorate neurological and cognitive deficits in vivo conditions via antiapoptotic and antioxidative properties.
5.10. Sodium Channel Blockers
The excessive activation of the voltage-gated sodium channel in TBI results in various types of cellular abnormalities. Huang et al. (2014) [99] suggested the therapeutic role of sodium channel blocker in the treatment of TBI experimentally induced in animals. Florence et al. (1997) [100] suggested that riluzole (sodium channel blocker) may be beneficial in the clinical treatment of TBI.
5.11. Mannitol
In reversing the acute brain swelling, sometimes mannitol showed the promising effect, but their effectiveness in clinical trials remains unclear [101]. Thus, mannitol could be developing as the promising agent in the treatment of TBI.
5.12. Erythropoietin (EPO)
Erythropoietin is the most widely recognized endogenous molecule which helps in stimulating the maturation, differentiation, survival of hematopoietic progenitor cells [2].
5.13. Bone Marrow Stromal Cells
A neuronal cell has limited to repair after injury. The progenitor cells are promising approaches for the treatment of TBI. However, the clinical use of embryonic stem cells is limited due to ethical concerns and other scientific problems. Thus, bone marrow stromal cells are the alternate source of stem cell replacement therapies [102]. In TBI, more research needed to develop bone marrow stromal cells for treatment of TBI.
5.14. Toll-like Receptor (TLRs) Inhibitors
Inhibition of TLR signaling pathways is also a promising approach in the treatment of TBI. Zhu et al. (2014) [103] suggested that post-injury, curcumin administration may improve the patient outcome by reducing the activation of macrophages and neuronal apoptosis through a mechanism involving the TLR4/MyD88/NF-κB signaling pathway in TBI.
6. CURRENT CHALLENGES
Various animal studies have shown a promising effect in TBI but often lack clinical relevance. In literature, the preclinical studies have evaluated the efficacy of various types of pharmacological agents against TBI but not conducted pharmacokinetic studies which are required to optimize or identify effective brain concentrations required.
In literature, most of the studies have conducted on one experimental model of TBI in one species of animals but to confirm the therapeutic effect, studies should be conducted across multiple experimental models and species. Further, pharmacokinetic, brain penetration and dose-response studies should be conducted in animals with optimum dosing and regimen.
Most of the drugs which showed a promising efficacy in an animal model of TBI, failed in clinical trials, targeted single mechanism such as calcium channel blockers. However, in secondary injury, multiple mechanisms are involved such as excitotoxicity, oxidative stress, caspase, calcium signaling pathways, etc. Thus, the drug that has multipotential effects on various secondary injury mechanisms could be an effective treatment for TBI.
CONCLUSION
TBI is the major problem affecting millions of peoples worldwide. It is a complex process which induces various neurological signaling pathways. The symptoms of TBI vary from individual to individual which makes diagnosis and treatment a challenging task. Thus, the continuing research efforts are required to understand the common underlying mechanism altered after TBI, which will provide the new therapeutic option.
ACKNOWLEDGEMENTS
Authors are thankful to Shri. Parveen Garg, Chairman, Indo-Soviet Friendship College of Pharmacy (ISFCP), Moga, for providing excellent research facility. Authors are also thankful to Dr. S. J. S. Flora, Director National Institute of Pharmaceutical Education and Research (NIPER), Raebareli, for their constant support and motivation.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Guerriero R.M., Giza C.C., Rotenberg A. Glutamate and GABA imbalance following traumatic brain injury. Curr. Neurol. Neurosci. Rep. 2015;15(5):27. doi: 10.1007/s11910-015-0545-1. [http://dx.doi.org/10.1007/s11910-015-0545-1]. [PMID: 25796572]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reis C., Gospodarev V., Reis H., Wilkinson M., Gaio J., Araujo C., Chen S., Zhang J.H. Traumatic brain injury and stem cell: Pathophysiology and update on recent treatment modalities. Stem Cells Int. 2017;2017:6392592. doi: 10.1155/2017/6392592. [http://dx.doi.org/10.1155/ 2017/6392592]. [PMID: 28852409]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmed S., Venigalla H., Mekala H.M., Dar S., Hassan M., Ayub S. Traumatic brain injury and neuropsychiatric complications. Indian J. Psychol. Med. 2017;39(2):114–121. doi: 10.4103/0253-7176.203129. [http://dx. doi.org/10.4103/0253-7176.203129]. [PMID: 28515545]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tanriverdi F., Kelestimur F. Neuroendocrine disturbances after brain damage: an important and often undiagnosed disorder. J. Clin. Med. 2015;4(5):847–857. doi: 10.3390/jcm4050847. [http://dx.doi.org/10.3390/ jcm4050847]. [PMID: 26239451]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pearn M.L., Niesman I.R., Egawa J., Sawada A., Almenar-Queralt A., Shah S.B., Duckworth J.L., Head B.P. Pathophysiology associated with traumatic brain injury: current treatments and potential novel therapeutics. Cell. Mol. Neurobiol. 2017;37(4):571–585. doi: 10.1007/s10571-016-0400-1. [http://dx.doi.org/10.1007/s10571-016-0400-1]. [PMID: 27383839]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cobb C.A., Cole M.P. Oxidative and nitrative stress in neurodegeneration. Neurobiol. Dis. 2015;84:4–21. doi: 10.1016/j.nbd.2015.04.020. [http://dx.doi.org/ 10.1016/j.nbd.2015.04.020]. [PMID: 26024962]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pearn M.L., Niesman I.R., Egawa J., Sawada A., Almenar-Queralt A., Shah S.B., Duckworth J.L., Head B.P. Pathophysiology associated with traumatic brain injury: current treatments and potential novel therapeutics. Cell. Mol. Neurobiol. 2017;37(4):571–585. doi: 10.1007/s10571-016-0400-1. [http://dx.doi.org/10.1007/s10571-016-0400-1]. [PMID: 27383839]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sinha V.D., Chakrabarty A. Quantitative research on traumatic brain injury in India: The travails and the new optimism. Neurol. India. 2017;65(2):261–262. doi: 10.4103/0028-3886.201852. [http://dx.doi.org/10.4103/0028-3886.201852]. [PMID: 28290385]. [DOI] [PubMed] [Google Scholar]
- 9.Marklund N. Rodent models of traumatic brain injury: methods and challenges. Injury Models of the Central Nervous System: Methods and Protocols, 2016:29–46. doi: 10.1007/978-1-4939-3816-2_3. [http://dx.doi.org/10.1007/978-1-4939-3816-2_3]. [DOI] [PubMed] [Google Scholar]
- 10.Bryan-Hancock C., Harrison J. The global burden of traumatic brain injury: Preliminary results from the global burden of disease project. Inj. Prev. 2010;16(Suppl. 1):A17–A17. [http://dx.doi. org/10.1136/ip.2010.029215.61]. [Google Scholar]
- 11.Lagraoui M., Sukumar G., Latoche J.R., Maynard S.K., Dalgard C.L., Schaefer B.C. Salsalate treatment following traumatic brain injury reduces inflammation and promotes a neuroprotective and neurogenic transcriptional response with concomitant functional recovery. Brain Behav. Immun. 2017;61:96–109. doi: 10.1016/j.bbi.2016.12.005. [http://dx.doi. org/10.1016/j.bbi.2016.12.005]. [PMID: 27939247]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marklund N. Rodent models of traumatic brain injury: methods and challenges. Injury Models of the Central Nervous System: Methods and Protocols, 2016:29–46. doi: 10.1007/978-1-4939-3816-2_3. [http://dx.doi.org/10.1007/978-1-4939-3816-2_3]. [DOI] [PubMed] [Google Scholar]
- 13.Traumatic brain injury-a neurobehavioural sequelae a review. Journal of evolution of medical and dental sciences-jemds. 2017;6(26):2192–2207. [Google Scholar]
- 14.Kaur P., Sharma S. Recent advances in pathophysiology of traumatic brain injury. Curr. Neuropharmacol. 2018;16(8):1224–1238. doi: 10.2174/1570159X15666170613083606. [PMID: 28606040]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lussier M.P., Sanz-Clemente A., Roche K.W. Dynamic regulation of N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors by posttranslational modifications. J. Biol. Chem. 2015;290(48):28596–28603. doi: 10.1074/jbc.R115.652750. [http://dx.doi.org/10.1074/jbc.R115.652750]. [PMID: 26453298]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schousboe A., Scafidi S., Bak L.K., Waagepetersen H.S., McKenna M.C. Glutamate and ATP at the Interface of Metabolism and Signaling in the Brain. 2014. Glutamate metabolism in the brain focusing on as trocytes; pp. 13–30. [http://dx.doi.org/10.1007/978-3-319-08894-5_2]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herbison A.E., Moenter S.M. Depolarising and hyperpolarising actions of GABA(A) receptor activation on gonadotrophin-releasing hormone neurones: towards an emerging consensus. J. Neuroendocrinol. 2011;23(7):557–569. doi: 10.1111/j.1365-2826.2011.02145.x. [http://dx.doi.org/10. 1111/j.1365-2826.2011.02145.x]. [PMID: 21518033]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shohami E., Biegon A. Novel approach to the role of NMDA receptors in traumatic brain injury. CNS & Neurological Disorders- Drug Targets (Formerly Current Drug Targets-CNS & Neurological Disorders), 2014;13(4):567–573. doi: 10.2174/18715273113126660196. [DOI] [PubMed] [Google Scholar]
- 19.Köles L., Kató E., Hanuska A., Zádori Z.S., Al-Khrasani M., Zelles T., Rubini P., Illes P. Modulation of excitatory neurotransmission by neuronal/glial signalling molecules: interplay between purinergic and glutamatergic systems. Purinergic Signal. 2016;12(1):1–24. doi: 10.1007/s11302-015-9480-5. [http://dx.doi.org/10.1007/s11302-015-9480-5]. [PMID: 26542977]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chao N., Li S.T. Synaptic and extrasynaptic glutamate signaling in ischemic stroke. Curr. Med. Chem. 2014;21(18):2043–2064. doi: 10.2174/0929867321666131228204533. [http://dx.doi.org/10.2174/0929867321666131228204533]. [PMID: 24372211]. [DOI] [PubMed] [Google Scholar]
- 21.Katayama Y., Becker D.P., Tamura T., Hovda D.A. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J. Neurosurg. 1990;73(6):889–900. doi: 10.3171/jns.1990.73.6.0889. [http://dx.doi.org/10.3171/jns.1990.73.6.0889]. [PMID: 1977896]. [DOI] [PubMed] [Google Scholar]
- 22.Hertz L. The glutamate–glutamine (GABA) cycle: importance of late postnatal development and potential reciprocal interactions between biosynthesis and degradation. Front. Endocrinol. (Lausanne) 2013;4:59. doi: 10.3389/fendo.2013.00059. [http://dx.doi.org/10.3389/fendo.2013. 00059]. [PMID: 23750153]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nani F., Bright D.P., Revilla-Sanchez R., Tretter V., Moss S.J., Smart T.G. Tyrosine phosphorylation of GABAA receptor γ2-subunit regulates tonic and phasic inhibition in the thalamus. J. Neurosci. 2013;33(31):12718–12727. doi: 10.1523/JNEUROSCI.0388-13.2013. [http://dx.doi.org/10.1523/ JNEUROSCI.0388-13.2013]. [PMID: 23904608]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X., Chen Y., Jenkins L.W., Kochanek P.M., Clark R.S. Bench-to-bedside review: Apoptosis/programmed cell death triggered by traumatic brain injury. Crit. Care. 2005;9(1):66–75. doi: 10.1186/cc2950. [http://dx.doi.org/10.1186/cc2950]. [PMID: 15693986]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park Y.H., Jeong M.S., Jang S.B. Structural insights of homotypic interaction domains in the ligand-receptor signal transduction of tumor necrosis factor (TNF). BMB Rep. 2016;49(3):159–166. doi: 10.5483/BMBRep.2016.49.3.205. [http://dx.doi.org/10.5483/BMBRep.2016.49.3.205]. [PMID: 26615973]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalimuthu S., Se-Kwon K. Cell survival and apoptosis signaling as therapeutic target for cancer: marine bioactive compounds. Int. J. Mol. Sci. 2013;14(2):2334–2354. doi: 10.3390/ijms14022334. [http://dx.doi.org/10.3390/ ijms14022334]. [PMID: 23348928]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belizário J., Vieira-Cordeiro L., Enns S. Necroptotic cell death signaling and execution pathway: lessons from knockout mice. Mediators Inflamm. 2015;2015:128076. doi: 10.1155/2015/128076. [http://dx.doi.org/10. 1155/2015/128076]. [PMID: 26491219]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhowmick S., D’Mello V., Ponery N., Abdul-Muneer P.M. Neurodegeneration and sensorimotor deficits in the mouse model of traumatic brain injury. Brain Sci. 2018;8(1):1–11. doi: 10.3390/brainsci8010011. [PMID: 29316623]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lorente L. Biomarkers associated with the outcome of traumatic brain injury patients. Brain Sci. 2017;7(11):142–153. doi: 10.3390/brainsci7110142. [http://dx. doi.org/10.3390/brainsci7110142]. [PMID: 29076989]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang C.Y., Lee Y.C., Li P.C., Liliang P.C., Lu K., Wang K.W., Chang L.C., Shiu L.Y., Chen M.F., Sun Y.T., Wang H.K. TDP-43 proteolysis is associated with astrocyte reactivity after traumatic brain injury in rodents. J. Neuroimmunol. 2017;313:61–68. doi: 10.1016/j.jneuroim.2017.10.011. [http://dx.doi.org/10.1016/j.jneuroim.2017.10.011]. [PMID: 29153610]. [DOI] [PubMed] [Google Scholar]
- 31.Qiu J., Whalen M.J., Lowenstein P., Fiskum G., Fahy B., Darwish R., Aarabi B., Yuan J., Moskowitz M.A. Upregulation of the Fas receptor death-inducing signaling complex after traumatic brain injury in mice and humans. J. Neurosci. 2002;22(9):3504–3511. doi: 10.1523/JNEUROSCI.22-09-03504.2002. [http://dx.doi.org/10.1523/JNEUROSCI.22-09-03504.2002]. [PMID: 11978827]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weber J.T. Altered calcium signaling following traumatic brain injury. Front. Pharmacol. 2012;3:60. doi: 10.3389/fphar.2012.00060. [http://dx.doi.org/10.3389/ fphar.2012.00060]. [PMID: 22518104]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chehab T. The role of calcium signalling in autophagy. 2018. [Google Scholar]
- 34.Nazıroğlu M., Şenol N., Ghazizadeh V., Yürüker V. Neuroprotection induced by N-acetylcysteine and selenium against traumatic brain injury-induced apoptosis and calcium entry in hippocampus of rat. Cell. Mol. Neurobiol. 2014;34(6):895–903. doi: 10.1007/s10571-014-0069-2. [http://dx. doi.org/10.1007/s10571-014-0069-2]. [PMID: 24842665]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abdul-Muneer P.M., Long M., Conte A.A., Santhakumar V., Pfister B.J. High Ca2+ influx during traumatic brain injury leads to caspase-1-dependent neuroinflammation and cell death. Mol. Neurobiol. 2017;54(6):3964–3975. doi: 10.1007/s12035-016-9949-4. [http://dx.doi.org/10.1007/s12035-016-9949-4]. [PMID: 27289225]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vasco V.R.L. Role of the phosphoinositide signal transduction pathway in the endometrium. Asian Pac. J. Reprod. 2012;1(3):247–252. [http://dx.doi.org/10.1016/S2305-0500(13)60086-X]. [Google Scholar]
- 37.Ryan M.J., Gross K.W., Hajduczok G. Calcium-dependent activation of phospholipase C by mechanical distension in renin-expressing As4. 1 cell. Am. J. Physiol. Endocrinol. Metab. 2000;279(4):823–829. doi: 10.1152/ajpendo.2000.279.4.E823. [http://dx.doi.org/10.1152/ajpendo.2000.279.4.E823]. [DOI] [PubMed] [Google Scholar]
- 38.Xu C., Bailly-Maitre B., Reed J.C. Endoplasmic reticulum stress: cell life and death decisions. J. Clin. Invest. 2005;115(10):2656–2664. doi: 10.1172/JCI26373. [http://dx.doi.org/10.1172/JCI26373]. [PMID: 16200199]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun G.Z., Gao F.F., Zhao Z.M., Sun H., Xu W., Wu L.W., He Y.C. Endoplasmic reticulum stress-induced apoptosis in the penumbra aggravates secondary damage in rats with traumatic brain injury. Neural Regen. Res. 2016;11(8):1260–1266. doi: 10.4103/1673-5374.189190. [http://dx.doi. org/10.4103/1673-5374.189190]. [PMID: 27651773]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi Z., Qiu W., Xiao G., Cheng J., Zhang N. Resveratrol attenuates cognitive deficits of traumatic brain injury by activating p38 signaling in the brain. Med. Sci. Monit. 2018;24:1097–1103. doi: 10.12659/MSM.909042. [http://dx.doi.org/10.12659/MSM.909042]. [PMID: 29467361]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Larner S.F., Hayes R.L., McKinsey D.M., Pike B.R., Wang K.K. Increased expression and processing of caspase-12 after traumatic brain injury in rats. J. Neurochem. 2004;88(1):78–90. doi: 10.1046/j.1471-4159.2003.02141.x. [http://dx.doi.org/10.1046/j.1471-4159.2003.02141.x]. [PMID: 14675152]. [DOI] [PubMed] [Google Scholar]
- 42.Weber J.T. Altered calcium signaling following traumatic brain injury. Front. Pharmacol. 2012;3:60. doi: 10.3389/fphar.2012.00060. [http://dx.doi.org/10.3389/ fphar.2012.00060]. [PMID: 22518104]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang J., Wang X., Vikash V., Ye Q., Wu D., Liu Y., Dong W. ROS and ROS-mediated cellular signaling. Oxid. Med. Cell. Longev. 2016;2016:4350965. doi: 10.1155/2016/4350965. [http://dx.doi.org/10.1155/2016/ 4350965]. [PMID: 26998193]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhattacharyya A., Chattopadhyay R., Mitra S., Crowe S.E. Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014;94(2):329–354. doi: 10.1152/physrev.00040.2012. [http://dx.doi.org/10.1152/physrev.00040.2012]. [PMID: 24692350]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar A., Sasmal D., Sharma N. An insight into deltamethrin induced apoptotic calcium, p53 and oxidative stress signalling pathways. Toxicol Environ Health Sci. 2015;7:25–34. [http://dx. doi.org/10.1007/s13530-015-0217-1]. [Google Scholar]
- 46.Lutton E.M., Razmpour R., Andrews A.M., Cannella L.A., Son Y.J., Shuvaev V.V., Muzykantov V.R., Ramirez S.H. Acute administration of catalase targeted to ICAM-1 attenuates neuropathology in experimental traumatic brain injury. Sci. Rep. 2017;7(1):3846. doi: 10.1038/s41598-017-03309-4. [http://dx.doi.org/10.1038/s41598-017-03309-4]. [PMID: 28630485]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng G., Kong R.H., Zhang L.M., Zhang J.N. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br. J. Pharmacol. 2012;167(4):699–719. doi: 10.1111/j.1476-5381.2012.02025.x. [http://dx.doi.org/10.1111/j.1476-5381.2012.02025.x]. [PMID: 23003569]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bains M., Hall E.D. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim. Biophys. Acta Mol. Basis Dis. 2012;1822(5):675–684. doi: 10.1016/j.bbadis.2011.10.017. [http://dx.doi.org/10.1016/j.bbadis.2011.10.017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clausen F., Lundqvist H., Ekmark S., Lewén A., Ebendal T., Hillered L. Oxygen free radical-dependent activation of extracellular signal-regulated kinase mediates apoptosis-like cell death after traumatic brain injury. J. Neurotrauma. 2004;21(9):1168–1182. doi: 10.1089/neu.2004.21.1168. [http://dx.doi.org/10.1089/neu.2004.21.1168]. [PMID: 15453987]. [DOI] [PubMed] [Google Scholar]
- 50.Huang Y.N., Yang L.Y., Greig N.H., Wang Y.C., Lai C.C., Wang J.Y. Neuroprotective effects of pifithrin-α against traumatic brain injury in the striatum through suppression of neuroinflammation, oxidative stress, autophagy, and apoptosis. Sci. Rep. 2018;8(1):2368. doi: 10.1038/s41598-018-19654-x. [http://dx.doi.org/10.1038/s41598-018-19654-x]. [PMID: 29402897]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anilkumar U., Prehn J.H. Anti-apoptotic BCL-2 family proteins in acute neural injury. Front. Cell. Neurosci. 2014;8:281. doi: 10.3389/fncel.2014.00281. [http://dx.doi.org/10.3389/fncel.2014.00281]. [PMID: 25324720]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Czabotar P.E., Lessene G., Strasser A., Adams J.M. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014;15(1):49–63. doi: 10.1038/nrm3722. [http://dx.doi.org/10.1038/nrm3722]. [PMID: 24355989]. [DOI] [PubMed] [Google Scholar]
- 53.Morrison R.S., Kinoshita Y. The role of p53 in neuronal cell death. Cell Death Differ. 2000;7(10):868–879. doi: 10.1038/sj.cdd.4400741. [http://dx.doi.org/ 10.1038/sj.cdd.4400741]. [PMID: 11279532]. [DOI] [PubMed] [Google Scholar]
- 54.Sabirzhanov B., Zhao Z., Stoica B.A., Loane D.J., Wu J., Borroto C., Dorsey S.G., Faden A.I. Downregulation of miR-23a and miR-27a following experimental traumatic brain injury induces neuronal cell death through activation of proapoptotic Bcl-2 proteins. J. Neurosci. 2014;34(30):10055–10071. doi: 10.1523/JNEUROSCI.1260-14.2014. [http://dx.doi.org/ 10.1523/JNEUROSCI.1260-14.2014]. [PMID: 25057207]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tehranian R., Rose M.E., Vagni V., Pickrell A.M., Griffith R.P., Liu H., Clark R.S., Dixon C.E., Kochanek P.M., Graham S.H. Disruption of Bax protein prevents neuronal cell death but produces cognitive impairment in mice following traumatic brain injury. J. Neurotrauma. 2008;25(7):755–767. doi: 10.1089/neu.2007.0441. [http://dx.doi.org/ 10.1089/neu.2007.0441]. [PMID: 18627254]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pisetsky D.S. The translocation of nuclear molecules during inflammation and cell death. Antioxid. Redox Signal. 2014;20(7):1117–1125. doi: 10.1089/ars.2012.5143. [http://dx.doi.org/10.1089/ars.2012.5143]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karve I.P., Taylor J.M., Crack P.J. The contribution of astrocytes and microglia to traumatic brain injury. Br. J. Pharmacol. 2016;173(4):692–702. doi: 10.1111/bph.13125. [http://dx.doi.org/10.1111/bph.13125]. [PMID: 25752446]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corrigan F., Mander K.A., Leonard A.V., Vink R. Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J. Neuroinflammation. 2016;13(1):264. doi: 10.1186/s12974-016-0738-9. [http://dx.doi.org/10.1186/s12974-016-0738-9]. [PMID: 27724914]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Atkins C.M., Oliva A.A., Jr, Alonso O.F., Pearse D.D., Bramlett H.M., Dietrich W.D. Modulation of the cAMP signaling pathway after traumatic brain injury. Exp. Neurol. 2007;208(1):145–158. doi: 10.1016/j.expneurol.2007.08.011. [http://dx.doi.org/10.1016/j.expneurol.2007.08.011]. [PMID: 17916353]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Donat C.K., Scott G., Gentleman S.M., Sastre M. Microglial activation in traumatic brain injury. Front. Aging Neurosci. 2017;9:208. doi: 10.3389/fnagi.2017.00208. [http://dx.doi.org/10.3389/fnagi.2017.00208]. [PMID: 28701948]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Don A.S.A., Tsang C.K., Kazdoba T.M., D’Arcangelo G., Young W., Zheng X.F. Targeting mTOR as a novel therapeutic strategy for traumatic CNS injuries. Drug Discov. Today. 2012;17(15-16):861–868. doi: 10.1016/j.drudis.2012.04.010. [http://dx.doi.org/10.1016/j.drudis.2012.04. 010]. [PMID: 22569182]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Garza-Lombó C., Gonsebatt M.E. Mammalian target of rapamycin: its role in early neural development and in adult and aged brain function. Front. Cell. Neurosci. 2016;10:157. doi: 10.3389/fncel.2016.00157. [http://dx.doi.org/ 10.3389/fncel.2016.00157]. [PMID: 27378854]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lake D., Corrêa S.A., Müller J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell. Mol. Life Sci. 2016;73(23):4397–4413. doi: 10.1007/s00018-016-2297-8. [http://dx.doi.org/10.1007/s00018-016-2297-8]. [PMID: 27342992]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Don A.S.A., Tsang C.K., Kazdoba T.M., D’Arcangelo G., Young W., Zheng X.F. Targeting mTOR as a novel therapeutic strategy for traumatic CNS injuries. Drug Discov. Today. 2012;17(15-16):861–868. doi: 10.1016/j.drudis.2012.04.010. [http://dx.doi.org/10.1016/j.drudis.2012.04. 010]. [PMID: 22569182]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang X., Seekaew P., Gao X., Chen J. Traumatic brain injury stimulates neural stem cell proliferation via mammalian target of rapamycin signaling pathway activation. eNeuro. 2016;3(5):1–14. doi: 10.1523/ENEURO.0162-16.2016. [http://dx.doi.org/10.1523/ENEURO.0162-16.2016]. [PMID: 27822507]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sun J., Nan G. The extracellular signal-regulated kinase 1/2 pathway in neurological diseases: A potential therapeutic target. Int. J. Mol. Med. 2017;39(6):1338–1346. doi: 10.3892/ijmm.2017.2962. [Review]. [http://dx.doi.org/ 10.3892/ijmm.2017.2962]. [PMID: 28440493]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Leisman G., Moustafa A.A., Shafir T. Thinking, walking, talking: integratory motor and cognitive brain function. Front. Public Health. 2016;4:94. doi: 10.3389/fpubh.2016.00094. [http://dx.doi.org/10.3389/fpubh.2016.00094]. [PMID: 27252937]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ahmed S., Venigalla H., Mekala H.M., Dar S., Hassan M., Ayub S. Traumatic brain injury and neuropsychiatric complications. Indian J. Psychol. Med. 2017;39(2):114–121. doi: 10.4103/0253-7176.203129. [http://dx. doi.org/10.4103/0253-7176.203129]. [PMID: 28515545]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Onwuchekwa C.R., Alazigha N.S. Computed tomography pattern of traumatic head injury in Niger Delta, Nigeria: A multicenter evaluation. Int. J. Crit. Illn. Inj. Sci. 2017;7(3):150–155. doi: 10.4103/IJCIIS.IJCIIS_6_17. [http://dx.doi.org/10.4103/IJCIIS.IJCIIS_6_17]. [PMID: 28971028]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nayebaghayee H., Afsharian T. Correlation between Glasgow Coma Scale and brain computed tomography-scan findings in head trauma patients. Asian J. Neurosurg. 2016;11(1):46–49. doi: 10.4103/1793-5482.165780. [http://dx.doi.org/10.4103/1793-5482.165780]. [PMID: 26889279]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Agoston D.V., Shutes-David A., Peskind E.R. Biofluid biomarkers of traumatic brain injury. Brain Inj. 2017;31(9):1195–1203. doi: 10.1080/02699052.2017.1357836. [http://dx.doi.org/10.1080/02699052.2017.1357836]. [PMID: 28981341]. [DOI] [PubMed] [Google Scholar]
- 72.Carney N., Totten A.M., O’Reilly C., Ullman J.S., Hawryluk G.W., Bell M.J., Bratton S.L., Chesnut R., Harris O.A., Kissoon N., Rubiano A.M., Shutter L., Tasker R.C., Vavilala M.S., Wilberger J., Wright D.W., Ghajar J. Guidelines for the management of severe traumatic brain injury. Neurosurgery. 2017;80(1):6–15. doi: 10.1227/NEU.0000000000001432. [PMID: 27654000]. [DOI] [PubMed] [Google Scholar]
- 73.Shirley R., Ord E.N., Work L.M. Oxidative stress and the use of antioxidants in stroke. Antioxidants. 2014;3(3):472–501. doi: 10.3390/antiox3030472. [http://dx.doi.org/10.3390/antiox3030472]. [PMID: 26785066]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lin C.J., Chen T.H., Yang L.Y., Shih C.M. Resveratrol protects astrocytes against traumatic brain injury through inhibiting apoptotic and autophagic cell death. Cell Death Dis. 2014;5(3):e1147. doi: 10.1038/cddis.2014.123. [http://dx.doi.org/10.1038/cddis.2014.123]. [PMID: 24675465]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Venegoni W., Shen Q., Thimmesch A.R., Bell M., Hiebert J.B., Pierce J.D. The use of antioxidants in the treatment of traumatic brain injury. J. Adv. Nurs. 2017;73(6):1331–1338. doi: 10.1111/jan.13259. [http://dx.doi. org/10.1111/jan.13259]. [PMID: 28103389]. [DOI] [PubMed] [Google Scholar]
- 76.Arifin M.Z., Faried A., Shahib M.N., Wiriadisastra K., Bisri T. Inhibition of activated NR2B gene- and caspase-3 protein-expression by glutathione following traumatic brain injury in a rat model. Asian J. Neurosurg. 2011;6(2):72–77. doi: 10.4103/1793-5482.92160. [http://dx.doi.org/ 10.4103/1793-5482.92160]. [PMID: 22347327]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pereira-Leite C., Nunes C., Jamal S.K., Cuccovia I.M., Reis S. Nonsteroidal anti‐inflammatory therapy: a journey toward safety. Med. Res. Rev. 2017;37(4):802–859. doi: 10.1002/med.21424. [http://dx.doi.org/10.1002/ med.21424]. [PMID: 28005273]. [DOI] [PubMed] [Google Scholar]
- 78.Thelin E.P., Hall C.E., Gupta K., Carpenter K.L.H., Chandran S., Hutchinson P.J., Patani R., Helmy A. Elucidating pro-inflammatory cytokine responses after traumatic brain injury in a human stem cell model. J. Neurotrauma. 2018;35(2):341–352. doi: 10.1089/neu.2017.5155. [http://dx.doi.org/10.1089/neu.2017.5155]. [PMID: 28978285]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wilson N.M., Gurney M.E., Dietrich W.D., Atkins C.M. Therapeutic benefits of phosphodiesterase 4B inhibition after traumatic brain injury. PLoS One. 2017;12(5):e0178013. doi: 10.1371/journal.pone.0178013. [http://dx.doi.org/ 10.1371/journal.pone.0178013]. [PMID: 28542295]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Garrido-Mesa N., Zarzuelo A., Gálvez J. Minocycline: far beyond an antibiotic. Br. J. Pharmacol. 2013;169(2):337–352. doi: 10.1111/bph.12139. [http://dx.doi.org/10.1111/bph.12139]. [PMID: 23441623]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lei B., Mace B., Dawson H.N., Warner D.S., Laskowitz D.T., James M.L. Anti-inflammatory effects of progesterone in lipopolysaccharide-stimulated BV-2 microglia. PLoS One. 2014;9(7):e103969. doi: 10.1371/journal.pone.0103969. [http://dx.doi.org/10.1371/journal.pone.0103969]. [PMID: 25080336]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Trippier P.C., Jansen Labby K., Hawker D.D., Mataka J.J., Silverman R.B. Target- and mechanism-based therapeutics for neurodegenerative diseases: strength in numbers. J. Med. Chem. 2013;56(8):3121–3147. doi: 10.1021/jm3015926. [http://dx.doi.org/10.1021/jm3015926]. [PMID: 23458846]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Haar C.V., Peterson T.C., Martens K.M., Hoane M.R. The use of nicotinamide as a treatment for experimental traumatic brain injury and stroke: A review and evaluation. Clin. Pharmacol. Biopharmaceut. S. 2013;1(2):1–8. [Google Scholar]
- 84.Xu X., Gao W., Cheng S., Yin D., Li F., Wu Y., Sun D., Zhou S., Wang D., Zhang Y., Jiang R., Zhang J. Anti-inflammatory and immunomodulatory mechanisms of atorvastatin in a murine model of traumatic brain injury. J. Neuroinflammation. 2017;14(1):167–182. doi: 10.1186/s12974-017-0934-2. [http://dx.doi.org/10.1186/s12974-017-0934-2]. [PMID: 28835272]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Siopi E., Cho A.H., Homsi S., Croci N., Plotkine M., Marchand-Leroux C., Jafarian-Tehrani M. Minocycline restores sAPPα levels and reduces the late histopathological consequences of traumatic brain injury in mice. J. Neurotrauma. 2011;28(10):2135–2143. doi: 10.1089/neu.2010.1738. [http://dx.doi.org/10.1089/neu.2010.1738]. [PMID: 21770756]. [DOI] [PubMed] [Google Scholar]
- 86.Wang C., Hu Z., Zou Y., Xiang M., Jiang Y., Botchway B.O.A., Huo X., Du X., Fang M. The post-therapeutic effect of rapamycin in mild traumatic brain-injured rats ensuing in the upregulation of autophagy and mitophagy. Cell Biol. Int. 2017;41(9):1039–1047. doi: 10.1002/cbin.10820. [http://dx.doi.org/10.1002/cbin.10820]. [PMID: 28685977]. [DOI] [PubMed] [Google Scholar]
- 87.Erlich S., Alexandrovich A., Shohami E., Pinkas-Kramarski R. Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol. Dis. 2007;26(1):86–93. doi: 10.1016/j.nbd.2006.12.003. [http://dx.doi.org/10. 1016/j.nbd.2006.12.003]. [PMID: 17270455]. [DOI] [PubMed] [Google Scholar]
- 88.Zhu X., Park J., Golinski J., Qiu J., Khuman J., Lee C.C., Lo E.H., Degterev A., Whalen M.J. Role of Akt and mammalian target of rapamycin in functional outcome after concussive brain injury in mice. J. Cereb. Blood Flow Metab. 2014;34(9):1531–1539. doi: 10.1038/jcbfm.2014.113. [http://dx.doi.org/10.1038/jcbfm.2014.113]. [PMID: 24938400]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shi G.D., OuYang Y.P., Shi J.G., Liu Y., Yuan W., Jia L.S. PTEN deletion prevents ischemic brain injury by activating the mTOR signaling pathway. Biochem. Biophys. Res. Commun. 2011;404(4):941–945. doi: 10.1016/j.bbrc.2010.12.085. [http://dx.doi.org/10.1016/j.bbrc.2010.12.085]. [PMID: 21185267]. [DOI] [PubMed] [Google Scholar]
- 90.You W., Wang Z., Li H., Shen H., Xu X., Jia G., Chen G. Inhibition of mammalian target of rapamycin attenuates early brain injury through modulating microglial polarization after experimental subarachnoid hemorrhage in rats. J. Neurol. Sci. 2016;367:224–231. doi: 10.1016/j.jns.2016.06.021. [http://dx.doi.org/10.1016/j.jns.2016.06.021]. [PMID: 27423593]. [DOI] [PubMed] [Google Scholar]
- 91.Gurkoff G., Shahlaie K., Lyeth B., Berman R. Voltage-gated calcium channel antagonists and traumatic brain injury. Pharmaceuticals (Basel) 2013;6(7):788–812. doi: 10.3390/ph6070788. [http://dx.doi.org/10.3390/ ph6070788]. [PMID: 24276315]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hoshide R., Cheung V., Marshall L., Kasper E., Chen C.C. Do corticosteroids play a role in the management of traumatic brain injury? Surg. Neurol. Int. 2016;7:84. doi: 10.4103/2152-7806.190439. [http://dx.doi.org/10.4103/2152-7806.190439]. [PMID: 27656315]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wu C., Sun D. GABA receptors in brain development, function, and injury. Metab. Brain Dis. 2015;30(2):367–379. doi: 10.1007/s11011-014-9560-1. [http://dx.doi. org/10.1007/s11011-014-9560-1]. [PMID: 24820774]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dutertre S., Becker C.M., Betz H. Inhibitory glycine receptors: an update. J. Biol. Chem. 2012;287(48):40216–40223. doi: 10.1074/jbc.R112.408229. [http://dx. doi.org/10.1074/jbc.R112.408229]. [PMID: 23038260]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dorsett C.R., McGuire J.L., DePasquale E.A., Gardner A.E., Floyd C.L., McCullumsmith R.E. Glutamate neurotransmission in rodent models of traumatic brain injury. J. Neurotrauma. 2017;34(2):263–272. doi: 10.1089/neu.2015.4373. [http://dx.doi.org/10.1089/neu.2015.4373]. [PMID: 27256113]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chunhua C., Chunhua X., Megumi S., Renyu L. Kappa opioid receptor agonist and brain ischemia. Transl. Perioper. Pain Med. 2014;1(2):27–34. [PMID: 25574482]. [PMC free article] [PubMed] [Google Scholar]
- 97.Plesnila N., von Baumgarten L., Retiounskaia M., Engel D., Ardeshiri A., Zimmermann R., Hoffmann F., Landshamer S., Wagner E., Culmsee C. Delayed neuronal death after brain trauma involves p53-dependent inhibition of NF-kappaB transcriptional activity. Cell Death Differ. 2007;14(8):1529–1541. doi: 10.1038/sj.cdd.4402159. [http://dx.doi. org/10.1038/sj.cdd.4402159]. [PMID: 17464322]. [DOI] [PubMed] [Google Scholar]
- 98.Yang L.Y., Greig N.H., Huang Y.N., Hsieh T.H., Tweedie D., Yu Q.S., Hoffer B.J., Luo Y., Kao Y.C., Wang J.Y. Post-traumatic administration of the p53 inactivator pifithrin-α oxygen analogue reduces hippocampal neuronal loss and improves cognitive deficits after experimental traumatic brain injury. Neurobiol. Dis. 2016;96:216–226. doi: 10.1016/j.nbd.2016.08.012. [http://dx.doi.org/10.1016/j.nbd.2016.08. 012]. [PMID: 27553877]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Huang X.J., Li W.P., Lin Y., Feng J.F., Jia F., Mao Q., Jiang J.Y. Blockage of the upregulation of voltage-gated sodium channel nav1. 3 improve outcomes after experimental traumatic brain injury. J. Neurotrauma. 2014;31(4):346–357. doi: 10.1089/neu.2013.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wahl F., Renou E., Mary V., Stutzmann J.M. Riluzole reduces brain lesions and improves neurological function in rats after a traumatic brain injury. Brain Res. 1997;756(1-2):247–255. doi: 10.1016/s0006-8993(97)00144-3. [http:// dx.doi.org/10.1016/S0006-8993(97)00144-3]. [PMID: 9187339]. [DOI] [PubMed] [Google Scholar]
- 101.Stocchetti N., Carbonara M., Citerio G., Ercole A., Skrifvars M.B., Smielewski P., Zoerle T., Menon D.K. Severe traumatic brain injury: targeted management in the intensive care unit. Lancet Neurol. 2017;16(6):452–464. doi: 10.1016/S1474-4422(17)30118-7. [http://dx.doi.org/10.1016/S1474-4422(17)30118-7]. [PMID: 28504109]. [DOI] [PubMed] [Google Scholar]
- 102.Xiong Y., Zhang Y., Mahmood A., Chopp M. Investigational agents for treatment of traumatic brain injury. Expert Opin. Investig. Drugs. 2015;24(6):743–760. doi: 10.1517/13543784.2015.1021919. [http://dx.doi.org/10.1517/ 13543784.2015.1021919]. [PMID: 25727893]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhu H.T., Bian C., Yuan J.C., Chu W.H., Xiang X., Chen F., Wang C.S., Feng H., Lin J.K. Curcumin attenuates acute inflammatory injury by inhibiting the TLR4/MyD88/NF-κB signaling pathway in experimental traumatic brain injury. J. Neuroinflammation. 2014;11(1):59. doi: 10.1186/1742-2094-11-59. [http://dx.doi.org/10.1186/1742-2094-11-59]. [PMID: 24669820]. [DOI] [PMC free article] [PubMed] [Google Scholar]